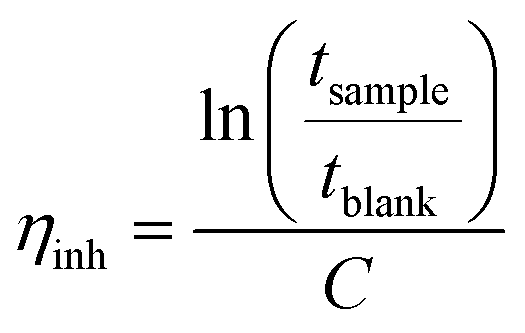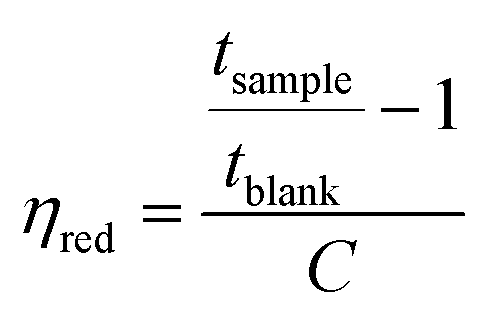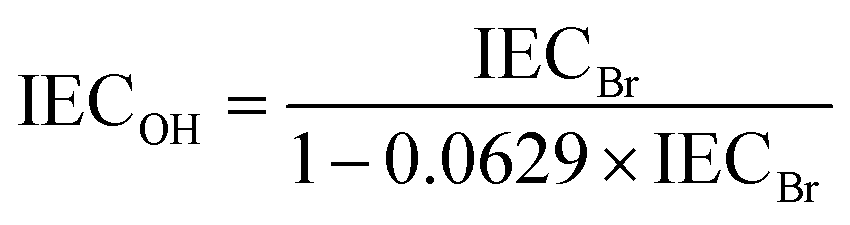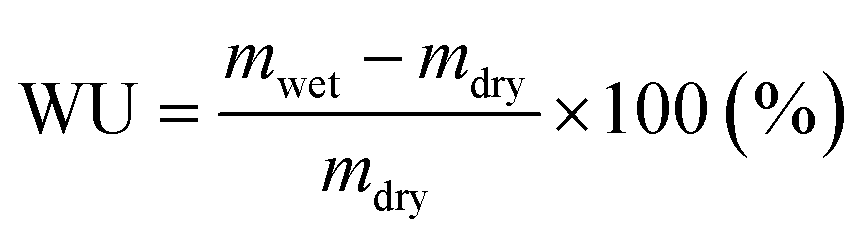DOI:
10.1039/C9TA05531B
(Paper)
J. Mater. Chem. A, 2019,
7, 15895-15906
Effects of the N-alicyclic cation and backbone structures on the performance of poly(terphenyl)-based hydroxide exchange membranes†
Received
24th May 2019
, Accepted 6th June 2019
First published on 6th June 2019
Abstract
Hydroxide ion conducting poly(terphenyl alkylene)s functionalized with piperidine-based quaternary ammonium cations were synthesized via superacid-catalyzed polyhydroxyalkylations. By employing different synthetic strategies, we have systematically varied the structures of the cation and the backbone polymer to study the effects on morphology, stability and hydroxide conductivity. Two monomers were initially prepared by attaching 4-benzylpiperidine groups to trifluororacetophenone and m-terphenyl, respectively, through Suzuki coupling reactions. Polymerizations followed by quaternizations were then carried out to obtain poly(terphenyl alkylene)s with approximately the same ionic contents. These contained either m- or p-terphenyl backbone units, and were tethered with monocyclic N,N-dimethylpiperidinium (DMP) or spirocyclic 6-azonia-spiro[5,5]undecane-6-ium (ASU) cations placed on either the stiff terphenyl or the more flexible alkylene units along the backbone. Polymer chain flexibility and functionalization with DMP cations were found to promote ionic clustering and conductivity. Hence, a membrane based on a m-terphenyl backbone tethered with DMP on pendant phenyl groups achieved a hydroxide conductivity of 146 mS cm−1 at 80 °C. While the thermal stability was significantly higher for ASU-functionalized HEMs, the alkaline stability was highest for the ones carrying DMP cations, which showed less than 5% ionic loss after 720 h in 2 M NaOH at 90 °C. After 168 h at 120 °C, 1H NMR analysis suggested that the DMP cation degraded by a combination of β-Hofmann elimination and methyl substitution. Overall, the results of the study demonstrated that the structural features of the present polymers provided high alkaline stability, most probably due to aryl ether-free backbones, and that all the β-protons of the DMP and ASU cations were placed in 6-membered rings.
1. Introduction
Research to develop thermochemically stable and highly conductive hydroxide exchange membranes (HEMs) is currently very intense. These efforts are mostly motivated by the prospects of the hydroxide exchange membrane fuel cell (HEMFC) that offer an attractive alternative to the more conventional proton exchange membrane fuel cell (PEMFC). By the transition from acidic to alkaline conditions, the HEMFC enables the use of electrocatalysts free from platinum group metals in the electrodes.1–5 However, HEMs are normally composed of organic polymers tethered with quaternary ammonium (QA) cations, and the presence of high concentrations of the strongly basic and nucleophilic hydroxide ion makes the development of durable and high-performing HEMs very challenging.
Over the last decade, a large number of studies of HEMs based on aromatic polymers such as polyethers, polysulfones, polyketones, polyphenylenes, and styrenic copolymers, tethered with QA cations, have been reported.4–16 Overall, the results indicate that the presence of diaryl ether bridges in the polymer backbone, as well as the introduction of QA cations in benzylic sites, may destabilize the backbone and introduce chain cleavage reactions.14–20 Hence, there is now a clear trend in the field to employ ether-free polymer backbones in the structural design of HEMs.13,21–26 In addition, the hydroxide ion is liable to react with QA cations which results in a loss of ion exchange capacity (IEC) and ionic conductivity.5,27–30 Depending on the structure of the cation, degradation may occur via, e.g., direct nucleophilic substitution by attack on an α-carbon, Hofmann β-elimination and different rearrangement reactions.27–29 In seminal work, Kreuer and Marino identified piperidine-based QA cations as extraordinarily alkali-stable after investigating a large number of different low-molecular weight model QA cations under highly basic conditions. Accordingly, N,N-dimethylpiperidinium (DMP) and the piperidine-based spirocyclic cation 6-azonia-spiro[5,5]undecane-6-ium (ASU) were found to possess approximately 21 and 26 times longer half-lives, respectively, than the conventional benzyltrimethyl QA cation in 6 M NaOH at 160 °C.29 The high resistance against substitution and elimination reactions of the former cations has been attributed to the low ring strain of the 6-membered ring and to the constrained conformations imposed by the ring structure. These features increase the transition state energy in both substitution and elimination reactions.29 Recently, the results of the ASU and DMP model compounds have inspired us25,26,31,32 and others24,33–40 to chemically design, prepare and study different HEMs tethered with piperidinium and spirocyclic QA cations. For example, we have prepared N-spirocyclic QA ionenes (spiro-ionenes) by cyclo-polycondensations involving dipiperidines.32 These materials showed excellent thermal and alkaline stability, with only ∼10% ionic loss after 336 h in 1 M KOD in D2O at 120 °C. In parallel, we have also prepared poly(arylene N,N-dimethylpiperidinium)s by Friedel–Crafts type polycondensation of N-methyl-4-piperidone and electron-rich biphenyl or p-terphenyl monomers via superelectrophilic activation in triflic acid, followed by quaternisation reactions with iodomethane.25 HEMs based on these polymers showed no structural degradation after storage in 2 M NaOH at 60 °C during 360 h, and a mere 5% ionic loss at 90 °C after the same period of time. Recent work by Yan et al. included fuel cell testing and confirmed the high alkaline stability of the poly(arylene N,N-dimethylpiperidinium)s.24 In an extension of our previous work on these materials, we have very recently cyclo-quaternized poly(biphenyl piperidine)s using 1,5-dibromopentane to form poly(biphenyl alkylene)s functionalized with bicyclic ASU cations.26 Alkaline testing up to 120 °C revealed that the ring of the ASU cation that was directly attached to the polymer backbone degraded significantly faster by Hofmann β-elimination than the second pendant ring. We hypothesized that the ring directly attached to the bulky and stiff polymer backbone may have significantly distorted bond angles and restricted conformational relaxation within the ASU cation, thus reducing the activation energy and activating Hofmann β-elimination reactions.26,32 Consequently, while piperidine-based QA cations possess high alkaline stability in general, their specific resistance against degradation seems to be sensitive to exactly how and where the cations are tethered to the polymer backbone.
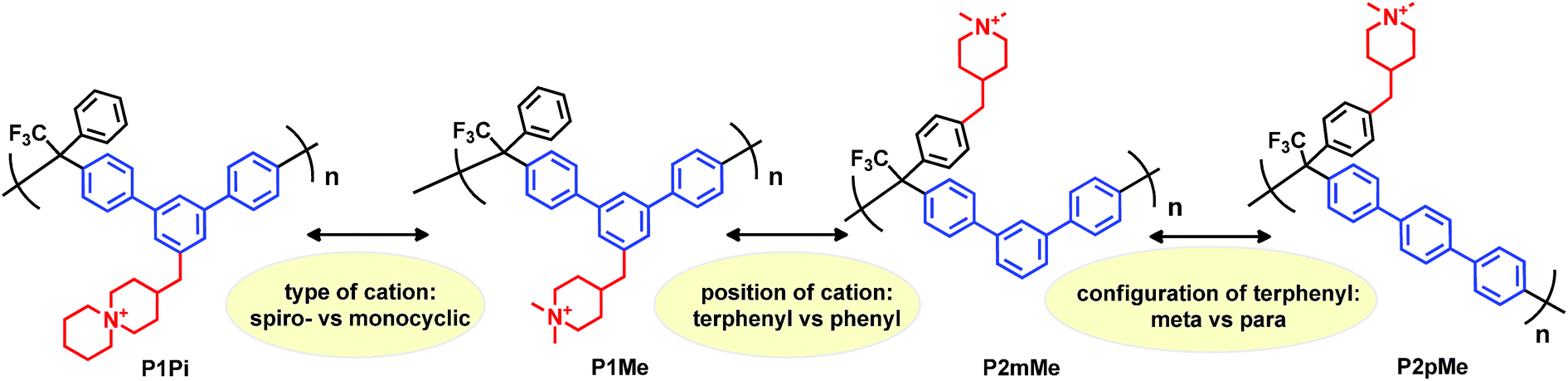
In the present work we have molecularly designed and synthesized a family of poly(terphenyl alkylene)s to systematically study how the type and placement of the piperidine-based cation, as well as the structure of the backbone polymer, influence the stability and properties of the corresponding HEMs (Scheme 1). Starting with poly(m-terphenyl alkylene) having ASU cations attached to the m-terphenyl units (P1Pi), we then switched the cation to DMP (P1Me), followed by a shift in the position of the cation to the more flexible pendant phenyl ring (P2mMe), and then finally a change in backbone configuration from m-terphenyl units to the stiffer p-terphenyl units (P2pMe). All the polymers in the series had approximately the same ionic content, which allowed a direct comparison of HEM properties. The membranes were characterized and compared with respect to morphology, water uptake, thermal and alkaline stability, and hydroxide conductivity.
