
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImmobilization of Co, Mn, Ni and Fe oxide co-catalysts on TiO2 for photocatalytic water splitting reactions†
Jasmin S.
Schubert
,
Janko
Popovic
,
Greta M.
Haselmann
,
Sreejith P.
Nandan
,
Jia
Wang
,
Ariane
Giesriegl
,
Alexey S.
Cherevan
 * and
Dominik
Eder
*
* and
Dominik
Eder
*
Institute of Materials Chemistry, Technical University of Vienna, Getreidemarkt 9, Vienna 1060, Austria. E-mail: alexey.cherevan@tuwien.ac.at; dominik.eder@tuwien.ac.at
First published on 18th July 2019
Abstract
Here we report a systematic study of a series of non-noble-metal co-catalysts based on Co, Mn, Ni and Fe oxides that were prepared by wet impregnation of the corresponding acetylacetonate precursors onto a model TiO2 substrate, followed by their oxidative decomposition. We analyze thermal evolution of the impregnated M(acac)x–TiO2 composites with a combination of analytical methods and reveal strong differences in the precursor decomposition onsets and the resulting product composition, compared to the case of pure M(acac)x precursors. Consequent electron microscopy analyses of the resulting MOx–TiO2 composites indicate the presence of small (1–5 nm) amorphous MOx nanoparticles that are homogeneously distributed on the surface of the substrate TiO2. Complementing Raman and photoluminescence (PL) spectra confirm pronounced effects of MOx deposition on the state of TiO2 substrate and suggest strong electronic communication between the components. The composites obtained at 350 °C were further tested towards sacrificial hydrogen evolution reaction (HER) and oxygen evolution reaction (OER) demonstrating the dynamic nature of the NiOx–TiO2 photocatalyst whose Ni0 active HER sites were generated in situ upon light exposure. In contrast, FeOx–TiO2, CoOx–TiO2, and NiOx–TiO2 were all active towards OER, featuring water oxidation ability in descending order, while XPS data of the samples after reaction indicate that partial oxidation of M species takes place during the course of the photocatalytic experiment. This work provides detailed insights on the wet chemistry-based preparation of MOx co-catalysts decorating oxide nanopowders including optimization of the thermal treatment, potential substrate effects and synergy as well as further prospects in photocatalysis.
Introduction
As of now, renewable energy systems are not mature enough to cover the ever-increasing world energy demands. As a result, the main energy production still comes from non-clean and non-renewable sources, such as fossil fuels, which further contribute to global warming. In addition, the intermittent nature of many renewable energy sources – like in the case of photovoltaics, whose performance depends on the sunlight availability – implies a need for alternative energy storage systems.1 H2 has been identified as a promising candidate potentially able to become a base chemical as well as an energy storage system (with its high energy density of 120 MJ kg−1 and a non-intermittent supply)2–4 for our future economy. However, given the fact that the majority of industrial H2 production is currently still based on natural gas reforming,5,6 renewable methods of H2 generation require further development and implementation.A prodigious approach for clean H2 production is water splitting, however, in order to induce this reaction using electro- or thermal catalysis, an extensive amount of energy has to be invested.7 One approach to circumvent this issue relies directly on the renewable energy of sunlight by means of photocatalysis. In this case, the light photons are used to break the water molecules assisted by a photocatalyst, a substance that generates photoexcited charge carriers, delivers them to the solid–liquid interface and catalyzes the redox half-reaction of water oxidation and reduction, the latter yielding the desired H2 product.8–10 Although the hydrogen evolution reaction (HER) is given much more attention in the community of heterogeneous photocatalysts,11 it is the oxygen evolution reaction (OER) that – being kinetically far more complicated (4 electron–4 proton transfer) – often becomes a limiting step for the overall process.
A well-suited model compound for photocatalytic water splitting studies is TiO2, as it features high chemical stability, shows no toxicity, is cheap, widely available and has a suitable band gap for both half-reactions.8,9,12–14 However, TiO2 has several drawbacks that keep it from becoming an efficient water splitting photocatalyst.13,14 These limitations include fast electron–hole recombination rates, poor and unselective catalytic sites on its surface, and a wide band gap that only allows absorption in the UV light range. A number of strategies have been explored to address these issues, such as the use of co-catalysts.11,13 These surface-attached species (mostly nanoparticles) provide new catalytic sites and simultaneously induce separation of photoexcited electrons and holes.15 The most widely used co-catalysts are Pt, Pd and IrO2, RuO2 for the photocatalytic reduction and oxidation of water, respectively. These are expensive materials based on rare noble metals.11,16,17 To achieve large scale industrial application of photocatalytic water splitting the development of new co-catalysts based on cheap and widely available elements remains an important issue.
With regard to novel abundant co-catalysts for photocatalytic water splitting, research efforts have focused on d-block transition metals of the fourth period, which are known for their excellent catalytic properties and applications in industry, research and nature.7,11,14,17 These elements – especially in their oxide form – can undergo quick and reversible redox shuttling, accept, accumulate and release electrons – conditions necessary to generate a self-recovering system. Besides this, their surface structure and chemistry can be varied through synthetic conditions (e.g. different oxides can be generated) allowing to further tune adsorption/desorption properties and thus their catalytic function – an aspect rarely investigated so far. For this reason, in this work we explore the nature of Ni, Mn, Co and Fe oxide based co-catalysts and directly compare their potential in both water splitting reactions (OER and HER).
Metal oxide co-catalysts are typically deposited on the photocatalyst surface by sol–gel processes or via the use of surfactants.18–25 These methods allow for a certain degree of control over the size and shape of the co-catalytic species;19 however, they are often limited when one-pot synthesis is desired. Wet impregnation routes, on the other hand, are simple and cost-effective: for example, by using metal salt impregnation followed by thermal decomposition.26–29 Although some previous works have reported the synthesis and application of such co-catalyst-loaded TiO2 powders in photocatalysis,11,14,17,30–37 many inconsistencies, originating from the wide variety of synthetic and calcination conditions, remain. Furthermore, the lack of comparative activity evaluations leave a gap in the fundamental understanding of their actual active state. Besides these, most of the co-catalysts have been tested solely for HER, and no data on OER performance have been reported.
In order to complement this knowledge gap, in this work, we used a wet impregnation route to prepare Mn, Co, Fe and Ni-based co-catalysts immobilized on TiO2 nanoparticles (NPs) and systematically investigated their prospects in photocatalytic water splitting reactions. We first analyzed the thermal evolution of pure metal salt precursors and the corresponding composites using thermogravimetric analysis (TGA) and in situ X-ray diffraction (XRD) to determine the optimal calcination temperature and product composition. We then elucidated the morphology, crystallinity and oxidation state of the prepared co-catalyst species with a combination of electron microscopy and diffraction as well as XRD, X-ray photoelectron spectroscopy (XPS) and attenuated total reflection Fourier-transform infrared spectroscopy (ATR-FTIR) analyses and correlated these characteristics with their performance towards HER and OER.
