
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrigin of the heat-induced improvement of catalytic activity and stability of MnOx electrocatalysts for water oxidation†
Miroslav V.
Abrashev
 *a,
Petko
Chernev
bc,
Paul
Kubella
b,
Mohammad Reza
Mohammadi
bd,
Chiara
Pasquini
b,
Holger
Dau
b and
Ivelina
Zaharieva
*b
*a,
Petko
Chernev
bc,
Paul
Kubella
b,
Mohammad Reza
Mohammadi
bd,
Chiara
Pasquini
b,
Holger
Dau
b and
Ivelina
Zaharieva
*b
aFaculty of Physics, University of Sofia “St. Kliment Ohridski”, 1164 Sofia, Bulgaria. E-mail: mvabr@phys.uni-sofia.bg
bFachbereich Physik, Freie Universität Berlin, Arnimallee 14, 14195 Berlin, Germany. E-mail: ivelina.zaharieva@physik.fu-berlin.de
cDepartment of Chemistry, Ångström Laboratory, Lägerhyddsvägen 1, 75120 Uppsala, Sweden
dDepartment of Physics, University of Sistan and Baluchestan, Zahedan, 98167-45845, Iran
First published on 18th June 2019
Abstract
Catalysis of the oxygen evolution reaction (OER) by earth-abundant materials in the near-neutral pH regime is of great interest as it is the key reaction for non-fossil fuel production. To address the pertinent stability problems and insufficiently understood structure–activity relations, we investigate the influence of moderate annealing (100–300 °C for 20 min) for two types of electrodeposited Mn oxide films with contrasting properties. Upon annealing, the originally inactive and structurally well-ordered Oxide 1 of birnessite type became as OER active as the non-heated Oxide 2, which has a highly disordered atomic structure. Oxide 2 also improved its activity upon heating, but more important is the stability improvement: the operation time increased by about two orders of magnitude (in 0.1 M KPi at pH 7). Aiming at atomistic understanding, electrochemical methods including quantitative analysis of impedance spectra, X-ray spectroscopy (XANES and EXAFS), and adapted optical spectroscopies (infrared, UV-vis and Raman) identified structure–reactivity relations. Oxide structures featuring both di-μ-oxo bridged Mn ions and (close to) linear mono-μ-oxo Mn3+–O–Mn4+ connectivity seem to be a prerequisite for OER activity. The latter motif likely stabilizes Mn3+ ions at higher potentials and promotes electron/hole hopping, a feature related to electrical conductivity and reflected in the strongly accelerated rates of Mn oxidation and O2 formation. Poor charge mobility, which may result from a low level of Mn3+ ions at high potentials, likely promotes inactivation after prolonged operation. Oxide structures related to the perovskite-like ζ-Mn2O3 were formed after the heating of Oxide 2 and could favour stabilization of Mn ions in oxidation states lower than +4. This rare phase was previously found only at high pressure (20 GPa) and temperature (1200 °C) and this is the first report where it was stable under ambient conditions.
Introduction
Oxidation of water to molecular oxygen is a key reaction in the production of chemical fuels for storage of renewable energy. As the process is mechanistically challenging, it requires high oxidation potentials; therefore efficient catalysts are needed. Mn based catalysts are particularly interesting due to their elemental and structural similarity to the biological catalyst which oxidizes water molecules as a step of the biological photosynthesis. The catalytic center operating in photosynthesising organisms, the CaMn4O5 cluster, has a well-defined structure, strongly restrained by the surrounding protein environment and highly preserved during evolution.1–3 In contrast, inorganic Mn oxides exist in more than 30 different stable polymorphs.4,5 The difference in oxide structures results in different catalytic activities,6–10 but so far, the catalytic centers have not been identified, which has hampered the understanding of the mechanism of the catalytic reaction and the development of efficient and stable catalysts.A simple solution for renewable energy production is electrochemical water splitting using electricity produced by solar cells. This setup requires the catalyst to be integrated into the anode of the (photo)electrochemical cell. In most cases of Mn oxides attached to an electrode and used in anodic water oxidation, the catalytically active phase was identified as (disordered) layered Mn oxide, birnessite.11–20 Recent studies show that the heating of electrodeposited Mn-oxide electrodes to moderately high temperatures often results in an increase of their catalytic activity.11,17,21,22 The improvement after heating to 90 °C was related to irreversible dehydration of the sample21 and formation of a small fraction of Mn3O4 (hausmannite)22 but further heating to 120 °C was suggested to rather degrade the catalytic performance.22 In another study, increased catalytic activity was found after heating to 210 °C and this was explained by the heat-induced amorphization of well-ordered MnOx layers.17
The stability of the Mn oxide catalysts for water oxidation reported in the literature so far is very low. The instability can be due to leaching of Mn ions into solution,16 or due to structural changes in the oxide material itself, or it can be related to changes in the contact between the oxide and the electrode surface induced by the applied potential. An understanding of the mechanisms of catalyst inactivation is required in order to develop stable and efficient catalysts for practical applications.
We study the effect of heating to moderate temperatures on the catalytic activity of electrodeposited Mn oxides with originally very different catalytic activities. The as-synthesised oxides were already investigated by X-ray absorption spectroscopy (XAS) in an attempt to identify the reasons for their contrasting catalytic properties.18,23 It was found that the catalytically active oxide has a highly disordered structure composed of a mixture of di-μ-(hydro)oxo bridged and mono-μ-(hydro)oxo bridged Mn3+/4+ ions. The second oxide had very low catalytic activity and a better ordered layered structure similar to the structure of birnessite.24 Now we study the effect of heating on both of them—the inactive oxide, deposited at constant potential (Oxide 1), and the catalytically active MnCat, deposited during cyclic voltammetry and used in this case as a reference system (Oxide 2). Combining the XAS structural investigations with electrochemical studies and Raman, infrared and UV-vis spectroscopy, we identify the structural features and material properties required for the catalytic activity and stable operation of the manganese oxide catalysts.
Results
Effect of heating on the electrocatalytic properties
Two oxides with different catalytic activities towards electrochemical water oxidation were deposited onto indium tin oxide (ITO) glass substrates using two very simple electrochemical techniques, described earlier.11 According to the previous experiments, electrodeposition at constant potential results in the formation of an inactive Mn oxide coating (Oxide 1), while deposition during gradual potential cycling results in a material with sizable catalytic activity (Oxide 2, MnCat). We studied the effect of heating on the two oxides up to 300 °C, as at higher temperatures the ITO substrate degrades. Fig. 1 compares the cyclic voltammograms (CVs) of both electrode types measured at room temperature (RT) after 20 min of heating.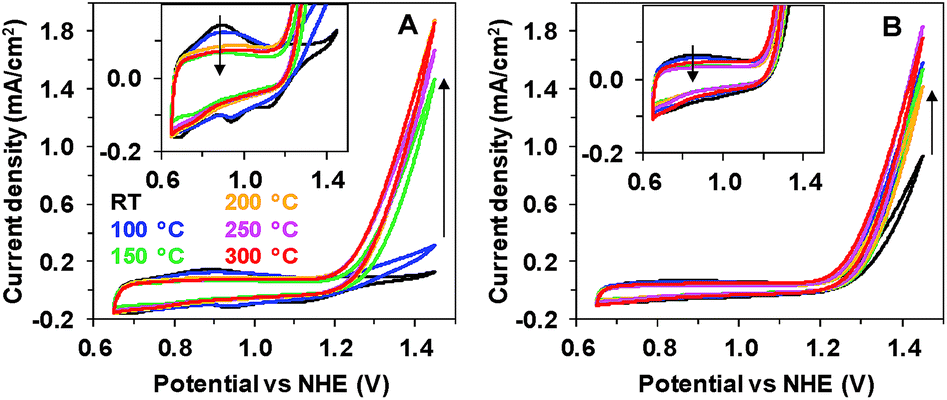 | ||
| Fig. 1 Effect of 20 min of heating on the activity of inactive Mn oxide (A, Oxide 1) and MnCat (B, Oxide 2). CVs at a scan rate of 20 mV s−1 were taken at room temperature in 0.1 M Mn-free KPi (pH 7). The second cycle is shown. The insets show an enlargement of the heat-induced changes in the non-catalytic regime. Arrows indicate the direction of changes with temperature increase. | ||
Heat treatment has a strong effect on Oxide 1, where it induces a catalytic activity comparable to that of Oxide 2 (Fig. 1A), while it affects only moderately the activity of Oxide 2 (Fig. 1B). For Oxide 1, heating above 150 °C results in a current density of about 1.5 mA cm−2 at 1.45 V at pH 7. In parallel, the well-defined peaks visible in the non-catalytic range (0.8–1.2 V) in the CV of Oxide 1 disappear, indicating structural amorphization, as proven also by X-ray absorption and Raman spectroscopy (see below). The peaks in the CV in the non-catalytic regime are likely due to Mn2+/Mn3+ and Mn3+/Mn4+ oxidation.25 As shown earlier, in the active MnCat (Oxide 2), these separate redox transitions are not readily resolved, which is likely explainable by extreme broadening of these redox transitions by electronic interactions of (different) Mn sites in the amorphous material.11
We also tested the effect of heating duration on the catalytic performance (ESI, Fig. S1†). A pronounced effect of the heating time on the CVs was observed only in the case of heating to 100 °C, where the temperature effect increases with the duration up to 80 min. At higher temperatures, 20 min was sufficient to reach the full effect in these thin films (approx. thickness of the Mn coating: 20 nm).11 At 300 °C, the catalytic performance even decreased for heating times longer than 20 min, which can be explained by the degradation of the ITO substrate. For the rest of the measurements presented in this study we chose 20 min as the heating time.
Heating induces changes also in other electrochemical characteristics like the Tafel slopes (see ESI Fig. S2†). To determine the Tafel slopes, we measured the currents after 90 s of equilibration at each potential, thus avoiding interference with the redox-currents observed during a CV scan. Similar to the CVs, the heating has little effect on Oxide 2, while for Oxide 1, heating at 100 °C already induces a strong decrease in the Tafel slope.
As shown in Fig. 2B, the short 20 min heating dramatically improves the stability of Oxide 2 (MnCat). For Oxide 2, the heating to temperatures in the interval between 150 and 250 °C results in stability for more than 24 h of operation in Mn2+-free buffer (without stirring) at a current density of 1 mA cm−2 and an overpotential of about 700–750 mV. The stability of Oxide 1 (Fig. 2A) is significantly lower (less than 5 h), but the overpotentials are comparable to those for the MnCat.