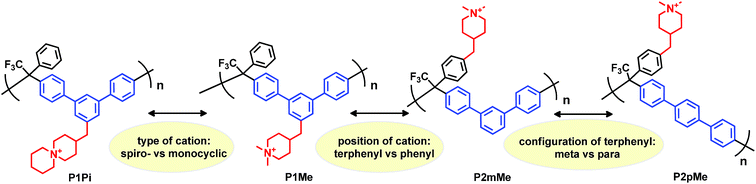 |
| | Scheme 1 Overview of the variations in the molecular design of the cationic polymers synthesized and investigated as HEMs. | |
2. Results and discussion
2.1 Monomer synthesis and characterization
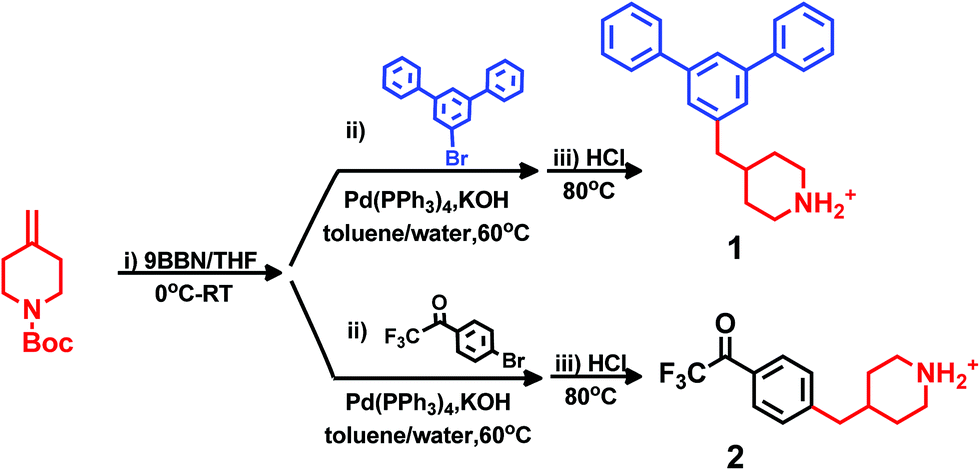
Friedel–Crafts type polycondensation of arenes (such as biphenyl, terphenyl and fluorene) and suitable ketones via hydroxyalkylation reactions in superacidic media is an attractive route to thermochemically and mechanically robust ether-free polymers suitable for the preparation of alkali-stable HEMs.24–26,41–45 In order to prepare the polymer structures depicted in Scheme 1 by this method, we initially synthesized two monomers: one m-terphenyl and one trifluoroacetophenone, each functionalized with a piperidine ring (Scheme 2). The piperidine ring can subsequently be quaternized by dimethylation to form the monocyclic DMP cation, or by cycloquaternization with 1,5-dibromopentane to form the spirocyclic ASU cation. Quite uniquely, all the β-hydrogens of the DMP and ASU cations are positioned in 6-membered rings which likely increases the resistance against Hofmann β-elimination. Access to the two monomers with m-terphenyl and trifluoroacetophenone moieties, respectively, enabled us to conveniently vary the type and position of the cation along the polymer backbone. Moreover, both the rigidity and the ionic content of the polymer can be controlled by a careful choice of the co-monomer.
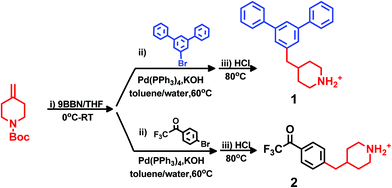 |
| | Scheme 2 Synthesis of m-terphenyl 1 and 2,2,2-trifluoroacetophenone 2 monomers containing the 4-benzylpiperidine moiety in three steps: (i) hydroboration, (ii) Suzuki coupling, and (iii) deprotection. | |
In general, the synthesis of monomers and polymers functionalized with DMP and ASU cations is challenging because of the need to attach the piperidine ring in the 4- (or possibly 3-) position instead of the more straightforward 1(N)-position which is accessible by Menshutkin reactions. The availability of commercial building blocks containing the piperidine ring is limited, thus restricting the possible synthetic strategies. Still, inspired by the work of Vice and co-workers,46 we have in three steps successfully synthesized one m-terphenyl (1) and one 2,2,2-trifluoroacetophenone (2) monomer, both bearing 4-benzyl-piperidine groups (Scheme 2). The synthesis was performed by hydroboration of N-Boc-4-methylene-piperidine with 9-BBN, followed by Suzuki coupling with 5′-bromo-m-terphenyl and 4′-bromo-2,2,2-trifluoroacetophenone, respectively. Final deprotection in aq. HCl yielded monomers 1 and 2, respectively, in total isolated yields between 60 and 75% without any prior optimization. The characteristics of the synthesis methods provide good possibilities for scale up.
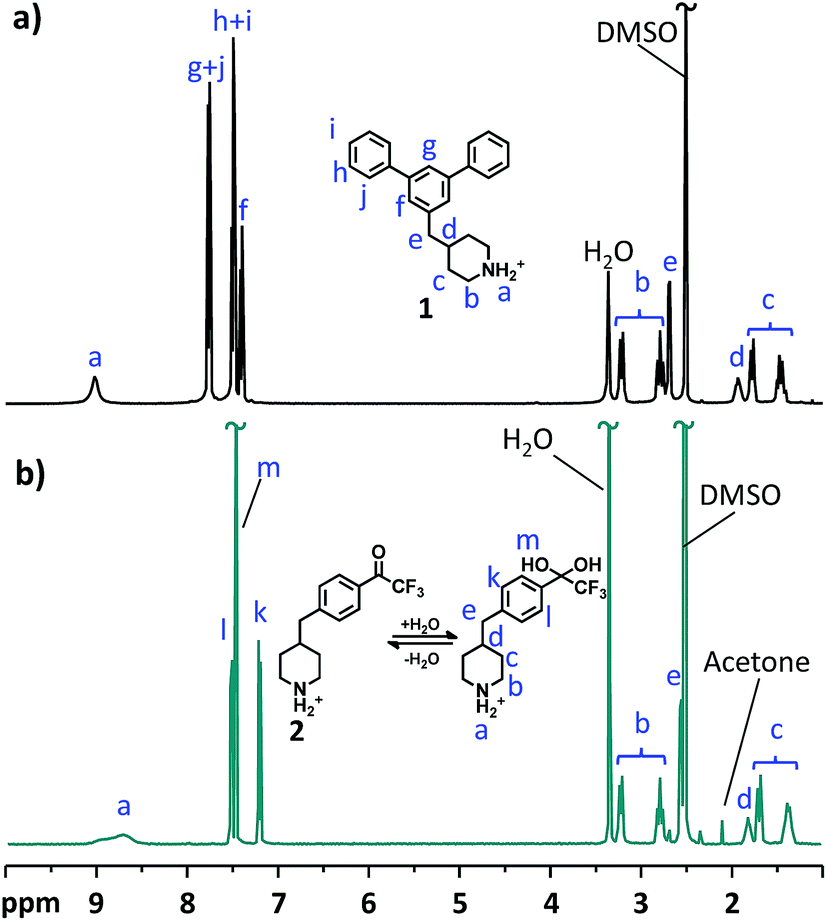
The structure and purity of the monomers were confirmed by 1H NMR spectroscopy by verification of signal positions and ratios (Fig. 1). As expected, the chemical shift and splitting of the signals corresponding to the protons of the 4-methylenepiperidine moiety were similar in the 1H NMR spectra of the two monomers. Both spectra showed distinct signals of the protonated secondary piperidinium protons (a) at above 8.5 ppm. The α (b) and β (c) protons in the piperidinium rings appeared as two pairs of signals at 2.8–3.3 and 1.3–1.8 ppm, respectively, and signals from the benzyl (e) and methine protons (d) emerged at 2.6–2.7 and 1.8–1.9 ppm, respectively. The signals from the aromatic protons appeared as three multiplets between 7.4 and 7.8 ppm in the spectrum of monomer 1, but as two doublets at 7.2 and 7.5 ppm in that of monomer 2. Notably, monomer 2 was found to exist entirely in the hydrated geminal diol form instead of the ketone form. This was indicated by the presence of the singlet at 7.48 ppm, corresponding to the two hydroxyl protons (m) of the gem-diol (Fig. 1b). The formation of the gem-diol was promoted by the strongly electron withdrawing trifluoromethyl group that destabilizes the carbonyl group.
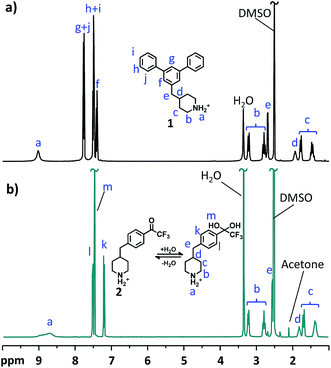 |
| | Fig. 1
1H NMR spectra of (a) m-terphenyl monomer 1 and (b) 2,2,2-trifluoroacetophenone monomer 2. | |
2.2 Polycondensations
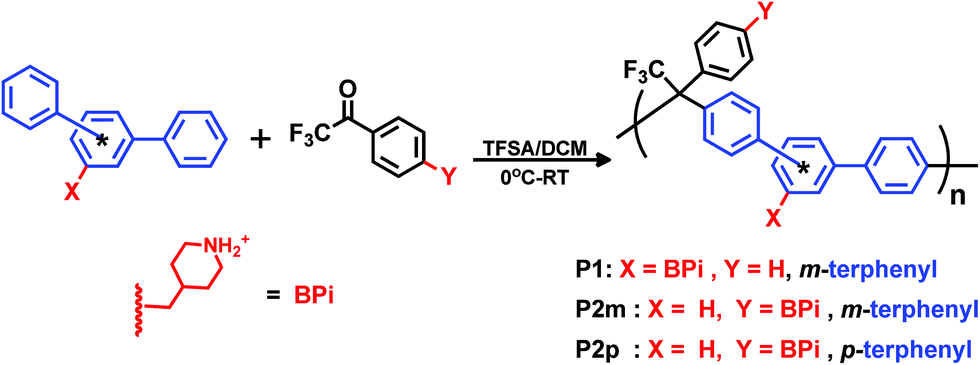
The monomers were employed in Friedel–Crafts type polycondensations with a mixture of TFSA and DCM as the superacidic reaction medium. Three different poly(arylene alkylene) precursors tethered with 4-methylene piperidine groups were synthesized. Polymer P1 was prepared by polycondensation of monomer 1 and 2,2,2-trifluoroacetophenone, while polymers P2m and P2p were produced in polycondensations of monomer 2 with m- and p-terphenyl, respectively (Scheme 3). Polycondensations by hydroxyalkylation reactions proceed in two consecutive steps.42 In the first step, the first arene monomer is added to the carbonyl monomer to form a hydroxyl-alkyl intermediate. This intermediate then condenses with the second incoming arene monomer in the second step. Since the first step is rate-determining, both the rate and the degree of polymerization are greatly enhanced by adding a small excess of the carbonyl monomer.42 This is in sharp contrast to the need for a strict stoichiometric ratio to reach high degrees of polymerization in conventional polycondensations. In the present work we consistently used a 10% molar excess of the carbonyl monomer.