Results and discussion
The metal oxide species were synthesized directly on the surface of TiO2 NPs following the wet impregnation – thermal decomposition method depicted in Fig. 1a. In a typical experiment, a given amount of TiO2 powder was dispersed in a solvent by ultrasonication, to which a pre-made solution of fully solubilized M(acac)x species (M = Fe, Mn, Co, Ni; acac = acetylacetonate) was slowly added. The solution was stirred until the solvent was completely evaporated, yielding the corresponding M(acac)x–TiO2 composites, which were converted into the respective MOx–TiO2 composites upon subsequent heat treatment. More details can be found in the Experimental section.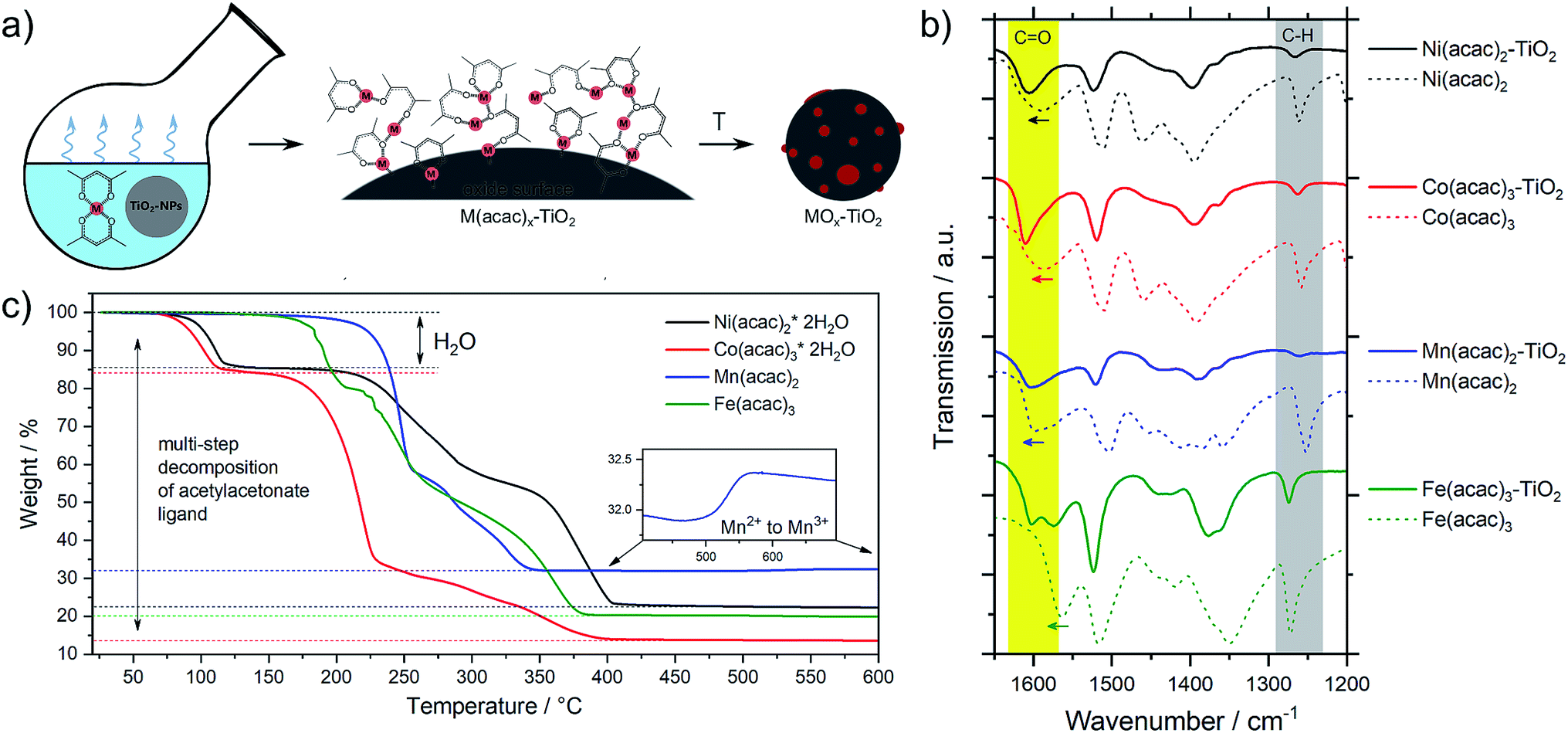 | ||
| Fig. 1 (a) Schematic description of the synthetic protocol, (b) FTIR spectra of the M(acac)x–TiO2 composites and the corresponding precursors and (c) TGA data of the pure M(acac)x precursor salts (heating rate: 5 °C min−1 in air; temperature range: 25 °C to 600 °C, M = Fe, Mn, Ni and Co). | ||
The presence of the M(acac)x salts in TiO2 powder after the impregnation can be confirmed by ATR-FTIR spectroscopy (Fig. S1†). The spectra of the composites constitute a superposition of the organic part (most pronounced are ligand-related IR bands) and the TiO2 substrate (broad absorption below 1000 cm−1), however, a closer look at the 1700–1000 cm−1 region (Fig. 1b) indicates that the original peaks of acetylacetonate species are shifted to higher wavenumbers, suggesting a strong binding (chemisorption) of the salt precursors with the substrate material. In addition, the formation of the composites is also apparent from the colour changes – brown, blue, green and red for the Mn, Co, Ni and Fe-based composites, respectively – of the originally white TiO2 powders observed after impregnation (Fig. S1†).
The composites have been further subjected to thermal treatment in ambient air to oxidatively decompose the metal precursors and yield the corresponding metal oxide co-catalysts on the TiO2 NPs. However, since there is no consensus in the literature as to which conditions generate which species,30,31,33,35,36,38–44 we systematically varied the calcination temperature and analysed thermal evolution of both, the pure M(acac)x precursors and the impregnated M(acac)x–TiO2 composites.
Thermal analysis
The TGA data were further complemented by in situ XRD using the same heating conditions. Fig. 2 shows that the thermal decompositions of all precursor salts coincide well with the formation of crystalline oxides (full data sets can be found in Fig. S2†).
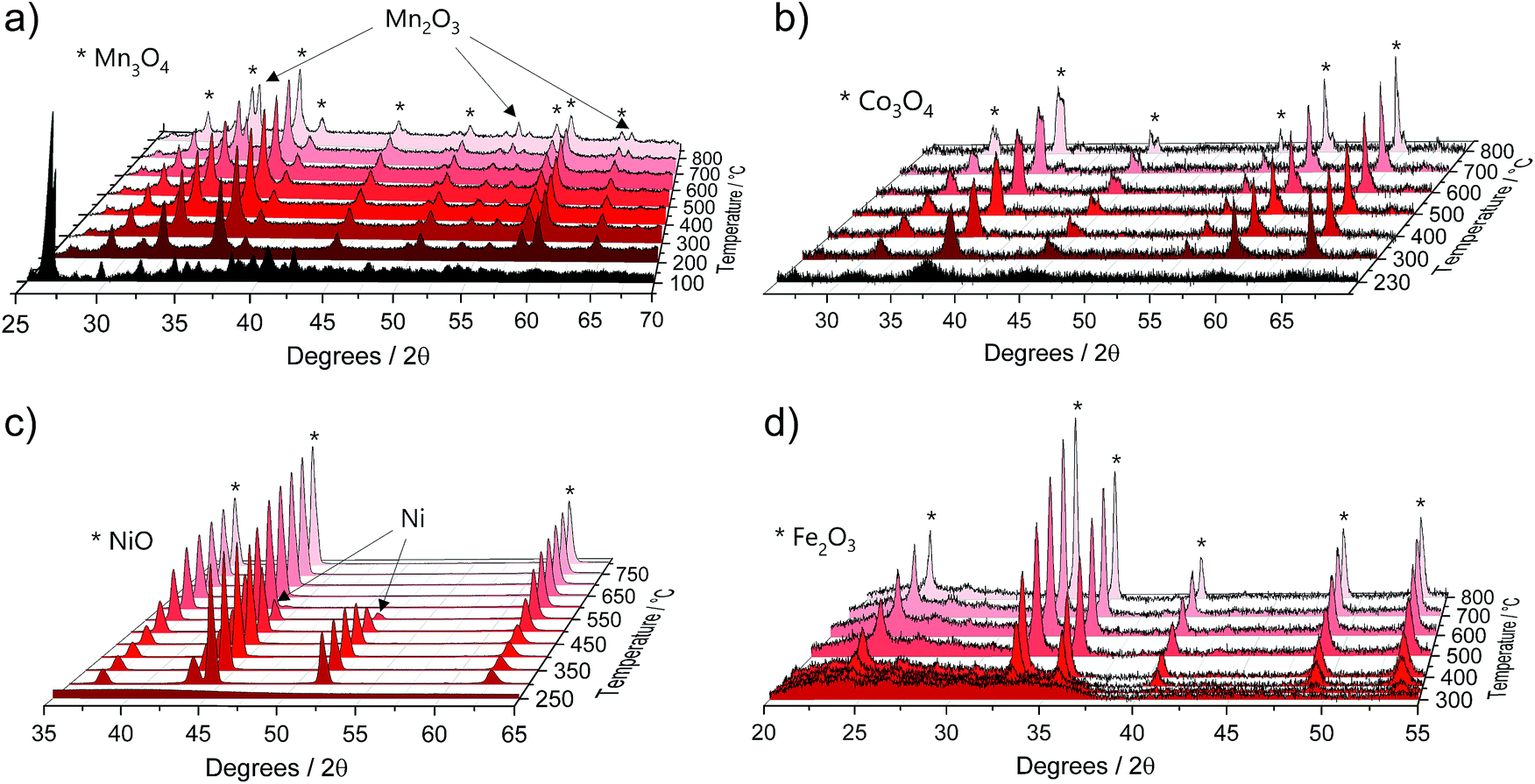 | ||
| Fig. 2 Selected in situ XRD spectra of the pure M(acac)x (M = Mn, Co, Ni and Fe) precursors acquired in air within a temperature range of 25 °C to 800 °C and heating rate of 5 °C min−1 for (a) Mn(acac)2, (b) Co(acac)3, (c) Ni(acac)2 and (d) Fe(acac)3. Full data sets can be found in Fig. S2.† | ||
Mn(acac)2. Fig. 2a shows the characteristic peaks of the original crystal structure of the precursor up to a temperature of about 150 °C, while at 200 °C all diffractions corresponding to the acetylacetonate have vanished, instead showing new diffractions corresponding to Mn3O4 (ICDD: 04-007-9641). Both, crystal phase and product composition remain unchanged up to temperatures of around 500 °C, above which the orthorhombic bixbyite Mn2O3 appears (ICDD: 04-007-0856), accompanied by corresponding Mn2+ oxidation. Both observations are in agreement with TGA (Fig. 1c). Interestingly, no apparent crystal growth is observed for the Mn3O4 particles upon calcination at high temperatures, as indicated by the absence of any peak narrowing. Instead, more and more of the Mn2+ is oxidized to Mn3+ and both oxide phases, Mn3O4 and Mn2O3, co-exist up to at least 800 °C.
Co(acac)3. The multi-step decomposition of acetylacetonate takes place between 200 and 400 °C according to TGA. The XRD results show that the original crystal structure of the salt degrades into an amorphous state already at 100 °C (see Fig. S2†) – temperature at which the removal of crystallization water takes place. At temperatures above 200 °C a broad diffraction at 36.8 degrees – corresponding to crystalline Co3O4 (ICDD: 04-014-7747) – appears and gets narrower up to 300 °C indicating gradual growth of the crystallites. The conversion of Co3+ species to the mixed-valent oxide is likely induced by the acetylacetonate species through radical formation in a similar way as has been suggested for Cu acetate.47 No apparent changes in crystal structure take place thereafter up to 800 °C; however, the splitting of the peaks at higher temperatures (>600 °C) may be related to the formation of non-stoichiometric oxides.
