
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxygen exchange kinetics on systematically doped ceria: a pulsed isotope exchange study †
Maximilian
Schaube
 ,
Rotraut
Merkle
* and
Joachim
Maier
,
Rotraut
Merkle
* and
Joachim
Maier
Max Planck Institute for Solid State Research, Stuttgart, Germany. E-mail: r.merkle@fkf.mpg.de
First published on 20th August 2019
Abstract
The importance of point defects for oxygen surface reaction kinetics on doped ceria is demonstrated by pulsed isotope exchange. The oxygen surface exchange reaction on 20 different Gd/Pr/Tb, and Nb single and co-doped ceria samples is studied from 500–850 °C in 10–0.1% O2 atmosphere. The highest rates are measured when both oxygen vacancies and redox-active centers are available. Gd-doping leads to exchange rates which are proportional to the concentrations of Gd and oxygen vacancies. Pr-doped ceria exhibits a much stronger variation of the exchange rate – by almost five orders of magnitude from 0.6 to 20 mol% Pr doping – emphasizing the importance of redox-active centers. The equilibrium exchange rates are low for Nb, Nb/Pr, and Nb/Gd co-doped ceria, emphasizing the important role of oxygen vacancies for oxygen dissociation and incorporation. The oxygen partial pressure dependence indicates that molecular oxygen species are involved in the rate-determining step (in addition to oxygen vacancies).
1 Introduction
Ceria (CeO2) based materials find an extremely wide range of application, e.g., exhaust emission control and soot oxidation in automotive, catalyst for steam reforming, water gas shift and oxidation reactions, solid electrolyte in solid oxide fuel cells (SOFC) (see, e.g., ref. 1–6). Furthermore, ceria-based materials are also very well suited for fundamental investigations of surface kinetics (where the (111), (110) and (100) facets dominate the morphology) for several reasons: (i) simple cubic fluorite structure for undoped as well as doped ceria, (ii) only one cation site (in contrast to e.g., perovskites, avoiding complications from varying cation site occupation), (iii) intrinsic redox activity of Ce, (iv) perceptible mobility of oxygen vacancies as well as oxygen interstitials, (v) large solubility of acceptor, donor and redox-active dopants and (vi) well-understood bulk defect chemistry that allows tuning of point defect concentrations over a large range.So far, isolated studies of the oxygen exchange kinetics were performed on Gd, Pr, Tb, and Nb doped ceria by oxygen isotope exchange line profiling (IELP),7–13 isothermal isotope exchange (IIE),14 isotope exchange gas analysis (IEGA)15 pulsed isotope exchange (PIE),16,17 electrochemical impedance spectroscopy (EIS)18,19 (ref. 19 refers to reducing conditions in H2 atmosphere), optical absorption spectroscopy,20,21 mass relaxation,22,23 and other methods.21,24,25
Regarding the reaction mechanism, experimental and ab initio studies indicate that on reduced ceria, adsorbed superoxide and peroxide is formed easily, that adsorption of oxygen in an oxygen vacancy is energetically favorable,26–29 and that this occurs via superoxide and peroxide intermediates. Nevertheless, actual dissociation of molecular oxygen species is studied only rarely. The work in ref. 28 indicate that even for a superoxide adsorbed in an oxygen vacancy still a perceptible dissociation barrier must be overcome. Point defects such as oxygen vacancies or redox-active dopants naturally represent catalytically active centers for surface reactions (see, e.g., ref. 30–36).
It is obvious that oxygen vacancies and electronic dopants are required to carry out oxygen incorporation. But which of these defects limits the reaction depends on the materials classes and has yet to be clarified. This knowledge may then serve to purposefully tune the materials properties for application in e.g., electrochemical devices or as catalyst.
Surprisingly, to the knowledge of the authors there is no experimental study that systematically investigates the oxygen surface exchange reaction on a variety of doped ceria materials and an extended range of oxygen partial pressures by the same method and identical sample preparation to elucidate the influence of acceptor/donor and mixed valence dopants on the oxygen exchange kinetics and reaction mechanism. In particular, the present investigation covers a large dopant concentration range from 0.6–22 mol% and allows for a direct comparison of dopants without and with redox-activity. The method of choice in the present study is pulsed isotope exchange:37 the sample is exposed to an isotope enriched oxygen pulse, and the resulting mixture of 32O2, 16O18O, and 36O2 isotopologues quantified by mass spectroscopy. This technique enables a fast screening of the oxygen incorporation reaction rate as a function of dopant content and oxygen partial pressure in the absence of any precious metals, and yields additional mechanistic information compared to IELP.37–41
2 Experimental
2.1 Sample preparation
In this study, ceria samples with 0.6 to 22 mol% of dopants are used and labelled by xAyBDC where x and y denote the content (in mol%) of dopants A and B, and DC abbreviates “doped ceria”. Single- and co-doped ceria was synthesized by a Pechini type approach42–44 using Me(NO3)3·H2O (Me: Ce, Gd, Pr, or Tb) (Reacton, Alfa Aesar, 99.99%) and NH4NbC2O4·H2O (Aldrich) in the desired molar ratios (the exact water content was determined by thermogravimetry). Citric acid (CA) (Roth, >99.5%) and ethylene glycol (EG) (Emsure, Merck) were added to a solution of 60 mL distilled water and 87 mmol metal salts with a few droplets of conc. HNO3 (Roth, p.a.) whereby the molar ratio of EG/CA is two and for EG/Me one. The mixture was stirred at 175 °C until a yellow resin was formed. After subsequent pyrolysis at T ≈ 250 °C in the glass beaker, the obtained powder was homogenized by dry milling in a zirconia mill for 45 min.For pulsed isotope exchange, 1 g powder at a time was compacted into dense pellets by spark plasma sintering (SPS, FCT-DP D 5/2, FCT Systeme) at 1000 °C for 3 min in a graphite mold with 10 mm diameter at 6 kN pressure. The pellets were calcined at 800 °C for 8 h and annealed at 1400 °C for 8 h in air with a heating/cooling rate of 100 °C h−1. Their density was determined using a 5 mL pycnometer with water as solvent. The pellets were crushed and sieved to a particle size between 60 and 100 μm, and fired again at 1400 °C for 8 h with 100 °C h−1 in air. The resulting particles showed a smooth surface (Fig. 2 in ESI†) and the particle size did not change.
2.2 Sample characterization
Phase purity and lattice parameters were examined by X-ray diffraction (Panalytical Empyrean) with Cu Kα radiation (40 kW, 40 mA) in Bragg-Brentano reflection geometry. The cation stoichiometry was checked by inductively coupled plasma optical emission spectroscopy (Spectro Ciros CCD, Spectro Analytical Instruments). The morphology of the particle surface was investigated by scanning electron microscopy (Merlin Gemini II, Carl Zeiss). BET surface area measurements were conducted by POROTEC GmbH (Thermo Scientific Surfer) using krypton as adsorption gas. Surface segregation of dopants was investigated by X-ray photoelectron spectroscopy (Kratos AXIS Ultra) with monochromatized Al Kα with a base pressure < 10−10 mbar. Spectra were obtained with analyzer pass energy of 20 eV and the analysis was performed with CasaXPS. Under the applied measurement conditions, a surface layer of approximately 4.5 nm thickness is probed.45 Segregation effects to grain boundaries were investigated by transmission electron microscopy (TEM) and energy-dispersive X-ray spectroscopy (EDX) with an ARM 200CF (JEOL) and a Centurio EDX detector (200 kV, probe size 1.2 Å). Thermogravimetry (TG, STA 449 C, Netzsch) of the oxygen non-stoichiometry of PDC was performed on powder samples (≈0.5 g) with oxygen partial pressure ranging between 830 and 0.1 mbar (total flow 60 mL min−1) and heating rates between 2 °C min−1 (high T and p(O2)) and 0.1 °C min−1 (small T and p(O2)) to ensure equilibration. Buoyancy correction was done by using a measurement of an empty Al2O3 crucible. The absolute value of O stoichiometry was obtained by assuming that the plateau at high T and low p(O2) corresponds to all Pr in 3+ oxidation state.2.3 Pulsed isotope exchange (PIE)
Pulsed isotope exchange17,37–39,41,46 was conducted in a vertical Al2O3 micro plug flow reactor (inner diameter 2 mm, length 300 mm, Friatec AG) loaded with 100 mg of sample particles, which were fixed by a 15 mg quartz wool plug. The bed height was always approximately 8 mm. The carrier gas – mixtures of 10%, 1% or 0.1% O2 in N2 – enters the reactor at the top, with a flow rate of 7 mL min−1 (Aera TC FC-R7800CD). The particle size (60 to 100 μm) and flow rate were chosen to keep the pressure drop across the packed bed below 20%. Prior to isotope exchange, the particles in the reactor were pre-treated by flushing with the appropriate carrier gas at 900 °C for 30 min with a heating and cooling rate of 2.5 °C min−1. An 0.5 mL injection loop was used to introduce an 18O enriched pulse of 10, 1 or 0.1% O2 mixed from 97 at% enriched 18O gas (Euriso-top) and 98 at% enriched 15N gas (Westfalen; in order to recognize the gas from the pulse loop and as internal standard) into the system by switching a 6-way-valve. At each measurement temperature the sample was equilibrated for 30 min in the carrier gas stream prior to isotope exchange pulses. The pulses were repeated three times with three minutes in-between. The pulse response was monitored by a quadrupole mass spectrometer (Balzers Prisma QME200, Pfeiffer Vacuum) at the exit of the reactor at m/z = 30 (15N2), 32 (16O2), 34 (18O16O) and 36 (18O2) with a resolution of five points per second. The quantification of oxygen isotopologues in the gas phase was achieved by normalizing the corresponding peak areas to the integrated signal of 15N2 (see ESI† for more information).3 Theory
3.1 Bulk defect chemistry
The overall oxygen incorporation into the bulk material can be expressed by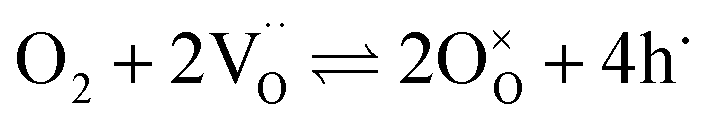 | (1) |
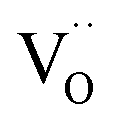 indicates oxygen vacancies,
indicates oxygen vacancies, 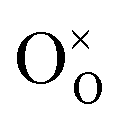 lattice oxygen, and h˙ holes. The defect concentrations are determined by a mass action constant
lattice oxygen, and h˙ holes. The defect concentrations are determined by a mass action constant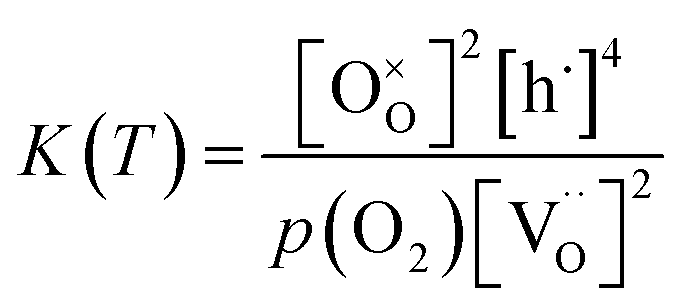 | (2) |
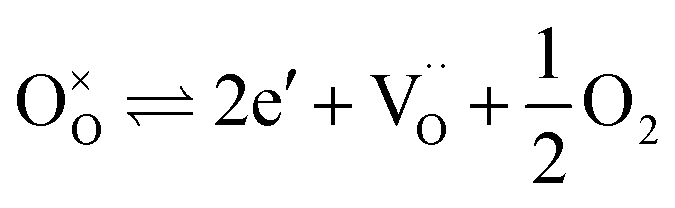 | (3) |
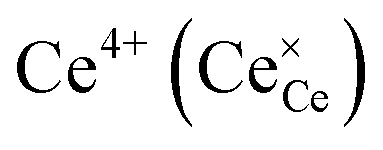 to
to 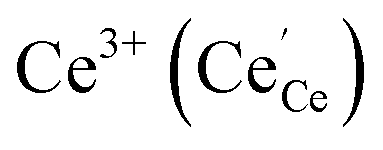 forming small polarons (cf.ref. 48). Typical values of δ at 800 °C range between 10−5 (1 bar O2) and 0.1 (10−20 bar O2).49 In this intrinsic regime, the electroneutrality condition is
forming small polarons (cf.ref. 48). Typical values of δ at 800 °C range between 10−5 (1 bar O2) and 0.1 (10−20 bar O2).49 In this intrinsic regime, the electroneutrality condition is 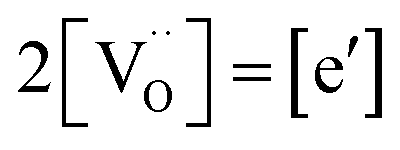 , and the oxygen partial pressure dependence of
, and the oxygen partial pressure dependence of 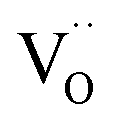 and
and 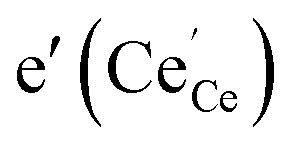 is
is 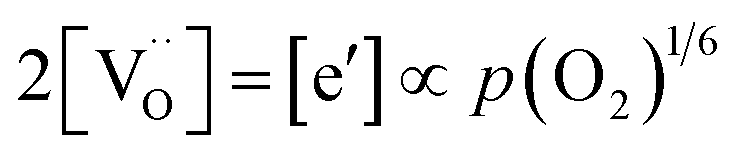 . Doping allows one to deliberately change the defect concentrations.
. Doping allows one to deliberately change the defect concentrations.
