
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A Ca2+ MOF combining highly efficient sorption and capability for voltammetric determination of heavy metal ions in aqueous media†
Anastasia D.
Pournara
a,
Antigoni
Margariti
b,
Georgios D.
Tarlas
b,
Andreas
Kourtelaris
c,
Valeri
Petkov
 d,
Christos
Kokkinos
e,
Anastasios
Economou
e,
Giannis S.
Papaefstathiou
*b and
Manolis J.
Manos
*a
d,
Christos
Kokkinos
e,
Anastasios
Economou
e,
Giannis S.
Papaefstathiou
*b and
Manolis J.
Manos
*a
aLaboratory of Inorganic Chemistry, Department of Chemistry, University of Ioannina, 45110 Ioannina, Greece. E-mail: emanos@cc.uoi.gr
bLaboratory of Inorganic Chemistry, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis, 157 71, Zografou, Greece. E-mail: gspapaef@chem.uoa.gr
cDepartment of Chemistry, University of Cyprus, 1678 Nicosia, Cyprus
dDepartment of Physics and Science of Advanced Materials Program, Central Michigan University, Mt. Pleasant, Michigan 48859, USA
eLaboratory of Analytical Chemistry, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis, 157 71, Zografou, Greece
First published on 30th May 2019
Abstract
We recently discovered that a Ca2+ two-dimensional framework, namely [Ca(H4L)(DMA)2]·2DMA (Ca-MOF), was capable of exchanging the Ca2+ ions with Cu2+ almost quantitatively in a matter of seconds in aqueous solution. Herein, we report that Ca-MOF exhibits the capability of both removal and voltammetric determination of heavy metal ions in aqueous media. Ca-MOF shows one of the highest Pb2+ sorption capacities (∼522 mg g−1) reported for MOFs. More importantly, a column filled with Ca-MOF (1% wt) particles dispersed in silica sand (99% wt) can quantitatively remove traces of Pb2+ (∼100 ppb) from a relatively large volume of a wastewater simulant solution (containing a large excess of competitive ions). Ca-MOF is also highly efficient for sorption of Cd2+, Ni2+ and Zn2+, even in the presence of several competitive cations. Actually, the Cd2+ sorption capacity (∼220 mg g−1) of Ca-MOF is one of the largest reported for MOFs. Furthermore, detailed Ni2+ and Zn2+ sorption studies of MOFs have not been described prior to this work. The mechanism of the M2+ (M2+ = Pb2+, Cd2+, Ni2+, Zn2+) exchange process was elucidated based on a series of spectroscopic, analytical and X-ray diffraction methods. In addition, a simple ready-to-use electrochemical sensor based on modified graphite paste with Ca-MOF was fabricated and successfully utilized for the determination of Pb2+, Cd2+, Cu2+ and Zn2+ at μg L−1 levels in aqueous solutions by anodic stripping voltammetry (ASV). Overall, this work demonstrates, for the first time, a dual function of a MOF as a sorbent and as an electrochemical sensor for heavy metal ions, thus opening a new window for materials with application in both environmental remediation and monitoring.
Introduction
Heavy metal ions, such as Pb2+, Cd2+, Cu2+, Ni2+, Zn2+etc., represent the major contaminants for water resources. They are easily accumulated in the environment and living organisms and are highly toxic even at low concentrations (with the exception of Zn2+ which in low concentration is essential for humans and other species).1 It is thus of vital importance to develop efficient and affordable methods for both the determination and the removal of these toxic species from water. Materials that can serve the dual function of sorption and detection-determination of heavy metal ions would be highly desirable; however, such materials are rare and are mainly limited to sorbents with the capability of luminescence sensing of heavy metal ions.2Metal–Organic Frameworks (MOFs), which are crystalline polymeric metal complexes based on metal ions or clusters interconnected via polytopic organic ligands, have attracted great attention thanks to their unique structural features and potential applications in various fields.3,4 MOFs combine a highly porous structure with tunable pore sizes and functionalities, thus being particularly promising for the sorption of pollutants. Several publications have described the use of MOFs as sorbents for ionic species. Among them, anion-exchange MOFs have been investigated in detail but MOFs with cation exchange properties are limited.5
Besides sorption, the detection of heavy metals is equally important. One of the most efficient, sensitive, and affordable techniques that can be combined with simple and portable instrumentation for the determination of heavy metal ions is anodic stripping voltammetry (ASV).6 Recently, there has been interest in the use of MOFs for such analytical purposes. Nevertheless, the development of MOF-modified electrodes for ASV of heavy metal ions is a challenging task and is still in its infancy.7–11
We recently described the synthesis and characterization of [Ca(H4L)(DMA)2]·2DMA (Ca-MOF, Fig. 1), where H6L is N,N′-bis(2,4-dicarboxyphenyl)-oxalamide and DMA = N,N-dimethylacetamide.12 This MOF was found capable of exchanging Ca2+ with Cu2+ within seconds, a property that enabled us to utilize Ca-MOF as a stationary phase in an ion-exchange column for the removal of Cu2+ from aqueous media. Interestingly, after the Cu2+ sorption the most expensive part of the MOF, i.e. the H6L ligand, was recycled.