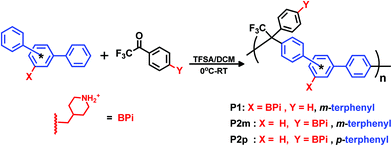 |
| | Scheme 3 Synthetic pathway to precursor polymers with different positions of secondary piperidinium cations and backbone configurations (meta or para) by superacid-catalyzed Friedel–Crafts type polycondensations. | |
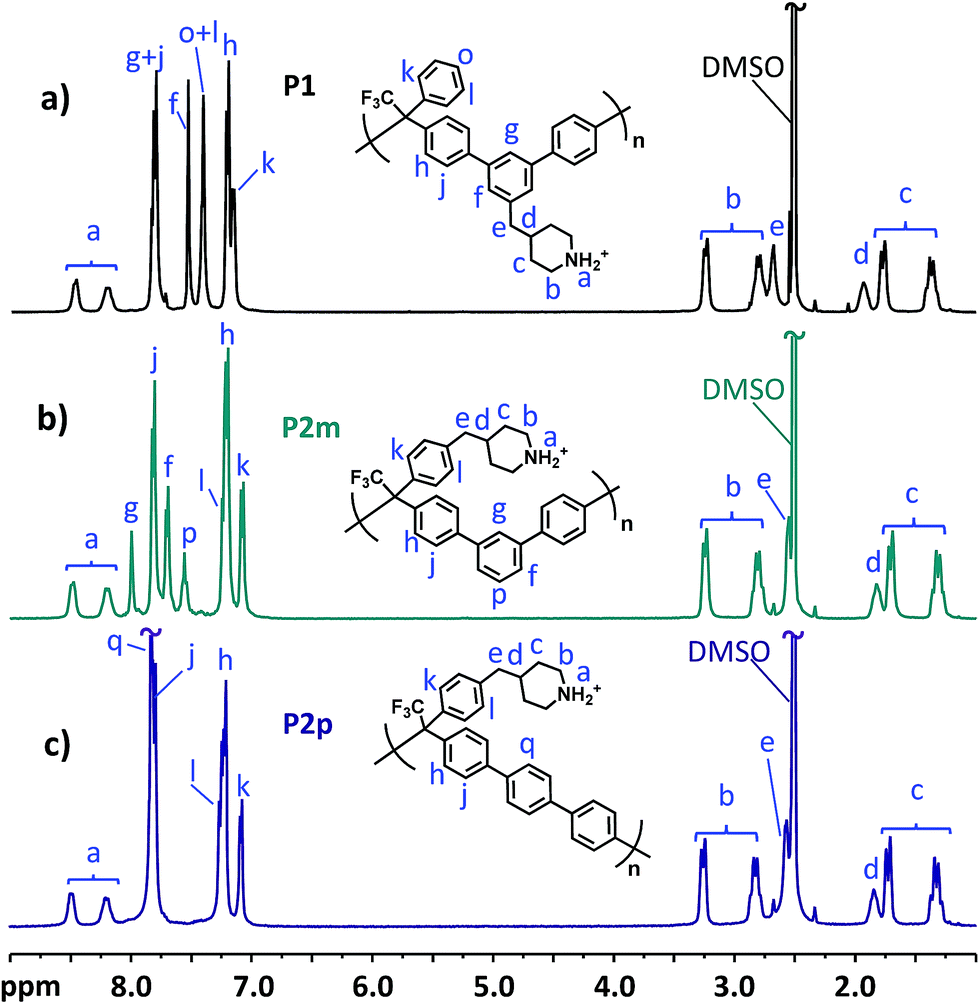
The molecular structure of the precursor polymers was confirmed by 1H NMR spectroscopy. To facilitate the NMR analysis, a small amount of TFA was added to adjust the pH of the polymer solutions, thereby shifting the water signal from 3.33 to above 10 ppm. As can be seen in Fig. 2, the most obvious difference in relation to the monomer spectra in Fig. 1 was observed in the aromatic region, while the signals b–e originating from the benzyl piperidinium moiety remained rather unchanged. Upon addition of TFA, the signals from the piperidinium protons (a) were split in a similar way to the α-protons (b). The molecular weight of the polymers was evaluated by dilute solution viscosimetry. The intrinsic viscosities of P1, P2m and P2p in 0.1 M LiBr in DMSO solution at 30 °C were measured to be 0.62, 0.26, and 0.57 g dL−1, respectively, suggesting a medium molecular weight. It is unclear to us why the molecular weight of P2m was markedly lower compared with those of the other polymers.
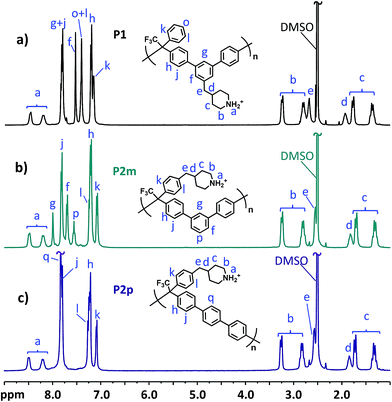 |
| | Fig. 2
1H NMR spectra of the precursor polymers: (a) P1, (b) P2m and (c) P2p recorded in DMSO-d6/TFA. TFA was added to shift the water signals (originally at ∼3.3 ppm) to above 10 ppm, revealing sample signals between 3.0 and 3.5 ppm. | |
2.3 Quaternizations
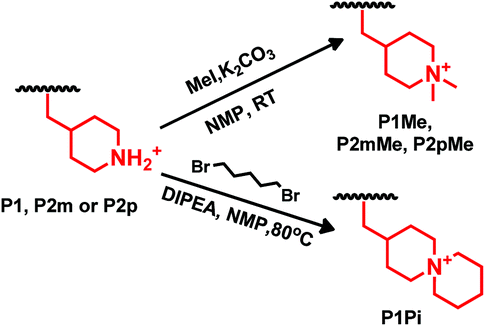
The secondary piperidine rings of the three precursor polymers P1, P2m and P2p were quaternized by dimethylation using methyl iodide as the methylation agent and K2CO3 as the catalyst to prepare polymers P1Me, P2mMe and P2pMe, respectively, functionalized with the DMP cation. In addition, precursor polymer P1 was functionalized with the ASU cation by cyclo-quaternization of the piperidine rings using 1,5-dibromopentane to prepare polymer P1Pi, following our previously reported procedure (Scheme 4).26 The polymer concentration was kept low (<2.5 wt%) throughout the cyclo-quaternization reaction to promote intramolecular cyclization and depress intermolecular crosslinking. Due to the lower reactivity of 1,5-dibromopentane in comparison with methyl iodide, the cyclo-quaternization was performed at 80 °C instead of 20 °C as in the dimethylation reaction. Furthermore, Hünig's base (N,N-diisopropylethylamine, DIPEA) was employed instead of K2CO3 as the base catalyst. DIPEA has higher solubility in the reaction solution but was found to react with the highly reactive methyl iodide,47 and could thus not be used in the dimethylation reaction.
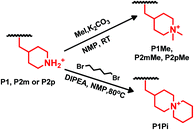 |
| | Scheme 4 Quaternizations by dimethylation using methyl iodide (upper) and by cyclo-quaternization using 1,5-dibromopentane (lower) to form the DMP and ASU cations, respectively. | |
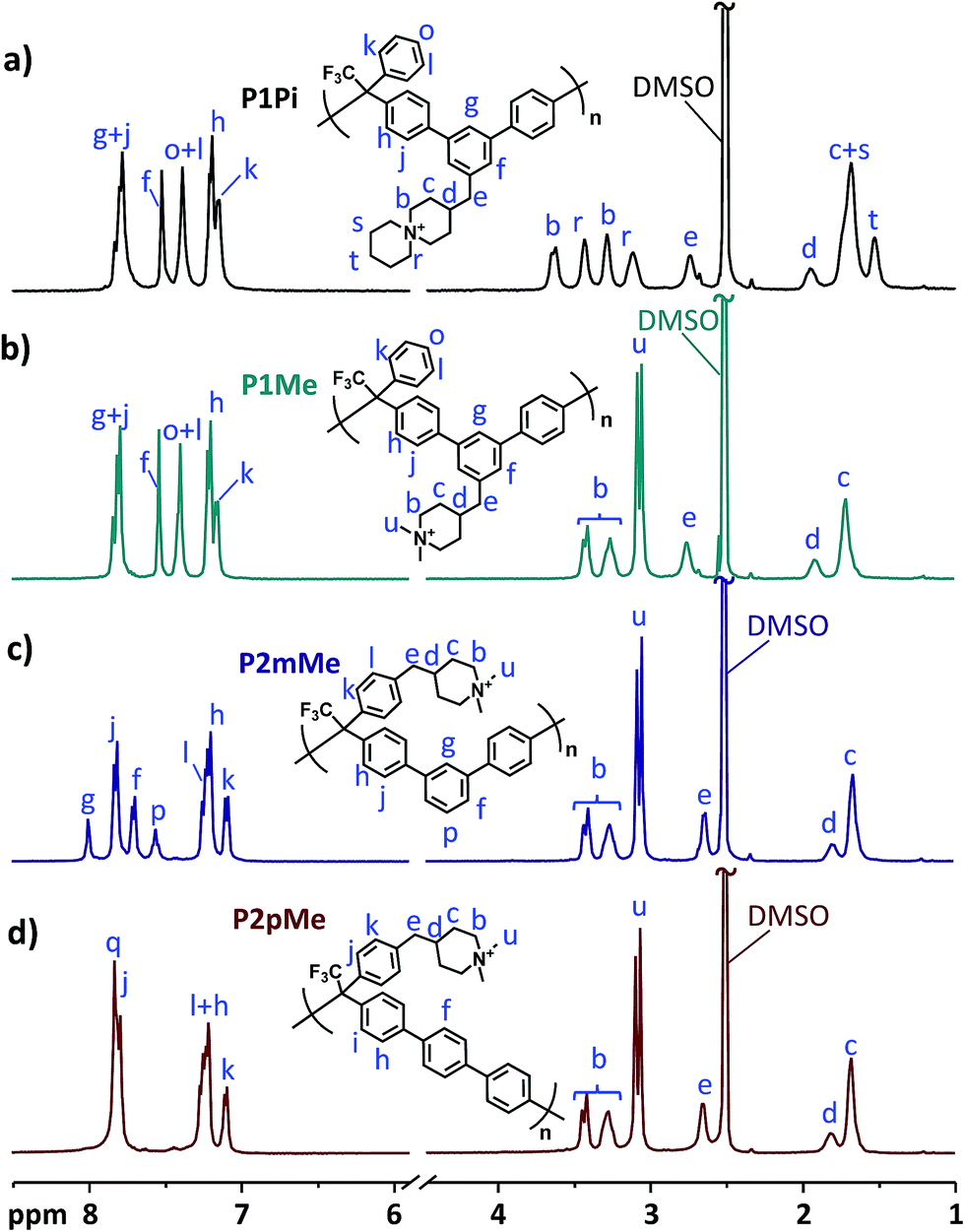
The success of the dimethylation and cyclo-quaternization reactions was confirmed by 1H NMR analysis of the polymers dissolved in DMSO-d6/TFA mixtures (Fig. 3). As expected, no significant changes in the aromatic region (7.0–8.0 ppm) were observed after the formation of the DMP and ASU cations. However, the signals corresponding to the secondary piperidinium protons (a) at 8.2–8.5 ppm completely disappeared and new signals originating from the methyl (u) and methylene (r, s and t) protons of the newly formed cyclic QA cations emerged at 3.1–3.5 ppm. In the transition from secondary piperidinium rings to quaternary ones, the chemical shifts of the methylene protons in the rings (b and c) also changed. As shown in Fig. 3, the splitting of both these signals was significantly reduced after the quaternization reactions. No signals corresponding to the methylene protons in the secondary piperidinium rings were observed and hence the quaternizations were considered to be quantitative within the limits of the 1H NMR method.