Oxygen vacancies can also be introduced by doping with an acceptor of fixed valence such as Gd3+, yielding an oxide ion conducting electrolyte material (see, e.g., ref. 50–52):
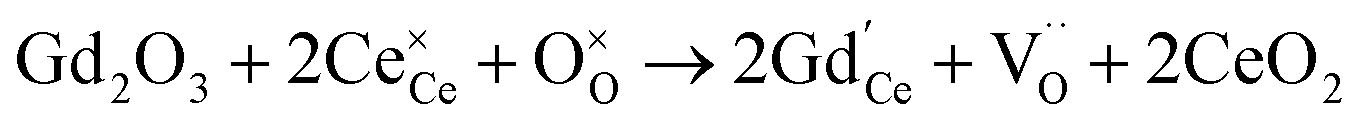 | (4) |
The amount of 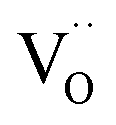 is fixed according to
is fixed according to 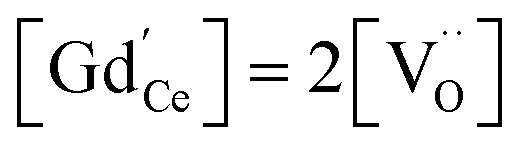 , and the oxygen partial pressure dependency in this case is found to be ±1/4 for holes
, and the oxygen partial pressure dependency in this case is found to be ±1/4 for holes 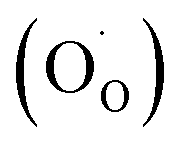 and excess electrons
and excess electrons 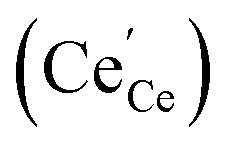 . It is important to note that
. It is important to note that 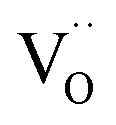 created via such acceptor doping do not lead to an enhanced conduction electron concentration (rather to an increased hole concentration owing to the equilibrium in eqn (1)). Since they can incorporate oxide ions,
created via such acceptor doping do not lead to an enhanced conduction electron concentration (rather to an increased hole concentration owing to the equilibrium in eqn (1)). Since they can incorporate oxide ions, 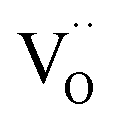 represent acidic centers31 (rather than Lewis basic centers as suggested in ref. 33).
represent acidic centers31 (rather than Lewis basic centers as suggested in ref. 33).
Donor doping, e.g., by Nb5+, leads to suppression of 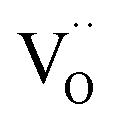 and formation of oxygen interstitials under oxidizing conditions,
and formation of oxygen interstitials under oxidizing conditions,
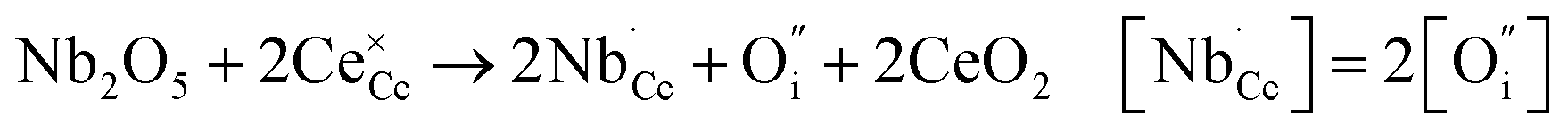 | (5) |
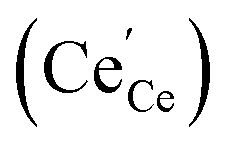 under reducing conditions13,53,54
under reducing conditions13,53,54 | (6) |
In the temperature and oxygen partial pressure regime probed by PIE, both defect compensations (5) and (6) occur simultaneously,54 but oxygen interstitials are the dominating defects.53 The solubility limit of Nb in ceria was found to be 3 mol%.55 For acceptor–donor co-doped materials with equal dopant concentration,  the oxygen defect concentration is minute.
the oxygen defect concentration is minute.
Praseodymium- as well as terbium-doped ceria show similar defect chemistry56 and much stronger redox activity compared to Ce. Thus the intrinsic incorporation reaction (1) is complemented by a redox reaction, e.g., in the case of Pr:
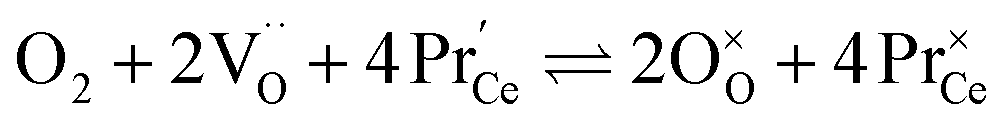 | (7) |
Such a deep acceptor situation involves electronic and ionic defects. A high amount of Pr-doping introduces increased electronic conductivity due to small polaron hopping between Pr3+/Pr4+ and Ce4+, making this material a mixed electronic and ionic conductor (MIEC).18,42,57,58Fig. 1 shows the 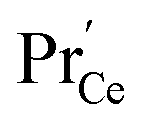 concentration for 2, 6 and 20PDC calculated from TG measurements according to eqn (7). In the regime of
concentration for 2, 6 and 20PDC calculated from TG measurements according to eqn (7). In the regime of 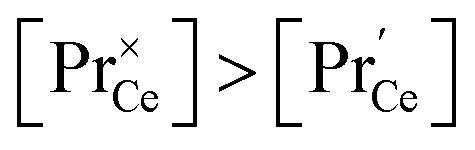 , the electroneutrality condition is
, the electroneutrality condition is 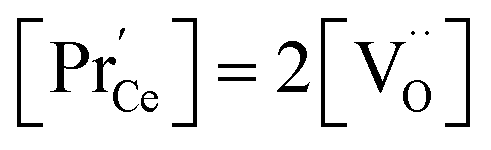 , and both concentrations scale with p(O2)−1/6. At higher T and/or lower p(O2), the dependence flattens and
, and both concentrations scale with p(O2)−1/6. At higher T and/or lower p(O2), the dependence flattens and 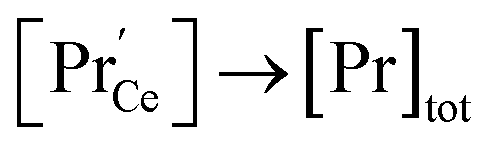 . Under the conditions of the PIE experiment (blue shaded area in Fig. 1), about 15–60% of the Pr is in the 3+ state, and a corresponding
. Under the conditions of the PIE experiment (blue shaded area in Fig. 1), about 15–60% of the Pr is in the 3+ state, and a corresponding 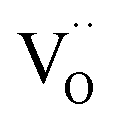 concentration is present. The actual
concentration is present. The actual 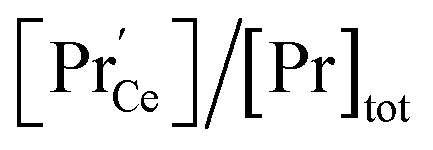 ratio varies a bit with p(O2) and T, but the dependence on [Pr]tot is small. Thus, overall the relation
ratio varies a bit with p(O2) and T, but the dependence on [Pr]tot is small. Thus, overall the relation 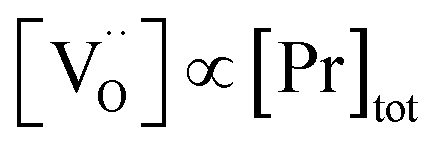 is a reasonably good approximation to describe the variation of
is a reasonably good approximation to describe the variation of 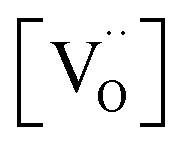 with [Pr]tot, which varies by 1.5 orders of magnitude between the lowest and highest doped samples.
with [Pr]tot, which varies by 1.5 orders of magnitude between the lowest and highest doped samples.
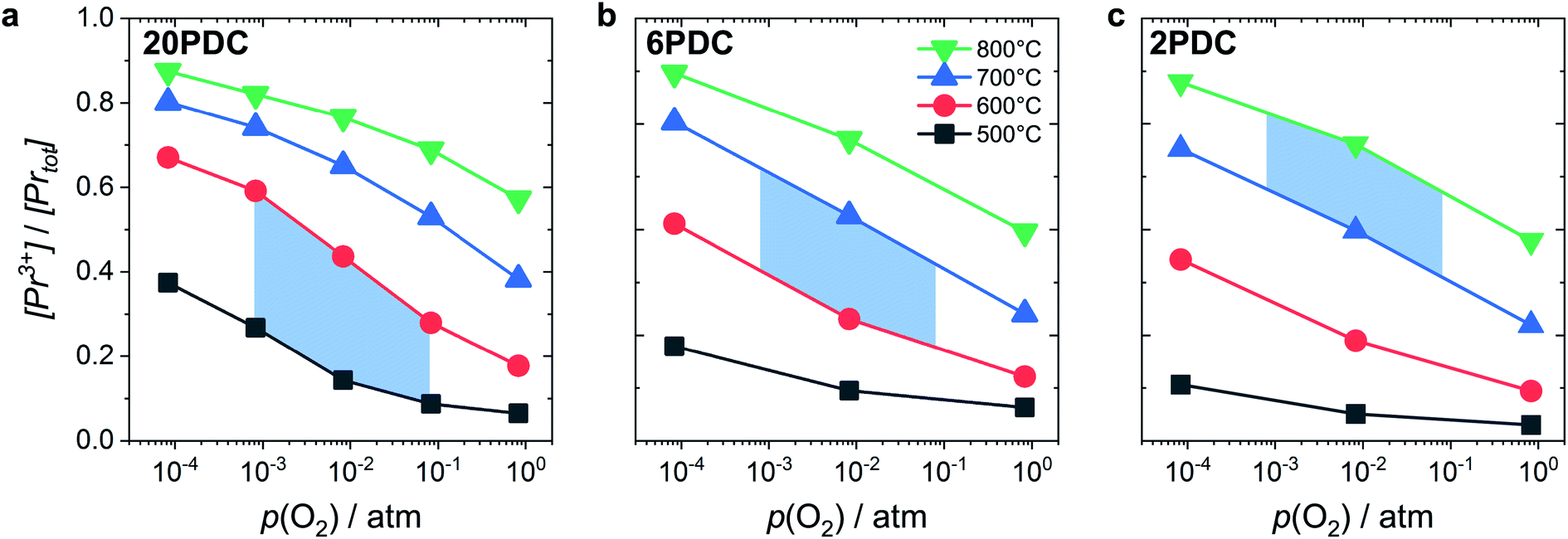 | ||
| Fig. 1 Ratio of Pr3+ relative to total Pr content as a function of oxygen partial pressure at different temperatures for 20PDC (a), 6PDC (b), and 2PDC (c). The blue shaded area indicates the typical conditions for the PIE measurements. | ||
3.2 Surface orientation and surface defect concentrations
The defect concentrations that are relevant for the oxygen exchange reaction are those in the surface layer. The most stable termination of CeO2 is (111), followed by (110) and (100).59 No surface energies for doped ceria could be retrieved, but at least for low dopant concentrations the same sequence of surface stabilities is highly probable. Since the particles used for PIE in the present investigation have been annealed at 1400 °C, it is reasonable to assume that the surface has largely reconstructed into the most stable terminations (in terms of free energy), yielding a similar distribution for undoped as well as doped samples. While under reducing conditions the dependence of the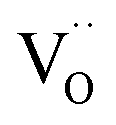 formation energy (ΔH0red) on the surface orientation is expected to lead to significantly different
formation energy (ΔH0red) on the surface orientation is expected to lead to significantly different 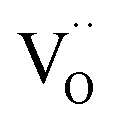 concentrations for the different surfaces,59,60 such differences are expected to be much less pronounced for acceptor doped samples where
concentrations for the different surfaces,59,60 such differences are expected to be much less pronounced for acceptor doped samples where 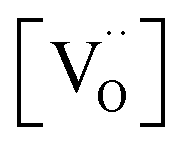 is largely fixed by the dopant concentration. Measurements of the orientation dependence of surface exchange rates are scarce. The challenges are, amongst others, the discrimination from strain effects and the fact that thermodynamically unstable surfaces must be prevented from reconstruction under the experimental conditions at elevated T (in turn, hypothetically very active but highly unstable surfaces will not contribute in a typical oxygen exchange experiment because they reconstruct). For epitaxial La0.7Sr0.3MnO3−δ perovskite films, the variation of surface rate constant between (001), (110), and (111) surface orientations has been found to be less than a factor of four.61,62
is largely fixed by the dopant concentration. Measurements of the orientation dependence of surface exchange rates are scarce. The challenges are, amongst others, the discrimination from strain effects and the fact that thermodynamically unstable surfaces must be prevented from reconstruction under the experimental conditions at elevated T (in turn, hypothetically very active but highly unstable surfaces will not contribute in a typical oxygen exchange experiment because they reconstruct). For epitaxial La0.7Sr0.3MnO3−δ perovskite films, the variation of surface rate constant between (001), (110), and (111) surface orientations has been found to be less than a factor of four.61,62
Since the surface represents a severe structural distortion, the absolute defect concentrations differ from the respective bulk values. Typically it is expected that, owing to the smaller number of bonds to cations to be broken, 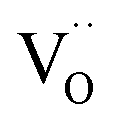 form thermodynamically more easily in the surface layer of an oxide. This is supported by DFT calculations, which yield a decreased
form thermodynamically more easily in the surface layer of an oxide. This is supported by DFT calculations, which yield a decreased 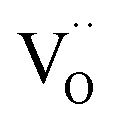 formation energy in the surface layer of ceria depending on the exposed facets, where ΔH0red increases in the order (110) < (100) < (111).60 Additionally, an excess surface charge and a subsurface space charge layer may appear. As long as the type of the majority carrier does not change relative to bulk and the surface charge (if present) is approximately p(O2) independent, the p(O2) dependence of the defects will be the same in bulk and surface layer.
formation energy in the surface layer of ceria depending on the exposed facets, where ΔH0red increases in the order (110) < (100) < (111).60 Additionally, an excess surface charge and a subsurface space charge layer may appear. As long as the type of the majority carrier does not change relative to bulk and the surface charge (if present) is approximately p(O2) independent, the p(O2) dependence of the defects will be the same in bulk and surface layer.