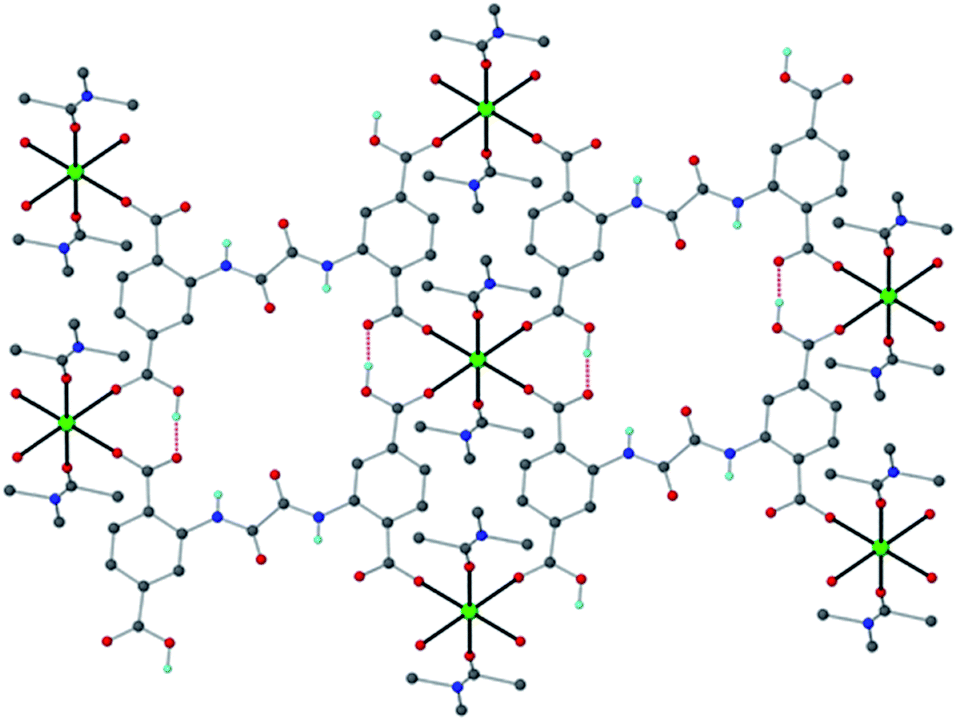 | ||
| Fig. 1 The structure of Ca-MOF. Several H atoms have been removed for clarity. Color code: Ca green, C grey, O red, N blue, and H cyan. | ||
Herein, for the first time, we show that a metal–organic material, namely Ca-MOF, shows the capability of both removal and voltammetric determination of heavy metal ions in aqueous media. Interestingly, Ca-MOF exhibits particularly high Pb2+ sorption capacity (∼522 mg g−1), which is among the highest reported for MOFs.3c,5f Importantly, a column filled with Ca-MOF (1% wt) particles dispersed in silica sand (99% wt) can eliminate traces of Pb2+ (∼100 ppb) from a wastewater simulant solution (containing a large excess of competitive ions). Ca-MOF also shows relatively high sorption capacities for Cd2+, Ni2+ and Zn2+ which are retained exceptionally well even in the presence of several competitive cations. A combination of spectroscopic and X-ray diffraction methods gave us insights into the structural characteristics of M2+ (M2+ = Pb2+, Cd2+, Ni2+, Zn2+)-loaded materials. We have also fabricated a simple ready-to-use electrochemical sensor based on modified graphite paste with Ca-MOF, which was successfully utilized for the determination of Pb2+, Cd2+, Cu2+ and Zn2+ at μg L−1 levels in water solutions by ASV. Overall, this work opens a new avenue for the development of materials that will be highly capable of both heavy metal ion remediation and monitoring in aqueous solutions.
Experimental
Materials
All reagents and solvents were commercially available and used as received.Synthesis of Ca-MOF
The synthesis of Ca-MOF was reported previously by us (it is also provided in the ESI†).12Synthesis of [Ni(H4L)(H2O)4]·2DMF (1)
An aqueous solution (10 mL) of Ni(SCN)2 (19 mg, 0.06 mmol) was added to a 10 mL DMF solution of H6L (25 mg, 0.06 mmol) resulting in a non-clear greenish solution. Upon addition of 80 μL of HCl (3 M) no change was observed. The solution was sealed in a glass vial and heated in an isotemp oven at 85 °C for 2 days. Small but well-shaped green needles were formed. The needles were collected by vacuum filtration, washed with DMF (5 mL), and dried in air for 1 h and then in a desiccator over silica gel overnight. Yield: 39.4 mg, 95%. Anal. calcd for C24H32N4O16Ni: C 41.70, H 4.67, N 8.11. Found: C 41.82, H 4.51, N 8.22. IR (KBr pellets, cm−1): 3462 brm, 3328 w, 3216 w, 3075 w, 2930 w, 2813 w, 2492 brw, 1935 brm, 1703 m, 1679 w, 1656 shw, 1627 m, 1579 w, 1575 w, 1511 s, 1446 m, 1418 w, 1371 s, 1307 sh, 1292 m, 1263 m, 1225 w, 1173 s, 1149 w, 1135 w, 1110 s, 1078 m, 1068 sh, 1028 w, 1005 w, 949 m, 915 m, 899 shw, 859 s, 813 m, 800 s, 765 s, 753 w, 678 s, 570 w, 545 w, 511 m, 503 sh, 406 w.Synthesis of [Pb2(H2L)]·2H2O (2)
An aqueous solution (10 mL) of PbCl2 (22 mg, 0.06 mmol) was added to a DMF solution (10 mL) of H6L (25 mg, 0.06 mmol) affording a white non-clear solution. The non-clear solution was then sealed in a glass vial and heated in an isotemp oven at 100 °C for 2 days leading to the formation of colorless plate like single crystals. The single crystals were collected via vacuum filtration, washed with DMF (5 mL) and dried in air for 1 h and then in a desiccator over silica gel overnight. Yield: 15.5 mg, 60%. Anal. calcd for C18H12N2O12Pb2: C 25.06, H 1.40, N 3.25. Found: C 25.17, H 1.31, N 3.33. IR (KBr pellets, cm−1): 3565 w, 3345 br, 3071 br, 1690 m, 1677 sh, 1616 w, 1593 w, 1540 sbr, 1506 s, 1449 s, 1396 w, 1366 s, 1345 sh, 1303 w, 1293 m, 1268 w, 1216 w, 1172 m, 1145 sh, 1080 w, 957 w, 923 w, 912 w, 868 w, 826 w, 813 w, 767 m, 696 w, 686 w, 570 m, 520 m, 486 m.Physical measurements