Ni(acac)2. The diffractograms in Fig. 2c indicate the formation of a mixed product consisting of metallic Ni (ICDD:04-010-6148) and NiO (ICDD:04-005-4393) in the temperature window of 220–400 °C, which is in agreement with the TGA in Fig. 1c. At temperatures above 250 °C Ni remains the major component up to 500 °C, after which the re-oxidized product starts to dominate the composition. This indicates that the presence of carbonaceous species in metal–organic salt precursors (e.g. acetylacetonate) can induce strong Ni2+ reduction to purely metallic species during the decomposition phase.47
Fe(acac)3. Fig. 2d shows that the decomposition proceeds in a way predicted by TGA. There is no reduction of the metal between 230 and 400 °C, while the single product of rhombohedral hematite Fe2O3 (ICDD: 04-002-7501) appears above 300 °C.
These results were further complemented by quasi in situ ATR-FTIR performed after subjecting the precursor salts to calcination at different temperatures. The spectra in Fig. S3† confirm that the decomposition of the original acetylacetonates and removal of the intermediate organic species for all composites finish at temperatures above 300 °C.
Interestingly, Fig. 3 not only shows large differences in the decomposition behavior (e.g. mechanism of acetylacetonate oxidation), it also indicates strong differences in the decomposition onsets and stability windows (up to 100 °C) for all precursors when impregnated on TiO2. For example, the Mn(acac)2 species do not undergo a two-step decomposition process within a narrow temperature interval anymore (Fig. 3a compared to Fig. 1c); instead we observe only one strong mass loss in the range between 200 and 250 °C. The final product of decomposition forms already above 250 °C, which is about 100 °C lower than for the pure precursor. In the case of the Co(acac)3–TiO2, the main weight loss takes place just below 300 °C and likely corresponds to the oxidation of acetylacetonate, which – in the case of the pure precursors – does not finish until 400 °C. Besides this, we see at least two additional minor weight losses at roughly 450 °C and 650 °C (see black arrows in Fig. 3b) for the composite with respect to the pure precursor salt.
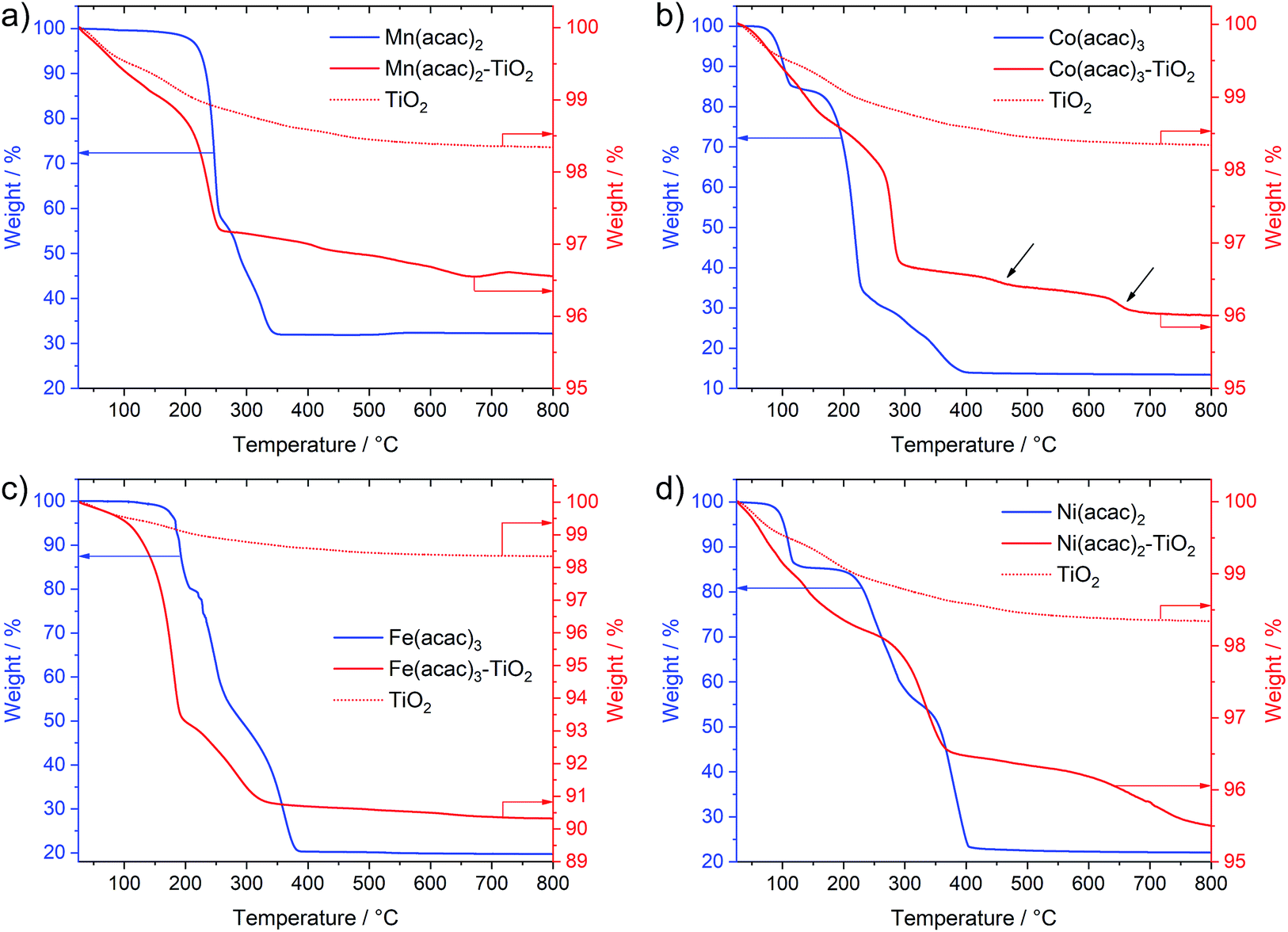 | ||
| Fig. 3 TGA data of the Mn(acac)2–TiO2 (a), Co(acac)3–TiO2 (b), Fe(acac)3–TiO2 (c) and Ni(acac)2–TiO2 (d) composites overlapped with that of the pure precursors and the TiO2 reference. The left y-axis corresponds to the weight loss of the pure precursors, the right y-axis corresponds to the weight loss of the composite and the reference TiO2. Data were recorded in air, heating rate 5 °C min−1 and in a temperature range from 25 °C to 800 °C. Note the differences in the scale of the y-axes. The minor weigh loss observed for the reference TiO2 in the range from 25 °C to 300 °C is attributed to the presence of physisorbed and chemisorbed species left from the synthesis and processing. | ||
Similar discrepancies can be observed for the other samples and indeed confirm different kinetics and stages for the decomposition of the immobilized M(acac)x precursors. More detailed investigations are required to gain full understanding for each individual composite. Our results further suggest that one needs to take care when applying heat treatment protocols, developed for the pure salts, unscrutinized to the corresponding nanocomposites.
After revealing thermal behavior of the impregnated M(acac)x–TiO2 composites, we have chosen calcination conditions for the thermal treatments aiming to fully decompose the acetylacetonate species, but avoid potential particle growth and possible crystallinity changes of the substrate TiO2. Considering differences between precursors, an optimum treatment of 350 °C that suited all M(acac)x–TiO2 samples was applied (see Experimental section) and yielded corresponding NiOx–TiO2, MnOx–TiO2, CoOx–TiO2 and FeOx–TiO2 nanocomposites that will be the focus of the following sections.
Morphological investigation
The calcined MOx–TiO2 composites were analysed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The SEM images in Fig. S4† confirm that the samples are composed of small nanoparticles (NPs) that are similar to the reference TiO2 (not shown), with no apparent changes in size and shape upon impregnation and calcination. There are also no signs of larger particles, thus we can exclude the formation and growth of unattached MOx. EDX further indicates a homogeneous distribution of the M elements over the TiO2 material on the microscale (Fig. S5†). Both observations suggest that no phase segregation upon calcination has occurred and the samples indeed are nanocomposites with M species dispersed across all TiO2 NPs.Fig. 4a and b show TEM image of the reference TiO2 NPs that range between 10 and 50 nm in size and constitute close-to-spherical highly crystalline particles with sharp edges. The electron diffraction (ED) pattern of the reference TiO2 shows diffuse rings that correspond well to polycrystalline, nanosized anatase and rutile particles (Fig. S6a†). In contrast, TEM images of the composites in Fig. 4 indicate strong changes in surface morphology of the substrate TiO2 and appearance of surface-attached species.
 | ||
| Fig. 4 TEM pictures of the TiO2 NPs (a and b) as well as MnOx–TiO2 (c), CoOx–TiO2 (d), FeOx–TiO2 (e) and NiOx–TiO2 (f) composites prepared at 350 °C. Arrows indicate examples of clearly visible MOx species. | ||
In the case of MnOx–TiO2 (Fig. 4c), the images clearly show the presence of dark spots with sizes between 2 and 5 nm, which likely correspond to the newly formed MnOx NPs. TEM of the CoOx–TiO2 composite in Fig. 4d indicates the presence of smaller 2–3 nm surface-attached CoOx NPs densely decoration the substrate. A similar morphology is found for NiOx–TiO2 (Fig. 4f) where the surface of TiO2 particles got covered with even smaller (1–2 nm) NiOx NPs of surprisingly uniform size and shape. For FeOx–TiO2 (Fig. 4e), the morphology of the deposits rather resembles a surface shell consisting of small particles sized below 5 nm.
