
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Demonstration of an azobenzene derivative based solar thermal energy storage system†
Zhihang
Wang
 a,
Raul
Losantos
b,
Diego
Sampedro
*b,
Masa-aki
Morikawa
cd,
Karl
Börjesson
e,
Nobuo
Kimizuka
*cd and
Kasper
Moth-Poulsen
*a
a,
Raul
Losantos
b,
Diego
Sampedro
*b,
Masa-aki
Morikawa
cd,
Karl
Börjesson
e,
Nobuo
Kimizuka
*cd and
Kasper
Moth-Poulsen
*a
aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, 41296 Gothenburg, Sweden. E-mail: kasper.moth-poulsen@chalmers.se
bDepartment of Chemistry, Centro de Investigación en Síntesis Química (CISQ), Universidad de La Rioja, Madre de Dios 53, E-26006 Logroño, La Rioja, Spain. E-mail: diego.sampedro@unirioja.es
cDepartment of Chemistry and Biochemistry, Graduate School of Engineering, Kyushu University, 744 Moto-oka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: kimizuka.nobuo.763@m.kyushu-u.ac.jp
dCenter for Molecular Systems (CMS), Kyushu University, 744 Moto-oka, Nishi-ku, Fukuoka 819-0395, Japan
eDepartment of Chemistry and Molecular Biology, University of Gothenburg, Kemigården 4, 41296 Gothenburg, Sweden
First published on 3rd June 2019
Abstract
Molecules capable of reversible storage of solar energy have recently attracted increasing interest, and are often referred to as molecular solar thermal energy storage (MOST) systems. Azobenzene derivatives have great potential as an active MOST candidate. However, an operating lab scale experiment including solar energy capture/storage and release has still not been demonstrated. In the present work, a liquid azobenzene derivative is tested comprehensively for this purpose. The system features several attractive properties, such as a long energy storage half-life (40 h) at room temperature, as well as an excellent robustness demonstrated by optically charging and discharging the molecule over 203 cycles without any sign of degradation (total operation time of 23 h). Successful measurements of solar energy storage under simulated sunlight in a microfluidic chip device have been achieved. The identification of two heterogeneous catalyst systems during testing enabled the construction of a fixed bed flow reactor demonstrating catalyzed back-conversion from cis to trans azobenzene at room temperature under flow conditions. The working mechanism of the more suitable catalytic candidate was rationalized by detailed density functional theory (DFT) calculations. Thus, this work provides detailed insights into the azobenzene based MOST candidate and identifies where the system has to be improved for future solar energy storage applications.
Introduction
The development of sustainable energy has been attracting increasing attention, due to the pressing environmental and social challenges linked to high dependency on fossil fuels in our modern society.1 As the most abundant energy source for Earth, the Sun can provide the energy needed for mankind for an entire year in only six hours.2 Various ways to take advantage of solar energy have been studied and developed over the last few decades, including photovoltaics,3 artificial photosynthesis,4 and solar thermal heating.5 In order to store sunlight for future use, molecular solar thermal storage techniques (MOST,6,7 also known as solar thermal fuels, STF8) focus on harvesting solar energy and storing it in photoswitchable materials. A parent molecule can be isomerized by solar irradiation to a metastable high energy photoisomer for a long storage period. When the energy is required, it can be thermally or catalytically back-converted to the parent state. Ideally, the stored energy should be released on demand as heat, thus operating as a closed-cycle system.In order to realize the MOST concept, several features concerning the charge and discharge process need to be considered.7,9,10 (1) Since more than 50% of sunlight is distributed from 300 nm to 800 nm, the parent molecules should be able to absorb broadly in this spectral region. (2) The quantum yield of the photoisomerisation reaction should be as close to unity as possible. (3) The storage half-life at room temperature should be long enough to fulfill application-relevant storage times, such as daily, monthly, yearly or even longer storage requirements. (4) The energy of the metastable photoisomer should be significantly higher than the ground state of the parent isomer. (5) The system should ideally be able to operate over an infinite number of charging and heat releasing cycles. (6) Using a heterogeneous catalyst, the photoisomer should easily release the stored energy as heat. (7) For energy collection, storage, and bulk heating applications, MOST materials need to be pumped between solar collector, storage reservoir and heat extraction devices. As a consequence, the MOST system should be a liquid or highly soluble solid.