Direct measurements of surface defect concentrations (preferably under conditions of well-defined T and p(O2)) are very challenging, thus only few data are available. For ceria-based materials, the following three observations have to be considered:
(i) For nanosized undoped ceria powder or ceramic samples, modified p(O2) dependencies for oxygen deficiency and/or n-type conductivity were found, which have been interpreted by pronounced defect association of e′ with 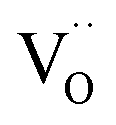 (which is equivalent to a less than doubly charged oxygen vacancy).63–65
(which is equivalent to a less than doubly charged oxygen vacancy).63–65
(ii) For thin films of undoped and Sm-doped ceria in strongly reducing atmosphere, ambient-pressure XPS indicated strongly enhanced surface 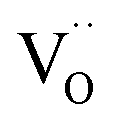 and
and 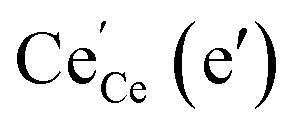 concentrations and correspondingly modified p(O2) dependencies.66–68
concentrations and correspondingly modified p(O2) dependencies.66–68
(iii) For a PDC thin film on a YSZ substrate, 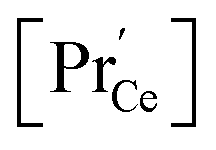 at the surface was measured by ambient-pressure XPS while the effective p(O2) was tuned by a d.c. bias, and an increased surface
at the surface was measured by ambient-pressure XPS while the effective p(O2) was tuned by a d.c. bias, and an increased surface 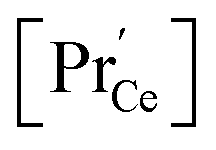 with decreased p(O2) dependence was found.69
with decreased p(O2) dependence was found.69
The relevance of observation (i) with modified defect charge caused by electron trapping in the oxygen vacancies is considered to be small for the present study; the acceptor doping leads to a high oxygen vacancy and low conduction electron concentration, so association cannot significantly alter the average oxygen valence state. The measurements of undoped and Sm-doped films in case (ii) yield strongly increased surface oxygen vacancy and  concentrations compared to bulk (in strongly reducing conditions), from which a decreased ceria reduction enthalpy by 1 eV was deduced. Such a decrease agrees well with earlier findings from DFT (decrease by 0.8 eV for (111) surface to 1.4 eV for (110) surface;60 and experiments on nanocrystalline ceria samples.70 Despite the more increased surface vacancy concentration, the excess
concentrations compared to bulk (in strongly reducing conditions), from which a decreased ceria reduction enthalpy by 1 eV was deduced. Such a decrease agrees well with earlier findings from DFT (decrease by 0.8 eV for (111) surface to 1.4 eV for (110) surface;60 and experiments on nanocrystalline ceria samples.70 Despite the more increased surface vacancy concentration, the excess 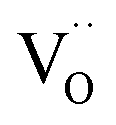 and
and 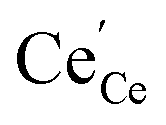 concentrations match almost exactly, so the charge of the surface layer is rather small.71 For comparably high p(O2) of 10−4 bar at 450 °C, the surface concentration of
concentrations match almost exactly, so the charge of the surface layer is rather small.71 For comparably high p(O2) of 10−4 bar at 450 °C, the surface concentration of 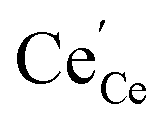 remains very low,71 much smaller than the acceptor concentrations used in the present work. Thus, we can reasonably assume that for the present GDC samples, bulk and surface layer are in the same defect chemical regime, with
remains very low,71 much smaller than the acceptor concentrations used in the present work. Thus, we can reasonably assume that for the present GDC samples, bulk and surface layer are in the same defect chemical regime, with 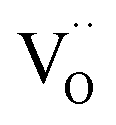 and
and 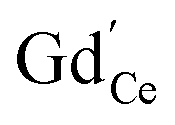 as majority defects. Based on this, a similar p(O2) dependence of the minority defect species
as majority defects. Based on this, a similar p(O2) dependence of the minority defect species  is expected in the present work as in GDC bulk, but with increased absolute values (at 700 °C, a decrease of ΔH0red by 1 eV corresponds to an increase of
is expected in the present work as in GDC bulk, but with increased absolute values (at 700 °C, a decrease of ΔH0red by 1 eV corresponds to an increase of  by a factor of approx. 400). Finding (iii), is not too surprising, since Pr is more easily reduced than Ce. However, the observation of a modified p(O2) dependence even under conditions where
by a factor of approx. 400). Finding (iii), is not too surprising, since Pr is more easily reduced than Ce. However, the observation of a modified p(O2) dependence even under conditions where  is relatively small remains unexplained in ref. 69.
is relatively small remains unexplained in ref. 69.
In the ESI† we discuss in more detail specific situations that can occur at the surface, and we derive the possible oxygen partial pressure dependences. Overall we conclude that in the present experimental conditions for GDC the surface 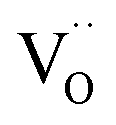 concentration is largely determined by the
concentration is largely determined by the 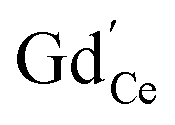 acceptor doping, and the p(O2) dependence of minority
acceptor doping, and the p(O2) dependence of minority  species is similar as in bulk. PDC might exhibit a smaller p(O2) dependence of
species is similar as in bulk. PDC might exhibit a smaller p(O2) dependence of  in the range of 0.1 instead of 1/6.
in the range of 0.1 instead of 1/6.
3.3 Calculation of reaction rates
During PIE measurements, the sample is in equilibrium with the gas phase p(O2), therefore the forward and back reaction rates of the oxygen exchange reaction are equal and characterized by the equilibrium exchange rate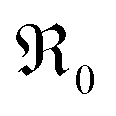 .72,73 In the PIE experiment,
.72,73 In the PIE experiment, 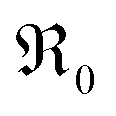 can be obtained from the isotope fraction before and after the reactor according to ref. 37.
can be obtained from the isotope fraction before and after the reactor according to ref. 37.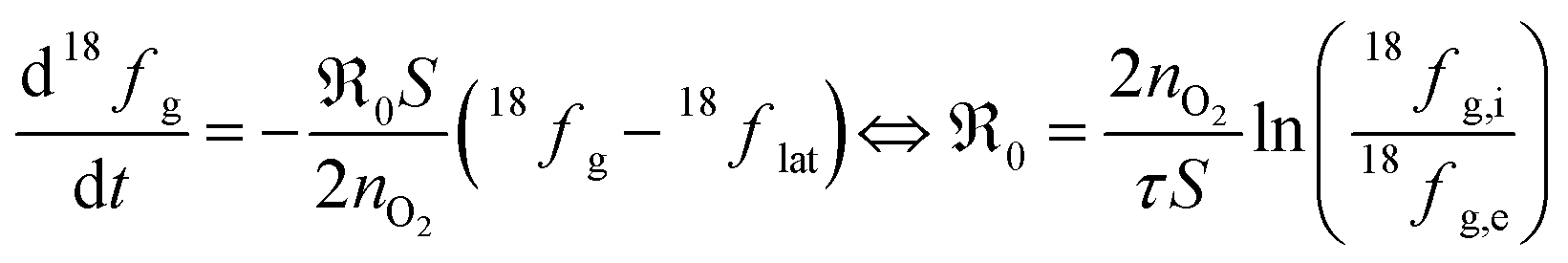 | (8) |
The surface area of the particles is denoted by S, nO2 is the molar amount of oxygen in the gas phase, t denotes time, and 18fg,i and 18fg,e are the gas phase fraction of 18O in the pulse at the inlet (97 at% 18O) and exit of the reactor (calculated according to 18f = 36f + 0.534f where 36f and 34f are the fractions of 36O2 and 34O2 in the gas phase). The mean residence time τ in the reactor was calculated with τ = V/ν (V is the gas phase volume in the reactor and ν the volume flow rate of oxygen passing through the reactor). 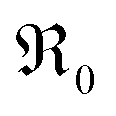 has the unit of oxygen atoms per time and area and is related to the effective rate constant of the surface exchange reaction k* [cm s−1] by39
has the unit of oxygen atoms per time and area and is related to the effective rate constant of the surface exchange reaction k* [cm s−1] by39
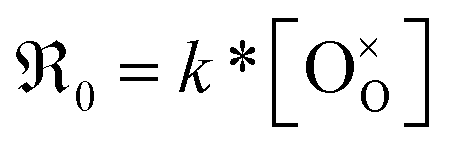 | (9) |
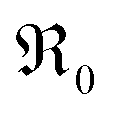 and k* exhibit the same p(O2) dependence). In the derivation of eqn (8), it is assumed that the concentration of 18O in the surface layer of the sample is negligible. This is fulfilled when bulk diffusion in the particle is fast compared to surface exchange, and the number of 18O atoms in the pulse is small compared to the total number of oxygen atoms in the particles.
and k* exhibit the same p(O2) dependence). In the derivation of eqn (8), it is assumed that the concentration of 18O in the surface layer of the sample is negligible. This is fulfilled when bulk diffusion in the particle is fast compared to surface exchange, and the number of 18O atoms in the pulse is small compared to the total number of oxygen atoms in the particles.
This analysis of 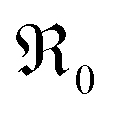 does not require the assumption of a specific reaction mechanism. From the variation of
does not require the assumption of a specific reaction mechanism. From the variation of 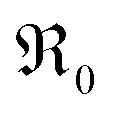 with p(O2) and dopant/defect concentrations several important conclusions can be drawn. If a dissociative-adsorption reaction is rate-determining, that reaction determines the overall exchange rate
with p(O2) and dopant/defect concentrations several important conclusions can be drawn. If a dissociative-adsorption reaction is rate-determining, that reaction determines the overall exchange rate 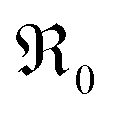 . For example, the oxygen incorporation may be determined by the rate of dissociative-adsorption (eqn (10), with a fast oxygen adsorption as a pre-equilibrium) which involves electronic defects. Due to microscopic reversibility principle, close to equilibrium the forward and backward reactions must have identical overall dependences on p(O2) and point defect concentrations. It suffices to analyze just the forward rate:
. For example, the oxygen incorporation may be determined by the rate of dissociative-adsorption (eqn (10), with a fast oxygen adsorption as a pre-equilibrium) which involves electronic defects. Due to microscopic reversibility principle, close to equilibrium the forward and backward reactions must have identical overall dependences on p(O2) and point defect concentrations. It suffices to analyze just the forward rate:
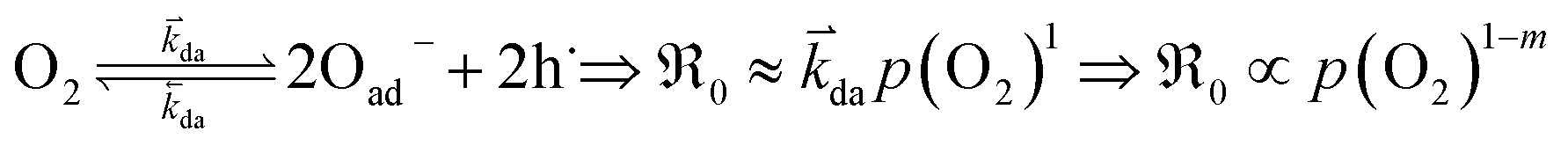 | (10) |
The exponent m denotes the influence of point defects on the overall oxygen partial pressure exponent n. Since molecular oxygen is involved, 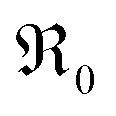 is proportional to p(O2)n with n ≤ 1. The exponent n can be lowered to 1 − m through the influence of point defects as exemplified in section 3 of the ESI,†e.g., if [h˙] ∝ p(O2)1/4 in case of GDC appear before the actual rate-determining elementary step (rds). A different p(O2) dependence can be obtained if instead incorporation of a single oxygen ad-atom is the rds (see eqn (12)). Then for the equilibrated dissociative-adsorption reaction, one can write the mass action law:
is proportional to p(O2)n with n ≤ 1. The exponent n can be lowered to 1 − m through the influence of point defects as exemplified in section 3 of the ESI,†e.g., if [h˙] ∝ p(O2)1/4 in case of GDC appear before the actual rate-determining elementary step (rds). A different p(O2) dependence can be obtained if instead incorporation of a single oxygen ad-atom is the rds (see eqn (12)). Then for the equilibrated dissociative-adsorption reaction, one can write the mass action law:
 | (11) |
The overall exchange rate 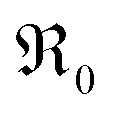 equals the rate of oxygen ad-atom incorporation. By considering eqn (11), one obtains that
equals the rate of oxygen ad-atom incorporation. By considering eqn (11), one obtains that 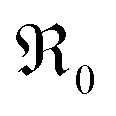 is proportional to p(O2)n with n ≤ 0.5:
is proportional to p(O2)n with n ≤ 0.5:
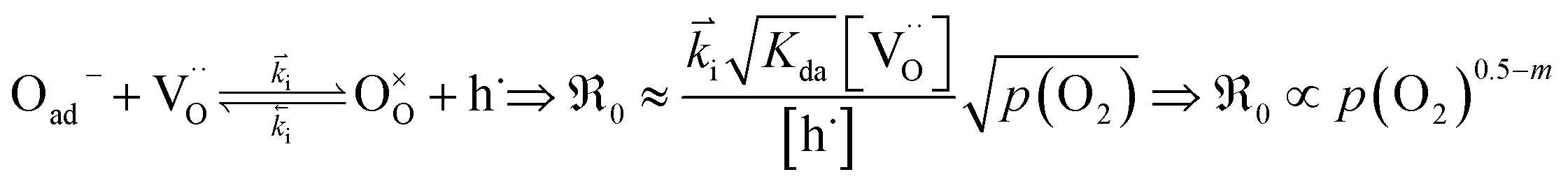 | (12) |
This concept is generally applicable and enables one to distinguish whether molecular or atomic oxygen is involved in the rds. We assume here ideal behavior; non-idealities might nevertheless be present at high dopant concentrations. However, sufficient quantitative data are lacking, and the effect on the p(O2) dependence of 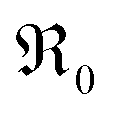 is expected to be rather modest.
is expected to be rather modest.
4 Results
4.1 Sample characterization
The SPS pellets showed densities between 95 and 98% of the theoretical value. The surface of the grains appears smooth without apparent porosity (ESI Fig. 2†). The mean grain size ranges from 1 to 4 μm, decreasing with increased doping concentration (Fig. 2a). The BET surface area of the 60–100 μm particles was measured to be 0.014 m2 g−1.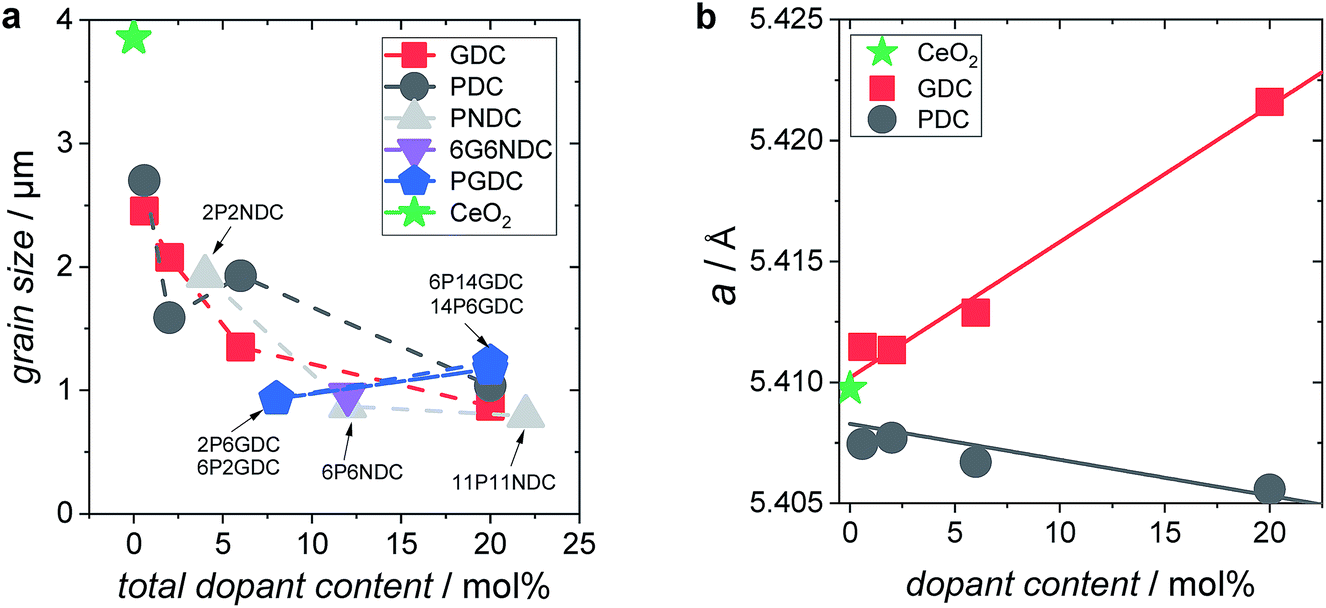 | ||
| Fig. 2 (a) Grain size of doped ceria samples as function of total dopant content. (b) Lattice parameter a as function of dopant content. | ||
XRD measurements on doped ceria particles confirmed a phase pure, cubic fluorite type crystal lattice (lattice parameters in ESI†). For Gd-doped ceria, the lattice parameter follows Vegard's law,74 indicating a solid solution with an increase corresponding to the larger ion radius of Gd3+ (Ce4+ 0.97 Å, Gd3+ 1.053 Å)75 (Fig. 2b). The slight decrease of the lattice constant for PDC indicates that the average Pr oxidation state is closer to 4+ rather than 3+ in the as-prepared samples (Pr4+ 0.96 Å, Pr3+ 1.126 Å)75 which is in accordance with the TG data in Fig. 1 and with defect model calculations.57
XPS measurements on 6GDC, 20GDC, 6PDC, 20PDC, and 6P6NDC (Fig. 3) revealed a moderate accumulation of dopants at the particle's surface of up to 30% relative to bulk values after the second annealing step at 1400 °C. The strength of the dopant segregation increases according to [Gd] < [Pr] < [Nb]. TEM/EDX analysis on 6PDC particles indicated also a Pr accumulation at the grain boundaries which is stronger than for the surface (ESI†).