Importantly, we can presently not exclude additional presence of atomic MOx species on TiO2 surface, but the majority of the visible MOx NPs appear to be uniform in size and homogeneously distributed on the surface sharing an intimate interface with the substrate.
Interaction with the matrix
To further elaborate on the state of the MOx in TiO2 matrix and on the differences between the composites, we have performed additional Raman and photoluminescence (PL) measurements.Fig. S7† shows Raman spectra of all as-prepared composites demonstrating that MnOx–TiO2, CoOx–TiO2 and FeOx–TiO2 samples feature strong peak shifts and peak broadening of the anatase-related Eg band with respect to the TiO2 reference (Table S1†). With regard to our composite systems, these can be assigned to the presence of surface-adsorbed species on TiO2, creation of O vacancies or even doping with M ions, which cannot be excluded based on ionic radii comparison. In contrast, Raman spectrum of the NiOx–TiO2 sample resembles that of the TiO2 suggesting no strong effect on TiO2 upon NiOx immobilization (detailed discussions of Raman data can be found in ESI†).
PL emission spectrum of the substrate TiO2 powder in Fig. S8† exhibits strong overlapping bands centred at 417, 432 and 461 nm. While the first peak (2.97 eV) can be assigned to band-to-band recombination of rutile component, the latter two (2.87 eV and 2.69 eV) can be attributed to sub-band gap emission characteristic for TiO2 NPs and related to shallow electronic states associated with e.g. oxygen vacancies, structural defects or dopants; in line with Raman results. Spectra of the composites still contain PL peaks of TiO2, however, they also feature new bands appearing at higher wavelength and characterized by broad emissions centred at 478 nm, 490 nm and 497 nm for the FeOx–TiO2, CoOx–TiO2 and NiOx–TiO2 respectively. The emergence of the additional emission bands at lower energies (2.60 eV, 2.53 eV and 2.49 eV) for all composites can be assigned to the appearance of new relaxation pathways for the carriers originally photoexcited in TiO2. Judging from TEM and Raman data, one such possibility could be that the electrons or holes are extracted by the surface-attached MOx species leading to new PL bands related to radiative recombination at MOx sites or the interface.
This result confirms active electronic communication between the components of the composites and further indicates the ability of the MOx species to facilitate charge separation in TiO2.
Crystallinity of the obtained MOx NPs
Further analysis of the MOx state and composition with HRTEM has proven to be complicated since the deposited NPs were mostly – with some rare exceptions found in TEM – of amorphous nature. This was further confirmed from the analysis of ED patterns (Fig. S6†) that indicate no additional diffraction spots corresponding to the expected MOx NPs.To elucidate more on the amorphous nature of the surface-deposited species in our MOx–TiO2 composites, we performed additional XRD measurements (see Fig. S9†). The data in Fig. S9a† shows no diffractions other than that of the TiO2 substrate for all calcined samples; however, this can also be explained by the low amounts of the MOx species, thus reaching the detection limit of the XRD technique. To further investigate this, we prepared a set of model samples where the impregnation was done using a much greater amounts of the precursor salts (up to 24.4 wt%) and diffractograms were recorded for the model composites before and after thermal treatment. Still, the XRD did not reveal any peak associated with newly formed oxide species even after calcination (Fig. S9b and c†), thus confirming the TEM and ED data and the conclusion that the MOx species in the final MOx–TiO2 composites are of amorphous nature regardless of the calcination protocol (see ESI for more details†). This amorphous nature of the generated NPs is surprising, because XRD of the pure precursors clearly indicated that the products of the acetylacetonate decomposition, obtained at this calcination temperature, were crystalline (see again Fig. 2). This different behaviour of the composites against the pure precursor is possibly related to surface effects, where TiO2 restricts the mobility of the surface-bound metallic species and thus prohibits bond rearrangement and formation of a crystalline lattice.
Confirming the oxidation state
Considering the strong differences in the thermal decomposition of the pure precursors and precursors immobilized on the surface of TiO2, we sought to verify the oxidation state and composition of the surface-deposited species (so far only suggested from in situ XRD of pure precursor salts) after thermal treatments using surface-sensitive XPS analyses. Measuring conditions and information about data treatment can be found in the Experimental section.Survey spectra of all composites are presented in Fig. S10† and reveal the presence of expected elements: Ti and O (mainly from the TiO2 substrate) as well as Co, Ni, Mn and Fe in the relevant composites. The characteristic C 1s signals correspond very well to adventitious carbon observed in the TiO2 reference indicating complete decomposition of the acetylacetonate ligand after the applied calcination.48
More importantly, Fig. 5 presents the detailed spectra of the Co 2p, Ni 2p, Mn 2p and Fe 2p regions and allows elucidating the oxidation state of the incorporated metals on the surface of TiO2. XPS analyses of these transition metals is quite complex due to multiplet splitting and different shake-up structures. Thus, the signal of a species can be deconvoluted into various different components that have been analysed in detail previously. This also means that the analysis of the oxidation state of these transition metals is not straight-forward and in some cases can only be seen as indicative. Our analysis is largely based on the work by Biesinger et al.49 Detailed fitting parameters can be found in Table S2.†
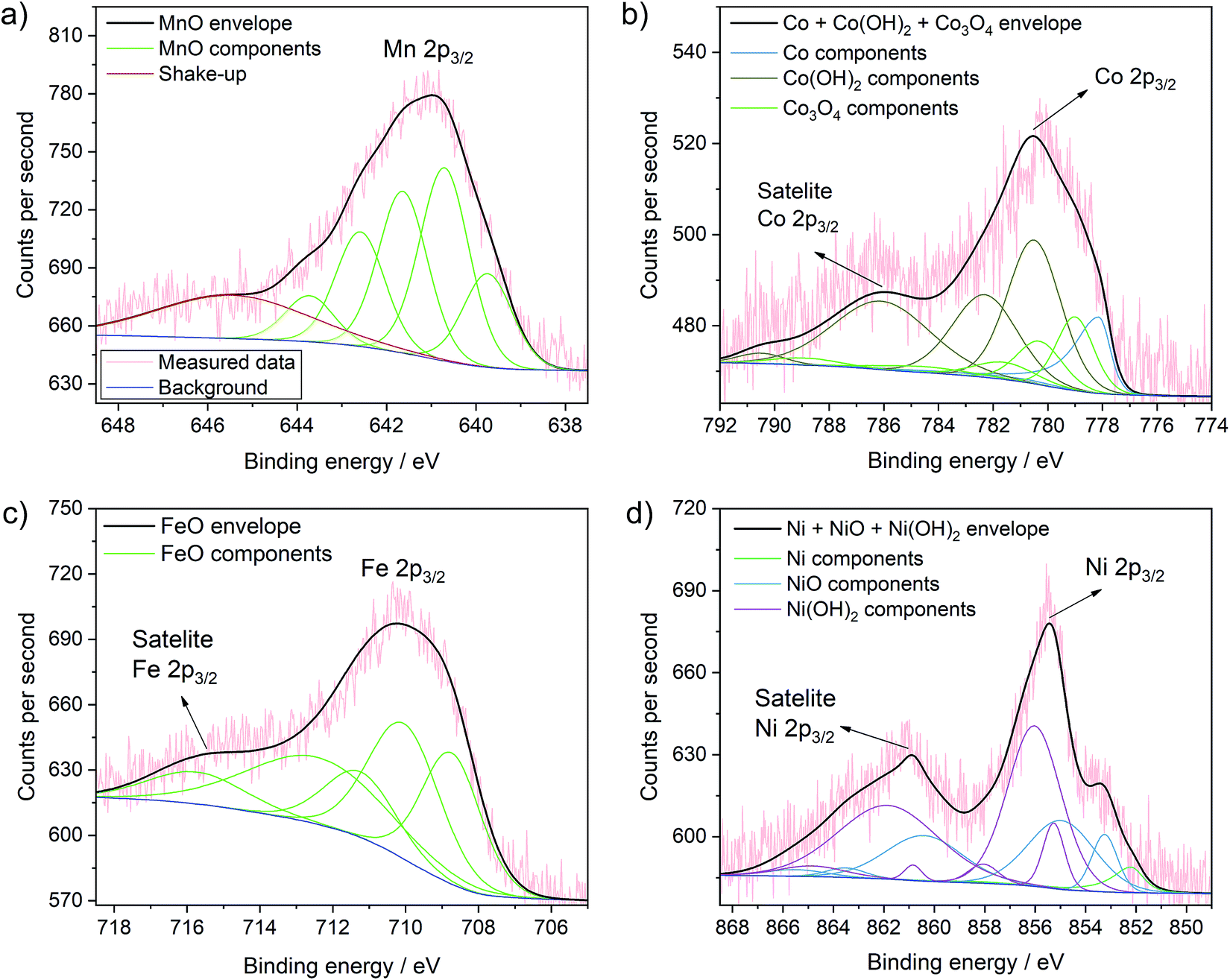 | ||
| Fig. 5 Detailed XPS spectra of the MnOx–TiO2 (a), CoOx–TiO2 (b), FeOx–TiO2 (c) and NiOx–TiO2 (d) samples with the corresponding fits of the different species and mixture of species. Further fits are shown in Fig. S11.† | ||
The Mn 2p spectrum most reliably confirms the presence of Mn2+ in the MnOx–TiO2 composite (Fig. 5a).49 We can further clearly exclude the presence of Mn0 species. Additionally fitting of Mn3+ – either from Mn2O3 as suggested by XRD, residual precursor salts or due to a partial hydroxylation, i.e. MnOOH – result in only a very low percentage (∼2%) of the overall signal, which indicates that the concentration of Mn3+ – if present at all – is negligible (Fig. S11a and b†). Furthermore, considering multiplet splitting, a Mn4+ species is not needed for peak deconvolution.49 Therefore, we can exclude the presence of Mn4+, in contrast to previous reports on Mn-doped TiO2.50,51 In conclusion, Mn2+ is the major component in the MnOx–TiO2 composite with a negligible contribution of Mn3+ due to Mn2O3 or MnOOH.