Microanalyses (C, H, and N) were performed with an EA 1108 Carlo Erba analyser. IR spectra were recorded on a Shimadzu FT/IR IRAffinity-1 spectrometer with samples prepared as KBr pellets. 1H-NMR spectra were recorded on a Bruker Avance 400 spectrometer operating at 400.13 MHz (1H). Samples were run in a 5 mm probe with DMSO-d6 as the internal lock and reference. Thermogravimetric analyses (TGA) were performed on a Mettler-Toledo TGA/DSC1 instrument under a N2 flow of 50 mL min−1 from room temperature to 800 °C with a heating rate of 10 °C min−1. PXRD diffraction patterns were recorded on a Bruker D8 Advance X-ray diffractometer (CuKα radiation, λ = 1.5418 Å). Energy dispersive spectroscopy (EDS) analyses were performed on a JEOL JSM-6390LV scanning electron microscope (SEM) equipped with an Oxford INCA PentaFET-x3 energy dispersive X-ray spectroscopy (EDS) detector. Data acquisition was performed with an accelerating voltage of 20 kV and 120 s accumulation time.Single crystal X-ray crystallography
The data for the single crystals of 1 and 2 were collected on an Oxford-Diffraction Supernova diffractometer, equipped with a CCD area detector utilizing Cu Kα (λ = 1.5418 Å) radiation. Suitable crystals were mounted on a Hampton cryoloop with Paratone-N oil and transferred to a goniostat where they were cooled for data collection. Empirical absorption corrections (multiscan based on symmetry-related measurements) were applied using CrysAlis RED software. The structures were solved by direct methods using SHELXS-97 and refined by full-matrix least-squares techniques on F2 with SHELXL-2016/4 (1) or SHELXL-2018/3 (ref. 13) (2) contained in the WinGX v2014.1 program package.14 The non-H atoms were treated anisotropically, whereas the methyl, aromatic H and NH hydrogen atoms were placed in calculated, ideal positions and refined as riding on their respective carbon or nitrogen atoms, and the rest of the H atoms were found in the difference map and refined isotropically. Complete crystal data and parameters for data collection and processing are reported in Table S1.† Selected interatomic distances and angles are given in the ESI (Table S2†).Pair distribution function (PDF) analysis
Samples were measured at 11-ID-C, Advanced Photon sources using X-rays with a wavelength of 0.1174 Å. Scettere 4d intensities were recorded with a 2D detector up to a wave vector of q = 25 A−1. The experimental diffraction patterns were converted to atomic pair distribution functions (PDFs).ASV procedure
The electrochemical measurements were conducted with a portable electrochemical potentiostat PGSTAT101 (Metrohm Autolab) connected to a laptop. Control and data manipulation (such as baseline correction of the voltammograms) were performed with NOVA 1.8 (Metrohm Autolab) software. Measurements were carried out in a 10 mL glass voltammetric cell equipped with a saturated calomel reference and a Pt wire auxiliary electrode. The square wave anodic stripping voltammetric (SWASV) determination of Pb2+, Cd2+ and Cu2+ was carried out in 0.10 mol L−1 acetate buffer (pH 4.5), while the SWASV analysis of Zn2+ was conducted in 1.0 × 10−3 mol L−1 KCl. The modified working electrode (WE) with Ca-MOF was immersed into the test solution and the preconcentration was carried out, for 240 s under stirring at −1.40 V in the case of Cd2+ and Cu2+ and at −1.60 V for Zn2+. After the accumulation of target metal ions on the WE surface, a SW voltammetric scan (frequency, 50 Hz; pulse height, 40 mV; step increment, 4 mV) was applied to the modified WE to oxidize the target metals and the voltammograms were recorded. Then, the WE was cleaned for traces of remaining accumulated metals for 20 s at 0.20 V.Preparation of the modified WE with Ca-MOF
The graphite paste electrode (GPE) was prepared by mixing 0.5 g graphite powder (grade #38, Fisher Scientific), 100 mg MOF and 0.20 g paraffin (Merck) in a mortar. The mixture was packed in a plastic syringe (internal radius: 2.4 mm). The electric contact was established by a copper wire passing through the paste. The surface of the WE was polished on a clean filter paper until a smooth surface was obtained. This simple fabrication procedure leads to the production of a ready-to-use-sensor, whose surface can be renewed via a slight pressure on the syringe plunger (Fig. S1†). Besides, modified WEs containing 25 and 50 mg Ca-MOF in 0.5 g graphite powder and a bare GPE were prepared. The modified WE with 100 mg MOF provided the highest sensitivity for all cations and was finally selected.Preparation of the column