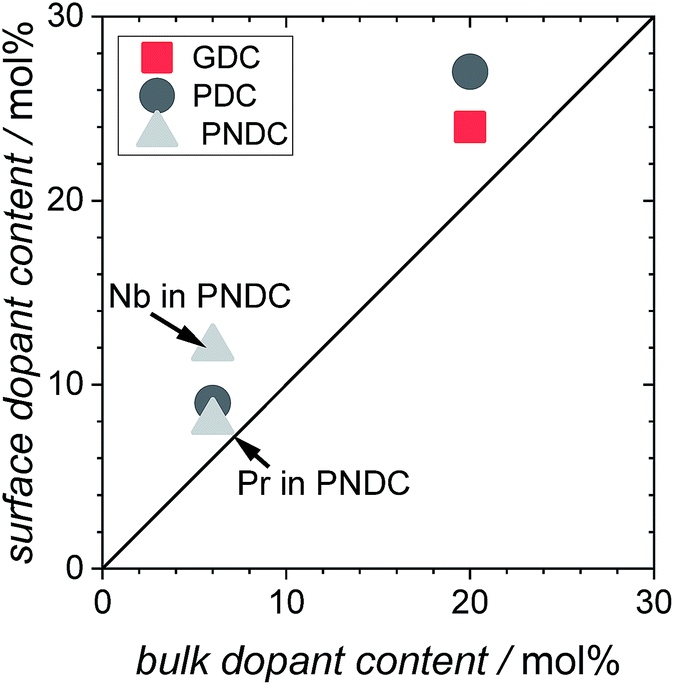 | ||
| Fig. 3 Dopant content at the surface of the PIE particles (double annealed at 1400 °C) plotted against the nominal dopant concentration. | ||
4.2 Pulsed isotope exchange
For all samples, the regime which was probed by PIE measurements is considered to be surface controlled. The characteristic lengths l0 = k*/D* (D* = oxygen tracer diffusion coefficient, k* = effective tracer exchange rate constant) calculated with data from ref. 8, 9 and 18 are typically 10–1000 times larger than the mean particle radius, which means that the concentration gradient of 18O inside the particles vanishes and the bulk diffusion does not affect the 18O exchange kinetics.76 Grain boundaries in acceptor-doped ceria are known to exhibit moderately blocking behavior for oxide ion diffusion, but in the temperature regime of the present PIE experiments, the blocking effect becomes small77,78 (see also conductivity data from impedance spectroscopy in the ESI†).In Fig. 4 the fractions of the molecular oxygen isotopologues in the pulse, measured at the exit of the packed bed reactor, are plotted against the reactor temperature. With increasing temperature, 18O starts to incorporate into the sample indicated by a decrease of 36f. Due to the fact that the oxygen stoichiometry is in equilibrium, the same amount of 16O needs to be released from the sample in the form of 32O2 or 34O2 depending on the underlying reaction mechanism.
 | ||
| Fig. 4 Molar oxygen isotopologues fractions (f) measured by mass spectrometry on the gas exiting the reactor; all measurements in 10% O2 atmosphere. | ||
Results obtained at temperatures above 800 °C should be interpreted with care, since in this regime the Al2O3 reactor itself showed oxygen exchange activity (Fig. 4, close symbols), mainly by isotopic scrambling at the surface as indicated by the high 34O2 fraction. Negative mole fractions for 32O2 originate from some unavoidable smearing of the 18O pulse: at the edges of the pulse, residual 32O2 can scramble at the reactor surface with 36O2 yielding 34O2, as indicated by peak splitting of the 34O2 MS signal. Undoped CeO2 (Fig. 4, green symbols) showed similar low oxygen exchange activity as the empty Al2O3 reactor. Thus, for undoped ceria and 2NDC no activation energies are given and only an upper limit of  can be indicated in Fig. 7.
can be indicated in Fig. 7.
All Gd or Pr/Tb single- and co-doped ceria samples measured in this study are more active for oxygen exchange than undoped CeO2. On 0.6GDC the oxygen incorporation begins at around 750 °C. This onset temperature shifts to lower values with increasing [Gd], so that for 20GDC the incorporation commences already at 600 °C. All GDC samples show a pronounced formation of 34O2, which points to a slow incorporation reaction rate relative to the rates of oxygen adsorption and dissociation.
The increase of the exchange activity on PDC samples is even more pronounced than on GDC. On 20PDC, oxygen incorporation was observed already at 500 °C. On 0.6PDC the conversion at high T is in the same low range as for undoped CeO2 and the empty reactor. The onset of perceptible oxygen conversion quickly shifty to lower T for higher Pr dopant contents. The absence of 34O2 at >2 mol% Pr or Tb suggests that dissociated oxygen species incorporate faster into the bulk compared to GDC with similar dopant content.
2NDC and co-doped 6P6NDC/6G6NDC were less active compared to GDC and PDC/TDC samples. Furthermore, the formation of 34O2 on PNDC was less distinct than for GNDC. Since Nb as donor dopant decreases the oxygen vacancy concentration or may even lead to oxygen interstitials, these results show that oxygen vacancies are crucial for the oxygen exchange reaction.
4.3 Analysis of the 18O gas phase fraction
From the measured overall isotope fractions 18fg,i and 18fg,e in the gas phase, the equilibrium exchange rates are calculated by eqn (8). According to Fig. 5, the temperature dependencies of follow Arrhenius behavior. Fig. 6 presents the extracted activation energies which range between 1.4–2.8 eV. Two distinct groups can be recognized: singly Pr/Tb and Gd doped ceria as well as Pr/Gd co-doped ceria showed Ea values ranging between 1.5–2.3 eV, while Nb doping led to high activation energies between 2.5 and 2.8 eV. Furthermore, with increasing total dopant concentration Ea tends to decrease slightly. However, since the measured Ea is a combination of the actual reaction barrier of the rds and the reaction enthalpies of fast preceding steps, the interpretation of this trend is not straightforward. The consistently higher Ea for the Nb co-doped samples might possibly arise from higher reaction barriers in absence of a high inherent
follow Arrhenius behavior. Fig. 6 presents the extracted activation energies which range between 1.4–2.8 eV. Two distinct groups can be recognized: singly Pr/Tb and Gd doped ceria as well as Pr/Gd co-doped ceria showed Ea values ranging between 1.5–2.3 eV, while Nb doping led to high activation energies between 2.5 and 2.8 eV. Furthermore, with increasing total dopant concentration Ea tends to decrease slightly. However, since the measured Ea is a combination of the actual reaction barrier of the rds and the reaction enthalpies of fast preceding steps, the interpretation of this trend is not straightforward. The consistently higher Ea for the Nb co-doped samples might possibly arise from higher reaction barriers in absence of a high inherent  , or from a higher effective T-dependence of the
, or from a higher effective T-dependence of the 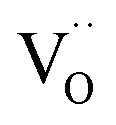 concentration than in PDC/TDC and GDC. It is interesting to note that while the prefactor of
concentration than in PDC/TDC and GDC. It is interesting to note that while the prefactor of 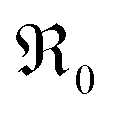 varies only moderately between 2% and 20% doping for the GDC samples, the increase of
varies only moderately between 2% and 20% doping for the GDC samples, the increase of 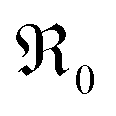 with increasing [Pr]tot is largely caused by an increased prefactor.
with increasing [Pr]tot is largely caused by an increased prefactor.
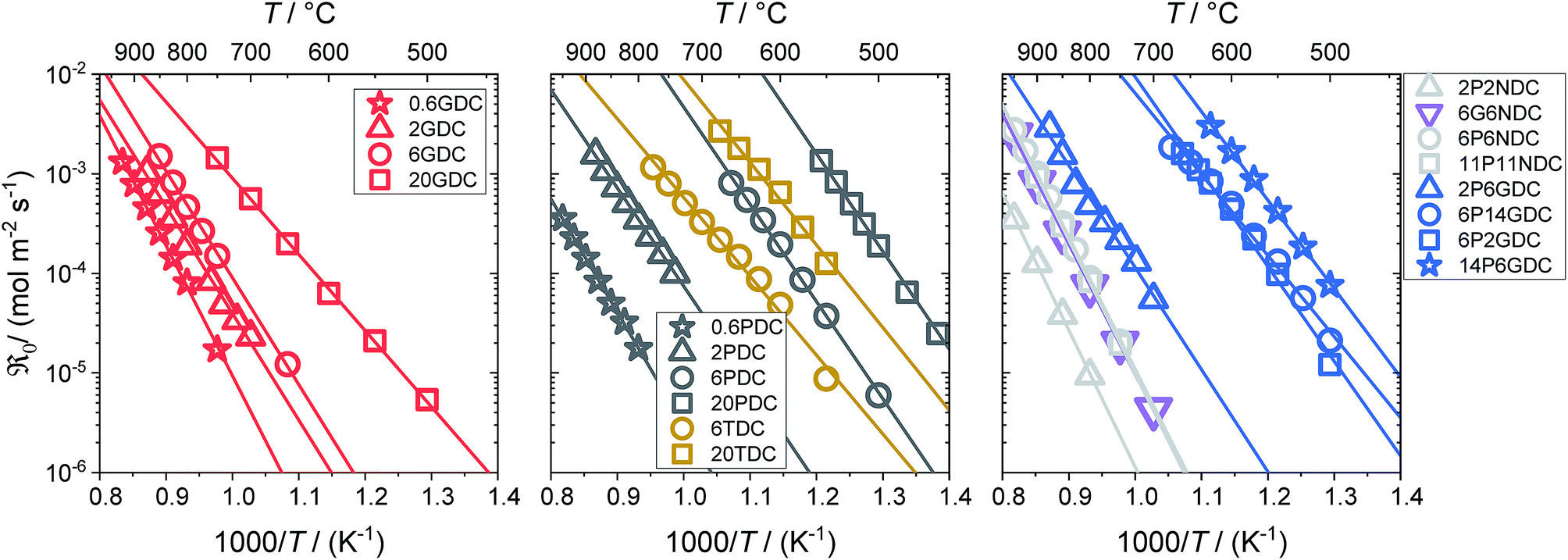 | ||
Fig. 5 Temperature dependence of the equilibrium exchange rate  for doped ceria samples, measured in 10% O2. for doped ceria samples, measured in 10% O2. | ||
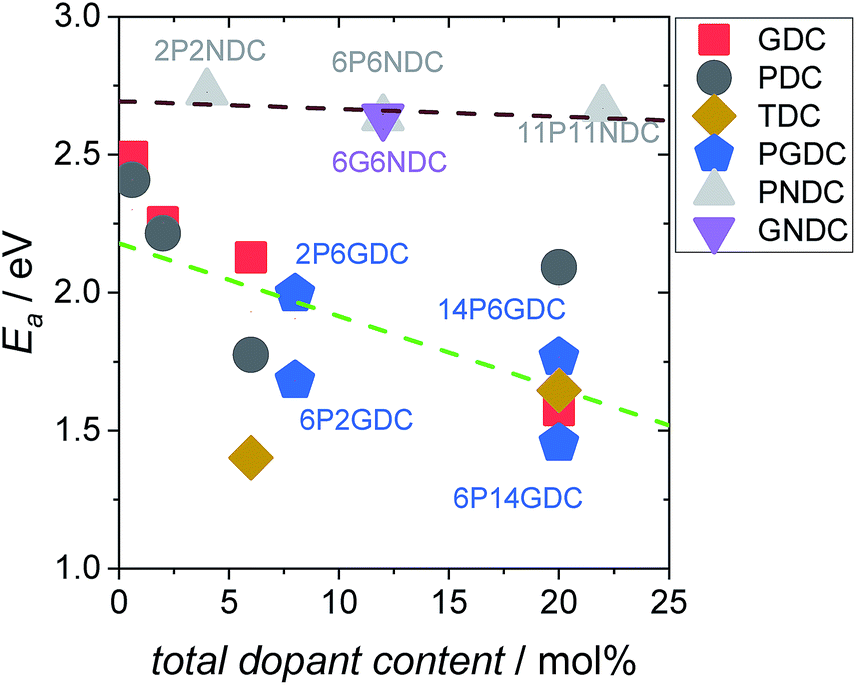 | ||
Fig. 6 Activation energies calculated from the temperature dependence of the equilibrium exchange rate  , in 10% O2. , in 10% O2. | ||
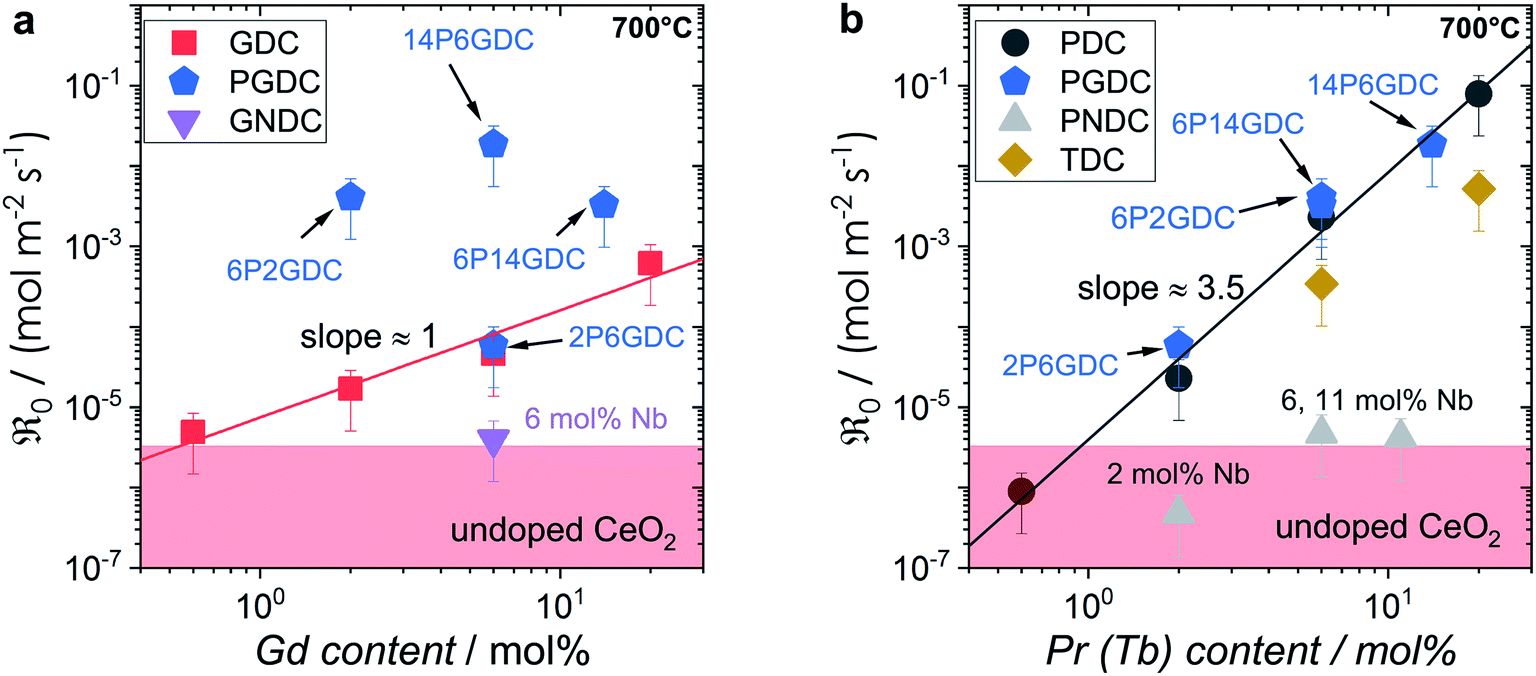 | ||
Fig. 7
 as a function of bulk (a) Gd and (b) Pr content (values measured in 10% oxygen atmosphere and inter/extrapolated to 700 °C). The shaded area indicates the activity range of undoped ceria and 2NDC. Values for 06GDC, 20GDC, 06PDC, 2PDC, 6PDC and 20PDC are mean values calculated from at least two measurements. as a function of bulk (a) Gd and (b) Pr content (values measured in 10% oxygen atmosphere and inter/extrapolated to 700 °C). The shaded area indicates the activity range of undoped ceria and 2NDC. Values for 06GDC, 20GDC, 06PDC, 2PDC, 6PDC and 20PDC are mean values calculated from at least two measurements. | ||
The trends of oxygen exchange rate with dopant type and concentration can best be compared at a fixed temperature (700 °C), as shown in Fig. 7. Since undoped CeO2 showed similar low oxygen exchange activity as the empty Al2O3 reactor no activation energies are given and only an upper limit of  can be indicated in Fig. 7.