The analysis of the Co 2p signal in Fig. 5b reveals that Co2+ and Co3+ species coexist in the CoOx–TiO2 composite, most likely in the form of oxides, mixed oxide or hydroxides. However, in contradiction to the pure salt XRD and TGA data, we cannot reliably exclude the formation of metallic Co species. The best fit was obtained for a combination of Co metal, Co(OH)2, and Co3O4.
In the case of FeOx–TiO2 sample, shown in Fig. 5c, the Fe 2p spectrum clearly indicates the presence of Fe2+ with a characteristic satellite at 714 eV.49 From these data, we can suggest that Fe3+ is only present as minority species, as we cannot observe pronounced characteristic satellite features at 719 and 735 eV. This supports our previous findings that the thermal evolution of Fe(acac)3 is strongly altered by the TiO2 substrate resulting in the formation of partially reduced FeIIO.
Finally, the XPS data for NiOx–TiO2 (Fig. 5d) show mainly Ni2+ in the form of NiO or Ni(OH)2. Although not required (Fig. S11c†), the addition of a Ni0 component improves the fit further and indicates the possible presence of a small fraction of metallic Ni in the final composition, which would also be expected from the data on the pure precursors.49,52
In summary, we have produced NiOx–TiO2, MnOx–TiO2, CoOx–TiO2 and FeOx–TiO2 composite samples with homogeneously distributed nanoparticles of uniform sizes (2–5 nm) as revealed by electron microscopy and EDX. The results further suggest that the MOx NPs are of amorphous nature, in contrast to the data on pure precursors, thus indicating substrate effects. XPS analyses shows that Co likely coexists in a mixture of Co2+ and Co3+, while major contributors of Mn and Fe species are of +2 oxidation state, again contrary to what has been observed for pure precursors. In the case of NiOx–TiO2, XPS additionally indicates a small proportion of metallic species, aside from Ni2+.
Photocatalytic water splitting
We investigated the performance of all samples for photocatalytic water splitting, namely, hydrogen evolution reaction (HER) and oxygen evolution reaction (OER), by means of sacrificial water splitting. To compare with literature, we used methanol as an electron donor to facilitate HER53 and AgNO3 as an electron acceptor to support OER.54,55 Detailed description of the experimental procedure can be found in Experimental section.Hydrogen evolution reaction
Fig. 6 shows H2 evolution profiles (on-line H2 evolution rate vs. time) of the as-prepared NiOx–TiO2, MnOx–TiO2, FeOx–TiO2 and CoOx–TiO2 composites from water–methanol mixtures under UV light. The results show that only NiOx–TiO2 was active towards HER. Hydrogen evolution rates of about 4 μmol h−1 were reached after 10 minutes of light exposure in our flow reactor (Fig. S12†). The rest of the composites showed no activity indicating incapability of the generated MnOx, FeOx and CoOx species to act as co-catalysts for H+ reduction, which is in stark contradiction to some of the literature reports.34,41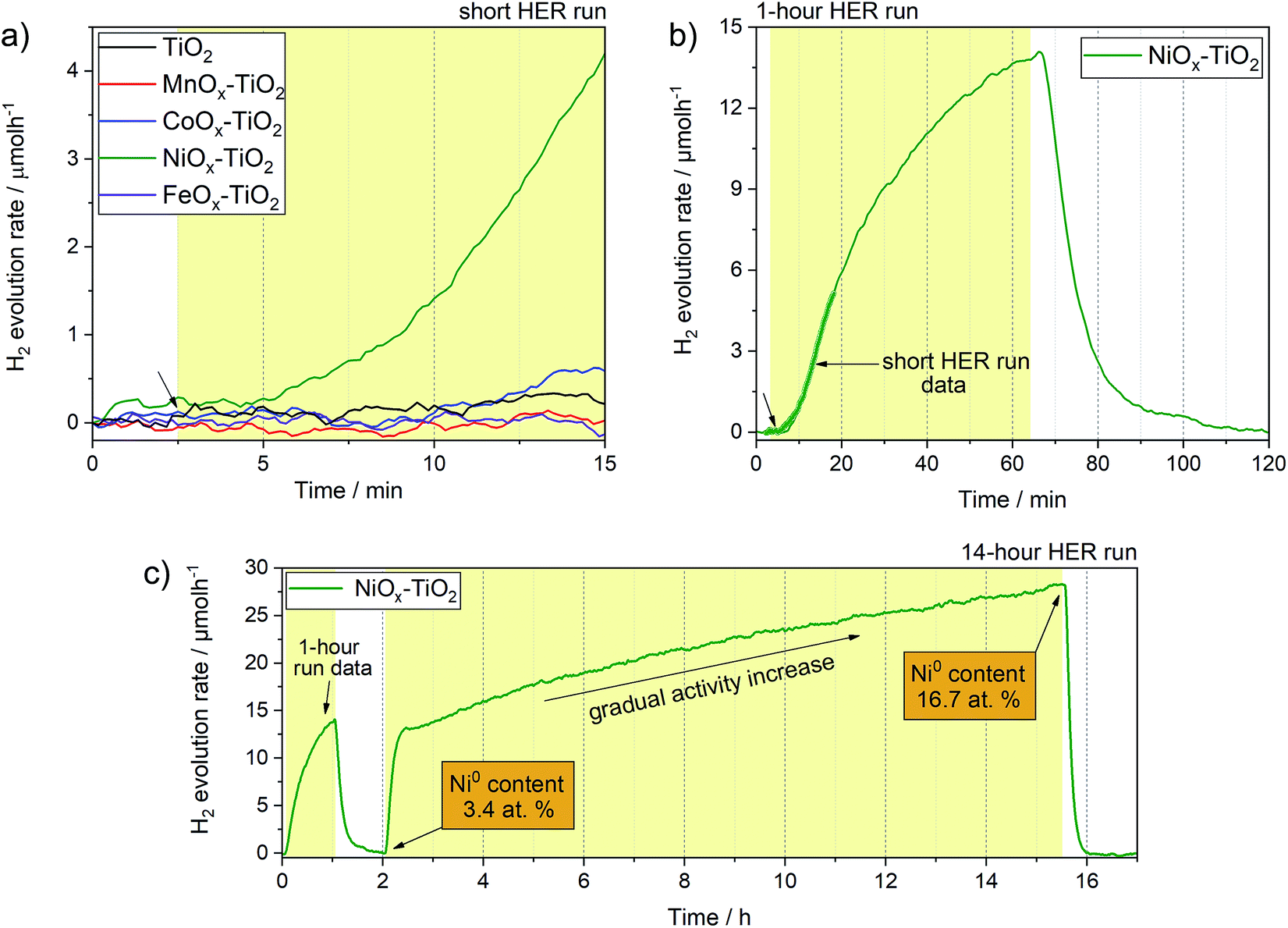 | ||
| Fig. 6 (a) Short run HER profiles of the NiOx–TiO2, MnOx–TiO2, FeOx–TiO2 and CoOx–TiO2 composites along with the reference TiO2 powder performed using flow-reactor. Arrow indicates the start of illumination, yellow box – illumination period. (b) 1 hour run HER profile of the active NiOx–TiO2 photocatalyst along with the corresponding short run data as well as (c) 14 hour run (long run) HER profile showing that the H2 production rate slowly increases with increasing illumination time. The 1 hour and 14 hour runs have been performed during the same experiment. The reactor was purged with Ar for 1 h in between the runs to remove the reaction products. | ||
We have further investigated the active NiOx–TiO2 system in long run HER experiments to identify the maximum activity and investigate the performance stability. Fig. 6b shows a HER profile over 60 minutes under illumination: interestingly, no stable evolution rate was reached within this period; instead, the H2 evolution rate continued to increase, reaching more than 13 μmol h−1 at the end of the illumination cycle. We purged the reactor with Ar to remove the reaction products and proceeded with UV illumination for another 14 h. Fig. 6c shows the resulting HER profile revealing two important observations: (a) the activity increased rapidly to a level of 13 μmol h−1 within the first hour of illumination, (b) the increase in activity continued in an almost linear fashion to reach rates as high as 27 μmol h−1 after 10 h.
This unusual behaviour indicates that either the catalytic sites of the NiOx–TiO2 system become more active or their number increases with time. Furthermore, it appears that this behaviour is triggered by light illumination. Although the Ni/NiO system has been investigated as co-catalyst for HER before,7,11,14,17,56 the question as to which Ni oxidation state is more active remains controversial. Our experimental setup is unique in that it allows for on-line activity detection within the first seconds of light illumination, and our data suggest that the increase in metallic Ni, which can gradually form from Ni2+ upon photoreduction, is likely the reason for the observed activity increase.57 This was confirmed by XPS measurements of the NiOx–TiO2 composite recovered immediately after the HER experiment, which revealed a substantial increase in the relative content of Ni0 compared to Ni2+ from 3.4 at% to 16.7 at% at the end of the illumination cycle (Table S3†). To the best of our knowledge, this has not been reported before. The results highlight the dynamics nature of this photocatalyst, and we emphasize that active species can form upon illumination not only during the first stages of a photocatalytic reaction, but even during extended periods of hours and days.