50 mg of Ca-MOF and 5 g of sand (50–70 mesh SiO2) were mixed using a mortar and pestle and filled in a glass column (0.7 cm ID column).Batch ion-exchange studies
A typical ion-exchange experiment of Ca-MOF with M2+ (M2+ = Pb2+, Cd2+, Ni2+, Zn2+) is as follows: in a solution of M2+ (0.36 mmol) in water (10 mL, pH was not adjusted), the compound Ca-MOF (50 mg, 0.06 mmol) was added as a solid. The mixture was kept under sonication and magnetic stirring for 1 h. Then, the solids were isolated by filtration, washed several times with water (5 × 10 mL) and acetone (1 × 5 mL) and dried in a desiccator over silica gel overnight. Yields: Ni-MOF: 33 mg, Zn-MOF: 28 mg, Cd-MOF: 30 mg, and Pb-MOF: 36 mg. IR (KBr pellets, cm−1): Ni-MOF: 3446 br, 2630 w, 2523 w, 1951 w, 1718 s, 1684 sh, 1627 s, 1599 sh, 1577 s, 1517 s, 1450 s, 1424 w, 1373 m, 1298 s, 1267 w, 1220 w, 1167 m, 1131 w, 1077 w, 948 w, 910 w, 862 m, 796 w, 759 s, 689 w, 569 w, 509 m. Zn-MOF: 3521 br, 2627 w, 2516 w, 1717 m, 1683 w, 1627 m, 1603 w, 1576 m, 1517 s, 1451 s, 1424 w, 1374 m, 1298 s, 1267 w, 1220 w, 1166 m, 1133 w, 1076 w, 948 w, 910 w, 862 m, 792 sh, 761 s, 691 w, 568 w, 510 m. Cd-MOF: 3501 br, 3071 br, 2496 br, 1683 s, 1630 w, 1601 w, 1573 s, 1517 s, 1450 s, 1424 w, 1366 w, 1294 m, 1263 w, 1220 w, 1165 w, 1129 w, 1070 w, 951 w, 906 w, 863 m, 789 w, 761 s, 688 w, 596 w, 567 m, 504 sh, 487 m. Pb-MOF: 3566 w, 3462 br, 3059 brw, 1955 w, 1689 m, 1658 sh, 1625 w, 1556 s, 1530 sh, 1507 s, 1448 s, 1410 w, 1391 w, 1361 brs, 1298 m, 1268 w, 1220 w, 1171 w, 1127 w, 1076 w, 951 w, 898 w, 864 m, 813 w, 803 w, 770 s, 688 m, 599 w, 569 m, 505 m, 483 s. EDS analysis of M-MOF (M2+ = Ni2+, Zn2+, Cd2+ and Pb2+) showed a M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ca weight or atomic % ratio of ∼100/0% with only traces of Ca detected in the case of Zn-MOF (Fig. S2†).
:Ca weight or atomic % ratio of ∼100/0% with only traces of Ca detected in the case of Zn-MOF (Fig. S2†).
The Pb2+ uptake from solutions of various concentrations (1–800 ppm, pH was not adjusted) was studied by the batch method at V:m ∼ 1000 mL g−1, room temperature and 10 min contact. These data were used for the determination of Pb2+ sorption isotherms. Variable pH ion exchange experiments and those with drinking water samples were also carried out with the batch method at V:m ratio ∼1000 mL g−1, room temperature and 10 min contact. For the determination of the sorption kinetics, Pb2+ ion-exchange experiments of various reaction times (1–60 min) have been performed. For each experiment, a 10 mL sample of Pb2+ solution (initial Pb2+ concentration = 1 ppm, pH = 7 ± 0.02) was added to each vial (containing 10 mg of Ca-MOF) and the mixtures were kept under magnetic stirring for the designated reaction times. The suspensions from the various reactions were filtered and the resulting solutions were analyzed for their Pb content with anodic stripping voltammetry, using a Trace Metal Analyzer (797 VA Computrace, Metrohm AG Ltd, Switzerland). More specifically, a three electrode system was used comprising a Hanging Mercury Drop Electrode (HMDE) as the working electrode, platinum (Pt) as the auxiliary electrode and Ag/AgCl/KCl (3 mol L−1) as the reference electrode. The analysis has been done following the AN-V-086 protocol developed by Metrohm (http://www.metrohm.com/en/applications/#). The same procedure was followed for the removal of Cd2+, Ni2+ and Zn2+ from aqueous solutions. Each sorption experiment has been done at least twice and the reported sorption data represent the average of sorption results from the different sorption experiments. The difference between the concentrations of metal ions determined from the different sorption experiments was <2%.