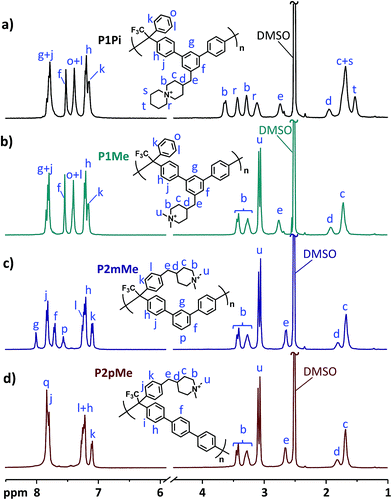 |
| | Fig. 3
1H NMR spectra of the quaternized polymers P1Pi (a), P1Me (b), P2mMe (c) and P2pMe (d) recorded in DMSO-d6/TFA. TFA was added to shift the water signals (originally at ∼3.3 ppm) to above 10 ppm, revealing sample signals between 3.0 and 3.5 ppm. | |
2.4 Membrane preparation and morphology
HEMs based on the quaternized polymers were prepared by solvent casting from 5 wt% solutions in DMSO. The resulting pale yellow membranes were transparent and flexible (Fig. 4). The HEMs based on P1Me, P2mMe and P2pMe, functionalized with the monocyclic DMP cation, had similar high flexibility. In contrast, the HEM based on P1Pi, functionalized with the bulky spirocyclic ASU cation, was less flexible, which may reflect a higher polymer chain stiffness of this polymer. We have previously found that HEMs functionalized with 5-azaspiro[4.5]decan-5-ium cations (6- plus 5-membered ring arrangement) were more flexible than the corresponding HEM tethered with the more bulky ASU cation (6- plus 6-membered ring arrangement).26 The IEC of the HEMs was verified by Mohr titrations of the HEMs in the bromide form. As shown in Table 1, the values were in good agreement with the theoretical values based on the chemical structure of the polymers as evaluated by 1H NMR spectroscopy.
 |
| | Fig. 4 Photographs of HEMs based on P2pMe (a and b) and P1Pi (c), indicating their color, transparency and flexibility. | |
Table 1 Properties of the HEMs
| HEM |
IEC (mequiv. g−1) |
WUb (wt%) |
λ
|
σ
(mS cm−1) |
T
d,95
(°C) |
q
max
(nm−1) |
d
max (nm) |
| Theoreticala |
Titrated |
|
Calculated from the chemical structure of the polymers in the bromide form (values for the hydroxide form within the parentheses).
Measured at 80 °C in the hydroxide form under fully hydrated conditions (immersed).
Measured by TGA under N2 at 10 °C min−1.
Measured by SAXS in the dry bromide form.
|
|
P1Pi
|
1.58(1.75) |
1.57 |
54 |
17 |
51 |
335 |
NA |
NA |
|
P1Me
|
1.69(1.89) |
1.71 |
99 |
29 |
107 |
282 |
2.5 |
2.5 |
|
P2mMe
|
1.69(1.89) |
1.73 |
103 |
30 |
146 |
269 |
2.5 |
2.5 |
|
P2pMe
|
1.69(1.89) |
1.82 |
73 |
21 |
103 |
275 |
2.5 |
2.5 |
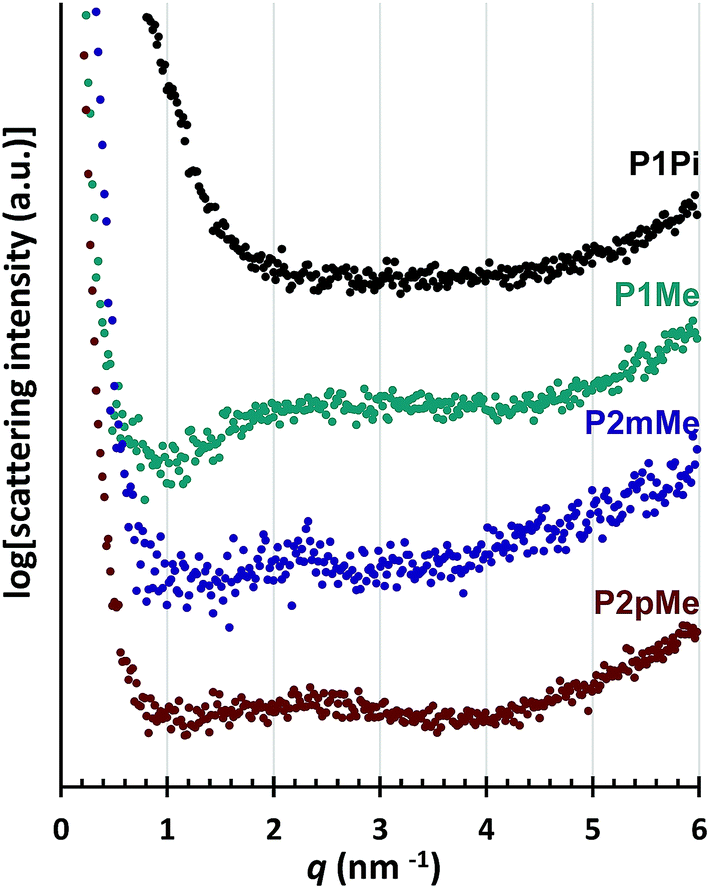
The morphology was studied by small angle X-ray scattering (SAXS) measurements of the dry HEMs in the bromide form. The SAXS profiles of P1Me, P2mMe and P2pMe (Fig. 5) displayed rather weak and broad scattering peaks, probably because of the high rigidity of the polymer backbones and the regularly separated cations. The scattering maxima at around q = 2.5 nm−1 corresponded to a characteristic distance of d ≈ 2.5 nm, which was in the same range as the calculated distance between two adjacent cations in the polymer chain. In contrast, no discernible ionomer peak was observed for P1Pi, most probably because the bulkiness of the ASU cations prevented effective assembly and clustering of these cations in the HEM. This observation was consistent with our previous findings concerning the ionic clustering in HEMs based on poly(arylene alkylene)s having N-alicyclic piperidinium cations directly attached to the backbone.25,26 The SAXS profile of HEMs functionalized with ASU cations showed no ionomer peaks26 while the profile of HEMs functionalized with DMP cations showed a weak ionomer peak at q = 2.6 nm−1.25 Hence, in general, it seems difficult for the ASU cations to phase-separate from the backbone polymer to form ionic clusters in HEMs. The phase-separation of both the ASU and DMP cations may be significantly improved if attached to the backbone via long flexible side chains.31
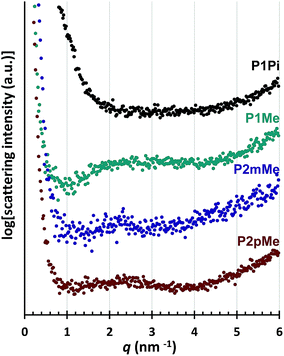 |
| | Fig. 5 SAXS profiles of dry HEMs in the bromide form. The data have been shifted vertically for clarity. | |
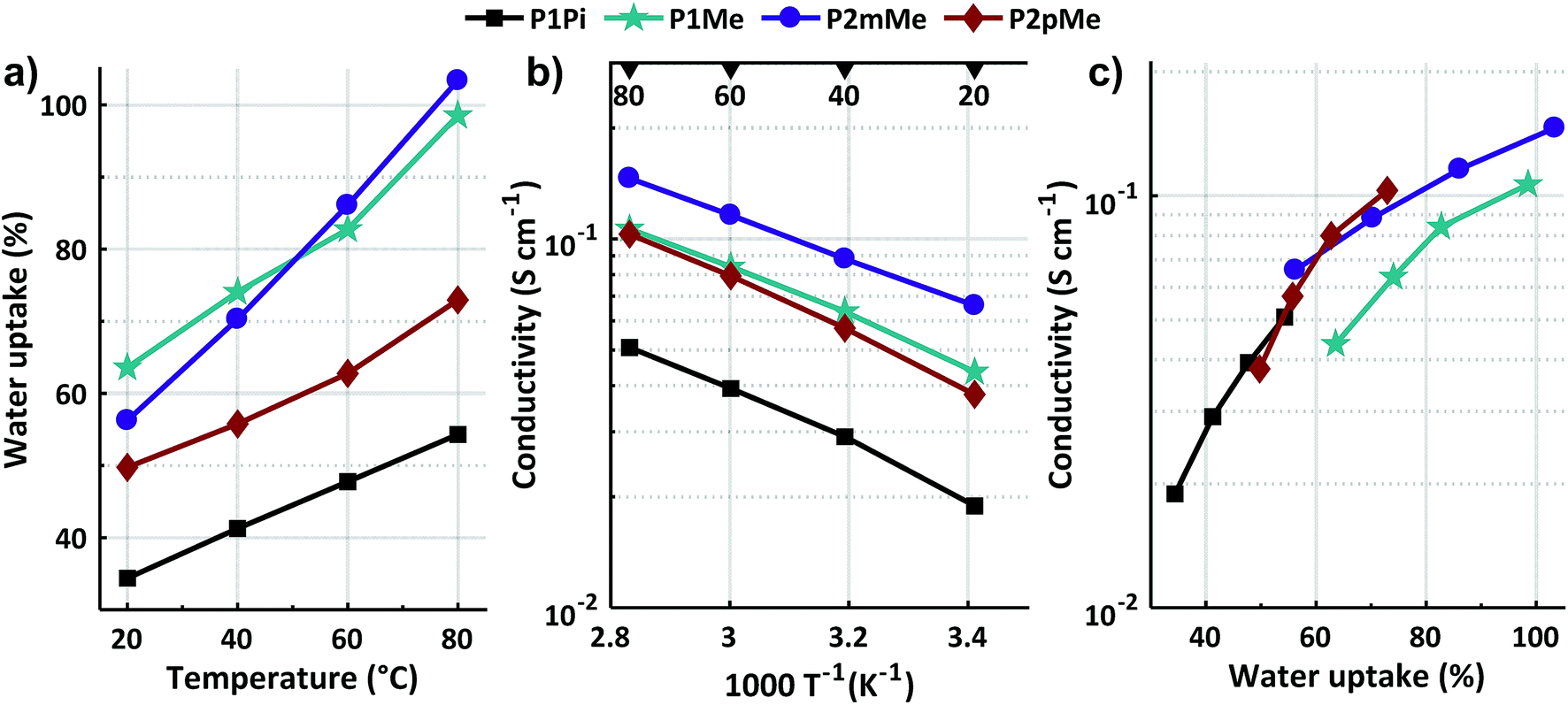
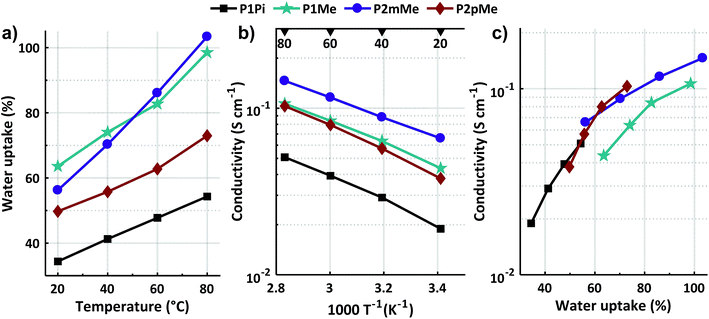
2.5 Water uptake and hydroxide conductivity
The water uptake and ionic conductivity (σ) of fully hydrated HEMs in the hydroxide form were determined between 20 and 80 °C by gravimetry and electrochemical impedance spectroscopy, respectively (Fig. 6a and b). As expected, both the water uptake and conductivity increased with temperature and IEC. However, the measured data were profoundly influenced by the structural variations of the polymers (Scheme 1). Membrane P1Pi with a low IEC and poorly clustered bulky spirocyclic ASU cations connected to m-terphenyl units along the backbone displayed the lowest water uptake and hydroxide conductivity of the structures in the present study. At 20 and 80 °C, this membrane reached 19 and 51 mS cm−1 at 34 and 54 wt% water uptake, respectively (Table 1). In membrane P1Me, the m-terphenyl based backbone structure was retained, but this membrane instead had the DMP cation connected to the m-terphenyl units. This resulted in a two-fold increase of both the water uptake and the hydroxide conductivity in relation to P1Pi. Consequently the λ value, i. e., [H2O]/[QA], of P1Me increased to 29, almost 70% higher than that of P1Pi. At 20 and 80 °C, membrane P1Me had hydroxide conductivities of 44 and 107 mS cm−1 at 63 and 99 wt% water uptake, respectively. Most probably, the smaller cation induced the more flexible structure of P1Me, thus allowing a higher water uptake and conductivity in relation to P1Pi. Membrane P2mMe had the same backbone structure as both P1Pi and P1Me, but instead had DMP cations connected to the pendant phenyl groups