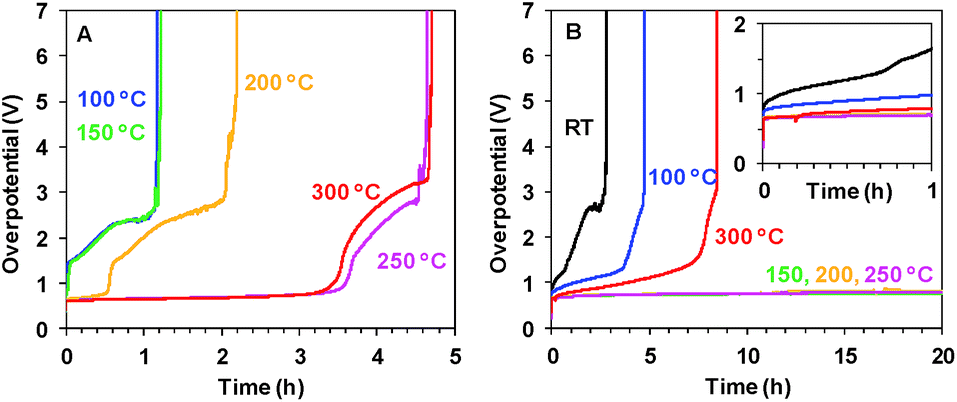 | ||
| Fig. 2 Chronopotentiometric measurements at 1 mA cm−2 in 0.1 M KPi (pH 7) for Oxide 1 (A) and MnCat, Oxide 2 (B) heated for 20 min at different temperatures. Note the different scales on the x axes in A and B. The inset in panel B shows the potential change during the first 1 h. | ||
Heat-induced structural changes
To answer the question whether the observed heat-induced changes in the activity and stability are due to structural changes in the Mn oxide materials, we applied X-ray absorption spectroscopy. This method is suitable for characterization of non-crystalline thin films and it has already been applied for structural characterization of non-heated Mn oxides both in their resting state as well as during operation.11,18,23Fig. 3A shows the X-ray absorption near-edge spectra (XANES) of Oxide 1 after 20 min of heating at different temperatures. The energy position of the X-ray absorption edge is related to the average oxidation state of Mn ions in the film, which can be determined using a calibration curve built from Mn oxides with known structures and oxidation states.7,26 The average Mn oxidation state of the as-deposited Oxide 1 was about 3.5 (Table 1). We note that in this study the oxidation state at room temperature was determined after drying the film in air for 20 min. The drying results in a slight decrease of the oxidation state in comparison to the previously reported oxidation state determined from XANES spectra taken after 30 min of incubation in KPi buffer and freezing the film with the buffer to preserve the natural resting state of the materials, where the Mn oxidation state for Oxide 1 was about 3.7.23 Heating at temperatures higher than RT results in a further decrease of the average Mn oxidation state of the material and this effect is observed up to 300 °C, where the oxidation state increases slightly again (Table 1). Interestingly, despite the different catalytic activity of Oxide 2, the Mn oxidation state of the dried films is similar at all temperatures (see Table 1 and ESI Fig. S3†), in agreement with recent findings that the total amount of Mn3+ ions in the resting state does not necessarily correlate with the catalytic activity.27 Notably, for Oxide 2, heating at 300 °C also induced a change in the edge shape, indicative of further structural rearrangements (ESI, Fig. S3†). In contrast, for Oxide 1, heating does not affect significantly the shape of the edge spectrum (Fig. 3A). When exposed to oxidizing potentials, non-heated Oxide 1 quickly reaches an all-Mn4+ oxidation state, while such a state in Oxide 2, as well as in Oxide 1 after heating, cannot be reached (Table 1, see also ESI Fig. S4†).
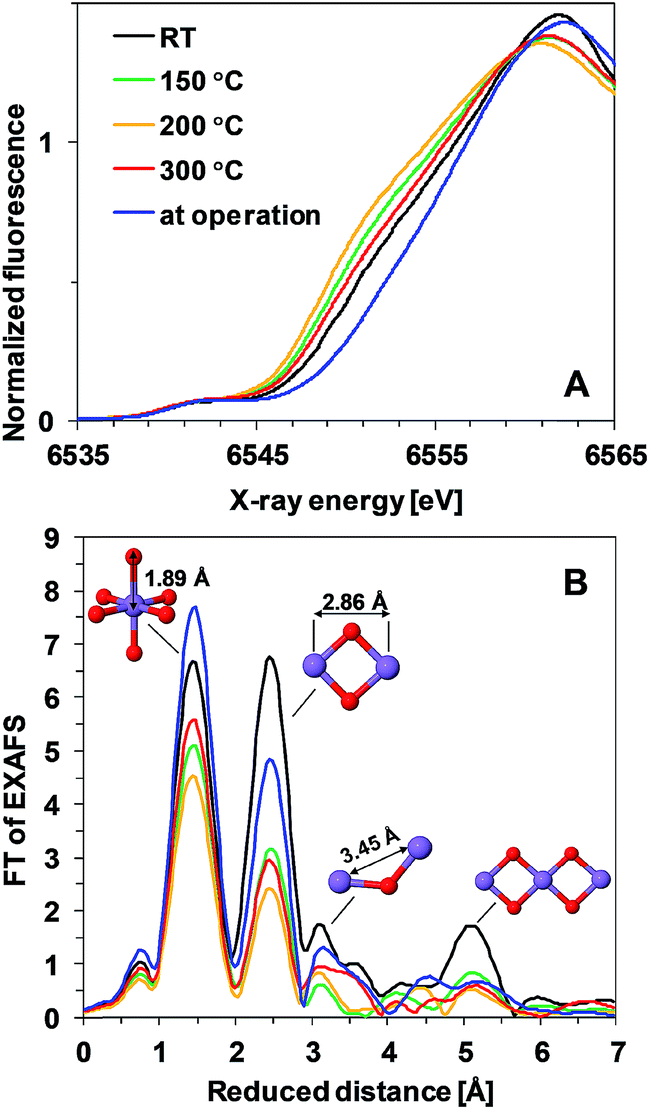 | ||
| Fig. 3 (A) XANES spectra and (B) Fourier transformed (FT) EXAFS spectra of Oxide 1 heated at different temperatures. In (B), the structural motifs and interatomic distances contributing to the peaks are indicated. The Mn film during operation (blue line) is the film heated to 150 °C and exposed to 1.35 V for 3 min. The resulting peak positions in the FT (reduced interatomic distances) do not take into account the energy dependence of the phase shift of the backscattered free-electron wave; real distances obtained from simulations are typically about 0.4 Å longer.28 | ||
Heat- and potential-induced structural changes in the thin layers can be best followed by analysis of the Extended X-ray Absorption Fine Structure (EXAFS). In the Fourier transformed (FT) EXAFS spectra, each peak could be related to a specific interatomic distance (structural motif), as shown in Fig. 3B for Oxide 1. The overall spectral shape is similar to the FT of EXAFS spectra recorded previously from catalytically active Mn oxides11,18,23 and indicates that heating does not induce any drastic changes in the oxide structure but decreases the degree of order (judged from the decreased amplitude of the FT peaks). The structure consists of predominantly edge-sharing Mn4+O6 octahedra that form a poorly ordered layered birnessite type structure. In this structure, missing Mn (or O) ions or defects for example corner-sharing octahedra contribute to the disorder of the material.
Precise information about interatomic distances (typical error lower than 0.02 Å) and number of atoms at these distances was obtained by simulations. The number of short Mn–Mn distances for one Mn atom, reflecting the number of edge-sharing MnO6 octahedra, is the only parameter where there are significant differences in the response of the two oxides to the heat and potential treatment (Fig. 4). For Oxide 2, even at RT the coordination number is lower than 2, showing a very strong deviation from perfectly ordered layers, where the expected coordination number is 6. Heating at temperatures below 300 °C increases the disorder even more. Exposure to oxidizing potentials leads to a slight increase in the number of short Mn–Mn distances, as also found earlier,18 but not above 2 per Mn ion. In contrast to Oxide 2, Oxide 1 clearly has more edge-sharing MnO6 octahedra, as visible also from the appearance in the Fourier transform of the peak at around 5 Å reduced distance which is related to the doubled 2.86 Å Mn–Mn distance found in a collinear arrangement of three edge-sharing MnO6 octahedra (Fig. 3B). As also reported previously, this clearly better ordered structure has almost no catalytic activity11,18 (see also Fig. 1). The heating results in a 2–3 times decrease in the number of short metal–metal distances and in the disappearance of the peak at 5 Å reduced distance, reflecting a higher degree of order in the starting material. These changes indicate that the annealing of Oxide 1 to moderate temperatures does not result in increased crystallinity, but in amorphization of the catalyst, together with a clear increase of the catalytic activity.
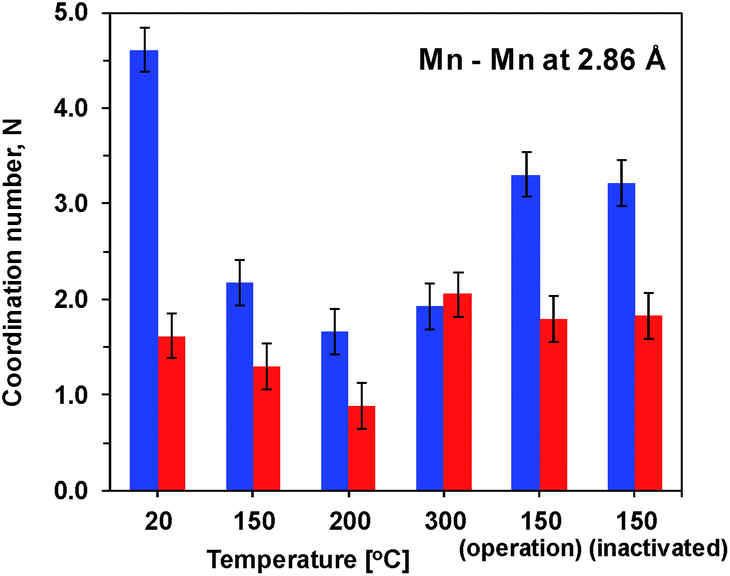 | ||
| Fig. 4 Number of short Mn–Mn distances for one Mn atom obtained by simulations of the EXAFS spectra. Blue – Mn film deposited at constant potential (Oxide 1). Red – MnCat (Oxide 2). The error bars correspond to a 68% confidence interval. | ||
To reflect the presence of the Mn3+ ions with their typical Jahn–Teller distortion, the first FT peak was simulated with two Mn–O distances (1.88 Å and 2.08 Å) and the number of oxygen atoms at these distances was fixed to 6 (sum of the two coordination numbers). Additionally, two Mn–Mn distances were used: 2.86 Å for the second FT peak, typical of the di-μ-oxo bridged Mn ions (edge-sharing Mn octahedra),24,29,30 and 3.45 Å for the third FT peak, a distance typical of the mono-μ-oxo bridged Mn ions (corner-sharing MnO6 octahedra (as it is in pyrolusite – MnO2 with a rutile-type structure)).24,29,30 The interatomic distances were varied during the simulations but kept the same for both oxides under all conditions (joint-fit approach; simulated spectra and parameters for the simulation are shown in ESI Fig. S5 and S6†).
The amplitude of the first FT peak, corresponding to the Mn–O distance (ESI, Fig. S5†) follows the trend observed for the oxidation state (Table 1). This relation is typical of Mn3+ containing samples, as due to the strong Jahn–Teller effect in the Mn3+ ions, two Mn–O distances are observed. Their overlap leads to an effective decrease of the FT peak in the EXAFS spectra proportional to the amount of Mn3+ ions.31 A larger difference between the two oxides with respect to the number of short Mn–O distances was observed only after heating at 300 °C (ESI, Fig. S6†), where as evident from the changes in the edge shape, for Oxide 2, formation of a different phase takes place. At this temperature also another increase in the number of short (2.86 Å) Mn–Mn distances is observed, more pronouncedly in Oxide 2.