Column sorption studies
These studies were performed with a wastewater simulant solution having the following composition: 3 mM NaHCO3, 0.143 mM NaNO3, 10−3 mM NaH2PO4·H2O, 5 × 10−2 mM NaF, 0.33 mM NaSiO3·5H2O, 0.99 mM CaCl2·2H2O, 5 mM MgSO4·7H2O, and 4.8 × 10−4 mM Pb2+.15 The pH of this solution was adjusted to 7. Several bed volumes of the solution were passed through the column and collected at the bottom in glass vials. The solutions were analysed with anodic stripping voltammetry.Ligand recycling studies
Ni-MOF (30 mg) were immersed in 5 mL HCl 6 M and the resulting white greenish slurry solution was left under stirring for 30 min. The resulting precipitate was centrifuged twice with the addition of H2O, filtered, washed extensively with H2O (5 × 5 mL) and dried in a desiccator over silica gel for 2 days. Yield: 17 mg, 72%. Zn-MOF (28 mg) was immersed in 5 mL HCl 6 M and the resulting white slurry solution was left under stirring for 30 min. The resulting precipitate was centrifuged twice with the addition of H2O, filtered, washed extensively with H2O (5 × 5 mL) and dried in a desiccator over silica gel for 2 days. Yield: 16 mg, 62%. Cd-MOF (30 mg) was immersed in 5 mL HCl 6 M and the resulting white greenish slurry solution was left under stirring for 30 min. The resulting precipitate was centrifuged twice with the addition of H2O, filtered, washed extensively with H2O (5 × 5 mL) and dried in a desiccator over silica gel for 2 days. Yield: 19 mg, 73%. Pb-MOF (36 mg) was immersed in 5 mL HCl 6 M and the resulting white greenish slurry solution was left under stirring for 30 min. The resulting precipitate was centrifuged twice with the addition of H2O, filtered, washed extensively with H2O (5 × 5 mL) and dried in a desiccator over silica gel for 2 days. Yield: 13 mg, 50%. The purity of the recycled ligands was confirmed by 1H-NMR (Fig. S3†).Similarly, recycling of the ligand can be performed for Pb-MOF in the ion-exchange column, as reported in our previous publication.12
Results and discussion
M2+ exchange studies of Ca-MOF
Our previous studies indicated that Ca-MOF is a very efficient sorbent for Cu2+,12 whereas this MOF shows very slow exchange for lanthanide ions.16 In the present study, we investigated the exchange properties of Ca-MOF for other divalent metal ions such as Ni2+, Zn2+, Cd2+ and Pb2+. The removal of Pb2+ and Cd2+ from water is of great interest due to the toxicity of these metal ions,17 whereas selective Ni2+ and Zn2+ capture is attractive as a recycling process for these metal ions existing in several types of waste (e.g. spent batteries and mine wastewater).18,19 In the following we provide detailed batch M2+ (Ni2+, Zn2+, Cd2+, and Pb2+) exchange studies as well as column Pb2+ sorption data.Batch sorption kinetics
At a first step of the Pb2+ sorption investigations, the sorption kinetics of the Pb2+ of Ca-MOF was determined through variable time sorption experiments. Fig. S4A† shows the variation of sorbed Pb2+ on Ca-MOF as a function of reaction time, with the initial Pb2+ concentration being 1 ppm. The kinetic investigations indicated that the capture of Pb2+ by Ca-MOF was remarkably fast. Interestingly, ∼95.6% of the initial Pb2+ content was removed within only 1 min of Ca-MOF/solution contact, while the time required to reach the sorption equilibrium for Pb was only 5 min. The kinetic data can be fitted very well with Ho and Mckay's pseudo-second-order equation shown below:where qt = the amount (mg g−1) of the ion sorbed by Ca-MOF at different reaction times (t), qe = the amount (mg g−1) of the metal ion sorbed in equilibrium, and k2 is the second-order rate constant [g(mg min)−1].20
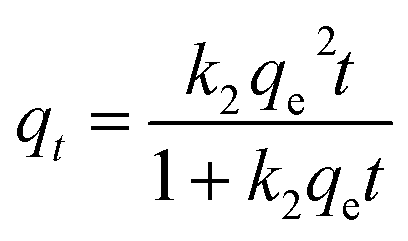
Likewise, the sorption of Ni2+ by Ca-MOF is quite fast, as revealed by the kinetic studies (Fig. S4B†). Specifically, the Ni2+ exchange equilibrium is achieved within 5 min. The best fitting of the kinetic data was achieved also with the Ho and Mckay's pseudo-second-order equation. The second order rate constants k2 for Ca-MOF were calculated to be 53.7 ± 5.2 and 6.13 ± 0.82 g(mg min)−1, for Pb2+ and Ni2+ sorption respectively, which indicate that Pb2+ exchange was completed significantly faster than the Ni2+ sorption process.