Based on our TGA, in situ XRD and XPS data, we further suggest that NiOx–TiO2 composites prepared at lower calcination temperatures (<350 °C) can offer a greater potential for photocatalytic HER as they preserve a higher fraction of metallic Ni species.
Oxygen evolution reaction
We have further tested the composites towards OER previously rarely reported for these series of co-catalysts. Again, we followed the typical conditions for sacrificial OER and used AgNO3 as an electron acceptor as well as a closed reactor system (see Experimental section and Fig. S12†). Fig. 7a reveals that three of the composite photocatalysts were active towards water oxidation, namely, FeOx–TiO2, CoOx–TiO2 and NiOx–TiO2, with the corresponding O2 evolution rates reaching 34.3, 31.4 and 19.9 μmol h−1 after 20 min of illumination, respectively (see Fig. 7b). Noteworthy, pure TiO2 powder without being loaded with any co-catalyst also exhibited respectable OER performance with the rate of 20.8 μmol h−1, suggesting that surface of bare TiO2 – with Ti4+ in d0 type configuration – also provides suitable sites for water oxidation.58 In contrast to other samples, the OER on TiO2 was completely suppressed in the case of the MnOx–TiO2 composite.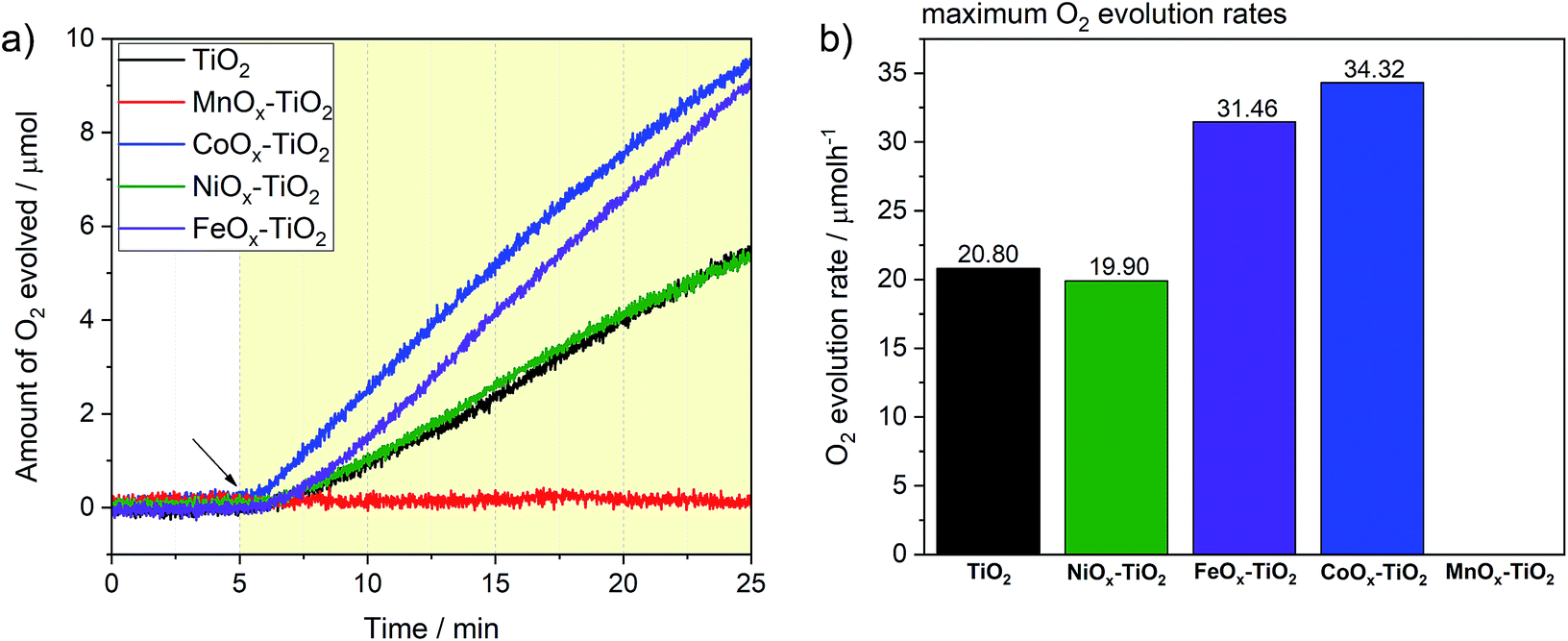 | ||
| Fig. 7 (a) OER evolution profiles for the NiOx–TiO2, MnOx–TiO2, FeOx–TiO2 and CoOx–TiO2 composites along with the reference TiO2 powder performed using a closed reactor. The arrow indicates start of illumination. The coloured area corresponds to the illumination period. (b) Bar chart presenting maxima of O2 evolution rates extracted from derivative analysis. | ||
Enhanced OER rates recorded in the presence of surface-attached CoOx and FeOx species can be ascribed to their active role in facilitating the charge separation and/or acting as water oxidation sites; in line with other literature reports where Co- and Fe-oxide-based co-catalysts have been shown to possess some photo- or electrocatalytic activity towards water oxidation.11,59–63 To further elaborate on the active state and stability of these OER-active co-catalysts, we performed XPS measurements after the OER experiments (see ESI†). Fig. S13a† clearly suggests that some of the initial Fe2+ species of the FeOx–TiO2 composite oxidized during the photocatalytic process resulting in a higher proportion of Fe3+, as can be seen from the appearance of the characteristic satellites at 719 and 737 eV. Nevertheless, judging from the stable rate of O2 evolution in Fig. 7a, we can suggest that no severe activation/deactivation process has taken place during the reaction and Fe2+ can still be considered as one of the OER-active components of the composite. With regard to CoOx–TiO2, XPS data of the sample after OER (Fig. S13b†) show that oxidation of the initially present Co species takes place during the photocatalytic reaction without any apparent activity loss. This suggests that high-oxidation state Co species are likely to be responsible for the OER performance. Elucidation of this question will require further dedicated investigations.
In the case of the NiOx–TiO2 composite, we observed no apparent increase in OER rate compared to bare TiO2, despite the fact that NiO – that is present in our composite – and various Ni oxyhydroxides are known for their excellent water oxidation performance.64,65 However, it has also been reported, that performance of Ni-based co-catalysts is strongly dependent on the calcination conditions and may need to be optimized to obtain desired OER rates. Nevertheless, the as-prepared NiOx–TiO2 composite containing both metallic Ni and NiO can be considered as a promising candidate for overall water splitting as it contains both HER and OER sites whose initial ratio can be controlled during the synthesis.
The rather negative result obtained for the MnOx–TiO2 case (Fig. 7b) can be related to well-known sensitivity of Mn-based oxides OER performance to their structure and Mn oxidation state.66 Our XPS data suggest that the MnOx co-catalyst NPs are mainly composed of Mn2+, while a number of literature reports suggest that the presence of Mn3+ and Mn4+ is required for the accumulation of the required oxidizing equivalents necessary to drive water oxidation.67,68 Judging from our TGA and in situ XRD data, we could suggest that much higher calcination temperatures (>600 °C) will be required to produce MnOx species with higher content of Mn in higher oxidation states, that could be of interest for photocatalytic OER.
Conclusions
In this contribution, we systematically studied a series of NiOx–TiO2, MnOx–TiO2, CoOx–TiO2 and FeOx–TiO2 photocatalysts prepared by wet impregnation of M(acac)x salts (M = Ni, Fe, Mn and Co) onto high surface area TiO2 NP substrate, followed by their oxidative decomposition. A combination of TGA, in situ XRD, ATR-FTIR as well as XPS revealed strong differences in thermal decomposition of the M(acac)x–TiO2 in comparison to pure M(acac)x salts. When loaded on TiO2, the M precursors undergo decomposition at substantially lower temperatures, via different mechanism and often result in the formation of otherwise unstable M species, such as Fe2+ in the case of FeOx–TiO2. Consequent SEM and TEM analyses along with EDX mappings showed that the morphology of the created MOx–TiO2 composites feature small (1–5 nm) MOx NPs homogeneously decorating the TiO2 NPs. In addition, XRD and ED suggested that the MOx co-catalysts prepared at 350 °C are of amorphous nature, while combined Raman and PL spectra indicate the possibility of M doping and strong electronic communication between the components of the composites.We further evaluated the potential of the composites towards photocatalytic water splitting reactions. We show that among all samples, NiOx–TiO2 was active towards hydrogen evolution reaction (HER) with metallic Ni species – gradually generated in situ upon light illumination – being the active site. In contrast, FeOx–TiO2, CoOx–TiO2, and NiOx–TiO2 were all active towards oxygen evolution reaction (OER), featuring water oxidation ability in descending order. XPS data of the OER-active samples after reaction indicated that mild oxidation of M species takes place during the course of the photocatalytic experiment. However, Fe2+ species could still be considered as one of the OER-active components of the FeOx–TiO2 composite.
This contribution provides a systematic study on the specifics of the wet chemistry-based synthesis of small MOx NPs directly onto the surface of TiO2, which can be expanded to other inorganic substrates. Besides, we show that some of the prepared catalysts are of interest for photocatalytic water splitting reactions and that their activity could be further controlled by modifying synthetic conditions.