The MOST concept has been represented by many potential candidates, including norbornadiene/quadricyclane derivatives,7,9–14 dihydroazulene/vinylheptafulvene couples,15–21 difulvalenediruthenium complexes,6 anthracene dimers,22 Dewar isomers23 and azobenzene derivatives.8,24–43 The last systems have been in the spotlight recently due to the broad absorption spectrum, high robustness, daily storage half-life, tunable energy density (J mol−1) of photoswitches, and the low synthetic cost.40–42AZO1 (see Fig. 1a) possesses many of the needed features described above; however it has not been tested in a full MOST operating cycle including energy capture, storage and release. Here, for the first time, we investigate the performance of AZO1 by studying its function through multiple energy storage and release cycles. In order to do so, we have identified and tested a heterogeneous catalyst system for the energy release step and constructed a flow system consisting of a solar collector and a fixed-bed catalytic converter. Furthermore, the functioning mechanism of the catalytic conversion is further proposed and rationalized using PCM-B3LYP-D3BJ/6-31G(d)+SDD(Cu) calculations.
 | ||
| Fig. 1 (a) Structure of AZO1 in the trans and cis state. (b) Absorption spectra of AZO1-trans (in blue) and its corresponding photoisomer AZO1-cis (in red). The sample (ca. 2 mg in 100 mL toluene) was converted with a 340 nm LED lamp. (c) Eyring plot of AZO1-cis. The half-life at 25 °C was determined as 36.3 h in toluene solution. (d) Optical cycling test of AZO1 in toluene. The figure shows 203 cycles in total. The absorbance was recorded at 350 nm. The charge and discharge processes were archived with a 340 nm LED (∼60 mW) over 100 s and a 455 nm LED (∼1020 mW) over 300 s, alternatively. | ||
Results and discussion
To characterize the physical properties of AZO1, it was first studied in toluene in terms of its absorptivity, half-life of energy storage and quantum yields of the photoisomerization reaction. Comparing these results with available data of AZO1 in methanol,43 the maximum in the absorption spectrum was observed to be slightly red shifted, indicating a low solvent polarity effect compared to, for instance, a DHA-MOST system18 (ca. 5 nm, εMax@350 nm = 2.6 × 104 M−1 cm−1, see Fig. 1b). Concerning the storage lifetime, a half-life from cis to trans state of 36.3 h in toluene can be extracted from the Eyring plot. The activation enthalpy was calculated as ΔH‡therm = 93.7 kJ mol−1, together with an activation entropy ΔS‡therm = −31.7 J mol−1 K−1 (S2, ESI†). The quantum yield of the trans to cis photoisomerisation reaction was determined to be 21% at 340 nm, using a literature procedure44 (S3, ESI†). In addition, it was observed that the AZO1-cis state can photoisomerise back to the trans form. The corresponding quantum yield of the cis to trans photoisomerisation was determined to be 23% at 455 nm, slightly higher than the reverse process. Due to this photo-induced two-way