Furthermore, the kinetics of Cd2+ and Zn2+ sorption by Ca-MOF was also investigated. The results indicated that the capture of Cd2+ by Ca-MOF was remarkably fast, as the equilibrium was reached within only 2 min of solution/MOF contact, with 96.5% of the initial Cd2+ amount (C0 = 1 ppm, pH = 7 ± 0.02) removed from the solution (Fig. S4C†). In the case of Zn2+ the ion exchange almost reached its equilibrium after 3 min with ∼88.2% removal capacity (Fig. S4D†). The kinetic data for Cd2+ and Zn2+ sorption can be very well fitted with Lagergren's first-order equation shown below:
| qt = qe[1 − exp(−KLt)] |
Effect of pH
Although the ion exchange studies for Ca-MOF were mainly performed at pH = 7 ± 0.02, the MOF material is capable of sorbing metal ions from solutions of a relatively wide pH range (2–8) (Fig. 2).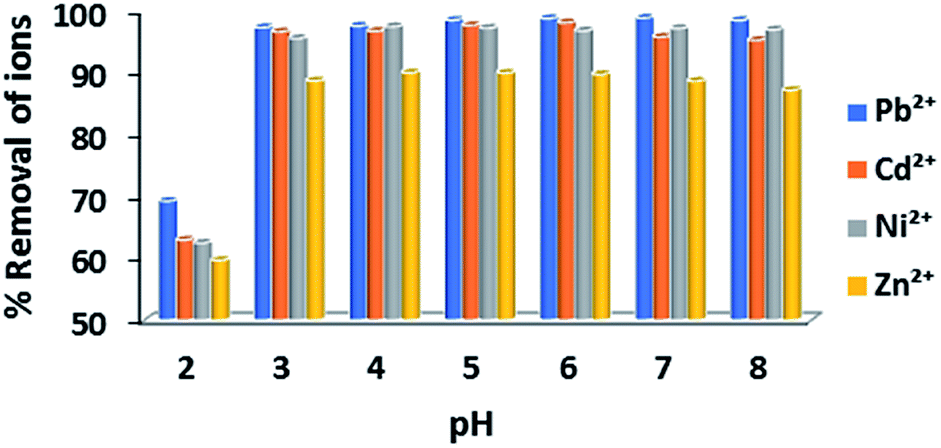 | ||
| Fig. 2 Percentage (%) sorption of Pb2+, Ni2+, Cd2+ and Zn2+ cations by Ca-MOF in the pH range of 2–8 (initial concentration of each metal ion = 1 ppm). | ||
Specifically, the removal capacities at pH = 3–8 (±0.02) were calculated to be ∼98.5, ∼97, ∼90 and ∼97.5% for Pb2+, Cd2+, Zn2+ and Ni2+ (initial cation concentration = 1 ppm), respectively, whereas Ca-MOF retains high removal capability for Pb2+ and Cd2+ even under highly acidic conditions (∼70 and 63% removal capacities at pH = 2, respectively).
Sorption thermodynamics
In order to better understand the equilibrium analysis of the sorption process, Langmuir, Freundlich and Langmuir–Freundlich isotherms were used to fit the sorption equilibrium data. The expressions of the three model equations are as follows:21

where q (mg g−1) is the amount of the cation sorbed at the equilibrium concentration Ce (ppm), qm is the maximum sorption capacity of the sorbent, b (L mg−1) is the Langmuir constant related to the free energy of the sorption, and KF and 1/n are the Freundlich constants. The parameters of Langmuir, Freundlich and Langmuir–Freundlich isotherms, found after the fitting of the isotherm cation sorption data (Fig. 3), are presented in Table 1.

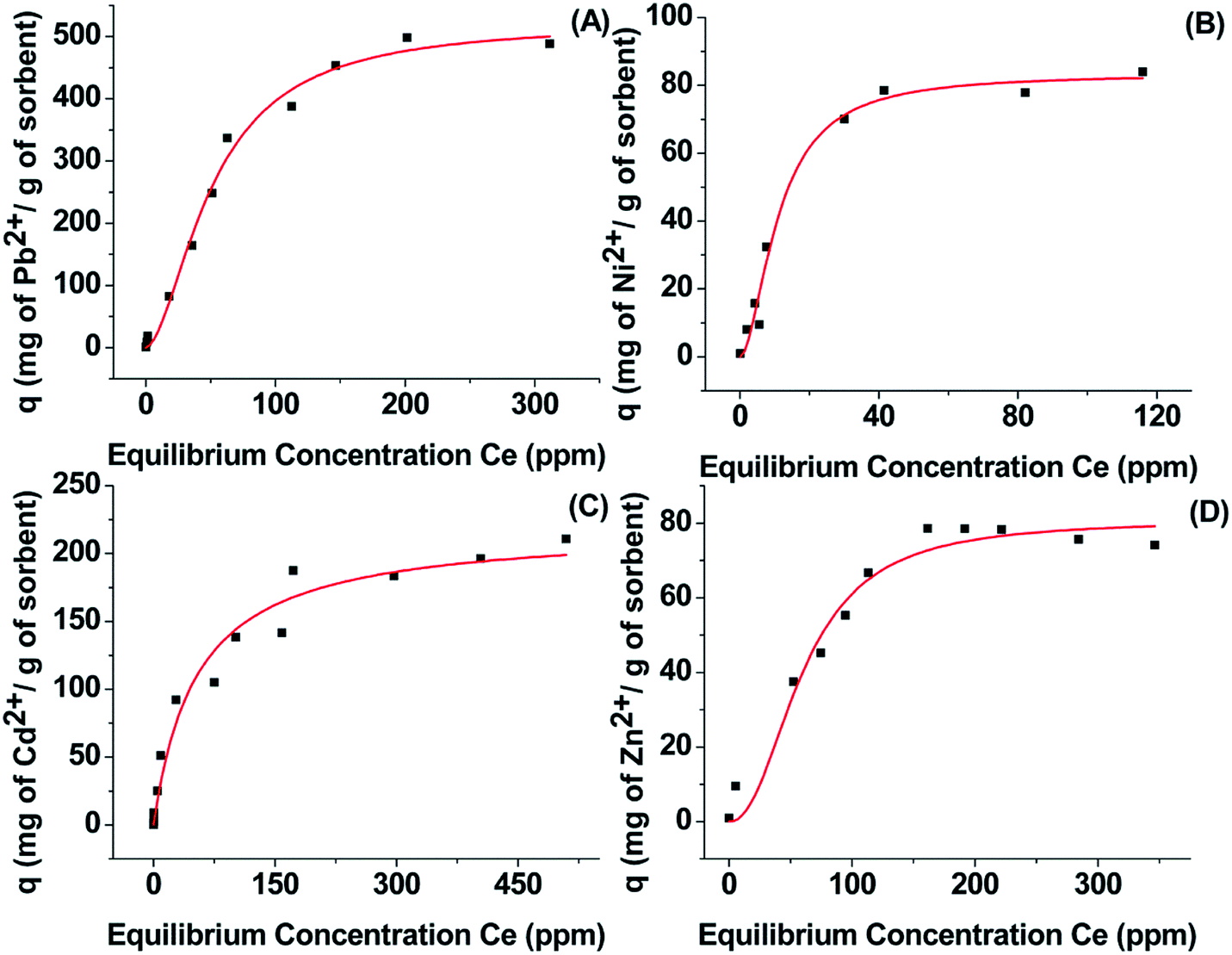 | ||