can be indicated in Fig. 7.
For GDC the condition 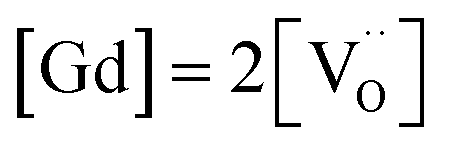 holds for the bulk. For the vacancy concentration at the surface, different absolute values but still an approximate proportionality to the bulk Gd concentration is expected (see Section 3.1). Fig. 7a shows that the exchange rate is approximately proportional to the bulk
holds for the bulk. For the vacancy concentration at the surface, different absolute values but still an approximate proportionality to the bulk Gd concentration is expected (see Section 3.1). Fig. 7a shows that the exchange rate is approximately proportional to the bulk 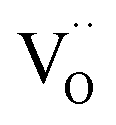 concentration
concentration 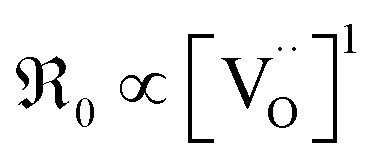 . This strongly indicates that one
. This strongly indicates that one 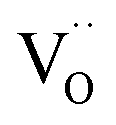 is involved before or in the rds step of the oxygen exchange reaction. The importance of
is involved before or in the rds step of the oxygen exchange reaction. The importance of 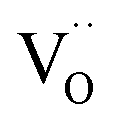 for the water splitting reaction on ceria has recently been emphasized by XPS measurements under near-ambient conditions.68
for the water splitting reaction on ceria has recently been emphasized by XPS measurements under near-ambient conditions.68
In contrast, for PDC 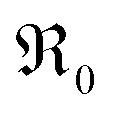 increases more strongly according to
increases more strongly according to 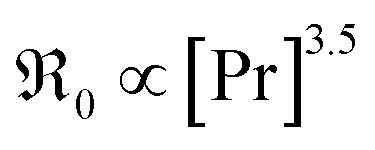 (Fig. 7b), which overall increases
(Fig. 7b), which overall increases 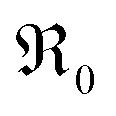 by almost 5 orders of magnitude relative to undoped ceria. Since rather extensive extrapolations are required to obtain
by almost 5 orders of magnitude relative to undoped ceria. Since rather extensive extrapolations are required to obtain 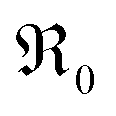 values for all PDC samples at one common T, the numerical value of this slope might carry some uncertainty. Nevertheless, such a strong dependence on Pr content suggests that in addition to the increased
values for all PDC samples at one common T, the numerical value of this slope might carry some uncertainty. Nevertheless, such a strong dependence on Pr content suggests that in addition to the increased 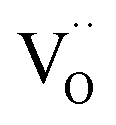 concentration (despite the mixed Pr3+/Pr4+ valence,
concentration (despite the mixed Pr3+/Pr4+ valence, 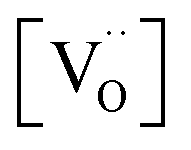 is roughly proportional to the overall Pr content, see Section 3.1), the inherent redox activity of Pr3+/Pr4+ itself also affects the exchange rate. Despite a small offset, TDC shows a similar strong increase of
is roughly proportional to the overall Pr content, see Section 3.1), the inherent redox activity of Pr3+/Pr4+ itself also affects the exchange rate. Despite a small offset, TDC shows a similar strong increase of 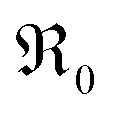 , indicating that redox-active dopants tend to activate the oxygen incorporation. This is in line with several observations in literature that mixed conducting fluorites or perovskites exhibit faster oxygen exchange than purely ionic or electronic conductors.38,79–84
, indicating that redox-active dopants tend to activate the oxygen incorporation. This is in line with several observations in literature that mixed conducting fluorites or perovskites exhibit faster oxygen exchange than purely ionic or electronic conductors.38,79–84
Similar trends as for the singly Gd or Pr doped samples are also found for Gd and Pr co-doped samples. While additional Gd doping of PDC samples (Fig. 7b) has almost no effect, Pr co-doping of GDC perceptibly increases  (Fig. 7a). Nb co-doping with Nb concentration matching that of Gd or Pr leads to exchange rates that are in the region of undoped ceria. This strongly supports the assignment that the rate increases with increasing [Gd] is related to the increased
(Fig. 7a). Nb co-doping with Nb concentration matching that of Gd or Pr leads to exchange rates that are in the region of undoped ceria. This strongly supports the assignment that the rate increases with increasing [Gd] is related to the increased 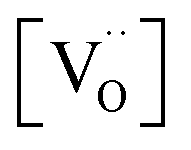 and not to some specific effect of the Gd cations.
and not to some specific effect of the Gd cations.
Further important information on the reaction mechanism is obtained from measurements of 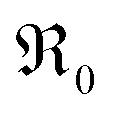 as function of oxygen partial pressure (Fig. 8). For all samples in the present study, the exponents n for the p(O2) dependence are always above 1/2. As discussed in Section 3.3, this means that molecular oxygen species are involved in the rds of oxygen incorporation.
as function of oxygen partial pressure (Fig. 8). For all samples in the present study, the exponents n for the p(O2) dependence are always above 1/2. As discussed in Section 3.3, this means that molecular oxygen species are involved in the rds of oxygen incorporation.
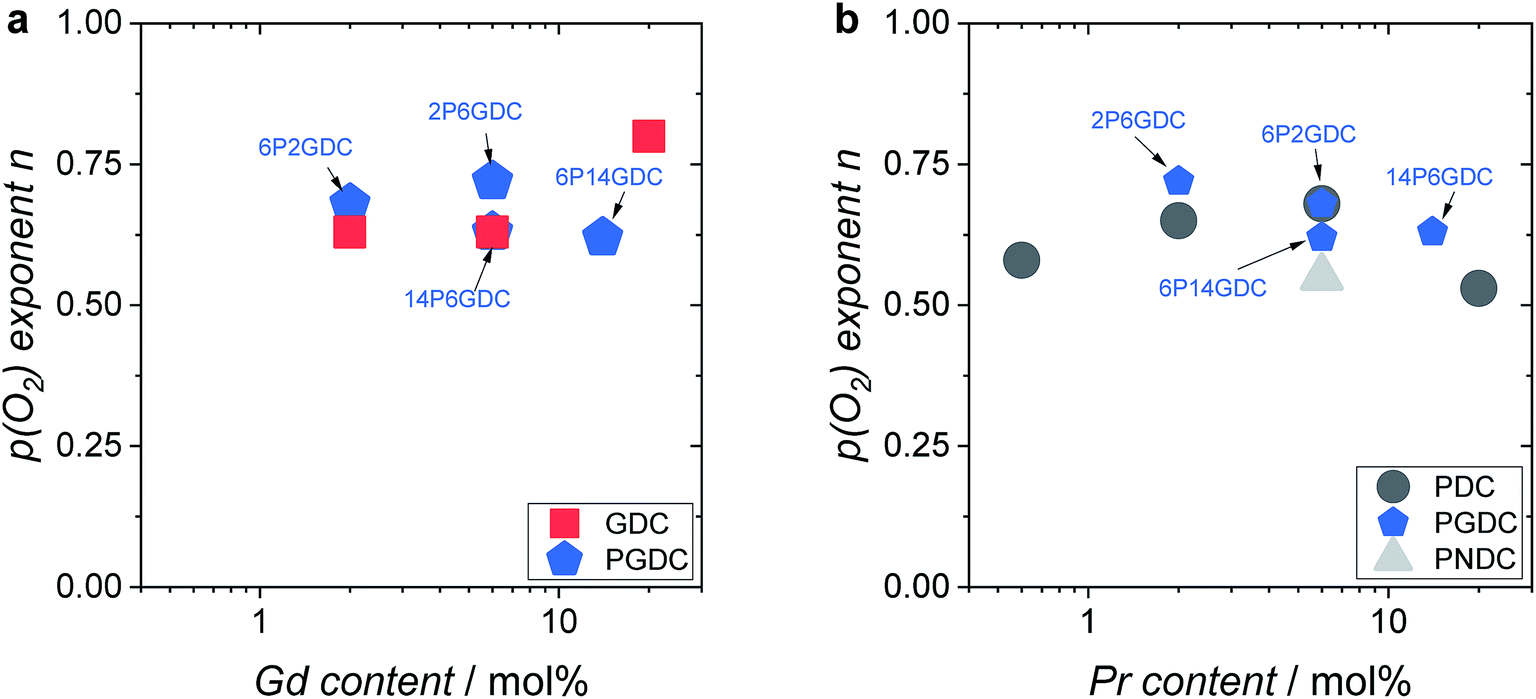 | ||
| Fig. 8 Oxygen partial pressure exponent n as function of (a) Gd and (b) Pr content. Measurement temperatures: 2GDC 810 °C, 6GDC 785 °C, 20GDC 625 °C, 2PDC 800 °C, 6PDC 650 °C, 20PDC 560 °C, 2P6GDC 775 °C, 6P2GDC, 6P14GDC 600 °C, 14P6GDC 575 °C, 2P2NDC 850 °C. The relative error for the exponents are sampler than the symbols. | ||
Let us summarize the key experimental observations regarding the oxygen exchange mechanism:
(i) On GDC,  and on PDC
and on PDC 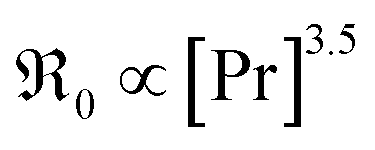 (with
(with 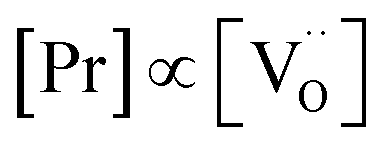 ), which indicates that at least one
), which indicates that at least one 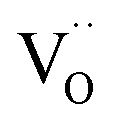 is involved in or before the rds.
is involved in or before the rds.
(ii) For both systems 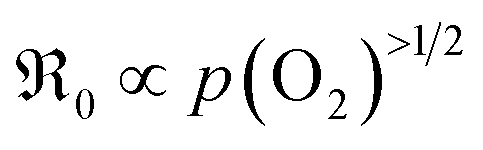 therefore, molecular oxygen species are involved in the rds.
therefore, molecular oxygen species are involved in the rds.
(iii) The fact that the exponent of the overall p(O2) dependencies is below one indicates that defects with negative p(O2) dependency are involved.
Based on this we can suggest for GDC the following mechanism depicted in Fig. 9 which is consistent with the measured data. From the observation that in addition to molecular oxygen species also 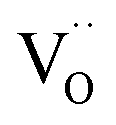 are involved in/before the rds, the dissociation without
are involved in/before the rds, the dissociation without 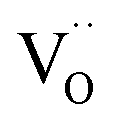 is most likely ruled out as a possible reaction mechanism for GDC as well as for PDC.
is most likely ruled out as a possible reaction mechanism for GDC as well as for PDC.