switch effect for AZO1, full conversion via full spectrum solar light can likely not be achieved and a band pass filter was used in the device conversion experiments.Good cyclability is one of the most important criteria of MOST systems. To investigate the robustness of AZO1, a solution of ca. 10−5 M in toluene was prepared without degassing. Two controllable LED light sources were turned on and off alternatingly (340 nm with 100 s irradiation time and 455 nm with 300 s irradiation time) to charge and discharge the molecules back and forth. After 203 complete cycles (with a total operation time of 22.6 h), no significant signs of degradation were observed, thus demonstrating high robustness of AZO1, even in the presence of oxygen (see Fig. 1c).
To further demonstrate the functionality of the AZO1 system in a real device, a continuous flow system was built. For this lab-scale conversion test under an AM1.5 solar simulator, diluted solutions of AZO1 were used to collect data by UV-vis spectroscopy. Two concentrations of AZO1 solutions were pumped individually through a microfluidic chip,20,21 with varying residence time (33.9 mm3 inner volume with a channel depth of 100 μm, see Fig. 2a). Since AZO1-cis can be back-converted by visible light, an optical UV transmitting band pass filter (<400 nm) was inserted in between the solar simulator and the microfluidic chip. The UV-Vis spectra of the AZO1 solution before and after pumping into the microfluidic chip were recorded. Eqn (1) was used to calculate the cis-to-trans conversion percentage (S4, ESI†):
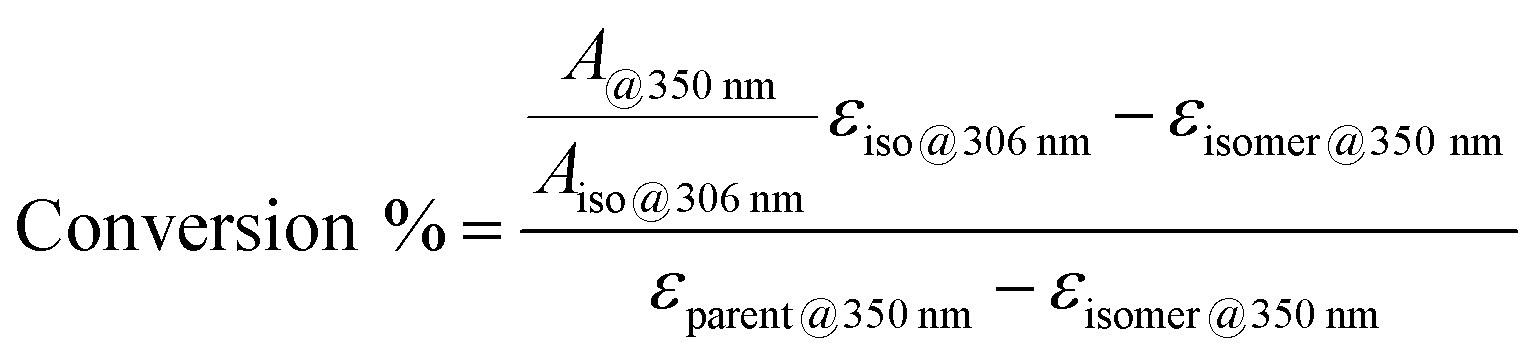 | (1) |
 | ||
| Fig. 2 (a) Experimental setup of AZO1 in a microfluidic chip device. Gray boxes 1 and 2 contain a flow UV-vis detection device connected to a portable spectrophotometer, 3, corresponding to a total volume of 33.9 mm3 quartz chip, with a 100 μm optical path length. (b) Experimental data of the two concentrations used: 2 × 10−4 M in green and 5 × 10−4 M in red. Conversion percentage of AZO1-trans with different residence times in the microfluidic chip. (c) Measured energy storage efficiency of the AZO1 compound in toluene. | ||
After solar capture/storage, energy release is the second fundamental process for the MOST concept. To estimate the adiabatic heat release as a function of concentration, a modified equation from a previous study11 was used (eqn (2)):
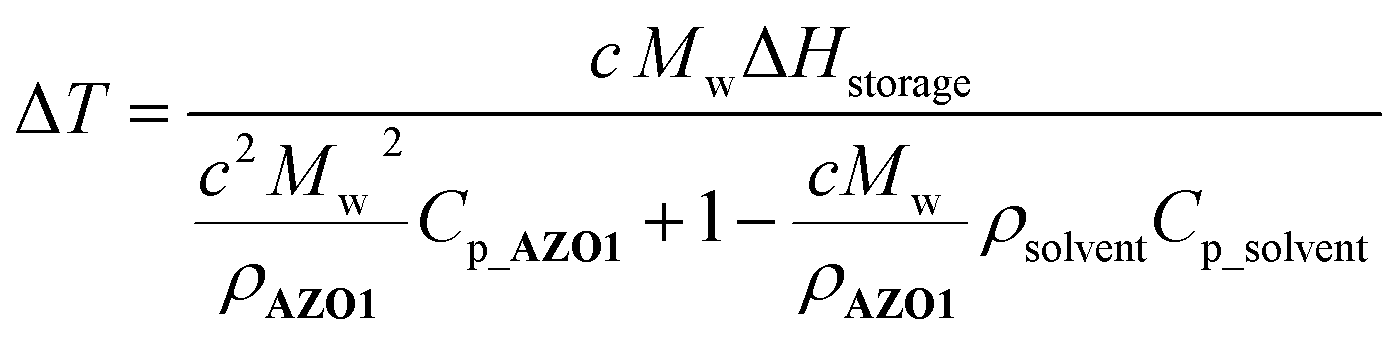 | (2) |
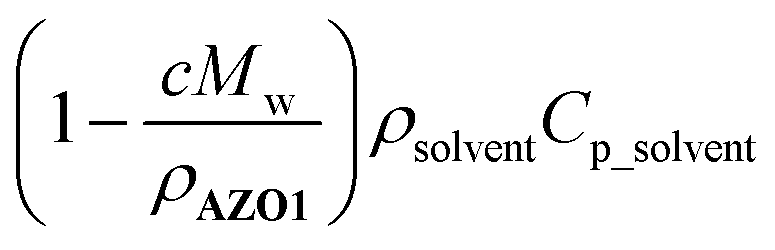 approaches zero. This correction is reasonable for all MOST heat release estimations with close to neat conditions. In the case of AZO1, the theoretical maximum temperature difference was calculated as ca. 226 °C for a fully charged neat sample (see S7, ESI†).
approaches zero. This correction is reasonable for all MOST heat release estimations with close to neat conditions. In the case of AZO1, the theoretical maximum temperature difference was calculated as ca. 226 °C for a fully charged neat sample (see S7, ESI†).