Similarly to the first peak, in the third peak (corner sharing MnO6 octahedra) there are no significant differences between the two oxides in their response to heat treatment. Under all experimental conditions, the number of mono-μ-oxo bridges in Oxide 2 remains higher than in Oxide 1, which is related to stronger deviation from the layered structure in MnCat.11,18
Functional investigations by impedance spectroscopy
We recorded potentiostatic impedance spectra at 9 different potentials in the catalytic and non-catalytic regime. A comparison between the impedance spectra recorded at 1.25 V (the onset of the catalytic wave) of Oxide 1 and Oxide 2 is shown in Fig. 5. The oxide films were equilibrated for 60 s at this potential before recording of the impedance spectra with a sinusoidal potential modulation of 10 mV. There are clear heat-induced changes observed in Oxide 1, but not in Oxide 2. In accordance with the structural changes observed by XAS, the impedance spectra show that heating to higher temperature converts the spectra of Oxide 1 to those of Oxide 2. Electrochemical impedance spectra for the two types of films after heating to different temperatures recorded at other potentials are shown in ESI Fig. S7–S11.†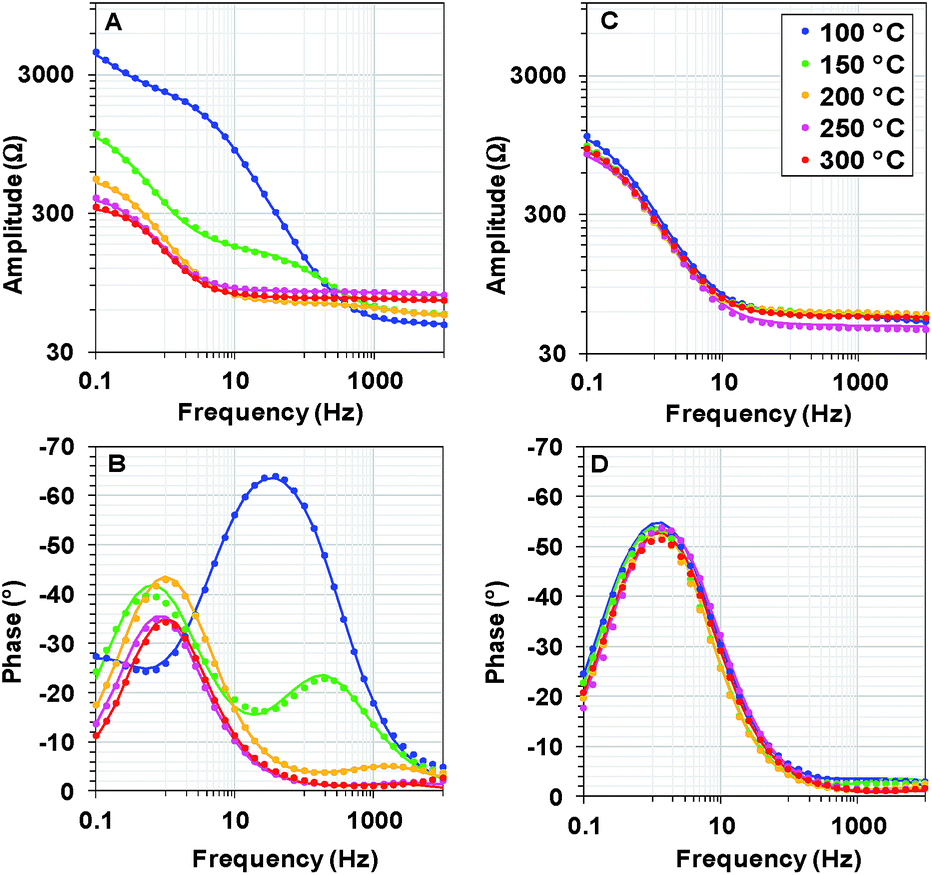 | ||
| Fig. 5 Bode plots for electrochemical impedance spectra obtained at 1.25 V vs. NHE (pH 7) after heating Oxide 1 (A and B) and MnCat, Oxide 2 (C and D) at different temperatures. The experimental data are presented as points, and the simulation results according to the model in the inset in Fig. 6B are indicated by solid lines. | ||
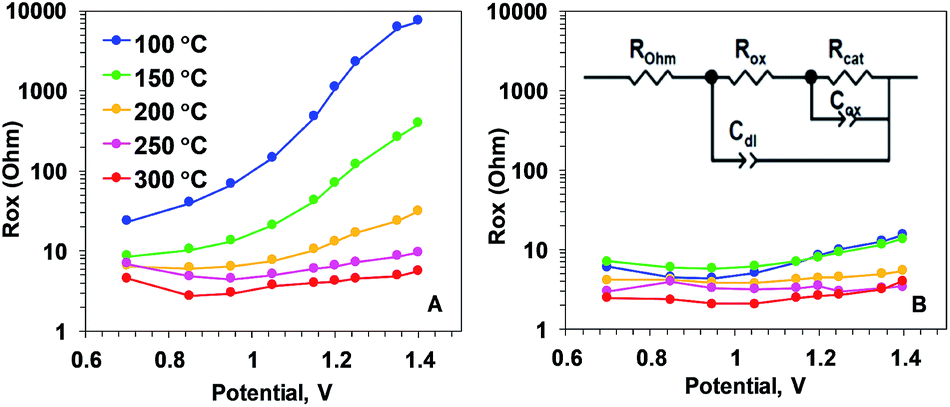 | ||
| Fig. 6 Resistance describing the oxidation state changes (Rox parameter) of Oxide 1 (A) and MnCat, Oxide 2 (B), obtained by simulations according to the equivalent circuit shown in the inset in panel B. ROhm – summed ohmic resistance of the electrolyte and ITO electrode; Cdl – double-layer capacitance, modelled as a constant-phase element; Cox – capacitance describing oxidation state changes of the catalyst film (in other contexts also denoted as pseudo-capacitance), modelled as a constant phase element; Rcat – catalytic resistance of the oxide (charge-transfer resistance). The full set of parameters for the simulations are shown in the ESI, Fig. S12.† | ||
The impedance spectra of the non-heated oxides were published earlier.18 We note that in the previous study, due to the lower stability of the materials, fresh films were used to record the spectra at each potential. Heating at 100 °C (see ESI Fig. S7†) results in essentially the same spectral shape for all potentials, but in this case, the stability of the oxide material increases, facilitating the recording of all spectra on the same film.
To understand which properties of the material change as a result of the heating, we modelled the impedance according to the equivalent circuit suggested in ref. 18. The circuit is shown as an inset in Fig. 6B. The model includes ohmic resistance (ROhm) of the electrolyte and electrode, double layer capacity at the oxide/electrolyte surface (Cdl), resistance (Rox) and capacitance (Cox), both related to the Mn oxidation state changes, and the ‘catalytic resistance’ (Rcat).
Our analysis showed that the clearest differences in the response of the two films to heating are observed in the Mn oxidation state change resistance, Rox (Fig. 6). For the as-deposited Oxide 2, Rox is weakly dependent on the potential and it decreases only slightly with the temperature (Fig. 6B). In contrast, for the non-heated, catalytically inactive Oxide 1, Rox strongly increases with the potential (see the non-heated oxide or the oxide after heating to 100 °C). As a result, the oxidation-state change resistance in the catalytic regime becomes comparable to or even higher than the catalytic resistance (see ESI Fig. S12†). Only 20 min of heating at temperatures above 150 °C results in a large decrease of Rox, especially at oxidizing potentials relevant for O2 evolution (Fig. 6A). This is clear evidence that the main effect of the heating is the drastic increase of the rate of oxidation state changes in the Mn ions. In addition, a heat-induced decrease of Rcat (inversely related to the rate of catalytic O–O bond formation) is observed in the originally inactive Oxide 1 (see ESI Fig. S12†).
The rest of the simulation parameters were clearly less influenced by the annealing (see ESI Fig. S12 and S13†). Interestingly, there was a slight heat-induced increase in Cox in Oxide 1 that could be interpreted as a moderate increase of the number of redox-active Mn ions. This result is in line with earlier findings that the increase of the electrocatalytic activity in first-row transition metal oxide films (like CoOx or NiOx) is related to increased capability of the metal ions to undergo redox changes upon changes in the applied potential.32–36 No clear effect of the annealing was observed on Cdl, suggesting that the roughness of the film surface was not modified by the heat treatment, in line with earlier observations.21 In general, Cdl for Oxide 2 is higher at all temperatures, which corresponds to the rougher surface (higher surface area) of this material.11 The data show that the heat treatment could result in a slight increase in ROhm, indicating the expected loss of conductivity of the ITO substrate. At the temperatures used in this study, this effect is rather low and it can be hidden in the normal variation in ROhm between the different ITO electrodes.
Optical spectroscopic techniques
The two Mn oxides – the inactive Oxide 1 and the catalytically active Oxide 2 (MnCat) – can also be optically distinguished.11 While the colour of the untreated MnCat films (Oxide 2) is rather greyish, that of the inactive Mn films (Oxide 1) is brownish. After the heat treatment, the colour of the Oxide 1 films becomes more similar to that of the Oxide 2 films, which is practically unchanged by the temperature. To measure these changes precisely, we recorded the optical transmission spectra of each film just before and just after 20 min of heating at different temperatures (Fig. 7A and B). The change in the absorbance (the difference between the spectra before and after heating) is shown in Fig. 7C and D. Due to the strong absorption capacity of the ITO/glass substrate in the UV region, the measurements are limited to 300 nm. After heating, the films become more transparent in the short wavelength region. The change is located at an energy of about 3.44 eV and is much larger in the Oxide 1 film compared to the Oxide 2 film. Earlier studies relate the absorption band near 3.44 eV to an electronic transition in the Mn3+O6 octahedron,37 but as the amount of Mn3+ ions increases after the temperature treatment, this cannot explain our results. Recently, Nocera and co-workers.38 reported a similar decrease of the absorption at 3.4 eV with activation of the Mn oxide and attributed this change to the decreased amount of octahedrally coordinated Mn4+ ions in the sample. For our case, this “static” picture is not sufficient to explain the results, as according to the XAS analysis, the Mn oxidation state in both oxides is similar for all heating temperatures while between the two oxides there is a clear difference in the absorption changes. For Oxide 2 (MnCat), the amplitude of the absorption changes is much smaller, and above 100 °C it does not follow a specific trend, while in Oxide 1 there is a clear increase of the absorption changes at least up to 200 °C. The magnitude of the absorption change roughly follows the change of the catalytic properties rather than the oxidation state of Mn ions (Fig. 7).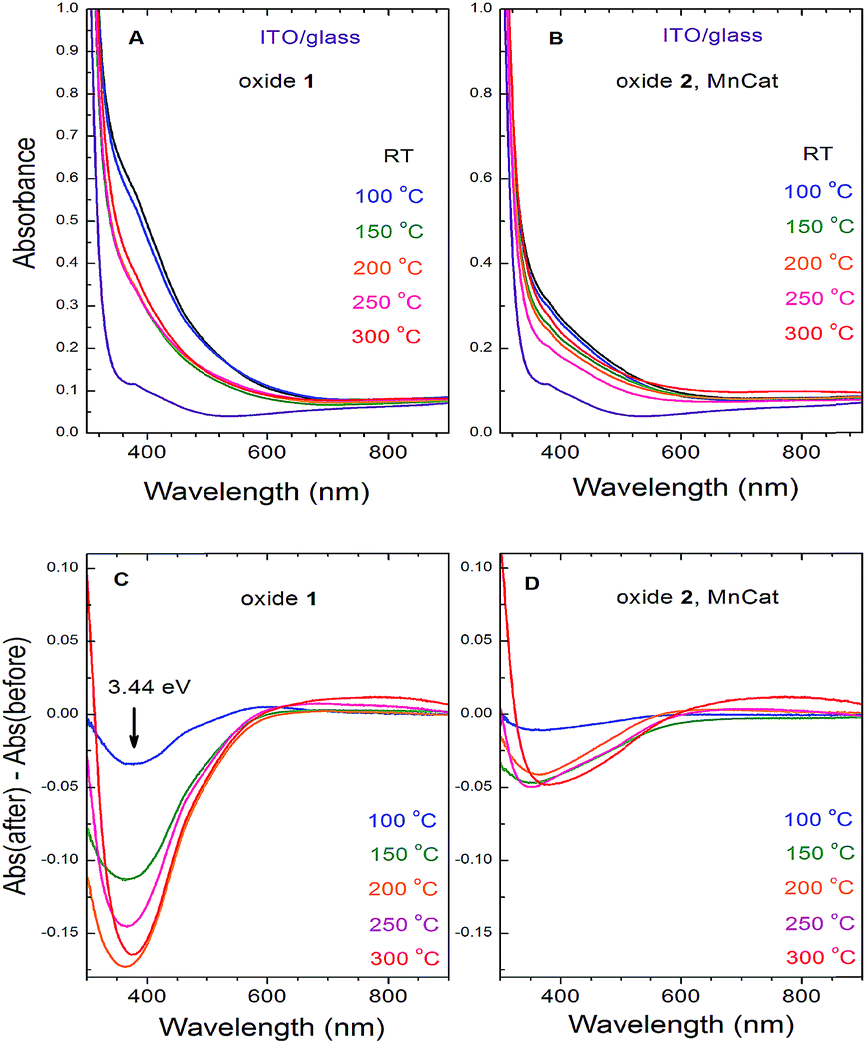 | ||
| Fig. 7 Optical absorbance spectra of the ITO/glass electrodes coated with the Oxide 1 (A) and Oxide 2, MnCat (B) films and the changes in their absorbance after the heat treatment for 20 min (Oxide 1 (C) and Oxide 2, MnCat (D)). | ||
In other studies where the optical properties of manganites with different Mn3+/Mn4+ ratios were investigated, it was observed that the decrease of the absorption coefficient in the 3–4 eV region follows the increase of the electrical conductivity of the samples.39,40 It was proposed that the absorption band in this area is due to an e1g(Mn3+) → e2g(Mn3+) interatomic electronic transition (hopping of an electron from a Mn3+ to another Mn3+ in the adjacent corner sharing MnO6 octahedron).41 This electronic transition is possible only in long-lived electronic states (low electrical conductivity). At high conductivity, when the frequency of the electron jumps is higher than that of the atomic vibrations, the Jahn–Teller splitting between the degenerate (in cubic approximation) e1g and e2g energy levels is lifted (it goes to zero) and this transition does not exist anymore. Based on these arguments, we interpret the heating-induced decrease of the magnitude of the absorption band at 3.44 eV observed in our samples as evidence for an increase of their electrical conductivity. It appears that the electrical conductivity is related to the catalytic properties. A decrease of absorption at around 390 nm in manganese oxides as a result of heating, coupled with a strong increase of the conductivity of the material, was also previously observed.42
Attenuated total reflection (ATR) detected FTIR spectra also show that heating induces larger changes in Oxide 1, making it similar to Oxide 2 (see ESI Fig. S14†). The ATR spectrum of the Oxide 2 film has only one broad band around 605 cm−1, which is typical of the IR-active stretching Mn–O vibration of the MnO6 octahedron in manganites,43 and it remains unchanged after 20 min of heating at different temperatures. The frequency of this band for Oxide 1, however, increases with the increase of the heating temperature (starting from 590 cm−1) and reaches that observed for the Oxide 2 films.