along the polymer backbone (Scheme 1). This arrangement had a significant positive effect on the conductivity, which at 80 °C increased by 186 and 36% compared to those of P1Pi and P1Me, respectively. Concurrently, the water uptake and λ value of P2mMe remained at the same level as those of P1Me. Hence, at 20 and 80 °C membrane P2mMe exhibited hydroxide conductivities of 66 and 146 mS cm−1 at 56 and 103 wt% water uptake, respectively. A possible explanation why P2mMe was a significantly more efficient hydroxide ion conductor than P1Me is that the position on the rather flexible pendant phenyl group away from the rigid backbone polymer provided higher local mobility. This typically promotes ionic clustering and the formation of efficient ion conducting water-filled channels in HEMs.9 In P2pMe, the type and placement of the cation were the same as in P2mMe. However, m-terphenyl units in the backbone polymer were exchanged by more inflexible p-terphenyl units (Scheme 1). This significantly increased the rigidity of the backbone polymer and, as expected, the visco-elastic properties of P2pMe resulting in a lower water uptake and conductivity than of P2mMe. Membrane P2pMe had a similar hydroxide conductivity as P1Me, but at a lower water uptake. Consequently, membrane P2pMe exhibited 38 and 103 mS cm−1 at 50 and 73 wt% water uptake at 20 and 80 °C, respectively.
 |
| | Fig. 6 Water uptake of fully hydrated HEMs in the hydroxide form as a function of temperature (a), hydroxide conductivity of fully hydrated HEMs as a function of T−1 (b) and hydroxide conductivity of fully hydrated HEMs as a function of water uptake (c) for the data measured between 20 and 80 °C. | |
Fig. 6c shows the relationship between the water uptake and conductivity measured between 20 and 80 °C. Comparing the isomeric polymer structures of P1Me, P2mMe and P2pMe, it is clear that the latter two had a higher ratio between their conductivity and water uptake than P1Me. This evidently showed their higher efficiency as hydroxide ion conductors and clearly demonstrated the advantage of placing the DMP cation on the flexible pendant phenyl group instead of the stiff terphenyl unit in the backbone polymer. Moreover, the data in Fig. 6c also indicate that the conductivity of P1Pi suffered because of the lower IEC value and water uptake of this membrane in relation to the other HEMs in the study.
The hydroxide conductivity of the HEMs of the present study compares quite favorably with previously reported data. For example, Bae et al. recently reported a conductivity of 112 mS cm−1 at 80 °C for an HEM based on a poly(m-terphenyl alkylene) having trimethyl QA cations attached via flexible pentyl spacers.48 This membrane had an IEC of 2.13 mequiv. g−1 and a water uptake of 70 wt%. Notably, the former value was higher than those of the present HEMs. In addition, we have previously reported a hydroxide conductivity of 118 mS cm−1 for an HEM based on a poly(arylene N,N-dimethylpiperidinium) with an IEC of 2.26 mequiv. g−1 and a water uptake of 141 wt%.49
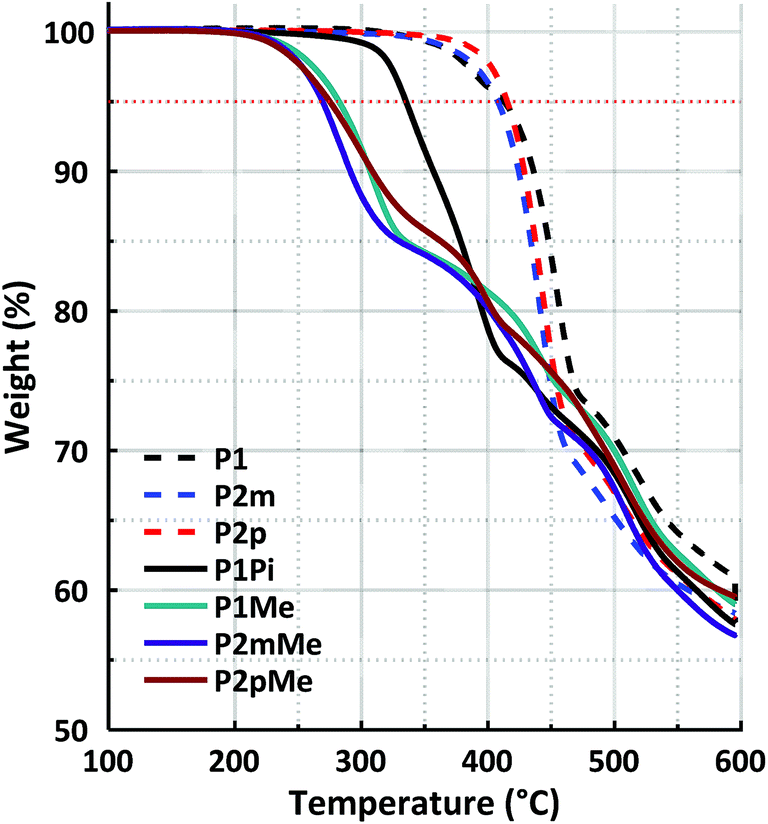
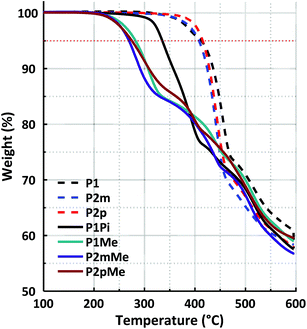
2.6 Thermal stability
The thermal stability of the precursor polymers in the protonated form with triflate counterions and the HEMs in the bromide form was investigated using thermogravimetric analysis (TGA) under a N2 atmosphere (Fig. 7). As expected, the precursor polymers had remarkably high thermal decomposition temperatures, all within a narrow temperature range. The thermal decomposition of P1, P2m and P2p occurred at Td,95 = 413, 408 and 415 °C, respectively. After quaternization, the thermal stability decreased sharply due to the presence of the QA cations. Membrane P1Pi with the rigid ASU cation decomposed at Td,95 = 335 °C, while the three HEMs functionalized with the more flexible DMP cation had Td,95 values between 269 and 282 °C. The thermal stability of the latter HEMs was similar to that of corresponding polymers with cyclic QAs directly attached to the polymer backbone. For example, we have previously found poly(p-terphenyl-N,N-dimethylpiperidinium) to decompose at Td,95 = 264 °C.25 In general, the increase in the polymer backbone rigidity by changing the position of the cation segments from the phenyl to the terphenyl units, or by replacing m-terphenyl units with p-terphenyl ones, had a positive effect on the thermal stability. Consequently, a comparison of the isomeric polymer structures of the present study showed that both P2pMe and P1Me had higher Td,95 values (275 and 282 °C, respectively) than P2mMe (269 °C).
 |
| | Fig. 7 TGA profiles of the protonated precursor polymers (triflate form) and the HEMs (bromide form). The red dotted line marks the 95% weight retention limit. | |
2.7 Alkaline stability
The thermochemical stability of the HEMs under the operating conditions of the electrochemical device is a crucial property that directly affects their performance and lifetime. The highly reactive hydroxide ions may readily attack and degrade QA cations by a number of different pathways. In the present case, the most plausible degradation pathways for the DMP and ASU cations are ring-opening Hofmann elimination, nucleophilic ring-opening substitution and nucleophilic substitution at a methyl group (Fig. S1†). All these reactions lead to the loss of the cationic charge by the formation of tertiary amines attached to the polymers.