Experimental section
Chemicals
All materials used for the syntheses were obtained from commercial suppliers. As such, P25 TiO2 from Degussa, Mn(acac)2 from Sigma-Aldrich (99.8% pure), Co(acac)3·3H2O from Fluka (99.9% pure), Ni(acac)2·2H2O from Sigma-Aldrich (99.8% pure) and Fe(acac)3 from Sigma-Aldrich (97% pure). The used solvent for the syntheses were deionized water and absolute ethanol (from Chem-Lab NV) and for photocatalytic experiments deionized water and HPLC-gradient grade methanol (from VWR).Synthesis of the composites
The metal oxide species were synthesized directly on the surface of TiO2 NPs following the wet impregnation – thermal decomposition method depicted in Fig. 1a. The general synthesis procedure used for all samples included (a) suspending the TiO2 powder (400 mg, 5 mmol) in 20 mL of ethanol; (b) stirring the suspension for 10 minutes; (c) adding the corresponding precursor salt: Mn(acac)2 (0.248 mmol, 62.7 mg), Co(acac)3 (0.297 mmol, 75.6 mg), Ni(acac)2 (0.113 mmol, 29.1 mg), or Fe(acac)3 (0.26 mmol, 92.0 mg); and (d) subjecting the resulting suspension to sonication to assist salt dissolution and homogenisation of the suspension components. The resulting mixture was left stirring at 60 °C until complete evaporation of the solvent. The remaining powder was then dried at 80 °C for 5 h and grinded. Afterwards, the samples were heat-treated at 350 °C for 10 h in ambient air to generate the corresponding oxides resulting in MOx–TiO2 composites.Characterisation methods
Scanning electron microscopy (SEM) images were acquired using FEI Quanta 250 FEG at 200 keV scanning electron microscope to obtain visual information on the morphology of the samples. Typically acceleration voltage of 2 kV and secondary electron detection mode were used. Energy dispersive X-ray spectroscopy (EDX) was performed using the SEM to obtain elemental maps.Transmission electron microscopy (TEM) images were obtained using FEI TECNAI F20 transmission electron microscope equipped with a field emission gun in bright field mode using 200 kV acceleration voltage. The sample was prepared from a suspension in ethanol without ultrasonication, using a copper holey carbon coated grids (Plano, 200 mesh).
X-ray diffraction (XRD) was performed using an XPERT III: PANalytical XPert Pro MPD (Θ–Θ Diffractometer) for the in situ experiments and an XPERT II: PANalytical XPert Pro MPD (Θ–Θ Diffractometer) for the ex situ experiments. The sample was placed on a sample holder and irradiated with a Cu X-ray source (8.04 keV, 1.5406 Å). The signal was acquired with Bragg–Brentano Θ/Θ-diffractometer geometry ranging from 5° to 80° degrees. The detector system was a semiconductor X'Celerator (2.1°) detector. The in situ experiment was performed under air flow and temperatures ranging from 25 °C to 800 °C.
The thermogravimetric (TGA) measurements were carried out on a PerkinElmer Thermogravimetric Analyser TGA 8000. The samples were placed into an Al2O3 crucible and heated with a dynamic method at a heating rate of 5 °C min−1 under air from 25 °C to 800 °C.
The chemical composition of the samples was obtained with X-ray photoelectron spectroscopy (XPS) using a custom-built SPECS XPS-spectrometer equipped with a monochromatised Al-Kα X-ray source (μ 350) and a hemispherical WAL-150 analyser (acceptance angle: 60°). All samples were mounted onto the sample holder using double-sided carbon tape. Pass energies of 100 eV and 30 eV and energy resolutions of 1 eV and 100 meV were used for survey and detail spectra, respectively (excitation energy: 1486.6 eV, beam energy and spot size: 70 W onto 400 μm, angle: 51° to sample surface normal, base pressure: 5 × 10−10 mbar, pressure during measurements: 2 × 10−9 mbar). To reduce charging effects, a broad-spot low energy electron source (SPECS FG 22 flood gun, 8–12 eV/15–30 μA) was used for charge compensation in some samples (containing Mn and Fe). Data analysis was performed using CASA XPS software, employing transmission corrections (as per the instrument vendor's specifications), Shirley backgrounds and Scofield sensitivity factors. Charge correction was applied so the adventitious carbon peak (C–C peak) was shifted to 284.8 eV binding energy (BE). All content values shown are in units of relative atomic percent (at%), where the detection limit in survey measurements usually lies around 0.1–1 at%, depending on the element. For the attenuated total reflection Fourier-transform infrared spectroscopy (ATR-FTIR) measurements a PerkinElmer FTIR Spectral UATR-TWO with a spectrum two Universal ATR (Single Reflection Diamond) accessory was used.
Fluorescence steady state measurements of the TiO2 and MOx–TiO2 NPs were carried out using PicoQuant FluoTime 300 spectrophotometer. For PL spectra, the excitation source was Coaxial UV-Xe arc lamp (ozone free – with 300 W power) coupled with a computer-controlled double-grating monochromator and the detection system comprised of PMA Hybrid 07 detector and a high resolution emission double monochromator. For all the measurements, the excitation wavelength was kept to be the 377 nm (corresponding to 3.29 eV photon energy), and the PL data was collected using the EasyTau2 software.
Raman measurements were conducted with LabRAM HR800 from Horiba. Ne:YAG diode was used as the 532 nm laser source and the characteristic Raman peak of Si at 520.8 cm−1 was used as the calibration peak. The laser intensity was keep at 5 mW.
Photocatalytic experiments
Hydrogen evolution experiments were performed using a top irradiation gas-flow slurry type home-made reactor equipped with a LED lamp light source centred at 365 nm (Thorlabs). In a single experiment, 10 mg of a powdered photocatalyst was dispersed in 50 mL 50 vol% MeOH–water solution by stirring. During the experiment, the reactor was continuously purged with argon (flow rate of 30 mL min−1, controlled with a mass flow controller from MCC-Instruments) to deliver the gaseous products to the online gas analyzer (X-Stream, Emerson Process Management) equipped with a thermal conductivity detector (TCD) for H2 quantification. The temperature of the reactor was kept constant through a water cooling system (Lauda). In a single experiment, the suspension was first stirred for 30 min in the dark, then illuminated for 15 min. A typical H2 evolution profile (e.g. in Fig. 6) obtained with our flow reactor includes an “induction” period (increasing H2 evolution rate during the first 5–10 min) that is due to the fact the H2 gas first needs to fill the dead volume (e.g. reactor volume, tubing volume) to reach the detector. When the illumination is stopped, the signal returns to its baseline. The H2 evolution rates were normalized by subtracting the H2 evolution rate measured in the blank experiment (no catalyst present in the MeOH–H2O mixture) as a result of UV-assisted MeOH oxidation (photo-reforming).Oxygen evolution experiments were performed in a home-made top illumination closed reactor using the same light source and water cooling system. The experiments were carried out using a 2 mL water solution containing 1 mg of a photocatalyst (dispersed via stirring) and AgNO3 (0.1 M) as electron scavenger. The reactor was first purged with Ar to remove the air and the O2 detection was started using a fibre-optic oxygen sensor (PyroScience) inserted in the reaction volume. The suspension was stirred for 30 min in the dark to allow for O2 signal stabilization followed by 20 minutes of UV illumination.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge facilities of the Vienna University of Technology for technical support and fruitful discussions: X-Ray Center (XRC) and especially Werner Artner and Klaudia Hradil; Analytical Instrumentation Center (AIC) and especially Markus Sauer and Annette Foelske-Schmitz; Electron Microscopy Center (USTEM) and especially Karin Whitmore and Johannes Bernardi.Notes and references
- H. Ibrahim, A. Ilinca and J. Perron, Renewable Sustainable Energy Rev., 2008, 12, 1221–1250 CrossRef CAS.
- K. T. Møller, T. R. Jensen, E. Akiba and H. Li, Prog. Nat. Sci.: Mater. Int., 2017, 27, 34–40 CrossRef.
- S. E. Hosseini and M. A. Wahid, Renewable Sustainable Energy Rev., 2016, 57, 850–866 CrossRef CAS.
- P. Millet, in Hydrogen Production, John Wiley & Sons, Ltd, 2015, pp. 63–116 Search PubMed.
- M. Serban, M. A. Lewis, C. L. Marshall and R. D. Doctor, Energy Fuels, 2003, 17, 705–713 CrossRef CAS.
- B. E. Logan, Environ. Sci. Technol., 2004, 38, 160A–167A CrossRef CAS PubMed.
- I. Roger, M. A. Shipman and M. D. Symes, Nat. Rev. Chem., 2017, 1, 0003 CrossRef CAS.
- Nanocomposites for Visible Light-induced Photocatalysis, ed. M. M. Khan, D. Pradhan and Y. Sohn, Springer International Publishing, 2017 Search PubMed.
- Photocatalysis,https://www.crcpress.com/Photocatalysis-Principles-and-Applications/Ameta-Ameta/p/book/9781482254938, accessed January 22, 2019.
- J. Schneider, D. Bahnemann, J. Ye, G. L. Puma and D. D. Dionysiou, Photocatalysis: Fundamentals and Perspectives, Royal Society of Chemistry, 2016 Search PubMed.
- X. Li, J. Yu, J. Low, Y. Fang, J. Xiao and X. Chen, J. Mater. Chem. A, 2015, 3, 2485–2534 RSC.
- A. O. Ibhadon and P. Fitzpatrick, Catalysts, 2013, 3, 189–218 CrossRef CAS.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef CAS.