We compared the ATR detected FTIR spectra of the samples, measured before and after testing the catalytic performance in 0.1 M KPi buffer (see ESI Fig. S15†). All the spectra measured after exposure to oxidizing potentials show a new strong absorption band near 1050 cm−1. This frequency is typical of the ν3 IR-active stretching vibration of PO4 tetrahedra.44 As the films were thoroughly rinsed with Milli-Q water after the electrochemical treatment, these results indicate that during the exposure to oxidizing potentials, (hydrogen)phosphate ions are strongly incorporated into the oxide structure in both oxides, with the relative increase of the amplitude of this band roughly following the catalytic activity. Again, no significant changes in the band near 600 cm−1 are observed.
The Raman spectra collected at room temperature for the two oxide films after the heat treatment at different temperatures are shown in Fig. 8. The strong background, increasing at higher shifts, originates from the ITO in the substrate (the spectrum of pure ITO/glass measured at different temperatures is shown in the ESI, Fig S16;† a strong background, likely related to the conducting properties of the ITO, is visible at room temperature and disappears at high temperatures). The spectra of both non-heated samples (labelled RT) consist of a broad band with an irregular shape near 600 cm−1. For the non-crystalline materials where there are no restrictions on the Raman activity of the atomic vibrations, we can expect that the spectra will follow the density of the vibrational states.
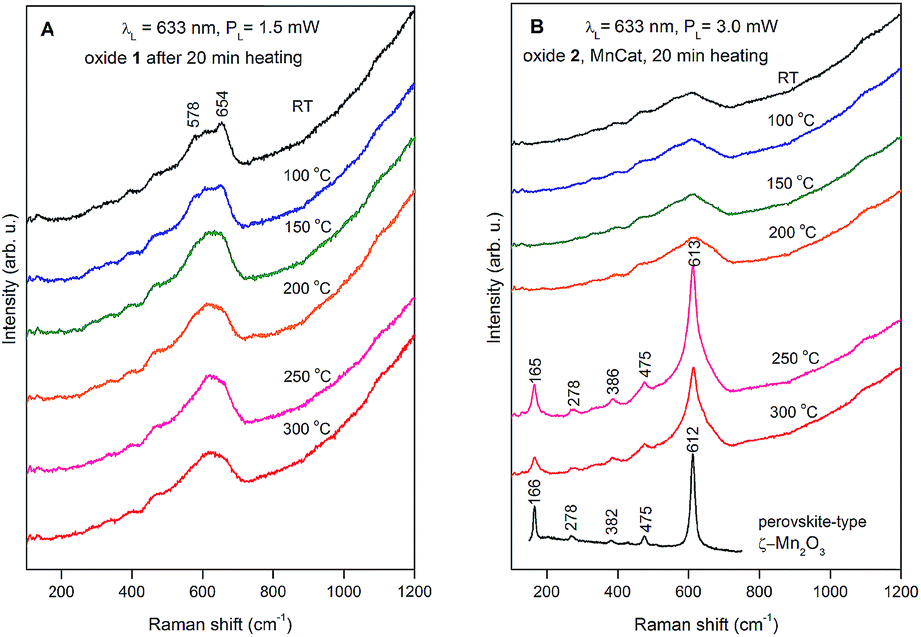 | ||
| Fig. 8 Raman spectra of Oxide 1 (A) and Oxide 2, MnCat (B) heated for 20 min at different temperatures. The used laser line and power are indicated in the figure. For better viewing, the spectra are shifted vertically. For comparison, the spectrum of ζ-Mn2O3 was added to panel B (redrawn with permission from ref. 45). | ||
The maximum near 600 cm−1 corresponds to the typical frequency of stretching oxygen vibrations in Mn–O bonds in Mn–O polyhedra. There are relatively subtle changes in the Raman spectra of Oxide 1 after heat treatment for 20 min at different temperatures. In order to understand the possible heat-induced structural changes in Oxide 1, we will try to explain the origin of the two small bumps at 578 and 654 cm−1, part of the broad band near 600 cm−1 in the Raman spectrum of the non-heated Oxide 1 (see Fig. 8A). We will comment at first on the lines observed in the Raman spectra of the MnO2 oxides with a complex tunnel or layered structure (Fig. 9A). It is very difficult and ambiguous to assign lines, observed in the Raman spectra of complex structures, to specific types of atomic vibrations. The problem arises because due to their large unit cell, theory predicts a large number of formally Raman-allowed normal modes, while the experimental spectra usually contain far fewer lines. Nevertheless, even in the case of poorly crystalline (or partially amorphous) materials, the frequency of a vibration is determined mainly by the local environment of the vibrating atoms. By comparing the spectra of structurally related reference oxides with a known crystal structure, some “characteristic” vibrations can be identified (Fig. 9A).
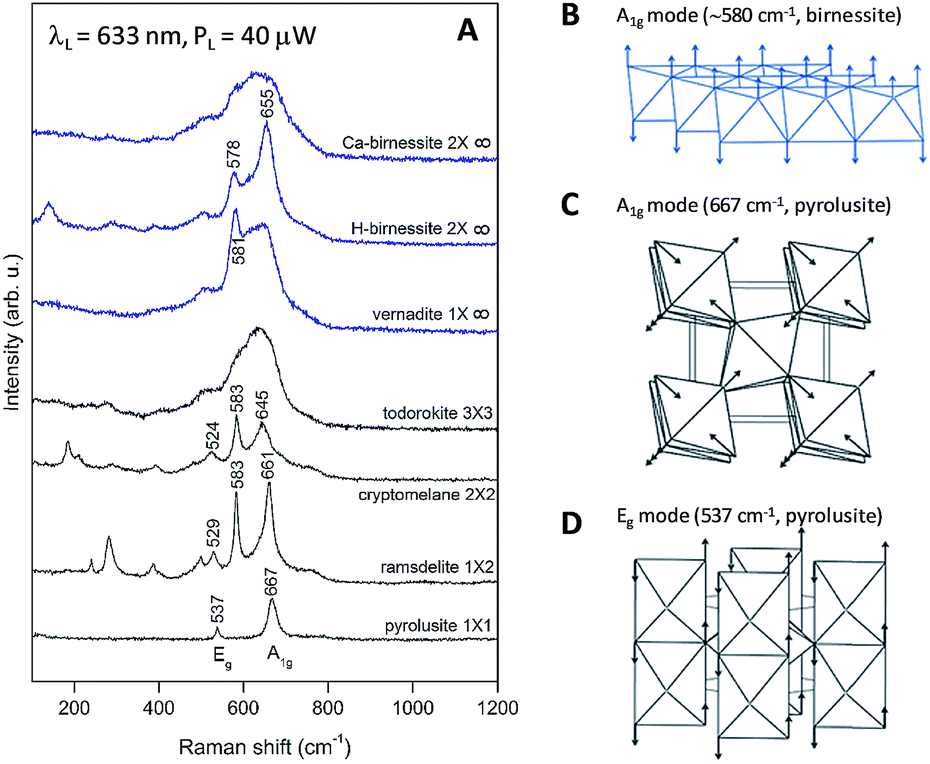 | ||
| Fig. 9 (A) Raman spectra of MnO2 oxides with a complex tunnel (black) or layered (blue) structure. The symbol mXn describes the size of the tunnels between the MnO2 chains/layers. (B) Shape of the characteristic vibration in layered MnO2 oxides along the hexagonal [001] direction (A1g mode). (C and D) Shape of the characteristic vibrations in pyrolusite (β-MnO2 with a rutile-type structure): A1g mode (667 cm−1, atomic vibrations are parallel to the tetragonal (001) plane) and Eg mode (537 cm−1, atomic vibrations are along the tetragonal [001] direction). | ||
In the case of pyrolusite (a structure containing edge-shared octahedral Mn–O chains with 1 × 1 tunnels), it is easy to make an assignment. This oxide has a rutile structure and there are only four Raman-active modes.46 The line at 537 cm−1 corresponds to the Eg mode (in-phase bending oxygen vibrations along the edge-shared octahedral Mn–O chains, Fig. 9D). The line at 667 cm−1 corresponds to the A1g mode (antistretching oxygen vibrations in a direction perpendicular to the chains, Fig. 9C).46 Similarly, ramsdellite (with 1 × 2 tunnels) and cryptomelane (with 2 × 2 tunnels) spectra contain two lines at close frequencies (at 529 and 661 cm−1 for ramsdellite and at 524 and 645 cm−1 for cryptomelane). They can be attributed to the same vibrations in the Mn–O chains. In addition, the spectra of ramsdellite and cryptomelane contain a new strong line at 583 cm−1. Its origin can be found by taking into account that these two structures contain a slab of edge-sharing MnO6 octahedra that connects the chains.47 This new line should correspond to some characteristic vibration of this layer. This line can be seen at close frequencies also in the spectra of layered MnO2 oxides vernadite (1 × ∞, at 581 cm−1) and H-birnessite (2 × ∞, at 578 cm−1). Analyzing an infinite trigonal layer containing MnO6 edge-sharing octahedra (an idealized birnessite), it can be shown that there is only one high-frequency Raman-active oxygen vibration (the oxygen atoms vibrate in a direction perpendicular to the layer, Fig. 9B). This vibration has A1g symmetry and it is “breathing” for the layer. Thus, we attribute the line at around 580 cm−1 to the MnO2 slabs (di-μ-oxo bridged Mn atoms, or edge-sharing MnO6 octahedra). In the spectra of birnessites, an additional line with variable intensity is observed at 655 cm−1. Because its frequency corresponds to the high-frequency end of the density of vibrational states, it should be the “breathing” oxygen vibration (usually the vibration with the highest frequency) for a single MnO6 octahedron. In the case of an ideally ordered material, it is not Raman-active, but due to the presence of a small amount of Jahn–Teller distorted Mn3+O6 octahedra and other types of defects it can be activated. Comparing the spectra of H-birnessite (Fig. 9A) and the non-heated Oxide 1 (Fig. 8A), it becomes obvious that they are very similar. Therefore, we can conclude that the crystal structure of the non-heated Oxide 1 can be that of a defect birnessite, in agreement with the XAS analysis. In the spectra of heated Oxide 1, the two bumps (the MnO6 layer-related one at 578 cm−1 and the single MnO6 octahedron-related one at 654 cm−1) disappear and the shape of the 600 cm−1 band become smoother – a sign of amorphization of the structure. From Fig. 9A it is seen that the Raman spectra of todorokite (3 × 3 tunnels) and Ca–birnessite (2 × ∞) samples are also very smooth – a sign of their low crystallinity.