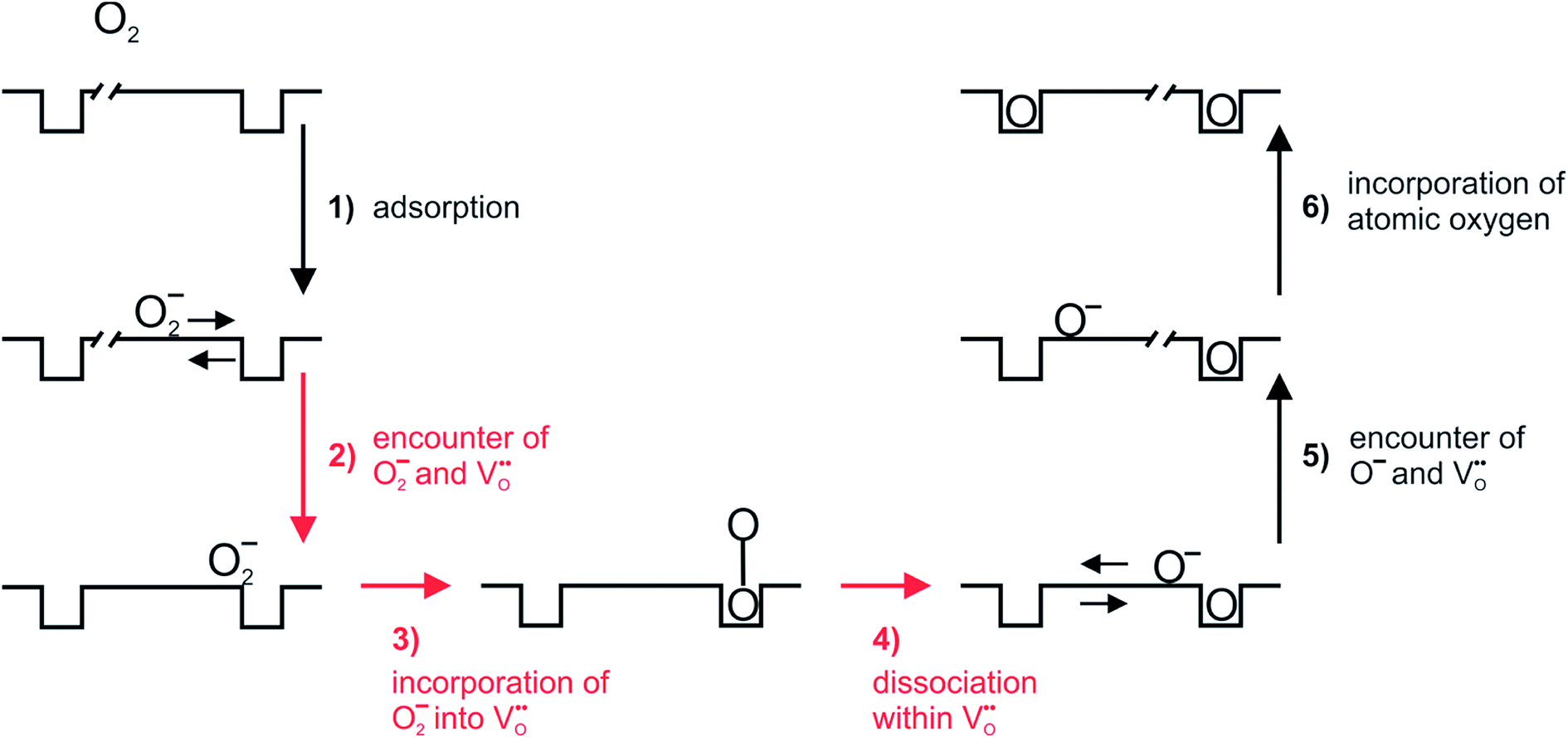 | ||
Fig. 9 Plausible reaction mechanism of oxygen exchange for GDC. The rate-determining step is expected to be within steps 2–4. The encounter in step 2 can occur by migration of O2− and/or 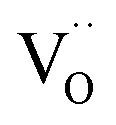 along the surface. The charges of the oxygen intermediates cannot be directly concluded; they are tentatively assigned. along the surface. The charges of the oxygen intermediates cannot be directly concluded; they are tentatively assigned. | ||
Adsorption including a first electron transfer is expected to be fast, since such chemisorption processes occur already at room temperature or below (see, e.g., ref. 26, 29 and 85). For reduced ceria26 and redox-active perovskites such as (La,Sr)MnO3±δ superoxide as well as peroxide species are formed.86 However, the latter are regarded as less probable for GDC (which has a large bandgap) under oxidizing conditions. On pre-reduced CeO2 nanocrystals, at 173 K a concentration of adsorbed molecular oxygen species of about 2–3 μmol O2 per m2 was found (corresponding to surface coverage of about 6%),29 but at the higher T and p(O2) of the PIE experiment significantly lower values are expected.
For perovskites87 as well as for ceria,28 DFT calculations indicate that the dissociation of molecular oxygen species is facilitated when it is first incorporated into an oxygen vacancy. Thus we consider oxygen exchange to proceed via an encounter of O2− and 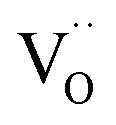 (step 2; at this stage we cannot distinguish which of these species migrates towards the other), incorporation of O2− into the
(step 2; at this stage we cannot distinguish which of these species migrates towards the other), incorporation of O2− into the 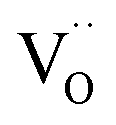 (step 3), and actual dissociation (step 4). For perovskites such as (La,Sr)MnO3±δ or Ba0.5Sr0.5Co0.8Fe0.2O3−δ there is evidence that rather a surface oxygen vacancy approaches the adsorbed oxygen species than vice versa.80,87
(step 3), and actual dissociation (step 4). For perovskites such as (La,Sr)MnO3±δ or Ba0.5Sr0.5Co0.8Fe0.2O3−δ there is evidence that rather a surface oxygen vacancy approaches the adsorbed oxygen species than vice versa.80,87
On the basis of the present experimental data, one of steps 2–4 is rate-determining. Since the overall p(O2) dependence indicates molecular oxygen species in the rds, the encounter of O− and 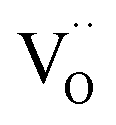 in step 5 cannot be limiting. This implies that either the concentration of atomic oxygen is higher than that of O2− (as observed for (La,Sr)MnO3±δ;87 leading to a shorter diffusion length to
in step 5 cannot be limiting. This implies that either the concentration of atomic oxygen is higher than that of O2− (as observed for (La,Sr)MnO3±δ;87 leading to a shorter diffusion length to 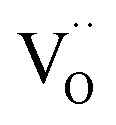 ), or that the mobile species is the adsorbed oxygen with a higher surface mobility for O− compared to O2−. The fact that the incorporation of oxygen into
), or that the mobile species is the adsorbed oxygen with a higher surface mobility for O− compared to O2−. The fact that the incorporation of oxygen into 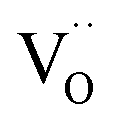 (step 6) is not the rds may indicate that also the similar step 3 is not limiting. The charges of the oxygen intermediates cannot be directly concluded from the measured p(O2) and
(step 6) is not the rds may indicate that also the similar step 3 is not limiting. The charges of the oxygen intermediates cannot be directly concluded from the measured p(O2) and 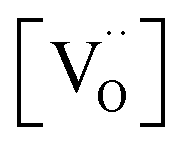 dependences, they are tentatively assigned (in particular for adsorbed atomic O species the charge may also depend on the actual surface termination).
dependences, they are tentatively assigned (in particular for adsorbed atomic O species the charge may also depend on the actual surface termination).
The overall p(O2) dependence of  contains also contributions from the involved point defects (a detailed deviation is found in the ESI†). Assuming that just one hole is involved in the fast pre-equilibrium in eqn (13) before the rds, one obtains for
contains also contributions from the involved point defects (a detailed deviation is found in the ESI†). Assuming that just one hole is involved in the fast pre-equilibrium in eqn (13) before the rds, one obtains for  :
:
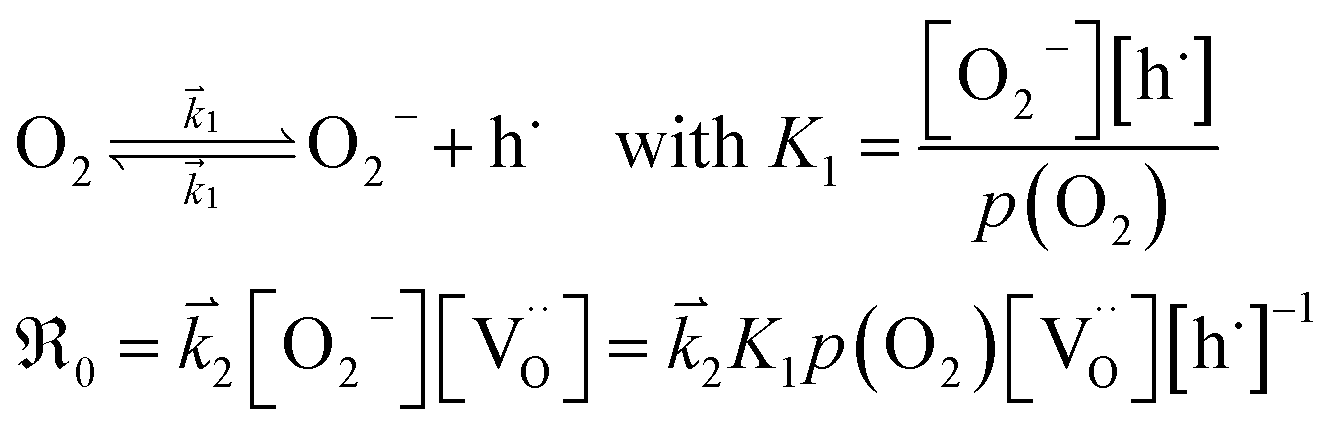 | (13) |
For GDC with fixed 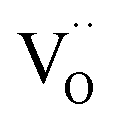 concentration, [h˙] ∝ p(O2)1/4 holds in the bulk, which yields
concentration, [h˙] ∝ p(O2)1/4 holds in the bulk, which yields 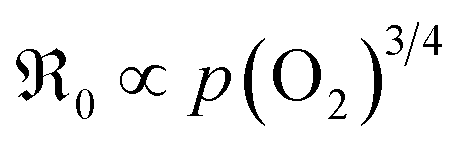 . This fits well to the measured overall p(O2) dependence showing an exponent of 0.6–0.8. From these values it follows that in addition to one
. This fits well to the measured overall p(O2) dependence showing an exponent of 0.6–0.8. From these values it follows that in addition to one 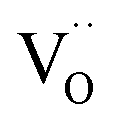 , at least one electron is consumed or one hole is produced before or in the rds. Regarding the dependence of
, at least one electron is consumed or one hole is produced before or in the rds. Regarding the dependence of 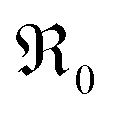 on
on 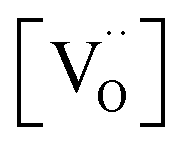 , one has to consider that the hole concentration depends on
, one has to consider that the hole concentration depends on 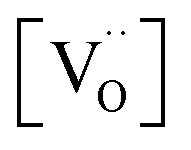 by eqn (2) according to
by eqn (2) according to 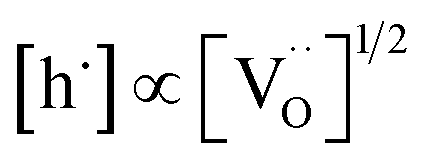 , resulting in
, resulting in 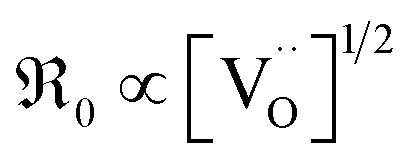 , in contrast to the measured
, in contrast to the measured 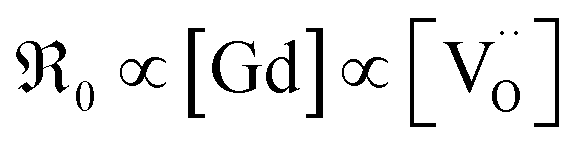 relation for GDC. A tentative consideration to resolve this discrepancy is that
relation for GDC. A tentative consideration to resolve this discrepancy is that 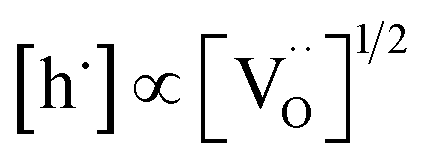 enters the rate expression as a consequence of the chemisorption equilibrium in eqn (13). If the system behaved ideally, K1 would be independent of [Gd] and
enters the rate expression as a consequence of the chemisorption equilibrium in eqn (13). If the system behaved ideally, K1 would be independent of [Gd] and 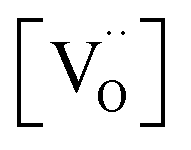 , but [O2−] would decrease with increasing [Gd] and
, but [O2−] would decrease with increasing [Gd] and 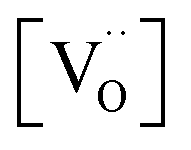 . However, the decreased effective cation charge with increasing Gd content might make the O2 chemisorption increasingly favorable (deviation from ideal behavior) such that overall the O2− concentration becomes approximately independent of
. However, the decreased effective cation charge with increasing Gd content might make the O2 chemisorption increasingly favorable (deviation from ideal behavior) such that overall the O2− concentration becomes approximately independent of 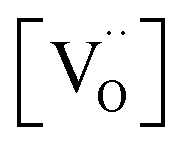 . Alternative mechanisms with two
. Alternative mechanisms with two 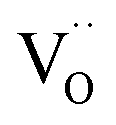 involved before or in the rds (leading to stronger
involved before or in the rds (leading to stronger 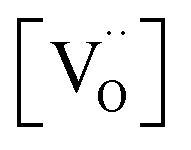 dependencies of
dependencies of 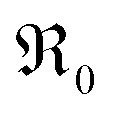 ) are considered less probable as they require the encounter of adsorbed oxygen with two
) are considered less probable as they require the encounter of adsorbed oxygen with two 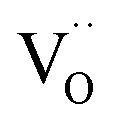 .
.