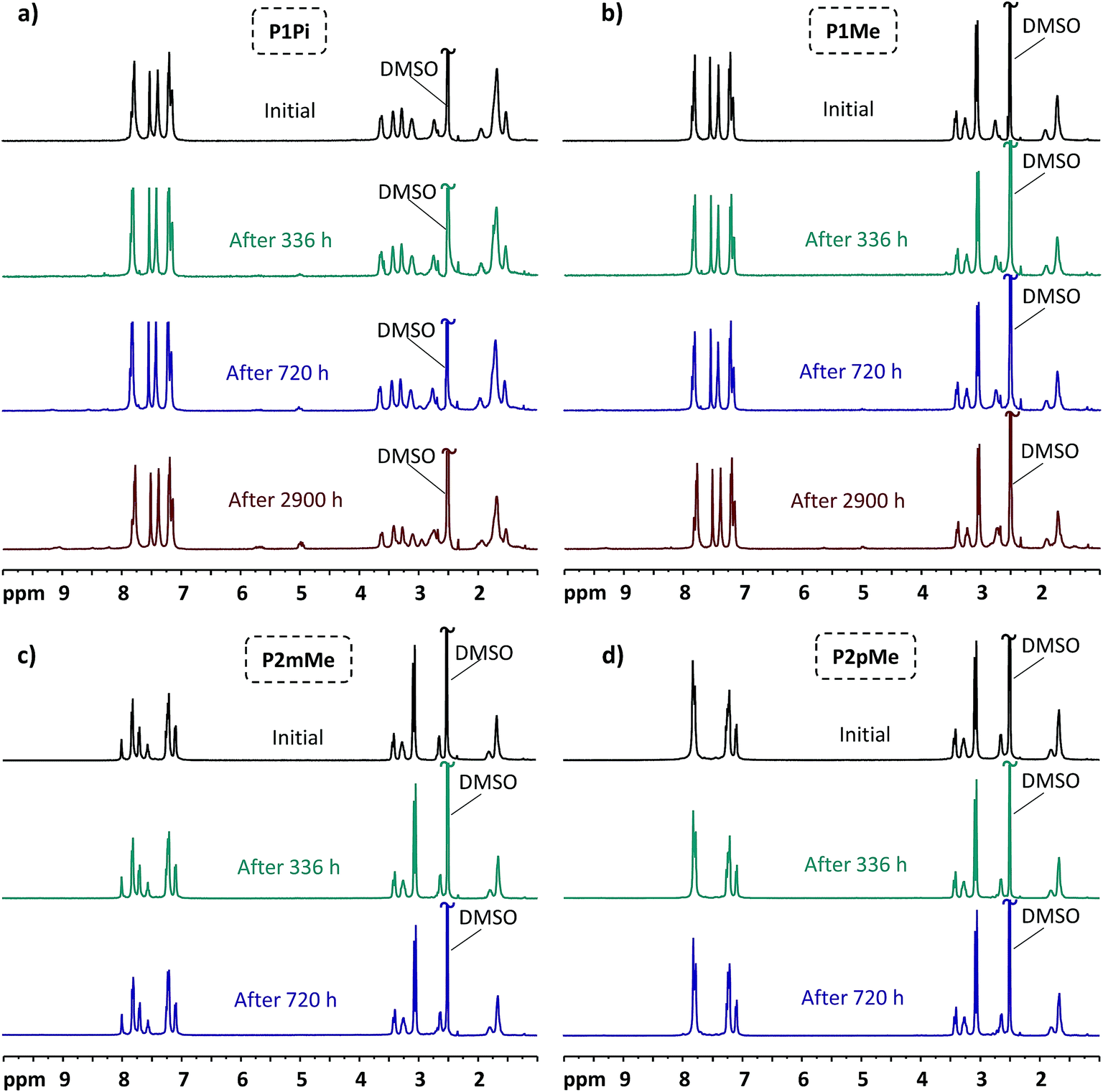
1H NMR spectroscopy is a powerful tool to detect membrane degradation products and study degradation mechanisms. The HEM samples of the present study were first stored during specific periods of time in 2 M aq. NaOH at 90 and 120 °C, respectively, and then exchanged to the bromide form, dried, dissolved in DMSO-d6 and finally analyzed by 1H NMR spectroscopy. By comparing 1H NMR spectra of the polymers before and after storage in alkaline solution, we have studied both the degradation degree and mechanisms of the polymer backbone and QA cations (Fig. 8). TFA was added to the polymer solutions in DMSO-d6 to shift the residual water signal from 3.33 to above 10 ppm during the analysis, revealing the signals originally overlapped by the water signal. Furthermore, all tertiary amine groups resulting from cationic loss were protonated by the highly acidic TFA and gave rise to signals above 9 ppm that were well separated from the original signals of the polymers. In addition, cationic loss via Hofmann elimination results in alkene products with distinct alkene and alkenyl signals at ∼4.9 and 5.6 ppm, respectively (Fig. S2 and S3†). Meanwhile, the excellent alkaline stability of the ether-free poly(terphenylene alkylene) backbone was confirmed since no apparent change in the 1H NMR signals in the aromatic region (7.0–8.0 ppm) were observed, and the HEMs retained their shape, transparency and flexibility after the alkaline stability test. Both the total cationic loss and the loss caused specifically by β-elimination were conveniently determined by comparing the intensity of the tertiary amine protons and the alkene/alkenyl signals, respectively, with the aromatic signals of the polymer backbone.
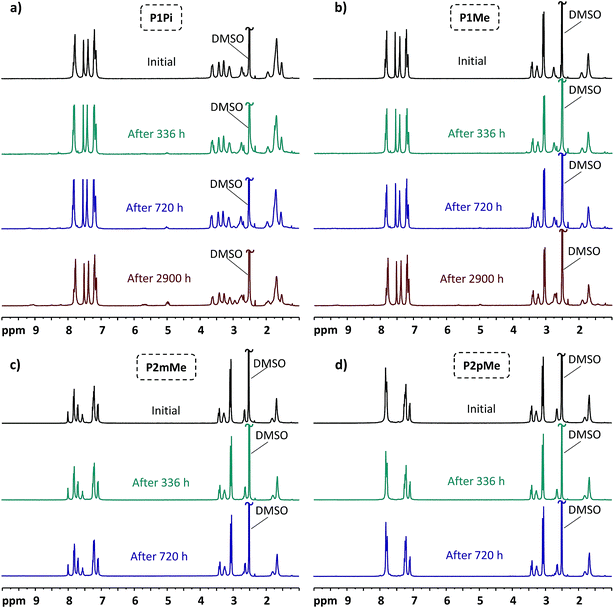 |
| | Fig. 8
1H NMR spectra of P1Pi (a), P1Me (b), P2mMe (c) and P2pMe (d) recorded in DMSO-d6/TFA before and after immersion in 2 M aq. NaOH at 90 °C for different periods of time. TFA was added to shift the water signals (originally at ∼3.3 ppm) to above 10 ppm, revealing sample signals between 3.0 and 3.5 ppm. | |
As can be seen in Fig. 8, the HEMs functionalized with the DMP cation (P1Me, P2mMe and P2pMe) were significantly more stable than P1Pi carrying the spirocyclic ASU cation after storage in alkaline solution at 90 °C. After 720 h storage, the total ionic loss of P1Pi reached approximately 10%. Meanwhile, the total ionic loss of the other three HEMs under the same conditions was less than 5%. When the storage time was extended to 2900 h, P1Pi recorded a total ionic loss of 27%, mainly via Hofmann elimination in both rings. In comparison, the membrane with the monocyclic arrangement, P1Me, also followed an alternative degradation pathway, most probably nucleophilic substitution at an α-methyl group. After 2900 h storage in alkaline solution at 90 °C, the ionic loss of P1Mevia Hofmann was 7%, while the total ionic loss was 13%. These findings agreed well with the results from our previous study, where the total ionic loss of a poly(arylene alkylene)-based HEM functionalized with DMP cations was found to be less than 10% after 720 h storage in 2 M aq. NaOH solution at 90 °C.25 Under the same conditions, the degree of degradation exceeded 20% for HEMs based on poly(arylene alkylene)s functionalized with ASU cations.26
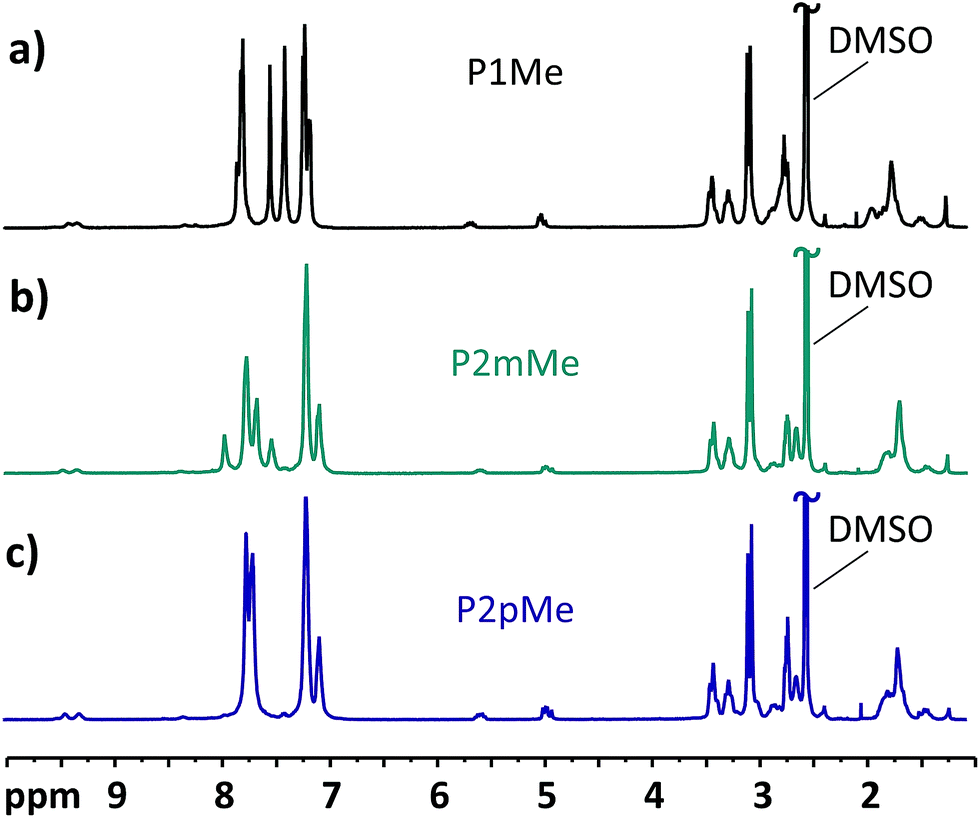
The ionic loss of the HEMs carrying DMP cations, i.e., membranes P1Me, P2mMe and P2pMe, was low and difficult to quantify after storage at 90 °C. In order to enable a comparison of the alkaline stability of these HEMs, the storage temperature was raised to 120 °C. The 1H NMR spectra of the polymers after 168 h storage in 2 M aq. NaOH at this temperature are displayed in Fig. 9. Under these harsher conditions, the cationic losses via Hofmann elimination for P1Me, P2mMe and P2pMe after 168 h increased to 16, 13 and 18%, respectively. At the same time, the total cationic losses were 33, 27 and 35%, respectively. These results hinted that the position of the cation on the more flexible pendant group as well as the increase in backbone flexibility had a positive effect on alkaline stability as P2mMe degraded notably less than both P1Me and P2pMe. However, since the arrangement with the DMP cation on the phenyl group still gives quite a rigid structure, the differences were small. The increased alkaline stability of P2mMe compared with that of P2pMe may also be partly caused by the higher water uptake and λ values of the former HEM.50 Hence, a higher flexibility of the polymer backbone and the pendant cations may facilitate strain relaxation of the alicyclic cationic rings, as well as a high λ value which will decrease the concentration of the harmful hydroxide ions.