In contrast to Oxide 1, there are large changes in the Raman spectra of Oxide 2 films (MnCat) heated at 250 °C and 300 °C. A spectrum consisting of narrow lines positioned at 165, 278, 386, 475, and 613 cm−1, indicating the formation of some crystalline phase, is observed. This spectrum does not coincide with the spectra of any of the well-known MnxOy oxides with a simple structure (rutile-type pyrolusite β-MnO2,48 bixbyite-type α-Mn2O3 or spinel-type hausmannite α-Mn3O4,49 see ESI Fig. S17†). The spectrum also differs from the spectra of MnO2 oxides with a complex tunnel or layered structure47 (Fig. 9A). Surprisingly, it is identical (both the frequency of all lines as well as their relative intensities) to the spectrum of the recently discovered perovskite-like ζ-Mn2O3 (narrow bandgap semiconductor), which is thermodynamically stable at very high pressures of about 20 GPa and temperatures of about 1200 °C.45,49 As we observe only subtle changes in the EXAFS spectrum of the Oxide 2 heated to 300 °C, we conclude that only a minor fraction (up to 20%) of the MnO2 is converted to ζ-Mn2O3 but this fraction dominates the Raman spectrum (the spectrum of the amorphous material has broad features with a low amplitude visible as a background). To illustrate the compatibility of the ζ-Mn2O3 structure with our EXAFS results, we repeated the EXAFS simulations including the shortest Mn–Mn distance of 3.04 Å observed in the ζ-Mn2O3 structure, and found that this distance is compatible with the experimental spectra (Fig. S18 and S19†). Additional support for the heat-induced formation of the crystalline phase in Oxide 2 is the change in the edge shape in the otherwise featureless XANES spectra (ESI, Fig. S3†).
To follow the appearance of the crystalline phase in Oxide 2, we measured Raman spectra during heating to 600 °C followed by a gradual cooling down (ESI, Fig. S20†). The final phase is the same, although in this case, the sample was heated to clearly higher temperatures and the entire cycle until returning again to room temperature took about 6 h. The formation of the crystalline phase can be followed during the cooling of the sample below 300 °C.
Stability of the heat-treated Mn oxide films
The stability of the Mn oxide electrocatalysts under catalytic conditions at pH 7 is typically very poor. Often data related to the stability of Mn oxide catalysts are missing or if available, the catalysts withstand the exposure to oxidizing potentials for less than a few hours at neutral pH if currents of 1 mA cm−2 or higher are maintained.16,27,50,51 Relatively stable electrochemical operation for Mn oxides (2 h at 1 mA cm−2 with 480 mV overpotential) was reported only for very thick (10 μm) layers of pre-synthesized Mn oxide powder, glued with a binder to an electrode.19 The Oxide 2, MnCat, used in this study (with a thickness of about 20 nm and incomplete coverage of the substrate, see ESI Fig. S21A†) was reported to operate for 2–3 hours at 0.1 mA cm−2 with an overpotential of about 0.5 V.11We carried out chronopotentiometric experiments to probe the ability of the electrodes after heating to sustain electrochemical water oxidation over extended time periods. Measurements extending over one week are shown in the ESI, Fig. S22.† To the best of our knowledge, this stable performance in neutral pH is significantly better than that of all other Mn oxide catalysts reported until now in the literature. More favourable experimental conditions (e.g. use of a co-solvent like 1 M MgSO4, or stirring, to lower the diffusion limitations) would likely decrease the overpotential.
For both films at all temperatures the inactivation is a multi-step process. First, the overpotentials increase by 1–2 V for about 1–2 h. The process is followed by a sharp complete inactivation (Fig. 2). The complete inactivation is not related to dissolution of the Mn oxide film, as the electrodes remain optically unchanged with the oxide still firmly attached to the electrode. The CVs however in all cases show that both the redox and catalytic activity is absent (see ESI Fig. S23†). As visible from the SEM images of these fully inactive electrodes, the ITO layer is damaged (ESI, Fig. S21B and C†) and the electrodes become non-conducting. The SEM images also show that the morphology of the Mn oxide film is itself unaffected by the long operation.
As found for the active MnCat, Oxide 2, the all-Mn4+ state cannot be reached even after full inactivation of the oxide, induced after prolonged exposure to high potential (chronopotentiostatic measurements at a current density of 5 mA cm−2 until a fully inactive state was reached). Despite the complete loss of activity observed after prolonged exposure to high oxidizing potentials, the edge shape does not change (ESI, Fig. S4†) and the Mn oxidation state remains almost unchanged, except for a small decrease in the case of Oxide 1 (Table 1). The latter effect can also be attributed to the short drying of the film after switching off the potential. Structural changes in the inactivated oxide were visible neither in the EXAFS (ESI, Fig. S5B and D and S6†) nor Raman spectra (ESI, Fig. S24†).
Some insight into the mechanism of inactivation comes from the potentiostatic impedance spectra of the Mn oxides, measured before the complete destruction of the ITO layer, when the plateau level of the overpotential (around 3 V) is reached (see ESI Fig. S25†). The impedance spectra of the semi-inactivated Oxide 2 are similar to those of the Oxide 1 heated to 150 °C, where only partial heat-induced activation is observed (compare ESI Fig. S25 and S8†). The potential-induced inactivation is related to a strong increase of the Rox parameter (see ESI Fig. S26†) and at potentials higher than 1.25 V vs. NHE, Rox becomes higher than the catalytic resistance, Rcat. At the same time, there is a strong potential-induced increase of the catalytic resistance, Rcat, itself. Notably, the change in the Mn oxidation state resistance is not related to the decrease of the ITO conductivity, as the ROhm parameter remains almost unchanged. Similar to the trend observed for heat treatment, the potential-induced increase in Rox (slowing down of Mn redox changes) is coupled to a decrease in the Cox parameter (number of redox-active Mn ions).
Discussion
We found that 20 min heat treatment of MnOx electrodes at moderate temperatures (up to 300 °C) has two favourable effects: on the one hand it increases the catalytic activity of the initially low-active electrodeposited Oxide 1 and on the other hand it significantly improves the stability of both Oxide 1 and the structurally related catalytically active Oxide 2 (MnCat). Heating of oxides in air (ambient O2), as has been done in this study, may result in Mn oxidation,7,8 but in our experiment it results in loss of oxygen and/or structural water and thus in partial decomposition of the oxide, related to a decrease of the mean oxidation state of the metal. This effect has already been observed21,22 and it is explainable assuming that several stable oxide structures with close formation energies exist in equilibrium, allowing decomposition of the high valence oxides into lower valence oxides to be easily driven by moderate temperature.52 The decrease of the average Mn oxidation state is paralleled by an increase of the Mn/O ratio and a decrease of the number of di-μ-oxo connected Mn ions at the expense of an increase of mono-μ-oxo bridges, as evident from XAS analysis (Table 1 and Fig. 3). In the structure of the originally inactive Oxide 1, the number of di-μ-oxo bridges is close to 6, the predicted coordination for the ideal layered birnessites composed exclusively of edge-sharing Mn+4O6 octahedra (di-μ3-oxo bridges).24 The heating induces a decrease of these short Mn–Mn distances to about 2 per Mn ion, and thus makes the structure deviate greatly from the layered oxide form and rather similar (but still not identical) to that of Oxide 2, deposited by a potential cycling protocol. On the other hand, Oxide 2 has a structure that is initially only weakly related to the layered oxides, containing an average of less than 2 di-μ-oxo bridges per Mn ion and a comparable number of mono-μ-oxo bridges (Fig. 4 and ESI Fig. S6†). We suggest that the favourable mean oxidation state of Mn ions (3.2, see Table 1) and the presence of a sufficient number of corner-shared MnO6 octahedra after heating to temperatures above 250 °C in Oxide 2 allow the crystallization of ζ-Mn2O3, an effect not observed in Oxide 1. The relation between these structural changes and the catalytic activity of Mn oxides is discussed in the following sections.Formation of a “catalytic site”
In all transition metal oxides, a short metal–metal distance of about 2.8 Å, which results from di-μ-oxo bridged metal ions, is shown to be a crucial requirement for catalytic activity towards water oxidation.9,34,53–56 A similar structural motif is also present in the native Photosystem II (PSII) catalytic center that is composed of one calcium and four manganese ions connected by bridging oxygens.57–59 However, as we have also shown previously, Mn oxides exhibiting such a short metal–metal distance exclusively are inactive towards water oxidation (non-heated Oxide 1 in this study; see also ref. 11 and 18). In the biological Mn4CaO5 catalytic center, a specific co-linear arrangement is found between Mn1–O5–Mn4 atoms.58 According to the recent femtosecond X-ray crystallography data, a long Mn–O distance of 2.7 Å exists between O5 and Mn1 in the resting state of the catalyst, corresponding to Mn1 occupying an undercoordinated (5-coordinated) site.58 During the catalytic cycle an additional O atom (oxo or OH group) is inserted between Mn1 and Mn4 resulting in coupled Mn1 oxidation and transition of Mn1 from 5- to 6-coordination, in line with previous experimental findings.60–65 Our previous XAS studies showed potential-induced structural changes in Mn oxides similar to the changes in the PSII catalytic center during operation.18 It is plausible to assume that a decreased number of saturated di-μ3-oxo bridges between Mn ions and increased deviation from the well-ordered layered oxide structure will favour the formation of similar co-linear arrangements of Mn–O–Mn atoms and may result in the formation of local structural motifs similar to the biological catalytic center. As suggested earlier, water oxidation should take place at such specific constellation of atoms, which may act as a catalytic site.18 In addition, a Mn–O–Mn angle close to 180° would have a strong impact on the electrical conductivity of the oxides, as discussed further below.Electron and proton conductivity of the oxides
Comparing the electrocatalytic activity towards water oxidation of two Mn oxides with different structures (layered-like Mn oxide containing a large number of di-μ-oxo bridges and 3D cross-linked Mn oxides with predominantly mono-μ-oxo bridged Mn ions), Bergmann et al.9 suggested that the better proton conductivity of the cross-linked Mn oxides rich in mono-μ-oxo bridges results in increased catalytic currents at high overpotentials, as compared to the oxides with a layered-like structure. Here, we propose that the electron conductivity of the materials also has a strong impact on the catalytic activity.The electrical conductivity of the thin oxides investigated in this study (with an approx. thickness of 20 nm) cannot be directly measured, inter alia, as they typically do not cover completely the conducting ITO substrate.11 Still, information about their electroconductivity can be accessed indirectly, e.g. via impedance measurements and optical spectroscopy. According to the decrease of the absorption band at around 3.4 eV observed in the optical transmission measurements (Fig. 7), both Oxide 1 and Oxide 2 become more conductive after the heat treatment, which is likely related to the increase in the catalytic activity. The heat-induced increase in the electrical conductivity of Mn oxides coupled with a decrease in the optical absorption at 3.18 eV has been measured before.42 We note that the mechanism of the electrical conductivity of Mn oxides is actually local and is due to an electron hopping between Mn ions in two adjacent (corner-shared) octahedra.41 Besides the presence of corner-sharing octahedra, the ratio of Mn3+ and Mn4+ ions is important for the conductivity.66 For a network of corner-shared MnO6 octahedra it was calculated that its electrical conductivity is highest for a value of 3.31 for the mean oxidation state of Mn ions.67
We propose that the electrical conductivity is important for catalytic activity not simply because of the better contact between the oxide and the electrode but it is likely related to the required ability of Mn ions to change their oxidation state during catalysis. In biological water oxidation, four electrons (and four protons) need to be removed from the Mn4CaO5 cluster before the chemical step of O–O bond formation.60 Assuming analogy in the catalytic mechanism of Mn oxides,68 the Mn oxidation should be faster than the catalytic reaction itself, which will allow for accumulation of oxidizing equivalents needed for the oxidation of the water molecule.18 In the framework of the model used to describe the electrochemical impedance spectra this is translated in Rox lower than Rcat as a prerequisite for the catalytic activity of the material. Rox higher than Rcat, as found in the Oxide 1 before heating, prohibits the catalytic activity.18 The results in our study, however, suggest that heat treatment induces a severe decrease of Rox thus allowing efficient water oxidation (Fig. 6).