Concerning related catalysts, several candidates, including mineral acids like perchloric acid, Cu(II) salts (CuCl2,Cu(OAc)2), gold nanoparticles which involve a redox mechanism, as well as electrocatalytic methods have been reported to induce the back-conversion of azobenzene derivatives.46–48 However, for a closed cycle system which can be operated in devices, a heterogeneous catalyst that can be fixed in a reaction centre is required. Based on this fact, a heterogeneous catalyst or an insoluble homogeneous catalyst needs to be developed. For AZO1, two potential catalysts, cobalt(II) phthalocyanine physisorbed on the surface of activated carbon (CoPc@C) and [Cu(CH3CN)4]PF6 fulfill the described physical properties and thus, they were tested individually. Both showed a positive effect on reducing the back-conversion half-life at room temperature. [Cu(CH3CN)4]PF6 has a very low solubility in toluene, being active for various MOST systems, including norbornadiene/quadricyclane derivatives11 and dihydroazulene/vinylheptafulvene couples.20,49 For the AZO1-cis compound, a back-conversion reaction rate of up to 30 s−1 was calculated at room temperature of 25 °C (up to 6 × 106 times higher than a reaction rate of 5 × 10−6 s−1 without the catalyst at 25 °C; see Fig. 3a, S7, ESI†). CoPc@C was produced following a reported procedure; however it had a risk of leaching from its solid support.11 Therefore, the [Cu(CH3CN)4]PF6 salt was chosen to be incorporated into the catalytic device for further testing. With this result in mind, a small-sized reaction centre was built. 5 mg of Cu(I) salt was inserted into a Teflon tube which has a 1 mm inner diameter (see Fig. 3b). 5 × 10−4 M 79% AZO1-cis solution from the microfluidic chip experiments was then flowed through the catalytic bed with a speed of 1 mL h−1. As result, 48% of AZO1-cis was successfully back-converted to the corresponding trans state. Thus, the described continuous fluidic chip experiments can achieve the requirements for the complete application of the MOST concept including photon capture/storage and energy release processes.
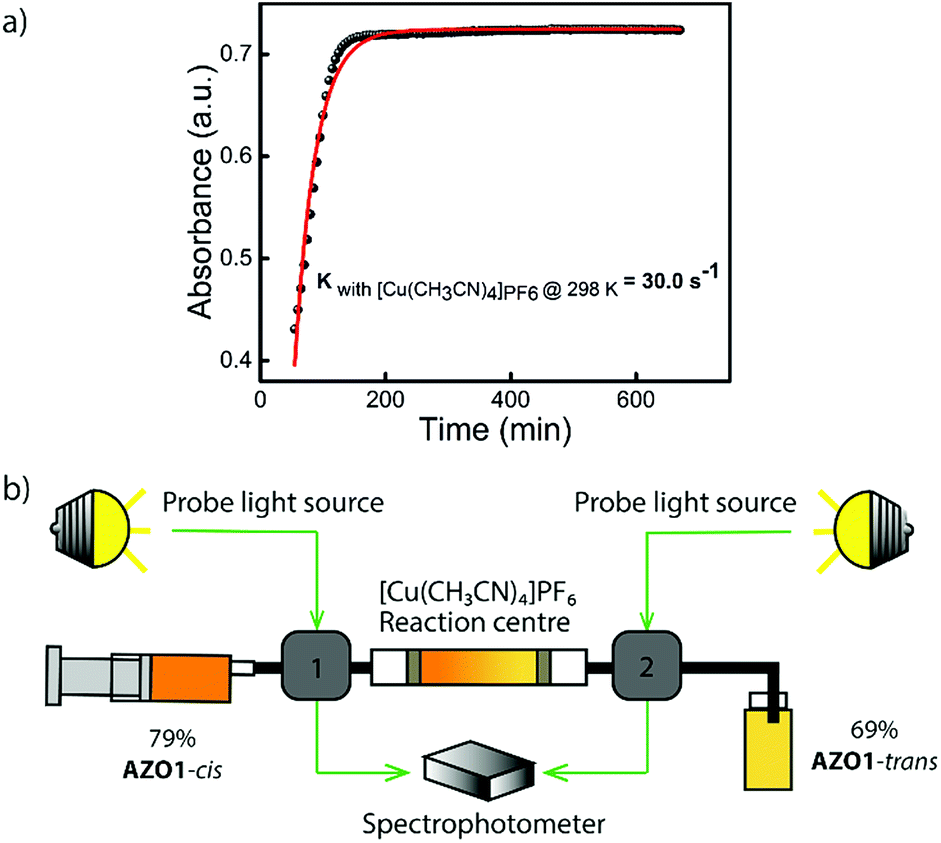 | ||
| Fig. 3 (a) Kinetics study of AZO1-cis in the presence of [Cu(CH3CN)4]PF6 in toluene at 25 °C. Black dots represent the experimental data, red curve corresponds to the exponential fitting. The reaction constant was calculated as 30.0 s−1 (b) Conceptual device demonstration of catalytic back-conversion of AZO1-cis. Around 5 mg of Cu(I) salt were loaded in to the reaction centre. A 5 × 10−4 M 79% AZO1-cis solution was flown through the Cu(I) reactor with a speed of 1 mL h−1. 69% of the AZO1-trans was obtained after the reaction centre, i.e. 48% of the AZO1-cis can be successfully back-converted to AZO1-trans state. | ||
To further understand the mechanism of back-conversion with [Cu(CH3CN)4]PF6, a detailed study within the framework of the density functional theory (DFT) was performed. A simplified AZO1 system was used replacing the ethylhexyl moiety with a methyl group at the PCM-B3LYP-D3BJ/6-31G(d)+SDD(Cu) level of theory (S8, ESI†). Among the different coordination options, the CH3CN ligands could be displaced by the new ligand (the cis isomer of the azobenzene moiety, in this case). Four different coordination possibilities were considered to ensure a good modelling of the experimental conditions. In all cases, the formation of the new complex is slightly endergonic (1–5 kcal mol−1). As the azobenzene derivative is not symmetric, two sets of coordination alternatives arise. Coordination through the nitrogen atom directly linked to the unsubstituted phenyl ring (path b, S8, ESI† and Fig. 4a) is favored (by ca. 2.3 kcal mol−1) as compared to the methoxyphenyl linked nitrogen atom (path a). Isomerization of azobenzenes is very dependent on substitution and reaction conditions.50 In this case, different pathways were considered and rotation and inversion mechanisms of the isomerization were explicitly evaluated. As in the not catalyzed thermal reaction,47 the inversion transition states (TS1a–TS1b) were found to be preferred.