| Fig. 3 Isotherm data for Pb2+ (A), Ni2+ (B), Cd2+ (C) and Zn2+ (D) sorption by Ca-MOF. The red lines represent the fitting of the data with the appropriate model (see the text for details). The model that best describes the sorption of the various metal ions by Ca-MOF was decided on the basis of optimum R2 values and the consistency between experimental and predicted (from the models) qmax values. | ||
| Metal ion | Langmuir | Freundlich | Langmuir–Freundlich | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| q m (mg g−1) | b (L mg−1) | R 2 | K F | n | R 2 | q m (mg g−1) | b (L mg−1) | n | R 2 | |
| Pb2+ | 667 ± 51 | 0.012 ± 0.002 | 0.98 | 40 ± 14 | 2.2 ± 0.3 | 0.93 | 522 ± 26 | 0.019 ± 0.002 | 1.7 ± 0.2 | 0.99 |
| Ni2+ | 98 ± 6 | 0.067 ± 0.014 | 0.97 | 15 ± 6 | 2.6 ± 0.6 | 0.85 | 84 ± 3 | 0.097 ± 0.009 | 1.6 ± 0.2 | 0.99 |
| Cd2+ | 220 ± 12 | 0.019 ± 0.004 | 0.97 | 24 ± 4 | 2.8 ± 0.2 | 0.97 | 301 ± 62 | 0.007 ± 0.005 | 0.6 ± 0.1 | 0.98 |
| Zn2+ | 99 ± 8 | 0.014 ± 0.003 | 0.96 | 10 ± 4 | 2.7 ± 0.5 | 0.90 | 81 ± 5 | 0.016 ± 0.002 | 2.2 ± 0.7 | 0.97 |
The best fitting of the isotherm data of Pb2+, Ni2+ and Zn2+ by Ca-MOF was achieved by using the Langmuir–Freundlich equation, which is a combination of the Langmuir and Freundlich isotherms.22 The isotherm data of Cd2+ sorption by Ca-MOF was fitted by using the Langmuir equation. The maximum sorption capacities were estimated to be 522 ± 26 mg Pb2+, 84 ± 3 mg Ni2+, 220 ± 12 mg Cd2+ and 81 ± 5 mg Zn2+ per g of Ca-MOF. A comparison of the sorption capacities of Ca-MOF with those of other heavy metal ion sorbents is provided below. The experimental Ni2+ and Zn2+ sorption capacities are close to those calculated for the exchange of one Ca2+ with one Ni2+ or Zn2+ (calculated qm for Ni2+ and Zn2+ exchange is 73 and 81 mg g−1 respectively). However, the Pb2+ exchange capacity determined from the isotherm sorption data was found to be two times higher than that calculated for the exchange of one Ca2+ with one Pb2+ (calculated value = 258 mg g−1). In addition, the experimentally found Cd2+ exchange capacity corresponds to the insertion of 1.5 Cd2+ per formula unit of the MOF. An explanation about these results is provided in a following section, where the mechanism of the metal ion sorption by Ca-MOF is discussed on the basis of various spectroscopic and X-ray diffraction data.