Interestingly, in oxides with low catalytic activity, Rox increases with the increase of the applied potential (Fig. 6). This effect might be explained by the potential-induced increase of the number of di-μ-oxo bridges between the Mn ions and thus hindered electron conductivity. This, however, is not the reason in this case, as the potential-induced increase of Rox is observed mostly in the non-heated Oxide 1 where, as evident from the XAS data published before, no significant potential-induced change in the number of di-μ-oxo bridges is detected due to the relatively rigid structure of the layered oxide.18 A more plausible explanation for the observed potential-induced increase of Rox is that in the inactive layered oxide most of the Mn ions are oxidized to Mn4+ already at potentials lower than the potentials relevant for water oxidation, as also found before.18 This hypothesis is supported by the sharp drop in Cox in the inactive Oxide 1 at potentials above 1.2 V vs. NHE. In contrast, the potential induces only a gradual decrease of Cox in catalytically active materials and the effect is smaller in amplitude (ESI, Fig. S12†).
Preventing the formation of an all-Mn4+ state
In earlier studies, it was shown that exposure of the non-heated Mn oxides to oxidizing potentials during the catalytic process results in an increase of the Mn oxidation state.11,18 For the catalytically active Oxide 2 this increase was to about 3.8, preserving a fraction of about 20% Mn ions in the +3 oxidation state even at relatively high oxidizing potentials. In contrast, exposure of the inactive Mn oxide film, deposited at constant potential, resulted in a quick formation of an all-Mn4+ state, which was proposed to be the reason for the absence of catalytic activity.17,18 As shown in Table 1, exposure of the heated Oxide 1 film to oxidizing potentials no longer results in the formation of an all-Mn4+ state, in line with previous observations.17 In Oxide 2 the heating results in a further increase of the amount of Mn3+ ions, stable at high oxidizing potential (Table 1).The importance of Mn3+ ions for the catalytic activity of Mn oxides has repeatedly been reported.6,11,69–77 It was proposed that the typically low catalytic activity, observed at neutral pH, is due to a decomposition of Mn3+ to Mn2+ and Mn4+ at pH lower than pH 9 and thus in the absence of surface associated Mn3+ ions that could act as precursors for water oxidation.71 The instability of (surface) Mn3+ species is often identified as the main reason for the reduced catalytic activity of MnOx as compared to alkaline pH.25,72,74 (We note that the Mn catalysts investigated here also show markedly higher catalytic activity and lower overpotentials when used in alkaline pH (see ESI Fig. S27†), but aiming at mechanistic understanding we focused the present study on pH 7 only). Recent results showed that required for catalytic activity are not just Mn3+ ions present in the resting state of the catalysts, but Mn3+ ions stable at oxidizing potentials high enough to drive the water oxidation reaction.17,18,27,38 Although the mid-point potential for water oxidation at pH 7 is 0.8 V vs. NHE, the onset of the catalytic activity is typically above 1.2 V.11 The potentials relevant to the water oxidation process clearly exceed the potential (about 1 V) needed to oxidize Mn2+ to Mn4+ ions in birnessite-like structures.38 The stabilization of Mn3+ ions is thus possible only via specific heat-induced structural changes in the oxide that can ensure locally specific atomic configurations able to host Mn3+ ions stable towards oxidation.18
The mean Mn oxidation state of both Oxide 1 and Oxide 2 in their resting state is similar (Table 1). Despite this, in the non-heated oxides the potential increase induces full oxidation of Mn3+ ions in Oxide 1, while in Oxide 2 a fraction of up to 20% Mn3+ ions remain stable, meaning that the local environment of the Mn3+ ions might be different as a result of different deposition procedures. Oxide 1 was deposited at constant oxidizing potential where a layered birnessite structure is formed.78 The presence of Mn3+ in this case is a result largely of the presence of defects in the structure of layered oxide.24 In the structure of Oxide 2 (MnCat), formation of Mn3+ was forced by potential cycling during the film deposition by lowering the potential during each cycle to −0.75 V, where below −0.4 V Mn3+ ions are produced by comproportionation reactions between Mn2+ and Mn4+ ions.38
Recently, Nocera and co-workers proposed that the stable Mn3+ ions in catalytically active Mn oxides occupy tetrahedral sites, in a structure analogous to Mn3O4 spinels.38 The heat-induced formation of perovskite-like ζ-Mn2O3 observed in this study provides experimental evidence for an alternative explanation of the presence of Mn3+ ions stable at high oxidizing potentials in the close to linear Mn–O–Mn motifs. The conclusion of Chan et al.38 for the existence of four-coordinated Mn3+ ions in an oxidized spinel-like structure is based mostly on EXAFS simulation, where a decrease of the Mn–O coordination number was found to correlate with the decrease of the Mn oxidation state. This result, however, may be imposed by the simulation approach where a single Mn–O distance was used. This could lead to potentially misleading results due to the strong Jahn–Teller distortion present in 6-coordinated Mn3+ ions which results in Mn–O bonds with different lengths. When simulating using a single Mn–O scattering path, the overlap of the two Mn–O distances results in an apparent decrease of the coordination number. In addition, an extended pre-edge feature (arising from electron transitions from 1s core levels to the empty 3d levels, hybridized by the O ligands) is expected in the XANES spectra for Mn3+ ions in the Td symmetry.79,80 Instead, the double peak feature associated with the crystal field splitting in octahedral coordination is visible under all experimental conditions.38 Notably, tetrahedral coordination for Mn3+ ions is also not observed in any of the large variety of stable Mn oxide polymorphs. Taking into account that the Mn ions occupying the tetrahedral sites in normal spinel structures are Mn2+ ions, and that inverted spinels, where the tetrahedral site is occupied by a cation in the +3 oxidation state, are reported for other transition metals, but not for Mn, we suggest that the Mn3+ ions are stabilized in a perovskite-like environment, as also implied by the formation of ζ-Mn2O3 when heating Oxide 2.
Conclusions
Studying the effect of heating at moderate temperatures of Mn oxides allowed us to identify characteristics required for OER activity and stability under operation conditions. Oxide structures featuring both structural motifs with di-μ-oxo bridged Mn ions and (close to) linear mixed-valent Mn3+/Mn4+ mono-μ-oxo Mn–O–Mn connectivity, seem to be a prerequisite for OER activity of Mn oxides. The presence of the latter motif likely stabilizes Mn3+ ions at higher potentials and thereby ensures high charge carrier mobility during operation at catalytic potentials, a feature related to the electrical conductivity of the oxide. An amorphous oxide structure that is close to the perovskite-like ζ-Mn2O3 could favour the stabilization of Mn ions in oxidation states lower than +4. Low charge carrier mobility and electrical conductivity are also likely related to inactivation of the oxides after prolonged operation, where degrading catalyst properties result in a greatly increased overpotential requirement and eventually the breakdown of the interface structure between the catalyst film and the ITO substrate electrode used here.Experimental section
Electrochemical measurements
In all electrochemical experiments, a single compartment three-electrode electrochemical cell was used with a saturated Ag/AgCl reference electrode (−0.197 V vs. NHE) and a platinum mesh (2 × 2 cm2, 90% purity) as a counter electrode. The working electrode was a 2 cm2 (2 cm × 1 cm) indium-tin oxide (ITO) coated glass plate (VisionTek Systems Ltd.) with an active geometric area of 1.5 cm2 (restricted with Kapton tape). The volume of the electrolyte was 50 mL. All electrochemical measurements were carried out in air at room temperature using potentiostat Interface 1000 (Gamry Instruments). No iR compensation was applied and the solution was not stirred during the measurements. All indicated potentials are referenced to the normal hydrogen electrode (vs. NHE).Manganese films were deposited on the ITO-covered surface of the electrode from aqueous solution of Mn2+ ions (0.5 mM MnII(CH3COO)2·4H2O (Fluka, ≥99%), in 18 MΩ cm Milli-Q water) as described earlier.11,18 Oxide 1 was deposited potentiostatically for 15 min at 1.35 V. Oxide 2 was deposited by 25 cyclic voltammetry (CV) sweeps, from 2.15 to −0.75 V vs. NHE with a scan rate of 100 mV s−1. After the deposition, the electrodes were rinsed with Milli-Q water, the Kapton tape was removed, and the electrodes were heated in an oven equipped with a Protherm PC442T temperature controller at ambient pressure and atmosphere.
The electrocatalytic activity of the electrodeposited Mn oxides was tested by cyclic voltammetry, chronoamperometry, chronopotentiometry and potentiostatic EIS (electrochemical impedance spectroscopy) experiments in 0.1 M phosphate buffer (pH 7, adjusted with 0.1 M KH2PO4 and 0.1 M K2HPO4 (Roth, ≥99%)). Cyclic voltammetry (CV) to test catalytic activity was performed from 0.9 to 1.45 V with a scan rate of 20 mV s−1. Tafel slopes were obtained by chronopotentiometric measurements, applying each potential for 3 min and considering only the currents averaged from the last 10 s at each potential step. EIS measurements were taken in the frequency range between 100 mHz and 10 kHz with a 10 mV modulation amplitude of the working-electrode potential. At each potential the films were equilibrated for 60 s before the EIS measurement.
X-ray absorption spectroscopy
X-ray absorption spectra were measured at the BESSY II synchrotron of the Helmholtz-Zentrum für Materialien und Energie (HZB, Berlin, Germany) at 20 K in a liquid-helium cryostat (Oxford Instruments) as described previously.31 The spectra were collected at the XPP/KMC-3 beamline in fluorescence mode using a 13-channel energy-resolving Ge detector (Canberra). The X-ray excitation energy was selected using a silicon (111) double-crystal monochromator. For all samples, KMnO4 powder was measured simultaneously in transmission mode and used as the energy calibration standard (the pre-edge maximum was set to 6543.3 eV). Other details about data analysis and simulations are provided elsewhere.31Raman spectroscopy measurements
Raman spectra were obtained using a micro-Raman spectrometer, LabRAM HR Visible (HORIBA Jobin Yvon). The 633 nm He–Ne laser line was used for excitation. A ×50 long working distance microscope objective was used both to focus the incident laser light onto a 2–3 μm spot and to collect the scattered light in backscattering geometry. To avoid any local overheating effects after tests the laser power was limited to fixed values, as shown in the figures, depending on the type of the sample (thin film or powder).ATR measurements
Attenuated total reflection (ATR) measurements in the infrared optical region were made using both a Tensor II (Bruker) and Vertex 80v (Bruker) equipped with a Platinum ATR Accessory.Optical absorption measurements
The optical absorption in the visible region was measured using an Agilent Cary60 UV-Vis spectrophotometer. The samples were measured at room temperature just before and immediately after their thermal heating.SEM measurements
Micrographs of the surfaces of the films were acquired at room temperature with a Hitachi SU 8030 microscope operated at 10 kV and 10 mA at a working distance of 8 mm. The microscope was equipped with a cold field emission cathode and an Oxford X-Max 80 mm2 EDX detector.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MVA thanks the Alexander von Humboldt Foundation, Bonn (Germany) for the research fellowship, ensuring his stay at Freie Universität Berlin (FUB), as well as the support by the Bulgarian Ministry of Education and Science under contract D01-155/2018 (INFRAMAT). The authors acknowledge support by the Bundesministerium für Bildung und Forschung (BMBF), through the IN SITU XAS project, 05K16KE2, and CO2EKAT project, 03SF0523B. We also thank the Deutsche Forschungsgemeinschaft (DFG) for financial support to the SPP1613, Grant No. DA402/7-1 and the collaborative research center on Protonation Dynamics in Protein Function (SFB 1078, project A4/Dau). Ivo Zizak from the Helmholtz-Zentrum Berlin provided excellent technical support at the XPP/KMC-3 beamline of the BESSY II synchrotron.Notes and references
- A. Williamson, B. Conlan, W. Hillier and T. Wydrzynski, The evolution of Photosystem II: insights into the past and future, Photosynth. Res., 2011, 107(1), 71–86 CrossRef CAS PubMed.
- C. Pagliano, G. Saracco and J. Barber, Structural, functional and auxiliary proteins of photosystem II, Photosynth. Res., 2013, 116(2–3), 167–188 CrossRef CAS PubMed.
- N. Nelson and C. F. Yocum, Structure and function of Photosystems I and II, Annu. Rev. Plant Biol., 2006, 57, 521–567 CrossRef CAS PubMed.
- J. E. Post, Manganese oxide minerals: Crystal structures and economic and environmental significance, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 3447–3454 CrossRef CAS PubMed.