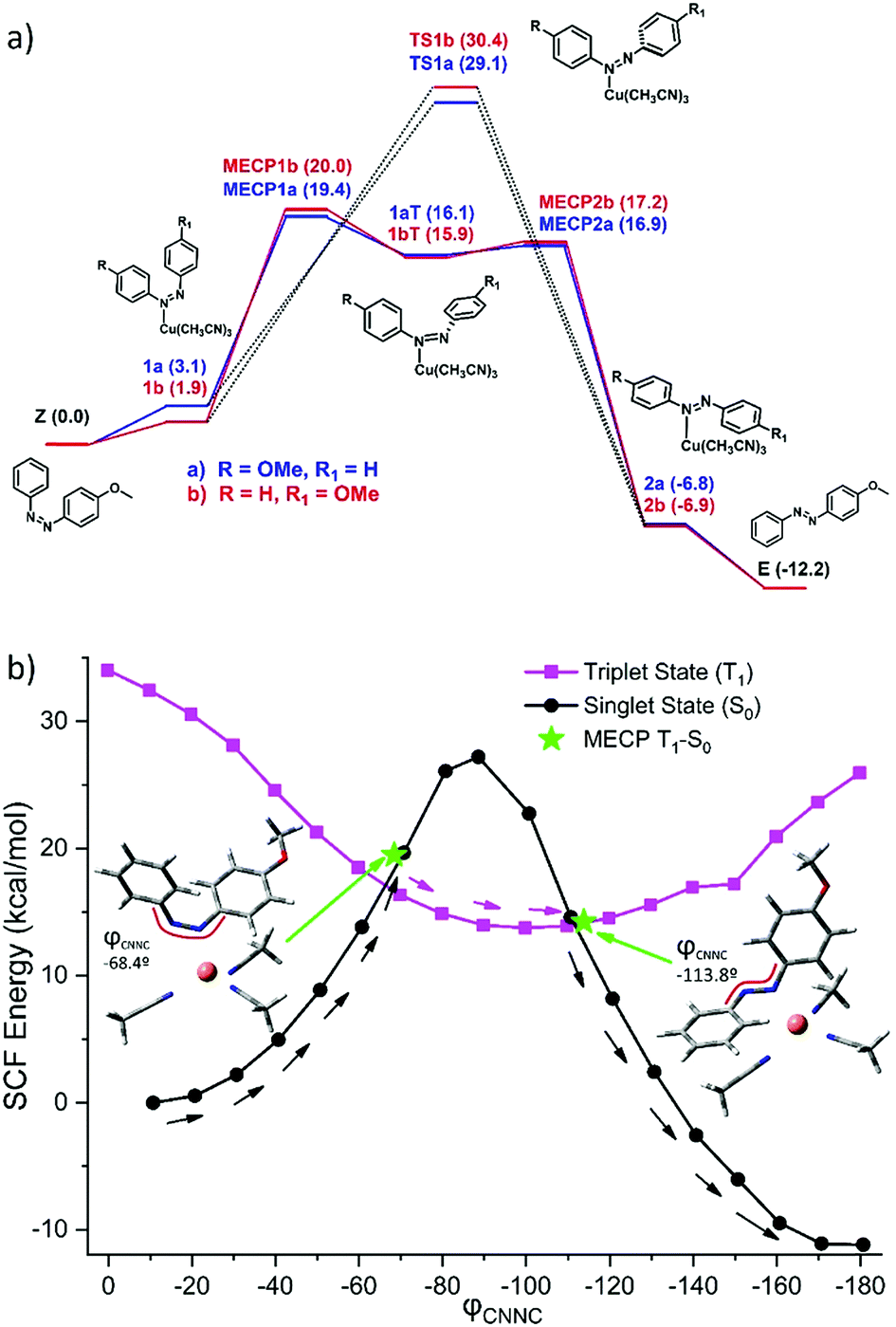 | ||
| Fig. 4 (a) Reaction paths for cis–trans isomerization. Coordination through the nitrogen atom directly linked to the phenyl ring (path b, in red) and to the methoxyphenyl nitrogen atom (path a, in blue). Solid lines are the S0–T1–S0 pathway (MECP1(a,b)-1(a,b)T-MECP2(a,b)). Dotted black lines are the singlet reaction path (TS1a–TS1b). Free energies referred to cis-AZO + Cu(CH3CN)4+. (b) Relaxed scan along CNNC rotation and MECPs. | ||
A relaxed scan along the rotation coordinate (CNNC dihedral) (see Fig. 4b) reveals a possible S0–T1–S0 mechanism accessible by coordination of the nitrogen atom of AZO1-cis to copper, which reveals a MLCT (metal-to-ligand charge transfer). In turn, this implies the participation of a π* orbital of the azobenzene allowing rotation along the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N with participation of the T1 state. At relative high torsion angles, the triplet state becomes less energetic and this path yields a barrier of around 20 kcal mol−1. This alternative mechanism allows for the bypassing of the more energetic inversion TS in the ground state (30 kcal mol−1) and it should be available at room temperature.