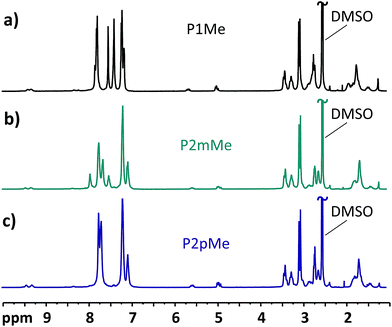 |
| | Fig. 9
1H NMR spectra of P1Me (a), P2mMe (b), and P2pMe (c) recorded in DMSO-d6/TFA before and after immersion in 2 M aq. NaOH at 120 °C during 168 h. TFA was added to shift the water signals (originally at ∼3.3 ppm) to above 10 ppm, revealing sample signals between 3.0 and 3.5 ppm. | |
3. Conclusions
A series of poly(terphenylene alkylene)s tethered with monocyclic DMP and spirocyclic ASU cations, respectively, were successfully prepared by superacid-mediated polycondensations of terphenyl and trifluoroketone monomers. The synthesis of the piperidine-functionalized m-terphenyl and trifluoroketone monomers allowed a systematic variation of the type and position of the cation and the rigidity of the backbone polymer to study the influence on HEM properties. Although all HEMs showed high alkaline stability, those functionalized with DMP cations showed the most attractive combination of high thermal and alkaline stability, high hydroxide conductivity and restricted water uptake. In particular, the placement of the DMP cation on the pendant phenyl group of the ketone monomer residues instead of on the more rigid backbone terphenyl units resulted in an improvement of both alkaline stability and hydroxide conductivity. Functionalization with the bulky ASU cation gave very high thermal stability, but poor ionic clustering and low conductivity. In addition, the ASU cation showed a lower alkaline stability than the DMP cation when compared on the same backbone and position. This might be explained by a larger degree of bond angle distortion of the former cation when attached to the rigid polymer backbone, which may facilitate degradation. The present study provides important structure–property relationships for the development of alkali-stable HEMs based on diaryl ether-free aromatic polymers carrying N-alicyclic cations having all β-hydrogens placed in 6-membered rings.
4. Experimental
4.1 Materials
1-N-Boc-4-Methylene-piperidine (96%, Fluorochem), 9-borabicyclo[3.3.1]nonane (9BBN, 0.5 M in THF, Sigma-Aldrich), tetrakis(triphenylphosphine)palladium(0) (99%, Sigma-Aldrich), 5′-bromo-m-terphenyl (98%, Fluorochem), 4′-bromo-2,2,2-trifluoroacetophenone (97%, Fluorochem), HCl (37%, VWR), m-terphenyl (99%, Alfa Aesar), p-terphenyl (99.5%, Sigma-Aldrich), 2,2,2-trifluoroacetophenone (TFAp, 99%, Sigma-Aldrich), 1,1,1-trifluoroacetone (TFAc, 99%, Sigma-Aldrich), 1,1,1-trifluoroacetic acid (TFA, 99%, Acros), triflic acid (TFSA, 99%, Acros), 1,5-dibromopentane (97%, Sigma-Aldrich), N,N-diisopropylethylamine (DIPEA, ≥99%, Sigma-Aldrich), methyliodide (99%, Sigma Aldrich), K2CO3 (99%, Sigma-Aldrich), toluene (reagent grade, VWR), isopropanol (IPA, reagent grade, VWR), diethyl ether (Et2O, reagent grade, VWR), N-methyl-2-pyrrolidone (NMP, reagent grade, Acros), dimethyl sulfoxide (DMSO, reagent grade, VWR), ethyl acetate (EtOAc, reagent grade, VWR), heptane (reagent grade, VWR), NaBr (99%, Sigma-Aldrich), ethanol (99.5%, Solveco), NaOH (99% pellets, VWR), KOH (99% pellets, VWR), CDCl3 (99.8 atom% D, Sigma-Aldrich) and DMSO-d6 (99.5 atom% D, Sigma-Aldrich) were all used as received. Dichloromethane was dried using an MBraun dry solvent dispenser system MB-SPS 800.
4.2 Polymer synthesis
Monomer synthesis.
Monomers 1 and 2 were synthesized in three steps as shown in Scheme 2. All steps except the deprotection using HCl were performed under a N2 atmosphere. Here follows a typical procedure for synthesis of monomer 2. 1-N-Boc-4-Methylene-piperidine (5.08 g, 25.8 mmol, 1 eq.) was added in a 100 ml one-neck round flask, and then degassed and cooled to 0 °C using an ice bath. A solution of 9BBN 0.5 M in THF (51.5 ml, 25.8 mmol, 1 eq.) was added and the white reaction mixture was stirred during 2 h before the ice bath was removed. The reaction mixture was stirred at RT for an additional 2 h, and then degassed and added dropwise to a degassed mixture of 4′-bromo-2,2,2-trifluoroacetophenone (6.52 g, 25.8 mmol, 1 eq.), KOH (2.60 g, 46.4 mmol, 1.8 eq.), Pd(PPh3)4 (0.431 g, 0.373 mmol, 0.0145 eq.), toluene (25 ml) and deionized (DI) water (45 ml) in a 250 ml two-neck round flask equipped with a condenser and N2 inlet. The mixture was stirred at 60 °C overnight and then extracted with EtOAc. The combined organic phase was washed with brine, then dried with MgSO4 and evaporated under reduced pressure until dry. The crude product was purified further by dry column vacuum chromatography,51 using an EtOAc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :heptane mixture (0:100 to 50:50) as the eluent. The still impure product was added to a 100 ml round flask containing HCl (conc., 25 ml) and DI water (25 ml) and then heated to 80 °C overnight. The solution was then cooled to 0 °C and filtered. The precipitate was recrystallized in water, yielding monomer 2 (in the hydrated form) as a white powder (5.0 g, total isolated yield 63%).
:heptane mixture (0:100 to 50:50) as the eluent. The still impure product was added to a 100 ml round flask containing HCl (conc., 25 ml) and DI water (25 ml) and then heated to 80 °C overnight. The solution was then cooled to 0 °C and filtered. The precipitate was recrystallized in water, yielding monomer 2 (in the hydrated form) as a white powder (5.0 g, total isolated yield 63%).
Polymerization.
The three precursor polymers P1, P2m and P2p were synthesized by polycondensation of either monomer 1 with 2,2,2-trifluoroacetophenone or of monomer 2 with m- or p-terphenyl, respectively, under a N2 atmosphere, using triflic acid as the catalyst (Scheme 3). Here follows the description of the synthesis of polymer P1 as an example. To a 25 ml two-neck round flask equipped with mechanical stirrer and N2 inlet monomer 1 in chloride form (1.00 g, 2.75 mmol, 1 eq.), 2,2,2-trifluoroacetophenone (0.424 ml, 3.02 mmol, 1.1 eq.) and anhydrous DCM (2.4 ml) were added. The mixture was cooled to 0 °C using an ice bath, followed by the dropwise addition of TFSA (2.4 ml, 27 mmol, 10 eq.). The mixture was stirred at 0 °C during 24 h. The highly viscous reaction mixture was then diluted with DMSO and precipitated in IPA. The precipitate was washed twice with diethyl ether, and then dried under vacuum, yielding polymer P1 (1.65 g, 94% isolated yield) as white powder.
Quaternization.
The cationic polymers P1Me, P2mMe and P2pMe were prepared by quaternization of the respective precursor polymers, using MeI in NMP and K2CO3 as the catalyst. The synthesis of P1Me is described here as an example. Polymer P1 (0.6 g, 0.95 mmol, 1 eq.), K2CO3 (0.39 g, 2.8 mmol, 3 eq.), NMP (10 ml) and MeI (0.29 ml, 4.7 mmol, 5 eq.) were added in a 50 ml one-neck round bottom flask. The bottle was covered in aluminium foil to prevent light-induced degradation of MeI and left stirring at room temperature for 48 h. The product was then precipitated in IPA, washed repeatedly with IPA and water, and finally dried under vacuum to give P1Me as a light yellow powder.
Polymer P1Pi was prepared by cyclo-quaternization of polymer P1 with 1,5-dibromopentane, using DIPEA as the catalyst. A solution of polymer P1 (0.6 g, 0.95 mmol, 1 eq.) in NMP (10 ml) was added dropwise into a 50 ml round bottom flask containing 1,5-dibromopentane (0.14 ml, 1.0 mmol, 1.1 eq.), DIPEA (0.82 ml, 4.7 mmol, 5 eq.) and NMP (14 ml) at 80 °C. Next, the reaction mixture was stirred at 80 °C during 24 h. The product was then precipitated in IPA, washed repeatedly with IPA and water and dried under vacuum to give P1Pi as a red brown powder.
Characterization.
The molecular structure of the polymers was confirmed from 1H NMR spectra recorded at 400 MHz using a Bruker DRX 400 spectrometer. The solvents used were either DMSO-d6 or a mixture of DMSO-d6 and TFA.
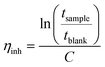
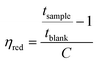
The intrinsic viscosity of the precursor polymers P1, P2m and P2p was measured at 30 °C using an Ubbelohde viscometer. Samples were dried at 50 °C under vacuum for at least 48 h, weighed, and dissolved in 0.1 M LiBr in DMSO solution (blank solution) to obtain stock solutions with a concentration between 0.83 and 1.29 g dL−1. These solutions were later diluted with the blank solution to reduce their concentrations. The resulting solutions were used immediately after preparation. The flow times of the blank solution through the capillary (tblank) and of the polymer solutions (tsample) were taken as the average of four measurements. The inherent (ηinh) and reduced (ηred) viscosities at four different concentrations were calculated as:
| |  | (1) |
| |  | (2) |
The intrinsic viscosity ([η]) was calculated as the average of the intersections of the linear regressions of ηinh and ηred with the y-axis.
4.3 Membrane preparation and characterization
Membrane preparation.
HEMs were cast from 5 wt% polymer solutions in DMSO at 80 °C. Approximately 0.15 g polymer was dissolved in DMSO to obtain a 3 g solution. This solution was filtered through a syringe-driven filter unit (ø = 25 mm, Fluoropore membrane, 5 μm) onto a Petri dish (ø = 50 mm) which was then placed in a ventilated casting oven at 80 °C for at least 24 h. Subsequently, the resulting HEM was immersed in 1 M aq. NaBr solution for at least 7 days to obtain the bromide form, and then washed thoroughly with DI water before storage in DI water. Membranes of the polymers functionalized with monocyclic piperidinium P1Me, P2mMe and P2pMe were flexible and almost colorless while the P1Pi membranes were darker in colour and less flexible.
HEMs in the hydroxide form were prepared by ion-exchange in 1 M aq. NaOH solution during at least 96 h. Next, the membrane was washed thoroughly with degassed DI water and stored in degassed DI water under nitrogen during 48–72 h before the measurement.
Small angle X-ray scattering.
Phase separation by ionic clustering was studied by small angle X-ray scattering (SAXS) measurements of the dry HEMs in the bromide form. Membrane samples were dried under vacuum prior to the measurements. Data were collected in the q-range 0.14–7.5 nm−1 using a SAXSLAB SAXS instrument (JJ X-ray Systems Aps, Denmark) equipped with a Pilatus detector.
Ion exchange capacity.
The IEC of the HEMs in the bromide (IECBr) form was determined by titration. The membranes were first dried at 50 °C under vacuum during 48 h and weighed to obtain their dry weights. The dry membranes were then immersed in 25 ml 0.2 M aq. NaNO3 solution at 40–50 °C for 7 days. Next the solutions were titrated with an aq. AgNO3 solution (approx. 0.01 M), using an aq. K2CrO4 solution (0.1 M) as the indicator. The IEC of the HEMs in the hydroxide form (IECOH) was calculated from the IECBr as| | 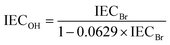 | (3) |
Thermal stability.