A striking feature for PDC is that 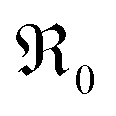 increases over-proportionally with [Pr]. It is reasonable to assume that a part of the dependence originates in one
increases over-proportionally with [Pr]. It is reasonable to assume that a part of the dependence originates in one 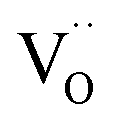 involved before/in the rds (as was found for GDC) which yields a contribution of
involved before/in the rds (as was found for GDC) which yields a contribution of 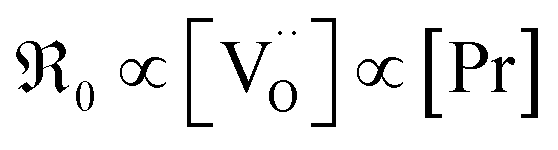 . The remaining dependence suggests a direct involvement of the redox couple Pr3+/Pr4+ in the oxygen exchange reaction. This is highly plausible as in PDC the n-type electronic carriers are localized in the form of
. The remaining dependence suggests a direct involvement of the redox couple Pr3+/Pr4+ in the oxygen exchange reaction. This is highly plausible as in PDC the n-type electronic carriers are localized in the form of 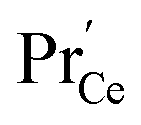 (in contrast to GDC where the n-type carriers
(in contrast to GDC where the n-type carriers 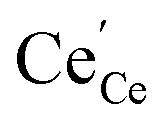 are independent of the dopant). Consequently, the formation of charged molecular oxygen adsorbates in the fast proceeding equilibrium should be formulated as
are independent of the dopant). Consequently, the formation of charged molecular oxygen adsorbates in the fast proceeding equilibrium should be formulated as
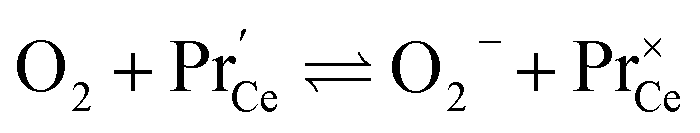 | (14) |
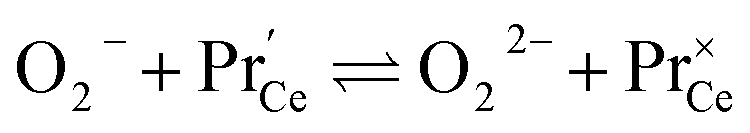 | (15) |
This means that the O2− and O22− adsorbate concentrations should scale with 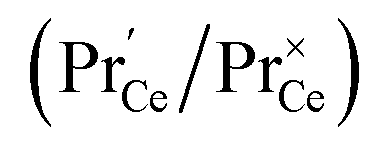 and
and 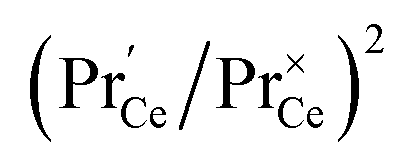 . It is not easy to estimate whether this ratio (in the surface layer of the sample) exhibits a pronounced systematic variation with [Pr]tot. On the other hand, one may expect that because of the possible valence change, sites close to Pr3+/4+ are preferred adsorption sites for O2− and O22−, which could result in adsorbate concentrations that systematically increase with [Pr]tot. This would then also increase the subsequent dissociation rate. Another aspect is that the character of the electronic defects changes drastically with [Pr]tot, from isolated Pr3+/4+ ions at low [Pr]tot to praseodymium states forming an impurity band with non-vanishing band width at high [Pr]tot.69 This may lead to more negative adsorption energies for O2− and O22− and further increases the dependency of
. It is not easy to estimate whether this ratio (in the surface layer of the sample) exhibits a pronounced systematic variation with [Pr]tot. On the other hand, one may expect that because of the possible valence change, sites close to Pr3+/4+ are preferred adsorption sites for O2− and O22−, which could result in adsorbate concentrations that systematically increase with [Pr]tot. This would then also increase the subsequent dissociation rate. Another aspect is that the character of the electronic defects changes drastically with [Pr]tot, from isolated Pr3+/4+ ions at low [Pr]tot to praseodymium states forming an impurity band with non-vanishing band width at high [Pr]tot.69 This may lead to more negative adsorption energies for O2− and O22− and further increases the dependency of 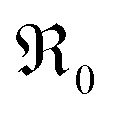 on [Pr]. The hypothesis of Pr band formation is supported by the strongly nonlinear increase of electronic conductivity (ESI Fig. 6,†cf. also 3 × 10−5 S cm−1 for 1PDC to 0.02 S cm−1 for 10PDC at 800 °C in air18). At present, we cannot draw a final conclusion about the atomistic origin of the strong [Pr]tot dependence of
on [Pr]. The hypothesis of Pr band formation is supported by the strongly nonlinear increase of electronic conductivity (ESI Fig. 6,†cf. also 3 × 10−5 S cm−1 for 1PDC to 0.02 S cm−1 for 10PDC at 800 °C in air18). At present, we cannot draw a final conclusion about the atomistic origin of the strong [Pr]tot dependence of 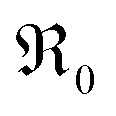 .
.
Another peculiarity of 6PDC and 20PDC (as well as of 6TDC and 20TDC) is that they show much lower formation of 34O2 compared to 6GDC, 20GDC despite higher equilibrium exchange rates. This might point towards a higher 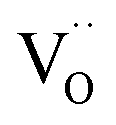 mobility in bulk but also between bulk and surface layer. According to defect chemical data, 10PDC57 has a moderately higher
mobility in bulk but also between bulk and surface layer. According to defect chemical data, 10PDC57 has a moderately higher  mobility than single crystal 10GDC (measured by 18O isotope exchange and SIMS line scan),7 which is in agreement with MD simulations.88 Nevertheless, this should be further confirmed in additional bulk diffusion measurements.
mobility than single crystal 10GDC (measured by 18O isotope exchange and SIMS line scan),7 which is in agreement with MD simulations.88 Nevertheless, this should be further confirmed in additional bulk diffusion measurements.
5 Comparison to literature
In Fig. 10a, tracer exchange coefficients k* obtained from literature for 10GDC to 31GDC7,8,11,15,16 are plotted versus temperature. Equilibrium exchange rates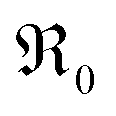 from the present study and from PIE measured by Yoo16 are converted according to
from the present study and from PIE measured by Yoo16 are converted according to 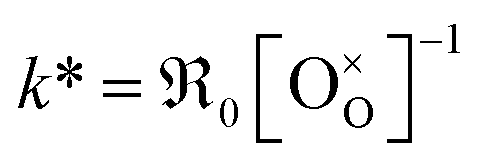 . The data from Yoo agree well with the present data, and both data sets connect reasonably well to isotope exchange line profile (IELP) results at lower T (ref. 11) (which, however, exhibit a decreased activation energy). Exchange rates determined by IELP in ref. 7 and 8 and isotope exchange gas phase analysis (IEGA)15 are two to three orders of magnitude smaller but show similar slopes at temperatures above 500 °C.
. The data from Yoo agree well with the present data, and both data sets connect reasonably well to isotope exchange line profile (IELP) results at lower T (ref. 11) (which, however, exhibit a decreased activation energy). Exchange rates determined by IELP in ref. 7 and 8 and isotope exchange gas phase analysis (IEGA)15 are two to three orders of magnitude smaller but show similar slopes at temperatures above 500 °C.
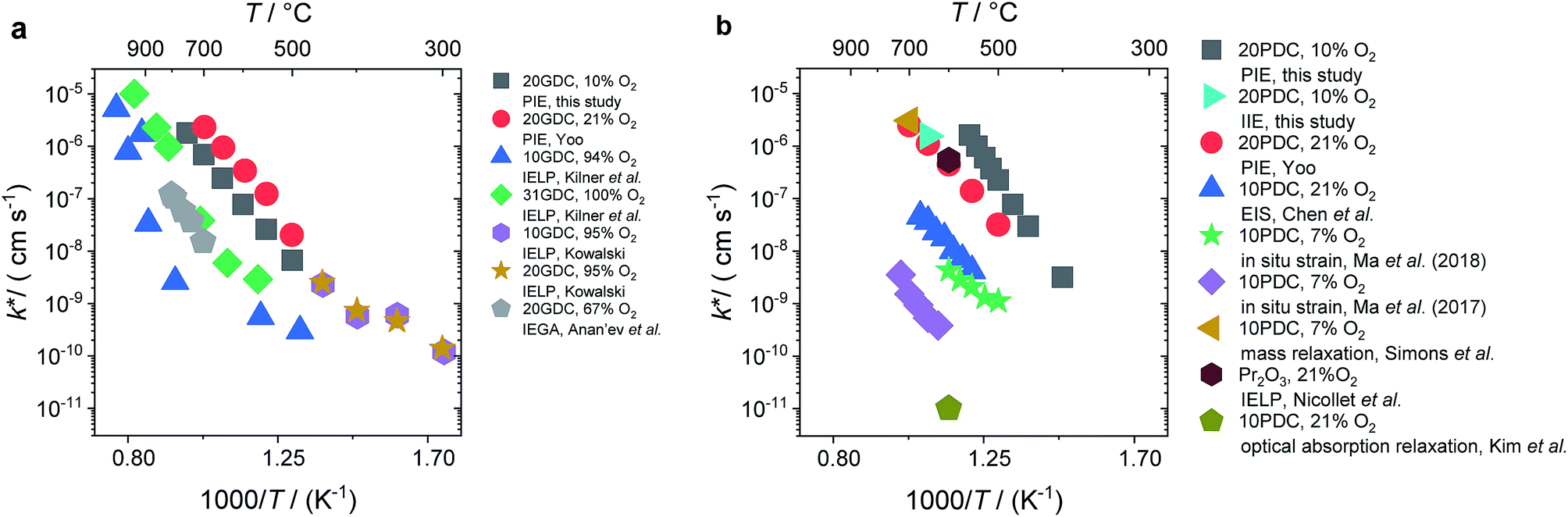 | ||
Fig. 10 Temperature dependence of k* (a) for GDC of this study and literature data.7,8,11,15,16 (b) For PDC of this study and literature data.12,16,18,23–25 The values k* were calculated by 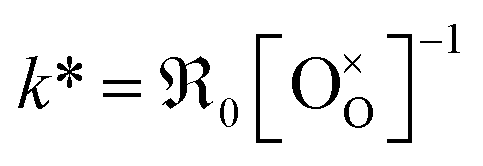 . . | ||
For PDC (Fig. 10b) the literature data scatter by almost six orders of magnitude. The present PIE data for 20PDC and isothermal isotope exchange (IIE) of the same 20PDC particles are of comparable magnitude. They are also similar to the 20PDC value of Yoo, to the exchange rate for 10PDC measured by mass relaxation of thin films deposited on a piezoelectric GaPO4 microbalance crystal23 (converted to k* using a mean thermodynamic factor of ω = 200, determined by TGA on 20PDC particles, T range: 500–700 °C), and to values for Pr6O11 from IELP.12 However, kq ≈ k* values from impedance spectroscopy of 10PDC films on YSZ substrates18 have a similar activation energy, but are about two orders of magnitude lower. The smallest exchange rates were found by Ma et al.24,25 by in situ strain measurements on 10PDC thin films on YSZ substrates and by Kim et al.21 by in situ optical absorption relaxation.
This comparison demonstrates that the surface exchange rates are extremely sensitive to experimental details. Since in this study the ceramic samples were never exposed to Ag, Pt, or other precious metals which are known to catalyze the oxygen exchange reaction, such an effect is not the origin of the high equilibrium exchange rates. Bucher et al.89 demonstrated that SiO2 from any source in the experimental set-up such as quartz glass (in particular when the gas phase is not absolutely dry) can severely decrease the surface exchange rate of mixed-conducting perovskites by formation of a thin glassy or silicate layer. Even stronger degradation by more than 1.5 orders or magnitude was found by Zhao et al.90 for 10PDC films, which might be related to the fact that ceria does not form silicates (nucleating only at specific sites), but instead silica probably homogeneously covers large parts of the surface.
6 Conclusions
For an extended set of ceria samples doped with Gd and Pr, the equilibrium oxygen exchange rate measured by pulsed isotope exchange correlates clearly with the dopant concentration. Together with the p(O2) dependence, this can be interpreted for GDC with one oxygen vacancy being involved before or in the rate-determining step, and molecular oxygen species in the rds. For PDC an even stronger increase with dopant concentration is observed, accelerating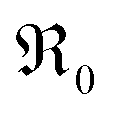 at 700 °C by five orders of magnitude for 20PDC compared to undoped CeO2. This is assigned to the formation of a Pr impurity band, which may facilitate electron transfer to adsorbed oxygen species and thus increase adsorbate coverage. Its closely related defect chemical behavior and very similar increase of
at 700 °C by five orders of magnitude for 20PDC compared to undoped CeO2. This is assigned to the formation of a Pr impurity band, which may facilitate electron transfer to adsorbed oxygen species and thus increase adsorbate coverage. Its closely related defect chemical behavior and very similar increase of 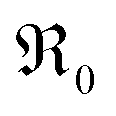 with Tb content indicate that the oxygen exchange reaction on Tb-doped ceria follows the same mechanism as for PDC. The observation that no 34O2 is formed on samples with a high Pr or Tb content might be related to enhanced oxygen diffusivity. The fact that co-doping with Nb decreases
with Tb content indicate that the oxygen exchange reaction on Tb-doped ceria follows the same mechanism as for PDC. The observation that no 34O2 is formed on samples with a high Pr or Tb content might be related to enhanced oxygen diffusivity. The fact that co-doping with Nb decreases 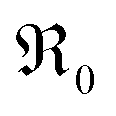 to the values of undoped CeO2 supports the interpretation that the effect of doping is to a significant part due to the role of
to the values of undoped CeO2 supports the interpretation that the effect of doping is to a significant part due to the role of 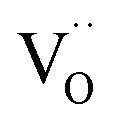 (with additional effects from electronic structure for Pr). The results impressively demonstrate the importance of point defects for surface reaction kinetics, with the highest rates achieved when
(with additional effects from electronic structure for Pr). The results impressively demonstrate the importance of point defects for surface reaction kinetics, with the highest rates achieved when 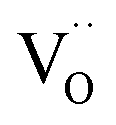 as well as redox-active centers are available. Furthermore, one has to keep in mind that surface reaction rates are much more sensitive to experimental details (e.g., surface poisoning or catalytically active impurities) than bulk properties. The present study demonstrates the importance to investigate an extended set of systematically doped samples by the same method in order to obtain clear correlations.
as well as redox-active centers are available. Furthermore, one has to keep in mind that surface reaction rates are much more sensitive to experimental details (e.g., surface poisoning or catalytically active impurities) than bulk properties. The present study demonstrates the importance to investigate an extended set of systematically doped samples by the same method in order to obtain clear correlations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank H. Hoier for performing XRD, K. Müller for XPS measurements, W. Sigle for TEM/EDX analysis, and A. Fuchs for SEM imaging.References
- Z. Gao, L. V. Mogni, E. C. Miller, J. G. Railsback and S. A. Barnett, Energy Environ. Sci., 2016, 9, 1602–1644 RSC.