N with participation of the T1 state. At relative high torsion angles, the triplet state becomes less energetic and this path yields a barrier of around 20 kcal mol−1. This alternative mechanism allows for the bypassing of the more energetic inversion TS in the ground state (30 kcal mol−1) and it should be available at room temperature.
Thus, copper coordination yields a decrease in the isomerization barrier of ca. 6 kcal mol−1 (computed barrier decreases from 25.4 kcal mol−1 for free azobenzene to 19.4 kcal mol−1). This relatively small decrease in the energy barrier is enough to allow the isomerization process to occur in a few minutes instead of some days, thus accelerating the isomerization by 4 orders of magnitude, resulting in good agreement with experiments (energy difference of 8 kcal mol−1, from 23.5 kcal mol−1 for the free AZO1 to 15.6 kcal mol−1 for the copper activated reaction). Overall, while [Cu(CH3CN)4]PF6 is useful to increase the reaction rate notably, there is still great potential for improvement in the design of the energy release step. This could be done, for instance, favoring the MLCT or stabilizing the MECP between singlet and triplet states to increase the efficiency of the crossing. In both cases, this could lead to a decrease in the energy barrier and a subsequent acceleration of the back-conversion.
Conclusions
An AZO1-MOST charging/discharging cycle has been successfully demonstrated, including the study of the photophysical properties and back-conversion of the photoisomer. The AZO1 system features strong absorption (εMax@350 nm = 2.6 × 104 M−1 cm−1) and long thermal half-life (36.3 h). The optical cyclability test for a diluted solution shows no significant degradation in the presence of oxygen, allowing the use of AZO1 in toluene in application. For MOST application purposes, the conversion quantum yield at 340 nm was determined to be 21% which is similar to the quantum yield of back-conversion of 23% at 455 nm. This implies that even diluted samples cannot be fully converted under sunlight. The estimated maximum energy storage efficiency for AZO1 in the neat state was calculated to be 0.88%. To experimentally demonstrate the functionality of this liquid azobenzene for MOST applications, conversion experiments were performed at different concentrations of AZO1 in a continuous microfluidic chip system. The measured maximum energy storage efficiency for a 5 × 10−4 M solution can reach close to 0.009%, close to reach the theoretical prediction of 0.02% at such a concentration. This, unfortunately, showed a strong photo-stationary effect between AZO-cis and AZO-trans, therefore highlights the need to further develop the azobenzene system by altering the optical properties.51–53 Further improvement to increase quantum yields and diminish the absorption of photoisomer, as well as differentiate the spectral difference between cis and trans states, would be certainly required. To estimate the theoretical heat release temperature difference, a corrected formula was introduced to account for the change from the diluted solution to the neat sample. A maximum temperature increase of 226 °C can be theoretically achieved by using eqn (2) with a correction term for a neat MOST sample. Furthermore, two new catalysts have been identified to allow the back-conversion of AZO1-cis and a reaction centre based on a Cu(I) salt has been prepared as a proof of concept for the energy release process. With a concentration of 5 × 10−4 M and a flow speed of 1 mL h−1, 48% of the cis state isomer was successfully converted back to the trans state. However, the reaction rate was still too low to be used for macroscopic heat release purposes. Finally, a detailed theoretical study of the mechanism has been proposed to further understand the discharge process using a Cu(I) salt. This could help in the future design of new azobenzene derivatives and the corresponding catalysts. In addition, results shown here could be extended to the control of azobenzene derivatives for different applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the K. & A. Wallenberg foundation and the Swedish Foundation for Strategic Research. D. S. and R. L. thank the Spanish Ministerio de Economía y Competitividad (MINECO)/Fondos Europeos para el Desarrollo Regional (FEDER) (CTQ2017-87372-P). R. L. thanks the Universidad de La Rioja for his fellowship. This work used the Beronia cluster (Universidad de La Rioja), which is supported by FEDER-MINECO grant number UNLR-094E-2C-225. Thanks a lot to Xin Wen and Sarah Lerch for all the fruitful discussions.Notes and references
- IPCC, Climate Change Synthesis Report Summary for Policymakers, 2014 Search PubMed.