The thermal decomposition of the HEMs in the bromide form and of precursor polymers in the protonated state with triflate counter ions was studied by thermogravimetric analysis (TGA) using a TGA Q500 (TA Instruments) during heating from 50 to 600 °C at 10 °C min−1 under a N2 atmosphere. In order to remove water residues, the samples were kept isothermally at 120 °C for 20 min prior to the analysis. The thermal decomposition temperature was reported at 5% weight loss (Td,95).
Water uptake.
The dry weight of the membranes (mdry,Br) in the bromide form was measured after 48 h drying at 50 °C under vacuum. By assuming that all Br− was exchanged to OH−, the dry weight of the HEMs in the hydroxide form (mdry) was calculated from mdry,Br as| | | mdry = mdry,Br × (1 − 0.0629 × IECBr) | (4) |
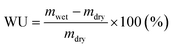
Subsequently, the HEMs were ion-exchanged to the hydroxide form. The weights of the hydrated membranes (mwet) were obtained after equilibration in DI water at 20, 40, 60 and 80 °C. Finally their water uptake was calculated as:
| |  | (5) |
Hydroxide conductivity.
The hydroxide ion conductivity of the hydrated HEMs was measured by electrochemical impedance spectroscopy using a two-probe set-up and a Novocontrol high-resolution dielectric analyzer V 1.01S. During the measurements, the membranes were kept hydrated in a closed cell filled with DI degassed water. The voltage amplitude was maintained at 50 mV while varying the frequency from 107 to 100 Hz in the temperature range of 20 to 80 °C.
Alkaline stability.
The alkaline stability of all HEMs at 90 and 120 °C was evaluated after immersion in 2 M aq. NaOH solution for different periods of time. After the immersion, the membranes were exchanged to the bromide form and dissolved in mixtures of DMSO-d6 and TFA before analysis by 1H NMR spectroscopy.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the Swedish Energy Agency (grants 45057-1 and 37806-3), the Swedish Research Council (grants 45397-1 and 2015-04820) and the Swedish Foundation for Strategic Research, SSF (grant EM16-0060), for financial support. We are also grateful to Peter Holmqvist for assistance with SAXS measurements and data treatment.
References
- G. He, Z. Li, J. Zhao, S. Wang, H. Wu, M. D. Guiver and Z. Jiang, Adv. Mater., 2015, 27(36), 5280–5295 CrossRef CAS PubMed.
- S. Gottesfeld, D. R. Dekel, M. Page, C. Bae, Y. Yan, P. Zelenay and Y. S. Kim, J. Power Sources, 2018, 375, 170–184 CrossRef CAS.
- G. Merle, M. Wessling and K. Nijmeijer, J. Membr. Sci., 2011, 377(1–2), 1–35 CrossRef CAS.
- M. A. Hickner, A. M. Herring and E. B. Coughlin, J. Polym. Sci., Part B: Polym. Phys., 2013, 51(24), 1727–1735 CrossRef CAS.
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7(10), 3135–3191 RSC.
- N. Li, Q. Zhang, C. Wang, Y. M. Lee and M. D. Guiver, Macromolecules, 2012, 45(5), 2411–2419 CrossRef CAS.
- G. Couture, A. Alaaeddine, F. Boschet and B. Ameduri, Prog. Polym. Sci., 2011, 36(11), 1521–1557 CrossRef CAS.
- C. Vogel and J. Meier-Haack, Desalination, 2014, 342, 156–174 CrossRef CAS.
- H. S. Dang, E. A. Weiber and P. Jannasch, J. Mater. Chem. A, 2015, 3(10), 5280–5284 RSC.
- J. L. Yan and M. A. Hickner, Macromolecules, 2010, 43(5), 2349–2356 CrossRef CAS.
- A. D. Mohanty, C. Y. Ryu, Y. S. Kim and C. Bae, Macromolecules, 2015, 48(19), 7085–7095 CrossRef CAS.
- H. Zarrin, J. Wu, M. Fowler and Z. W. Chen, J. Membr. Sci., 2012, 394, 193–201 CrossRef.
- R. Akiyama, N. Yokota and K. Miyatake, Macromolecules, 2019, 52(5), 2131–2138 CrossRef CAS.
- S. Miyanishi and T. Yamaguchi, Phys. Chem. Chem. Phys., 2016, 18(17), 12009–12023 RSC.
- T. H. Pham and P. Jannasch, ACS Macro Lett., 2015, 4(12), 1370–1375 CrossRef CAS.
- A. D. Mohanty, S. E. Tignor, J. A. Krause, Y. K. Choe and C. Bae, Macromolecules, 2016, 49(9), 3361–3372 CrossRef CAS.
- C. G. Arges and V. Ramani, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(7), 2490–2495 CrossRef CAS PubMed.
- A. Amel, L. Zhu, M. Hickner and Y. Ein-Eli, J. Electrochem. Soc., 2014, 161(5), F615–F621 CrossRef CAS.
- C. Fujimoto, D.-S. Kim, M. Hibbs, D. Wrobleski and Y. S. Kim, J. Membr. Sci., 2012, 423–424, 438–449 CrossRef CAS.
- Y.-K. Choe, C. Fujimoto, K.-S. Lee, L. T. Dalton, K. Ayers, N. J. Henson and Y. S. Kim, Chem. Mater., 2014, 26(19), 5675–5682 CrossRef CAS.
- W.-H. Lee, A. D. Mohanty and C. Bae, ACS Macro Lett., 2015, 4(4), 453–457 CrossRef CAS.
- M. Ozawa, T. Kimura, K. Otsuji, R. Akiyama, J. Miyake, M. Uchida, J. Inukai and K. Miyatake, ACS Omega, 2018, 3(11), 16143–16149 CrossRef CAS.
- A. G. Wright and S. Holdcroft, ACS Macro Lett., 2014, 3(5), 444–447 CrossRef CAS.
- J. Wang, Y. Zhao, B. P. Setzler, S. Rojas-Carbonell, C. Ben Yehuda, A. Amel, M. Page, L. Wang, K. Hu, L. Shi, S. Gottesfeld, B. Xu and Y. Yan, Nat. Energy, 2019, 4, 392–398 CrossRef CAS.
- J. S. Olsson, T. H. Pham and P. Jannasch, Adv. Funct. Mater., 2018, 28(2), 1702758 CrossRef.
- T. H. Pham, J. S. Olsson and P. Jannasch, J. Mater. Chem. A, 2018, 6(34), 16537–16547 RSC.
- S. Chempath, B. R. Einsla, L. R. Pratt, C. S. Macomber, J. M. Boncella, J. A. Rau and B. S. Pivovar, J. Phys. Chem. C, 2008, 112(9), 3179–3182 CrossRef CAS.
- J. B. Edson, C. S. Macomber, B. S. Pivovar and J. M. Boncella, J. Membr. Sci., 2012, 399–400, 49–59 CrossRef CAS.
- M. G. Marino and K. D. Kreuer, ChemSusChem, 2015, 8(3), 513–523 CrossRef CAS PubMed.
- D. Chen and M. A. Hickner, ACS Appl. Mater. Interfaces, 2012, 4(11), 5775–5781 CrossRef CAS PubMed.
- H.-S. Dang and P. Jannasch, J. Mater. Chem. A, 2016, 4(30), 11924–11938 RSC.
- T. H. Pham, J. S. Olsson and P. Jannasch, J. Am. Chem. Soc., 2017, 139(8), 2888–2891 CrossRef CAS PubMed.
- L. Gu, H. Dong, Z. Sun, Y. Li and F. Yan, RSC Adv., 2016, 6(97), 94387–94398 RSC.
- D. J. Strasser, B. J. Graziano and D. M. Knauss, J. Mater. Chem. A, 2017, 5(20), 9627–9640 RSC.
- H. Peng, Q. Li, M. Hu, L. Xiao, J. Lu and L. Zhuang, J. Power Sources, 2018, 390, 165–167 CrossRef CAS.
- N. Chen, C. Long, Y. Li, C. Lu and H. Zhu, ACS Appl. Mater. Interfaces, 2018, 10(18), 15720–15732 CrossRef CAS PubMed.
- X. Chu, Y. Shi, L. Liu, Y. Huang and N. Li, J. Mater. Chem. A, 2019, 7(13), 7717–7727 RSC.
- F. Gong, R. Wang, X. Chen, P. Chen, Z. An and S. Zhang, Polym. Chem., 2017, 8(29), 4207–4219 RSC.
- C. Vogel, H. Komber and J. Meier-Haack, React. Funct. Polym., 2017, 117, 34–42 CrossRef CAS.
- Y. Yang, Y. Xu, N. Ye, D. Zhang, J. Yang and R. He, J. Electrochem. Soc., 2018, 165(5), F350–F356 CrossRef CAS.
- W.-H. Lee, Y. S. Kim and C. Bae, ACS Macro Lett., 2015, 4(8), 814–818 CrossRef CAS.
- M. T. Guzmán-Gutiérrez, D. R. Nieto, S. Fomine, S. L. Morales, M. G. Zolotukhin, M. C. G. Hernandez, H. Kricheldorf and E. S. Wilks, Macromolecules, 2011, 44(2), 194–202 CrossRef.
- M. Zolotukhin, S. Fomine, R. Salcedo and L. Khalilov, Chem. Commun., 2004,(8), 1030–1031 RSC.
- A. M. Diaz, M. G. Zolotukhin, S. Fomine, R. Salcedo, O. Manero, G. Cedillo, V. M. Velasco, M. T. Guzman, D. Fritsch and A. F. Khalizov, Macromol. Rapid Commun., 2007, 28(2), 183–187 CrossRef CAS.
- A. R. Cruz, M. G. Zolotukhin, S. L. Morales, J. Cardenas, G. Cedillo, S. Fomine, M. Salmon and M. P. Carreón-Castro, Chem. Commun., 2009,(29), 4408–4410 RSC.
- S. Vice, T. Bara, A. Bauer, C. A. Evans, J. Ford, H. Josien, S. McCombie, M. Miller, D. Nazareno, A. Palani and J. Tagat, J. Org. Chem., 2001, 66(7), 2487–2492 CrossRef CAS PubMed.
- J. Su, Y. Wang, J. Lin, J. Liang, J. Sun and X. Zou, Dalton Trans., 2013, 42(5), 1360–1363 RSC.
- W.-H. Lee, E. J. Park, J. Han, D. W. Shin, Y. S. Kim and C. Bae, ACS Macro Lett., 2017, 6(5), 566–570 CrossRef CAS.
- J. S. Olsson, T. H. Pham and P. Jannasch, J. Membr. Sci., 2019, 578, 183–195 CrossRef CAS.
- D. R. Dekel, M. Amar, S. Willdorf, M. Kosa, S. Dhara and C. E. Diesendruck, Chem. Mater., 2017, 29(10), 4425–4431 CrossRef CAS.
- D. S. Pedersen and C. Rosenbohm, Synthesis, 2001, 2001(16), 2431–2434 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta05531b |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Joel S.
Olsson
,
Joel S.
Olsson