
An electron-donating strategy to guide the construction of MOF photocatalysts toward co-catalyst-free highly efficient photocatalytic H2 evolution†

Abstract
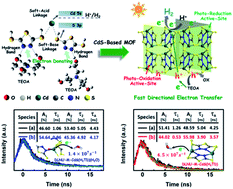
Herein, an interesting electron-donating strategy is proposed to guide the construction of novel CdS-based MOFs for co-catalyst-free photocatalytic H2 evolution in the presence of triethanolamine as the hole-scavenger. The optimally designed CdS-based MOF exhibits an exceptional H2-evolution rate of 26.1 mmol g−1 h−1 which is almost the highest rate among the reported co-catalyst-free MOF systems.
 Please wait while we load your content...
Please wait while we load your content...