- T. Jafari, E. Moharreri, A. S. Amin, R. Miao, W. Song and S. L. Suib, Molecules, 2016, 21, 900 CrossRef PubMed.
- Z. Zhang and J. T. Yates, Chem. Rev., 2012, 112, 5520–5551 CrossRef CAS PubMed.
- T. Kawai and T. Sakata, J. Chem. Soc., Chem. Commun., 1980, 694–695 RSC.
- B. M. Hunter, H. B. Gray and A. M. Müller, Earth-Abundant Heterogeneous Water Oxidation Catalysts, Chem. Rev., 2016, 116(22), 14120–14136 Search PubMed.
- Sol-Gel Derived Nanomaterials and It's Applications, https://www.researchgate.net/publication/288630167_Sol-Gel_Derived_Nanomaterials_and_It's_Applications_A_Review, accessed January 22, 2019.
- R. Nagarajan and T. Alan Hatton, Nanoparticles: Synthesis, Stabilization, Passivation, and Functionalization, American Chemical Society, 2008, vol. 996, pp. i–v Search PubMed.
- H. Chen, X. Y. Liu, X. D. Hao and Y. X. Zhang, Ceram. Int., 2016, 42, 19425–19428 CrossRef CAS.
- P. Praveen, G. Viruthagiri, S. Mugundan and N. Shanmugam, Spectrochim. Acta, Part A, 2014, 120, 548–557 CrossRef CAS PubMed.
- W. Xin, D. Zhu, G. Liu, Y. Hua and W. Zhou, Synthesis and Characterization of Mn–C–Co-doped Nanoparticles and Photocatalytic Degradation of Methyl Orange Dye under Sunlight Irradiation, https://www.hindawi.com/journals/ijp/2012/767905/, accessed January 22, 2019 Search PubMed.
- C. Karunakaran, P. Vinayagamoorthy and J. Jayabharathi, Superlattices Microstruct., 2013, 64, 569–580 CrossRef CAS.
- Y. Wan, Z. Xu, W. Chao and J. Zhang, J. Exp. Nanosci., 2013, 8, 782–787 CrossRef.
- W. Liu, Z. Liu, G. Wang, X. Sun, Y. Li and J. Liu, Sci. China Mater., 2017, 60, 438–448 CrossRef CAS.
- S. G. Babu, P. Karthik, M. C. John, S. K. Lakhera, M. Ashokkumar, J. Khim and B. Neppolian, Ultrason. Sonochem., 2019, 50, 218–223 CrossRef CAS PubMed.
- A. M. Antolín, S. Contreras, F. Medina and D. Tichit, Top. Catal., 2017, 60, 1156–1170 CrossRef.
- T. Harifi and M. Montazer, Appl. Catal., A, 2014, 473, 104–115 CrossRef CAS.
- J. Lasek, Y.-H. Yu and J. C. S. Wu, Environ. Technol., 2012, 33, 2133–2141 CrossRef CAS PubMed.
- J.-D. Lin, S. Yan, Q.-D. Huang, M.-T. Fan, Y.-Z. Yuan, T. T.-Y. Tan and D.-W. Liao, Appl. Surf. Sci., 2014, 309, 188–193 CrossRef CAS.
- J. Yu, Y. Hai and B. Cheng, J. Phys. Chem. C, 2011, 115, 4953–4958 CrossRef CAS.
- A. T. Montoya and E. G. Gillan, ACS Omega, 2018, 3, 2947–2955 CrossRef CAS.
- P. D. Tran, L. Xi, S. K. Batabyal, L. H. Wong, J. Barber and J. S. C. Loo, Phys. Chem. Chem. Phys., 2012, 14, 11596–11599 RSC.
- J. Si, S. Xiao, Y. Wang, L. Zhu, X. Xia, Z. Huang and Y. Gao, Nanoscale, 2018, 10, 2596–2602 RSC.
- H. Tada, Q. Jin, H. Nishijima, H. Yamamoto, M. Fujishima, S. Okuoka, T. Hattori, Y. Sumida and H. Kobayashi, Angew. Chem., Int. Ed., 2011, 50, 3501–3505 CrossRef CAS PubMed.
- H. Yu, H. Irie, Y. Shimodaira, Y. Hosogi, Y. Kuroda, M. Miyauchi and K. Hashimoto, J. Phys. Chem. C, 2010, 114, 16481–16487 CrossRef CAS.
- A. Li, T. Wang, X. Chang, W. Cai, P. Zhang, J. Zhang and J. Gong, Chem. Sci., 2016, 7, 890–895 RSC.
- M. Higashi, K. Domen and R. Abe, J. Am. Chem. Soc., 2012, 134, 6968–6971 CrossRef CAS PubMed.
- A. Singh, S. L. Y. Chang, R. K. Hocking, U. Bach and L. Spiccia, Energy Environ. Sci., 2013, 6, 579–586 RSC.
- W.-T. Chen, A. Chan, D. Sun-Waterhouse, T. Moriga, H. Idriss and G. I. N. Waterhouse, J. Catal., 2015, 326, 43–53 CrossRef CAS.
- W. Wang, S. Liu, L. Nie, B. Cheng and J. Yu, Phys. Chem. Chem. Phys., 2013, 15, 12033–12039 RSC.
- H. Yu, J. Tian, F. Chen, P. Wang and X. Wang, Sci. Rep., 2015, 5, 13083 CrossRef CAS PubMed.
- Z. Yan, X. Yu, Y. Zhang, H. Jia, Z. Sun and P. Du, Appl. Catal., B, 2014, 160–161, 173–178 CrossRef CAS.
- M. Nolan, A. Iwaszuk and H. Tada, Aust. J. Chem., 2012, 65, 624–632 CrossRef CAS.
- B. Pal and M. Sharon, Thin Solid Films, 2000, 379, 83–88 CrossRef CAS.
- S. A. Sadeek, An. Asoc. Quim. Argent., 2005, 93, 165–176 CAS.
- Z. Lin, D. Han and S. Li, J. Therm. Anal. Calorim., 2012, 107, 471–475 CrossRef CAS.
- T. L. Barr and S. Seal, J. Vac. Sci. Technol., A, 1995, 13, 1239–1246 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson, R. St and C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- M. Chandra Sekhar, B. Purusottam Reddy, S. V. Prabhakar Vattikuti, G. Shanmugam, C.-H. Ahn and S.-H. Park, J. Cluster Sci., 2018, 29, 1255–1267 CrossRef CAS.
- Q. R. Deng, X. H. Xia, M. L. Guo, Y. Gao and G. Shao, Mater. Lett., 2011, 65, 2051–2054 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, L. W. M. Lau, A. Gerson and R. S. C. Smart, Surf. Interface Anal., 2009, 41, 324–332 CrossRef CAS.
- J. K. Stolarczyk, S. Bhattacharyya, L. Polavarapu and J. Feldmann, ACS Catal., 2018, 8, 3602–3635 CrossRef CAS.
- R. Niishiro, R. Konta, H. Kato, W.-J. Chun, K. Asakura and A. Kudo, J. Phys. Chem. C, 2007, 111, 17420–17426 CrossRef CAS.
- G. Hitoki, T. Takata, J. N. Kondo, M. Hara, H. Kobayashi and K. Domen, Chem. Commun., 2002, 1698–1699 RSC.
- W. Zhang, Y. Li, X. Zeng and S. Peng, Sci. Rep., 2015, 5, 10589 CrossRef CAS PubMed.
- I. Majeed, M. A. Nadeem, E. Hussain, G. I. N. Waterhouse, A. Badshah, A. Iqbal, M. A. Nadeem and H. Idriss, ChemCatChem, 2016, 8, 3146–3155 CrossRef CAS.
- J. Sato, N. Saito, H. Nishiyama and Y. Inoue, J. Phys. Chem. B, 2001, 105, 6061–6063 CrossRef CAS.
- F. Meng, J. Li, S. K. Cushing, J. Bright, M. Zhi, J. D. Rowley, Z. Hong, A. Manivannan, A. D. Bristow and N. Wu, ACS Catal., 2013, 3, 746–751 CrossRef CAS.
- N. Zhang, J. Shi, S. S. Mao and L. Guo, Chem. Commun., 2014, 50, 2002–2004 RSC.
- D. M. Jang, I. H. Kwak, E. L. Kwon, C. S. Jung, H. S. Im, K. Park and J. Park, J. Phys. Chem. C, 2015, 119, 1921–1927 CrossRef CAS.
- M. Okazaki, Y. Wang, T. Yokoi and K. Maeda, J. Phys. Chem. C, 2019, 123, 10429–10434 CrossRef CAS.
- M. S. Burke, S. Zou, L. J. Enman, J. E. Kellon, C. A. Gabor, E. Pledger and S. W. Boettcher, J. Phys. Chem. Lett., 2015, 6, 3737–3742 CrossRef CAS PubMed.
- H. Kato and A. Kudo, J. Phys. Chem. B, 2001, 105, 4285–4292 CrossRef CAS.
- C. K. Mavrokefalos and G. R. Patzke, Inorganics, 2019, 7, 29 CrossRef.
- R. Pokhrel, M. K. Goetz, S. E. Shaner, X. Wu and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 8384–8387 CrossRef CAS PubMed.
- M. Wiechen, M. M. Najafpour, S. I. Allakhverdiev and L. Spiccia, Energy Environ. Sci., 2014, 7, 2203–2212 RSC.
- P. Kurz, Top. Curr. Chem., 2016, 371, 49–72 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta05637h |
| This journal is © The Royal Society of Chemistry 2019 |