- R. Perez and M. Perez, The International Energy Agency SHCP Solar Update, 2009, vol. 50, pp. 2–3 Search PubMed.
- K. Mertens, Photovoltaics: Fundamentals, Technology and Practice, Wiley, 2014 Search PubMed.
- F. Collings and C. Critchley, Artificial Photosynthesis: from Basic Biology and Industrial Application, Wiley, 2005 Search PubMed.
- S. M. Hasnain, Energy Convers. Manage., 1998, 39, 1127–1138 CrossRef CAS.
- K. Moth-Poulsen, D. Ćoso, K. Börjesson, N. Vinokurov, S. K. Meier, A. Majumdar, K. P. C. Vollhardt and R. A. Segalman, Energy Environ. Sci., 2012, 5, 8534–8537 RSC.
- Z.-i. Yoshida, J. Photochem., 1985, 29, 27–40 CrossRef CAS.
- L. Dong, Y. Feng, L. Wang and W. Feng, Chem. Soc. Rev., 2018, 47, 7339–7368 RSC.
- V. A. Bren ’, A. D. Dubonosov, V. I. Minkin and V. A. Chernoivanov, Russ. Chem. Rev., 1991, 60, 913–948 Search PubMed.
- K. Börjesson, A. Lennartson and K. Moth-Poulsen, ACS Sustainable Chem. Eng., 2013, 1, 585–590 CrossRef.
- Z. Wang, A. Roffey, R. Losantos, A. Lennartson, M. Jevric, A. U. Petersen, M. Quant, A. Dreos, X. Wen, D. Sampedro, K. Börjesson and K. Moth-Poulsen, Energy Environ. Sci., 2019, 12, 187–193 RSC.
- A. Dreos, Z. Wang, J. Udmark, A. Ström, P. Erhart, K. Börjesson, M. B. Nielsen and K. Moth-Poulsen, Adv. Energy Mater., 2018, 8, 1703401 CrossRef.
- M. Jevric, A. U. Petersen, M. Mansø, S. K. Singh, Z. Wang, A. Dreos, C. Sumby, M. B. Nielsen, K. Börjesson, P. Erhart and K. Moth-Poulsen, Chem.–Eur. J., 2018, 24, 12767–12772 CrossRef CAS PubMed.
- A. U. Petersen, M. Jevric and K. Moth-Poulsen, Eur. J. Org. Chem., 2018, 32, 4465–4474 CrossRef.
- A. Vlasceanu, S. L. Broman, A. S. Hansen, A. B. Skov, M. Cacciarini, A. Kadziola, H. G. Kjaergaard, K. V. Mikkelsen and M. B. Nielsen, Chem.–Eur. J., 2016, 22, 10796–10800 CrossRef CAS PubMed.
- M. Cacciarini, A. B. Skov, M. Jevric, A. S. Hansen, J. Elm, H. G. Kjaergaard, K. V. Mikkelsen and M. B. Nielsen, Chem.–Eur. J., 2015, 21, 7454–7461 CrossRef CAS PubMed.
- M. Cacciarini, M. Jevric, J. Elm, A. U. Petersen, K. V. Mikkelsen and M. B. Nielsen, RSC Adv., 2016, 6, 49003–49010 RSC.
- O. Schalk, S. L. Broman, M. Å. Petersen, D. V. Khakhulin, R. Y. Brogaard, M. B. Nielsen, A. E. Boguslavskiy, A. Stolow and T. I. Sølling, J. Phys. Chem. A, 2013, 117, 3340–3347 CrossRef CAS PubMed.
- A. B. Skov, S. L. Broman, A. S. Gertsen, J. Elm, M. Jevric, M. Cacciarini, A. Kadziola, K. V. Mikkelsen and M. B. Nielsen, Chem.–Eur. J., 2016, 22, 14567–14575 CrossRef CAS PubMed.
- Z. Wang, J. Udmark, K. Börjesson, R. Rodrigues, A. Roffey, M. Abrahamsson, M. B. Nielsen and K. Moth-Poulsen, ChemSusChem, 2017, 10, 3049–3055 CrossRef CAS PubMed.