- S. L. Suib, Porous manganese oxide octahedral molecular sieves and octahedral layered materials, Acc. Chem. Res., 2008, 41(4), 479–487 CrossRef CAS PubMed.
- D. M. Robinson, Y. B. Go, M. Mui, G. Gardner, Z. Zhang, D. Mastrogiovanni, E. Garfunkel, J. Li, M. Greenblatt and G. C. Dismukes, Photochemical water oxidation by crystalline polymorphs of manganese oxides: Structural requirements for catalysis, J. Am. Chem. Soc., 2013, 135(9), 3494–3501 CrossRef CAS PubMed.
- I. Zaharieva, M. M. Najafpour, M. Wiechen, M. Haumann, P. Kurz and H. Dau, Synthetic manganese-calcium oxides mimic the water-oxidizing complex of photosynthesis functionally and structurally, Energy Environ. Sci., 2011, 4(7), 2400–2408 RSC.
- E. Baktash, I. Zaharieva, M. Schroder, C. Goebel, H. Dau and A. Thomas, Cyanamide route to calcium manganese oxide foams for water oxidation, Dalton Trans., 2013, 16920–16929 RSC.
- A. Bergmann, I. Zaharieva, H. Dau and P. Strasser, Electrochemical water splitting by layered and 3D cross-linked manganese oxides: correlating structural motifs and catalytic activity, Energy Environ. Sci., 2013, 6(9), 2745–2755 RSC.
- R. Pokhrel, M. K. Goetz, S. E. Shaner, X. Wu and S. S. Stahl, The “Best Catalyst” for Water Oxidation Depends on the Oxidation Method Employed: A Case Study of Manganese Oxides, J. Am. Chem. Soc., 2015, 137(26), 8384–8387 CrossRef CAS PubMed.
- I. Zaharieva, P. Chernev, M. Risch, K. Klingan, M. Kohlhoff, A. Fischer and H. Dau, Electrosynthesis, functional and structural characterization of a water-oxidizing manganese oxide, Energy Environ. Sci., 2012, 5, 7081–7089 RSC.
- F. Zhou, A. Izgorodin, R. K. Hocking, L. Spiccia and D. R. MacFarlane, Electrodeposited MnOx films from ionic liquid for electrocatalytic water oxidation, Adv. Energy Mater., 2012, 2(8), 1013–1021 CrossRef CAS.
- R. K. Hocking, R. Brimblecombe, L.-Y. Chang, A. Singh, M. H. Cheah, C. Glover, W. H. Casey and L. Spiccia, Water-oxidation catalysis by manganese in a geochemical-like cycle, Nat. Chem., 2011, 3(6), 461–466 CrossRef CAS PubMed.
- Y. Gorlin, B. Lassalle-Kaiser, J. D. Benck, S. Gul, S. M. Webb, V. K. Yachandra, J. Yano and T. F. Jaramillo, In situ X-ray absorption spectroscopy investigation of a bifunctional manganese oxide catalyst with high activity for electrochemical water oxidation and oxygen reduction, J. Am. Chem. Soc., 2013, 135(23), 8525–8534 CrossRef CAS PubMed.
- M. Huynh, D. K. Bediako, Y. Liu and D. G. Nocera, Nucleation and growth mechanisms of an electrodeposited manganese oxide oxygen evolution catalyst, J. Phys. Chem. C, 2014, 118(30), 17142–17152 CrossRef CAS.
- M. Huynh, D. K. Bediako and D. G. Nocera, A Functionally Stable Manganese Oxide Oxygen Evolution Catalyst in Acid, J. Am. Chem. Soc., 2014, 136(16), 6002–6010 CrossRef CAS PubMed.
- D. Gonzalez-Flores, I. Zaharieva, J. Heidkamp, P. Chernev, E. Martinez-Moreno, C. Pasquini, M. R. Mohammadi, K. Klingan, U. Gernet, A. Fischer and H. Dau, Electrosynthesis of biomimetic manganese-calcium oxides for water oxidation catalysis - atomic structure and functionality, Chemsuschem, 2016, 9(4), 379–387 CrossRef CAS PubMed.
- I. Zaharieva, D. González-Flores, B. Asfari, C. Pasquini, M. R. Mohammadi, K. Klingan, I. Zizak, S. Loos, P. Chernev and H. Dau, Water oxidation catalysis – role of redox and structural dynamics in biological photosynthesis and inorganic manganese oxides, Energy Environ. Sci., 2016, 9, 2433–2443 RSC.
- S. Y. Lee, D. Gonzalez-Flores, J. Ohms, T. Trost, H. Dau, I. Zaharieva and P. Kurz, Screen-printed calcium-birnessite electrodes for water oxidation at neutral pH and an “Electrochemical Harriman series”, ChemSusChem, 2014, 7(12), 3442–3451 CrossRef CAS PubMed.
- L. Xi, C. Schwanke, J. Xiao, F. F. Abdi, I. Zaharieva and K. M. Lange, In situ L-edge XAS study of a manganese oxide water oxidation catalyst, J. Phys. Chem. C, 2017, 121(22), 12003–12009 CrossRef CAS.
- F. Zhou, A. Izgorodin, R. K. Hocking, V. Armel, L. Spiccia and D. R. MacFarlane, Improvement of catalytic water oxidation on MnOx films by heat treatment, ChemSusChem, 2013, 6(4), 643–651 CrossRef CAS PubMed.
- M. Khan, J. Xiao, F. Zhou, M. Yablonskikh, D. R. MacFarlane, L. Spiccia and E. F. Aziz, On the origin of the improvement of electrodeposited MnOx films in water oxidation catalysis induced by heat treatment, ChemSusChem, 2015, 8(11), 1980–1985 CrossRef CAS PubMed.
- G. Mattioli, I. Zaharieva, H. Dau and L. Guidoni, Atomistic texture of amorphous manganese oxides for electrochemical water splitting revealed by ab initio calculations combined with X-ray spectroscopy, J. Am. Chem. Soc., 2015, 137(32), 10254–10267 CrossRef CAS.
- T. G. Spiro, J. R. Bargar, G. Sposito and B. M. Tebo, Bacteriogenic manganese oxides, Acc. Chem. Res., 2010, 43(1), 2–9 CrossRef CAS PubMed.
- K. Jin, H. Seo, T. Hayashi, M. Balamurugan, D. Jeong, Y. K. Go, J. S. Hong, K. H. Cho, H. Kakizaki, N. Bonnet-Mercier, M. G. Kim, S. H. Kim, R. Nakamura and K. T. Nam, Mechanistic investigation of water oxidation catalyzed by uniform, assembled MnO nanoparticles, J. Am. Chem. Soc., 2017, 139(6), 2277–2285 CrossRef CAS PubMed.
- H. Dau, P. Liebisch and M. Haumann, X-ray absorption spectroscopy to analyze nuclear geometry and electronic structure of biological metal centers—potential and questions examined with special focus on the tetra-nuclear manganese complex of oxygenic photosynthesis, Anal. Bioanal. Chem., 2003, 376(5), 562–583 CrossRef CAS PubMed.
- P. F. Smith, B. J. Deibert, S. Kaushik, G. Gardner, S. Hwang, H. Wang, J. F. Al-Sharab, E. Garfunkel, L. Fabris, J. Li and G. C. Dismukes, Coordination geometry and oxidation state requirements of corner-sharing MnO6 octahedra for water oxidation catalysis: An investigation of manganite (γ-MnOOH), ACS Catal., 2016, 6(3), 2089–2099 CrossRef CAS.
- B. Teo, EXAFS: Basic Principles and Data Analysis, Springer Verlag, Berlin, Germany, 1986 Search PubMed.
- A.-C. Gaillot, D. Flot, V. A. Drits, A. Manceau, M. Burghammer and B. Lanson, Structure of synthetic K-rich birnessite obtained by high-temperature decomposition of KMnO4. I. Two-layer polytype from 800 °C experiment, Chem. Mater., 2003, 15, 4666–4678 CrossRef CAS.
- I. Saratovsky, P. G. Wightman, P. A. Pasten, J.-F. Gaillard and K. R. Poeppelmeier, Manganese oxides: Parallels between abiotic and biotic structures, J. Am. Chem. Soc., 2006, 128, 11188–11198 CrossRef CAS PubMed.
- M. Wiechen, I. Zaharieva, H. Dau and P. Kurz, Layered manganese oxides for water-oxidation: alkaline earth cations influence catalytic activity in a photosystem II-like fashion, Chem. Sci., 2012, 3(7), 2330–2339 RSC.
- M. Risch, K. Klingan, F. Ringleb, P. Chernev, I. Zaharieva, A. Fischer and H. Dau, Water oxidation by electrodeposited cobalt oxides - role of anions and redox-inert cations in structure and function of the amorphous catalyst, ChemSusChem, 2012, 5(3), 542–549 CrossRef CAS PubMed.
- D. Gonzalez-Flores, I. Sanchez, I. Zaharieva, K. Klingan, J. Heidkamp, P. Chernev, P. W. Menezes, M. Driess, H. Dau and M. L. Montero, Heterogeneous water oxidation: surface activity versus amorphization activation in cobalt phosphate catalysts, Angew. Chem., Int. Ed., 2015, 54(8), 2472–2476 CrossRef CAS PubMed.
- M. Risch, F. Ringleb, M. Kohlhoff, P. Bogdanoff, P. Chernev, I. Zaharieva and H. Dau, Water oxidation by amorphous cobalt-based oxides: in situ tracking of redox transitions and mode of catalysis, Energy Environ. Sci., 2015, 8(2), 661–674 RSC.
- M. Gorlin, J. F. de Araujo, H. Schmies, D. Bernsmeier, S. Dresp, M. Gliech, Z. Jusys, P. Chernev, R. Kraehnert, H. Dau and P. Strasser, Tracking Catalyst Redox States and Reaction Dynamics in Ni-Fe Oxyhydroxide Oxygen Evolution Reaction Electrocatalysts: The Role of Catalyst Support and Electrolyte pH, J. Am. Chem. Soc., 2017, 139(5), 2070–2082 CrossRef PubMed.
- D. González-Flores, K. Klingan, P. Chernev, S. Loos, M. R. Mohammadi, C. Pasquini, P. Kubella, I. Zaharieva, R. D. L. Smith and H. Dau, Nickel-iron catalysts for electrochemical water oxidation – redox synergism investigated by in situ X-ray spectroscopy with millisecond time resolution, Sustainable Energy Fuels, 2018, 2(9), 1986–1994 RSC.
- D. M. Sherman, The electronic-structures of manganese oxide minerals, Am. Mineral., 1984, 69(7–8), 788–799 CAS.
- Z. Morgan Chan, D. A. Kitchaev, J. Nelson Weker, C. Schnedermann, K. Lim, G. Ceder, W. Tumas, M. F. Toney and D. G. Nocera, Electrochemical trapping of metastable Mn3+ ions for activation of MnO2 oxygen evolution catalysts, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(23), E5261–E5268 CrossRef PubMed.
- H. J. Lee, J. H. Jung, Y. S. Lee, J. S. Ahn, T. W. Noh, K. H. Kim and S. W. Cheong, Optical properties of a Nd0.7Sr0.3MnO3 single crystal, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 60(8), 5251–5257 CrossRef CAS.
- Y. Moritomo, A. Machida, K. Matsuda, M. Ichida and A. Nakamura, Magnetization-dependent behaviors of interband transitions between the exchange-split bands in doped manganite films, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56(9), 5088–5091 CrossRef CAS.
- J. H. Jung, K. H. Kim, T. W. Noh, E. J. Choi and J. Yu, Midgap states of La1-xCaxMnO3: Doping-dependent optical-conductivity studies, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57(18), R11043–R11046 CrossRef CAS.
- Z.-R. Tian, W. Tong, J.-Y. Wang, N.-G. Duan, V. V. Krishnan and S. L. Suib, Manganese oxide mesoporous structures: Mixed-valent semiconducting catalysts, Science, 1997, 276(5314), 926–930 CrossRef CAS.
- M. V. Abrashev, A. P. Litvinchuk, M. N. Iliev, R. L. Meng, V. N. Popov, V. G. Ivanov, R. A. Chakalov and C. Thomsen, Comparative study of optical phonons in the rhombohedrally distorted perovskites LaAlO3 and LaMnO3, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(6), 4146–4153 CrossRef CAS.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds Part A: Theory and Applications in Inorganic Chemistry, John Wiley & Sons Inc, Hoboken, New Jersey, 2009 Search PubMed.