- J. A. Kilner, Solid State Ionics, 2000, 129, 13–23 CrossRef CAS.
- J. Paier, C. Penschke and J. Sauer, Chem. Rev., 2013, 113, 3949–3985 CrossRef CAS PubMed.
- M. Melchionna and P. Fornasiero, Mater. Today, 2014, 17, 349–357 CrossRef CAS.
- R. J. Gorte, J. M. Vohs and S. McIntosh, Solid State Ionics, 2004, 175, 1–6 CrossRef CAS.
- A. Trovarelli, C. de Leitenburg, M. Boaro and G. Dolcetti, Catal. Today, 1999, 50, 353–367 CrossRef CAS.
- P. S. Manning, J. D. Sirman and J. A. Kilner, Solid State Ionics, 1996, 93, 125–132 CrossRef CAS.
- E. Ruiz-Trejo, J. D. Sirman, Y. M. Baikov and J. A. Kilner, Solid State Ionics, 1998, 113–115, 565–569 CrossRef CAS.
- J. A. Lane and J. A. Kilner, Solid State Ionics, 2000, 136, 927–932 CrossRef.
- J. Rutman, M. Kilo, S. Weber and I. Riess, Solid State Ionics, 2014, 265, 29–37 CrossRef CAS.
- K. Kowalski, Defect Diffus. Forum, 2009, 289–292, 769–774 CAS.
- C. Nicollet, A. Flura, V. Vibhu, A. Rougier, J.-M. Bassat and J.-C. Grenier, Int. J. Hydrogen Energy, 2016, 41, 15538–15544 CrossRef CAS.
- K. Yashiro, T. Suzuki, A. Kaimai, H. Matsumoto, Y. Nigara, T. Kawada, J. Mizusaki, J. Sfeir and J. Van herle, Solid State Ionics, 2004, 175, 341–344 CrossRef CAS.
- E. N. Armstrong, K. L. Duncan, D. J. Oh, J. F. Weaver and E. D. Wachsman, J. Electrochem. Soc., 2011, 158, B492–B499 CrossRef CAS.
- M. V. Anan'ev, E. K. Kurumchin, G. K. Vdovin and N. M. Bershitskaya, Russ. J. Electrochem., 2012, 48, 871–878 CrossRef.
- C.-Y. Yoo, PhD thesis, Universiteit Twente, Twente, The Netherlands, 2012.
- M. Balaguer, C.-Y. Yoo, H. J. M. Bouwmeester and J. M. Serra, J. Mater. Chem. A, 2013, 1, 10234–10242 RSC.
- D. Chen, S. R. Bishop and H. L. Tuller, J. Electroceram., 2012, 28, 62–69 CrossRef CAS.
- A. Karthikeyan and S. Ramanathan, Appl. Phys. Lett., 2008, 92, 243109 CrossRef.
- S. R. Bishop, J. J. Kim, N. Thompson and H. L. Tuller, in Ionic and Mixed Conducting Ceramics 8, ed. M. B. Mogensen, T. M. Gur, X. D. Zhou, T. Armstrong and H. Yokokawa, Electrochemical Soc Inc, Pennington, 2012, vol. 45, pp. 491–495 Search PubMed.
- J. J. Kim, S. R. Bishop, N. J. Thompson and H. L. Tuller, ECS Trans., 2013, 57, 1979–1984 CrossRef.
- M. Katsuki, S. Wang, K. Yasumoto and M. Dokiya, Solid State Ionics, 2002, 154–155, 589–595 CrossRef CAS.
- P. Simons, H. I. Ji, T. C. Davenport and S. M. Haile, J. Am. Ceram. Soc., 2017, 100, 1161–1171 CrossRef CAS.
- Y. Ma and J. D. Nicholas, ECS Trans., 2017, 78, 395–403 CrossRef CAS.
- Y. Ma and J. D. Nicholas, Phys. Chem. Chem. Phys., 2018, 20, 27350–27360 RSC.
- Y. M. Choi, H. Abernathy, H. T. Chen, M. C. Lin and M. L. Liu, ChemPhysChem, 2006, 7, 1957–1963 CrossRef CAS PubMed.
- M. Huang and S. Fabris, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 4 Search PubMed.
- H.-T. Chen, J.-G. Chang, H.-L. Chen and S.-P. Ju, J. Comput. Chem., 2009, 30, 2433–2442 CrossRef CAS PubMed.
- Z. Wu, M. Li, J. Howe, H. M. Meyer and S. H. Overbury, Langmuir, 2010, 26, 16595–16606 CrossRef CAS PubMed.
- P. J. Gellings and H. J. M. Bouwmeester, Catal. Today, 2000, 58, 1–53 CrossRef CAS.
- J. Maier, Chem.–Eur. J., 2001, 7, 4762–4770 CrossRef CAS.
- R. Merkle and J. Maier, Top. Catal., 2006, 38, 141–145 CrossRef CAS.
- E. W. McFarland and H. Metiu, Chem. Rev., 2013, 113, 4391–4427 CrossRef CAS PubMed.
- N. Nilius and H.-J. Freund, Acc. Chem. Res., 2015, 48, 1532–1539 CrossRef CAS PubMed.
- G. Simkovich and C. Wagner, J. Catal., 1962, 1, 521–525 CrossRef CAS.
- P. Murugaraj and J. Maier, Solid State Ionics, 1989, 32–33, 993–999 CrossRef CAS.
- H. J. M. Bouwmeester, C. Song, J. Zhu, J. Yi, M. v. S. Annaland and B. A. Boukamp, Phys. Chem. Chem. Phys., 2009, 11, 9640–9643 RSC.
- C.-Y. Yoo and H. J. M. Bouwmeester, Phys. Chem. Chem. Phys., 2012, 14, 11759–11765 RSC.
- C.-Y. Yoo, B. A. Boukamp and H. J. M. Bouwmeester, J. Solid State Electrochem., 2011, 15, 231–236 CrossRef CAS.
- I. Riess, Solid State Ionics, 2015, 280, 51–65 CrossRef CAS.
- C.-Y. Yoo, B. A. Boukamp and H. J. M. Bouwmeester, Solid State Ionics, 2014, 262, 668–671 CrossRef CAS.
- S. R. Bishop, T. S. Stefanik and H. L. Tuller, Phys. Chem. Chem. Phys., 2011, 13, 10165–10173 RSC.
- L. W. Tai and P. A. Lessing, J. Mater. Res., 1992, 7, 511–519 CrossRef CAS.
- L. W. Tai and P. A. Lessing, J. Mater. Res., 1992, 7, 502–510 CrossRef CAS.
- C. J. Powell and A. Jablonski, NIST electron effective-attenuation-length database, Version 1.3, SRD 82, National Institute of Standards and Technology, Gaithersburg, 2011 Search PubMed.
- S. Saher, S. Naqash, B. A. Boukamp, B. Hu, C. Xia and H. J. M. Bouwmeester, J. Mater. Chem. A, 2017, 5, 4991–4999 RSC.
- F. A. Kröger and H. J. Vink, in Solid State Physics, ed. F. Seitz and D. Turnbull, Academic Press, 1956, vol. 3, pp. 307–435 Search PubMed.
- H. L. Tuller and A. S. Nowick, J. Phys. Chem. Solids, 1977, 38, 859–867 CrossRef CAS.
- I. Riess, H. Janczikowski and J. Nölting, J. Appl. Phys., 1987, 61, 4931–4933 CrossRef CAS.
- B. Zachau-Christiansen, T. Jacobsen and S. Skaarup, Solid State Ionics, 1996, 86–88, 725–731 CrossRef CAS.
- S. Wang, H. Inaba, H. Tagawa and T. Hashimoto, J. Electrochem. Soc., 1997, 144, 4076–4080 CrossRef CAS.
- B. C. H. Steele, Solid State Ionics, 2000, 129, 95–110 CrossRef CAS.
- M. C. Göbel, G. Gregori and J. Maier, Solid State Ionics, 2012, 215, 45–51 CrossRef.
- I. K. Naik and T. Y. Tien, J. Electrochem. Soc., 1979, 126, 562–566 CrossRef CAS.
- T. Kolodiazhnyi, H. Sakurai, A. A. Belik and O. V. Gornostaeva, Acta Mater., 2016, 113, 116–123 CrossRef CAS.
- C. Chatzichristodoulou, P. V. Hendriksen and A. Hagen, J. Electrochem. Soc., 2010, 157, B299–B307 CrossRef CAS.
- S. R. Bishop, T. S. Stefanik and H. L. Tuller, J. Mater. Res., 2012, 27, 2009 CrossRef CAS.
- C. Chatzichristodoulou and P. V. Hendriksen, J. Electrochem. Soc., 2010, 157, B481–B489 CrossRef CAS.
- M. Nolan, S. Grigoleit, D. C. Sayle, S. C. Parker and G. W. Watson, Surf. Sci., 2005, 576, 217–229 CrossRef CAS.
- M. Nolan, J. E. Fearon and G. W. Watson, Solid State Ionics, 2006, 177, 3069–3074 CrossRef CAS.
- L. Yan, K. R. Balasubramaniam, S. Wang, H. Du and P. A. Salvador, Solid State Ionics, 2011, 194, 9–16 CrossRef CAS.
- L. Zhang, S. Wang, H. Huang, Y. Li, Y. Lu and C. Xia, J. Electrochem. Soc., 2017, 164, F610–F615 CrossRef CAS.
- O. Porat, H. L. Tuller, E. B. Lavik and Y. M. Chiang, in Nanophase and Nanocomposite Materials Ii, ed. S. Komarneni, J. C. Parker and H. J. Wollenberger, Materials Research Society, Warrendale, 1997, vol. 457, pp. 99–103 Search PubMed.
- P. Knauth and H. L. Tuller, J. Eur. Ceram. Soc., 1999, 19, 831–836 CrossRef CAS.
- S. Kim, R. Merkle and J. Maier, Surf. Sci., 2004, 549, 196–202 CrossRef CAS.
- C. B. Gopal, F. E. Gabaly, A. H. McDaniel and W. C. Chueh, Adv. Mater., 2016, 28, 4692–4697 CrossRef CAS PubMed.
- W. C. Chueh, A. H. McDaniel, M. E. Grass, Y. Hao, N. Jabeen, Z. Liu, S. M. Haile, K. F. McCarty, H. Bluhm and F. El Gabaly, Chem. Mater., 2012, 24, 1876–1882 CrossRef CAS.
- Z. A. Feng, F. El Gabaly, X. Ye, Z.-X. Shen and W. C. Chueh, Nat. Commun., 2014, 5, 4374 CrossRef CAS PubMed.
- Q. Lu, G. Vardar, M. Jansen, S. R. Bishop, I. Waluyo, H. L. Tuller and B. Yildiz, Chem. Mater., 2018, 30, 2600–2606 CrossRef CAS.
- G. Zhou, P. R. Shah, T. Montini, P. Fornasiero and R. J. Gorte, Surf. Sci., 2007, 601, 2512–2519 CrossRef CAS.
- C. Balaji Gopal, M. García-Melchor, S. C. Lee, Y. Shi, A. Shavorskiy, M. Monti, Z. Guan, R. Sinclair, H. Bluhm, A. Vojvodic and W. C. Chueh, Nat. Commun., 2017, 8, 15360 CrossRef CAS PubMed.
- J. Maier, Solid State Ionics, 2000, 135, 575–588 CrossRef CAS.
- J. Maier, Solid State Ionics, 1998, 112, 197–228 CrossRef CAS.
- L. Vegard, Z. Phys., 1921, 5, 17–26 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- N. Knoblauch, L. Dorrer, P. Fielitz, M. Schmucker and G. Borchardt, Phys. Chem. Chem. Phys., 2015, 17, 5849–5860 RSC.
- T. S. Zhang, J. Ma, H. Cheng and S. H. Chan, Mater. Res. Bull., 2006, 41, 563–568 CrossRef CAS.
- H. J. Avila-Paredes, K. Choi, C. T. Chen and S. Kim, J. Mater. Chem., 2009, 19, 4837–4842 RSC.
- E. N. Armstrong, K. L. Duncan and E. D. Wachsman, Phys. Chem. Chem. Phys., 2013, 15, 2298–2308 RSC.
- L. Wang, R. Merkle, Y. A. Mastrikov, E. A. Kotomin and J. Maier, J. Mater. Res., 2012, 27, 2000–2008 CrossRef CAS.
- M. M. Kuklja, E. A. Kotomin, R. Merkle, Y. A. Mastrikov and J. Maier, Phys. Chem. Chem. Phys., 2013, 15, 5443–5471 RSC.
- A. Rothschild, W. Menesklou, H. L. Tuller and E. Ivers-Tiffée, Chem. Mater., 2006, 18, 3651–3659 CrossRef CAS.
- V. Metlenko, W. Jung, S. R. Bishop, H. L. Tuller and R. A. De Souza, Phys. Chem. Chem. Phys., 2016, 18, 29495–29505 RSC.
- A. Schmid, G. M. Rupp and J. Fleig, Chem. Mater., 2018, 30, 4242–4252 CrossRef CAS PubMed.
- C. Yang, X. Yu, S. Heißler, P. G. Weidler, A. Nefedov, Y. Wang, C. Wöll, T. Kropp, J. Paier and J. Sauer, Angew. Chem., Int. Ed., 2017, 56, 16399–16404 CrossRef CAS PubMed.
- Y. A. Mastrikov, R. Merkle, E. A. Kotomin, M. M. Kuklja and J. Maier, J. Mater. Chem. A, 2018, 6, 11929–11940 RSC.
- Y. A. Mastrikov, R. Merkle, E. Heifets, E. A. Kotomin and J. Maier, J. Phys. Chem. C, 2010, 114, 3017–3027 CrossRef CAS.
- P. P. Dholabhai, J. B. Adams, P. A. Crozier and R. Sharma, J. Mater. Chem., 2011, 21, 18991–18997 RSC.
- E. Bucher, W. Sitte, F. Klauser and E. Bertel, Solid State Ionics, 2011, 191, 61–67 CrossRef CAS.
- L. Zhao, N. H. Perry, T. Daio, K. Sasaki and S. R. Bishop, Chem. Mater., 2015, 27, 3065–3070 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The supplementary contains details of PIE quantification, a discussion of the p(O2) dependence of surface defect concentrations, additional experimental data (lattice parameters, XRD pattern, SEM, EDX at grain boundaries, electrochemical impedance spectroscopy, isothermal isotope exchange, and exponential prefactors). See DOI: 10.1039/c9ta05908c |
| This journal is © The Royal Society of Chemistry 2019 |