- M. H. Hansen, S. T. Olsena, K. O. Sylvester-Hvidb and K. V. Mikkelsen, Chem. Phys., 2019, 519, 92–100 CrossRef CAS.
- R. R. Islangulov and F. N. Castellano, Angew. Chem., Int. Ed., 2006, 45, 5957–5959 CrossRef CAS PubMed.
- K. Edel, X. Yang, J. S. A. Ishibashi, A. N. Lamm, C. Maichle-Mössmer, Z. X. Giustra, S. Liu and H. F. Bettinger, Angew. Chem., Int. Ed., 2018, 57, 5296–5300 CrossRef CAS PubMed.
- H. Taoda, K. Hayakawa, K. Kawase and H. Yamakita, J. Chem. Eng. Jpn., 1987, 3, 265–270 CrossRef.
- A. Natansohn and P. Rochon, Chem. Rev., 2002, 102, 4139–4175 CrossRef CAS PubMed.
- K. G. Yager and C. J. Barrett, J. Photochem. Photobiol., A, 2006, 182, 250–261 CrossRef CAS.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- J. Olmsted III, J. Lawrence and G. G. Yee, Sol. Energy, 1983, 3, 271–274 CrossRef.
- H. Taoda, K. Hayama, K. Kawase and H. Yamashita, J. Chem. Eng. Jpn., 1987, 20, 265–270 CrossRef CAS.
- A. Natansohn and P. Rochon, Chem. Rev., 2002, 102, 4139–4175 CrossRef CAS PubMed.
- K. G. Yager and C. J. Barrett, J. Photochem. Photobiol., A, 2006, 182, 250–261 CrossRef CAS.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- M. Irie, Bull. Chem. Soc. Jpn., 2008, 81, 917–926 CrossRef CAS.
- A. M. Kolpak and J. C. Grossman, Nano Lett., 2011, 11, 3156–3162 CrossRef CAS PubMed.
- A. M. Kolpak and J. C. Grossman, J. Phys. Chem., 2013, 138, 034303 CrossRef PubMed.
- T. J. Kucharski, N. Ferralis, A. M. Kolpak, J. O. Zheng, D. G. Nocera and J. C. Grossman, Nat. Chem., 2014, 6, 441–447 CrossRef CAS PubMed.
- I. Gui, K. Sawyer and R. Prasher, Science, 2012, 335, 1454–1455 CrossRef PubMed.
- L. C. Branco and F. Pina, Chem. Commun., 2009, 6204–6206 RSC.
- S. L. Zhang, Q. Zhang and Y. Deng, Chem. Commun., 2011, 47, 6641–6646 RSC.
- G. D Han, S. S. Park, Y. Liu, D. Zhitomirsky, E. Cho, M. Dinca and J. C. Grossman, J. Mater. Chem. A, 2016, 4, 16157–16165 RSC.
- D. Zhitomirsky and J. C. Grossman, ACS Appl. Mater. Interfaces, 2016, 8, 26319–26325 CrossRef CAS PubMed.
- D. Zhitomirsky, E. Cho and J. C. Grossman, Adv. Energy Mater., 2016, 6, 1502006 CrossRef.
- K. Masutani, M.-a. Morikawa and N. Kimizuka, Chem. Commun., 2014, 50, 15803–15806 RSC.
- K. Stranius and K. Börjesson, Sci. Rep., 2017, 7, 41145 CrossRef CAS PubMed.
- J. A. Bouwstra, V. V. De Leeuw and J. C. Van Miltenburg, J. Chem. Thermodyn., 1985, 17, 685–695 CrossRef CAS.
- E. Titov, L. Lysyakova, N. Lomadze, A. V. Kabashin, P. Saalfrank and S. Santer, J. Phys. Chem. C, 2015, 119(30), 17369–17377 CrossRef CAS.
- A. Cembran, F. Bernardi, M. Garavelli, L. Gagliardi and G. Orlandi, J. Am. Chem. Soc., 2004, 126, 3234–3243 CrossRef CAS.
- A. Goulet-Hanssens, M. Utecht, D. Mutruc, E. Titov, J. Schwarz, L. Grubert, D. Bléger, P. Saalfrank and S. Hecht, J. Am. Chem. Soc., 2017, 139, 335–341 CrossRef CAS PubMed.
- M. Cacciarini, A. Vlasceanu, M. Jevric and M. B. Nielsen, Chem. Commun., 2017, 53, 5874 RSC.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41(5), 1809–1825 RSC.
- C. García-Iriepa, M. Marazzi, L. M. Frutos and D. Sampedro, RSC Adv., 2013, 3, 6241–6266 RSC.
- H. M. Dhammika Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- J. Moreno, L. Grubert, J. Schwarz, D. Bléger and S. Hecht, Chem.–Eur. J., 2017, 23, 14090–14095 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta04905c |
| This journal is © The Royal Society of Chemistry 2019 |