- S. V. Ovsyannikov, A. M. Abakumov, A. A. Tsirlin, W. Schnelle, R. Egoavil, J. Verbeeck, G. Van Tendeloo, K. V. Glazyrin, M. Hanfland and L. Dubrovinsky, Perovskite-like Mn2O3: A path to new manganites, Angew. Chem., Int. Ed., 2013, 52(5), 1494–1498 CrossRef CAS.
- O. Frank, M. Zukalova, B. Laskova, J. Kürti, J. Koltai and L. Kavan, Raman spectra of titanium dioxide (anatase, rutile) with identified oxygen isotopes (16, 17, 18), Phys. Chem. Chem. Phys., 2012, 14(42), 14567–14572 RSC.
- Q. Feng, H. Kanoh and K. Ooi, Manganese oxide porous crystals, J. Mater. Chem., 1999, 9(2), 319–333 RSC.
- C. Julien, M. Massot, S. Rangan, M. Lemal and D. Guyomard, Study of structural defects in γ-MnO2 by Raman spectroscopy, J. Raman Spectrosc., 2002, 33(4), 223–228 CrossRef CAS.
- S. V. Ovsyannikov, A. E. Karkin, N. V. Morozova, V. V. Shchennikov, E. Bykova, A. M. Abakumov, A. A. Tsirlin, K. V. Glazyrin and L. Dubrovinsky, A hard oxide semiconductor with a direct and narrow bandgap and switchable p–n electrical conduction, Adv. Mater., 2014, 26(48), 8185–8191 CrossRef CAS PubMed.
- M. Fekete, R. K. Hocking, S. L. Y. Chang, C. Italiano, A. F. Patti, F. Arena and L. Spiccia, Highly active screen-printed electrocatalysts for water oxidation based on β-manganese oxide, Energy Environ. Sci., 2013, 6(7), 2222–2232 RSC.
- C. E. Frey, F. Kwok, D. Gonzales-Flores, J. Ohms, K. A. Cooley, H. Dau, I. Zaharieva, T. N. Walter, H. Simchi, S. E. Mohney and P. Kurz, Evaporated manganese films as a starting point for the preparation of thin-layer MnOx water-oxidation anodes, Sustainable Energy Fuels, 2017, 1(5), 1162–1170 RSC.
- L. D'Amario, R. Jiang, U. B. Cappel, E. A. Gibson, G. Boschloo, H. Rensmo, L. Sun, L. Hammarström and H. Tian, Chemical and physical reduction of high valence Ni states in mesoporous NiO film for solar cell application, ACS Appl. Mater. Interfaces, 2017, 9(39), 33470–33477 CrossRef PubMed.
- M. Risch, F. Ringleb, V. Khare, P. Chernev, I. Zaharieva and H. Dau, Characterisation of a water-oxidizing Co-film by XAFS, J. Phys.: Conf. Ser., 2009, 190, 012167 CrossRef.
- M. W. Kanan, J. Yano, Y. Surendranath, M. Dinca, V. K. Yachandra and D. G. Nocera, Structure and valency of a cobalt-phosphate water oxidation catalyst determined by in situ X-ray spectroscopy, J. Am. Chem. Soc., 2010, 132(39), 13692–13701 CrossRef CAS PubMed.
- M. Risch, K. Klingan, J. Heidkamp, D. Ehrenberg, P. Chernev, I. Zaharieva and H. Dau, Nickel-oxido structure of a water-oxidizing catalyst film, Chem. Commun., 2011, 47(43), 11912–11914 RSC.
- D. Shevchenko, M. F. Anderlund, S. Styring, H. Dau, I. Zaharieva and A. Thapper, Water oxidation by manganese oxides formed from tetranuclear precursor complexes: the influence of phosphate on structure and activity, Phys. Chem. Chem. Phys., 2014, 16(24), 11965–11975 RSC.
- M. Suga, F. Akita, K. Hirata, G. Ueno, H. Murakami, Y. Nakajima, T. Shimizu, K. Yamashita, M. Yamamoto, H. Ago and J.-R. Shen, Native structure of photosystem II at 1.95 Å resolution viewed by femtosecond X-ray pulses, Nature, 2015, 517(7532), 99–103 CrossRef CAS PubMed.
- J. Kern, R. Chatterjee, I. D. Young, F. D. Fuller, L. Lassalle, M. Ibrahim, S. Gul, T. Fransson, A. S. Brewster, R. Alonso-Mori, R. Hussein, M. Zhang, L. Douthit, C. de Lichtenberg, M. H. Cheah, D. Shevela, J. Wersig, I. Seuffert, D. Sokaras, E. Pastor, C. Weninger, T. Kroll, R. G. Sierra, P. Aller, A. Butryn, A. M. Orville, M. N. Liang, A. Batyuk, J. E. Koglin, S. Carbajo, S. Boutet, N. W. Moriarty, J. M. Holton, H. Dobbek, P. D. Adams, U. Bergmann, N. K. Sauter, A. Zouni, J. Messinger, J. Yano and V. K. Yachandra, Structures of the intermediates of Kok's photosynthetic water oxidation clock, Nature, 2018, 563(7731), 421–425 CrossRef CAS PubMed.
- J.-R. Shen, K. Kawakami and H. Koike, Purification and crystallization of oxygen-evolving photosystem II core complex from thermophilic cyanobacteria, Methods Mol. Biol., 2011, 684, 41–51 CrossRef CAS PubMed.
- H. Dau and M. Haumann, The manganese complex of photosystem II in its reaction cycle—basic framework and possible realization at the atomic level, Coord. Chem. Rev., 2008, 252(3–4), 273–295 CrossRef CAS.
- M. Haumann, C. Müller, P. Liebisch, L. Iuzzolino, J. Dittmer, M. Grabolle, T. Neisius, W. Meyer-Klaucke and H. Dau, Structural and oxidation state changes of the photosystem II manganese complex in four transitions of the water oxidation cycle (S0 → S1, S1 → S2, S2 → S3, and S3,4 → S0) characterized by X-ray absorption spectroscopy at 20 K and room temperature, Biochemistry, 2005, 44(6), 1894–1908 CrossRef CAS PubMed.
- V. Krewald, M. Retegan, N. Cox, J. Messinger, W. Lubitz, S. DeBeer, F. Neese and D. A. Pantazis, Metal oxidation states in biological water splitting, Chem. Sci., 2015, 6(3), 1676–1695 RSC.
- M. Retegan, V. Krewald, F. Mamedov, F. Neese, W. Lubitz, N. Cox and D. A. Pantazis, A five-coordinate Mn(IV) intermediate in biological water oxidation: spectroscopic signature and a pivot mechanism for water binding, Chem. Sci., 2016, 7(1), 72–84 RSC.
- T. Lohmiller, V. Krewald, M. P. Navarro, M. Retegan, L. Rapatskiy, M. M. Nowaczyk, A. Boussac, F. Neese, W. Lubitz, D. A. Pantazis and N. Cox, Structure, ligands and substrate coordination of the oxygen-evolving complex of photosystem II in the S2 state: a combined EPR and DFT study, Phys. Chem. Chem. Phys., 2014, 16(24), 11877–11892 RSC.
- I. Zaharieva, P. Chernev, G. Berggren, M. Anderlund, S. Styring, H. Dau and M. Haumann, Room-Temperature Energy-Sampling Kbeta X-ray Emission Spectroscopy of the Mn4Ca Complex of Photosynthesis Reveals Three Manganese-Centered Oxidation Steps and Suggests a Coordination Change Prior to O2 Formation, Biochemistry, 2016, 55(30), 4197–4211 CrossRef CAS.
- R. N. De Guzman, A. Awaluddin, Y.-F. Shen, Z. R. Tian, S. L. Suib, S. Ching and C.-L. O'Young, Electrical resistivity measurements on manganese oxides with layer and tunnel structures: Birnessites, todorokites, and cryptomelanes, Chem. Mater., 1995, 7(7), 1286–1292 CrossRef CAS.
- J. B. Goodenough, Theory of the role of covalence in the perovskite-type manganites [La, M(II)]MnO3, Phys. Rev., 1955, 100(2), 564–573 CrossRef CAS.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, The mechanism of water oxidation: from electrolysis via homogeneous to biological catalysis, ChemCatChem, 2010, 2(7), 724–761 CrossRef CAS.
- C. H. Kuo, I. M. Mosa, A. S. Poyraz, S. Biswas, A. M. E-Sawy, W. Q. Song, Z. Luo, S. Y. Chen, J. F. Rusling, J. He and S. L. Suib, Robust Mesoporous Manganese Oxide Catalysts for Water Oxidation, ACS Catal., 2015, 5(3), 1693–1699 CrossRef CAS.
- A. Indra, P. W. Menezes, F. Schuster and M. Driess, Significant role of Mn(III) sites in eg1 configuration in manganese oxide catalysts for efficient artificial water oxidation, J. Photochem. Photobiol., B, 2015, 152(Part A), 156–161 CrossRef CAS.
- T. Takashima, K. Hashimoto and R. Nakamura, Mechanisms of pH-dependent activity for water oxidation to molecular oxygen by MnO2 electrocatalysts, J. Am. Chem. Soc., 2012, 134(3), 1519–1527 CrossRef CAS.
- T. Takashima, K. Hashimoto and R. Nakamura, Inhibition of charge disproportionation of MnO2 electrocatalysts for efficient water oxidation under neutral conditions, J. Am. Chem. Soc., 2012, 134(44), 18153–18156 CrossRef CAS.
- M. M. Najafpour, D. J. Sedigh, S. M. Hosseini and I. Zaharieva, Treated Nanolayered Mn Oxide by Oxidizable Compounds: A Strategy To Improve the Catalytic Activity toward Water Oxidation, Inorg. Chem., 2016, 55(17), 8827–8832 CrossRef PubMed.
- A. Yamaguchi, R. Inuzuka, T. Takashima, T. Hayashi, K. Hashimoto and R. Nakamura, Regulating proton-coupled electron transfer for efficient water splitting by manganese oxides at neutral pH, Nat. Commun., 2014, 5, 4256 CrossRef CAS PubMed.
- T. Takashima, A. Yamaguchi, K. Hashimoto, H. Irie and R. Nakamura, In situ UV-vis absorption spectra of intermediate species for oxygen-evolution reaction on the Surface of MnO2 in neutral and alkaline media, Electrochemistry, 2014, 82(5), 325–327 CrossRef CAS.
- I. G. McKendry, S. K. Kondaveeti, S. L. Shumlas, D. R. Strongin and M. J. Zdilla, Decoration of the layered manganese oxide birnessite with Mn(II/III) gives a new water oxidation catalyst with fifty-fold turnover number enhancement, Dalton Trans., 2015, 44(29), 12981–12984 RSC.
- X. Li, J. Liu, Y. Zhao, H. Zhang, F. Du, C. Lin, T. Zhao and Y. Sun, Significance of surface trivalent manganese in the electrocatalytic activity of water oxidation in undoped and doped MnO2 nanowires, ChemCatChem, 2015, 7(12), 1848–1856 CrossRef CAS.
- B. A. Pinaud, Z. Chen, D. N. Abram and T. F. Jaramillo, Thin films of sodium birnessite-type MnO2: optical properties, electronic band structure, and solar photoelectrochemistry, J. Phys. Chem. C, 2011, 115(23), 11830–11838 CrossRef CAS.
- F. Farges, Ab initio and experimental pre-edge investigations of the Mn K-edge XANES in oxide-type materials, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71(15), 155109 CrossRef.
- A. Manceau, A. I. Gorshkov and V. A. Drits, Structural Chemistry of Mn, Fe, Co, and Ni in Manganese Hydrous Oxides: 1. Information from Xanes Spectroscopy, Am. Mineral., 1992, 77(11–12), 1133–1143 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures of the results from electrochemical experiments, XAS measurements, infrared and Raman spectroscopy, and SEM images. See DOI: 10.1039/c9ta05108b |
| This journal is © The Royal Society of Chemistry 2019 |