The affinity of Ca-MOF for the above cations can be expressed in terms of the distribution coefficient Kd which is given by the following equation:
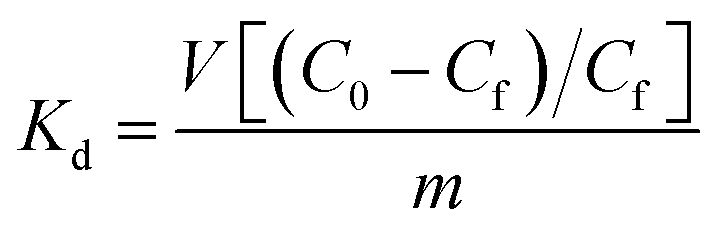
Selectivity studies
In order to explore the selectivity of Ca-MOF for heavy metal ions vs. common competitive cations such as alkali and alkaline earth metal ions, individual sorption experiments for each metal ion were performed in natural spring water (containing 93.1 ppm Ca2+, 1.9 ppm Mg2+, 2.6 ppm Na+ and 0.7 ppm K+). The sorption capacities of Ca-MOF were calculated to be 98.3%, 98%, 89.4%, 75% and 50% for Pb2+, Cd2+, Ni2+, Cu2+ and Zn2+, respectively (Table 2). As a matter of fact, the sorption capacity of Ca-MOF seems not to be influenced significantly in the presence of the competitive cations, with the exception of Zn2+, making it an excellent sorbent and also promising for the remediation of real-world wastewater.| Sample | C 0 (ppb) | C e (ppb) | % Removal |
|---|---|---|---|
| a Natural spring water with Ca2+: 93.1 ppm, Mg2+: 1.9 ppm, K+: 0.7 ppm, and Na+: 2.6 ppm. b Distilled water. c Initial concentration of each ion. | |||
| Pb2+a | 1000 | 17 | 98.3 |
| Cd2+a | 1000 | 20 | 98.0 |
| Ni2+a | 1000 | 106 | 89.4 |
| Cu2+a | 1000 | 250 | 75.0 |
| Zn2+a | 1000 | 500 | 50.0 |
| Mixture of ionsb | 1000c | 20 (Pb), 20 (Cd), 450 (Ni), 370 (Cu), 600 (Zn) | 98 (Pb), 98 (Cd), 55 (Ni), 63 (Cu), 40 (Zn) |
| Mixture of ionsa | 1000c | 30 (Pb), 30 (Cd), 250 (Ni), 370 (Cu), 1000 (Zn) | 97 (Pb), 97 (Cd), 75 (Ni), 63 (Cu), 0 (Zn) |
As a next step of the selectivity studies, we decided to investigate the selectivity of Ca-MOF towards specific heavy metal ions (Table 2). Thus, an aqueous solution containing 1 ppm each of Pb2+, Ni2+, Cd2+, Zn2+ and Cu2+ was treated with Ca-MOF. Ca-MOF showed excellent Pb2+ and Cd2+ removal capacities (∼98%), whereas the sorption capacities for Cu2+, Ni2+ and Zn2+ were found to be moderate (63%, 55% and 40% respectively). This result indicates a significant selectivity of Ca-MOF for Pb2+ and Cd2+ in comparison to the other heavy metal ions. Further selectivity studies were conducted in natural spring water intentionally contaminated with a mixture of the aforementioned cations (Table 2). The removal capacities for Pb2+, Cd2+, and Cu2+ were found similar (97%, 97%, and 63% respectively) to those in the experiments involving distilled water. Unexpectedly, the Ni2+ removal capacity (∼75%) was found somewhat higher compared to that determined in the experiments with distilled water and in addition, no Zn2+ sorption capacity was observed. Overall, the selectivity investigations indicated Ca-MOF as a highly efficient sorbent for Pb2+ and Cd2+, whereas it shows moderate selectivity for Ni2+ and Cu2+ and relatively low selectivity for Zn2+.
Recycling studies
As also reported in our previous publication,12Ca-MOF cannot be regenerated after the sorption of the various metal ions. However, after the ion exchange process it is possible to recover the most expensive part of the sorbent, i.e. the organic ligand. This recycling process involves treatment of the metal ion-loaded MOF with acidic solution, which results in the decomposition of the MOF through dissolution of the metal ion and protonation of the H4L2− ligand. The resulted H6L compound is insoluble under acidic conditions and can be easily isolated in a pure form and high yield (Fig. S3†). Details of the recycling process are given the in the Experimental section. We should also note that during the course of our studies (a period > 2 years), we have used recycled ligand samples to prepare new batches of Ca-MOF. Such MOF samples have been used for sorption experiments, showing similar results to those obtained with Ca-MOF batches synthesized with freshly prepared ligands.Column ion exchange study
Batch ion exchange studies revealed remarkably high sorption capacities of Ca-MOF towards the examined heavy metals. However, industrial wastewater processes require continuous bed flow ion exchange columns. Ca-MOF is isolated in the form of relatively large particle-crystals (size 100–300 μm, see Fig. S6†), thus fulfilling an important criterion for a sorbent to be used as a stationary phase in ion exchange columns.12 The final step in our investigations was the study of the column Pb2+ sorption properties of the Ca-MOF material. We chose Pb2+ among the other heavy metal ions for these studies because of the highly efficient Pb2+ sorption properties of Ca-MOF and the urgent need to find suitable sorbents to address the pollution of water resources by Pb2+ representing one of the most important environmental issues.As in previous studies,12 the stationary phase in the column used for Pb2+ sorption consisted of Ca-MOF and silica sand (Ca-MOF:sand mass ratio = 1:100). In order to investigate the capability of this column to sorb Pb2+ under realistic conditions, we conducted a column sorption study with a ground water simulant solution (its composition is given in the Experimental section) containing traces of Pb2+ (initial Pb2+ concentration ∼ 100 ppb). Note that this solution contained a series of competitive cations with concentrations up to 104 times higher than that of Pb2+. The results indicated that ∼1.2 L of the solution passed through the column exhibited a concentration <10 ppb. i.e. below the allowed USA-EPA and EU levels for Pb2+ in water (Fig. 4).
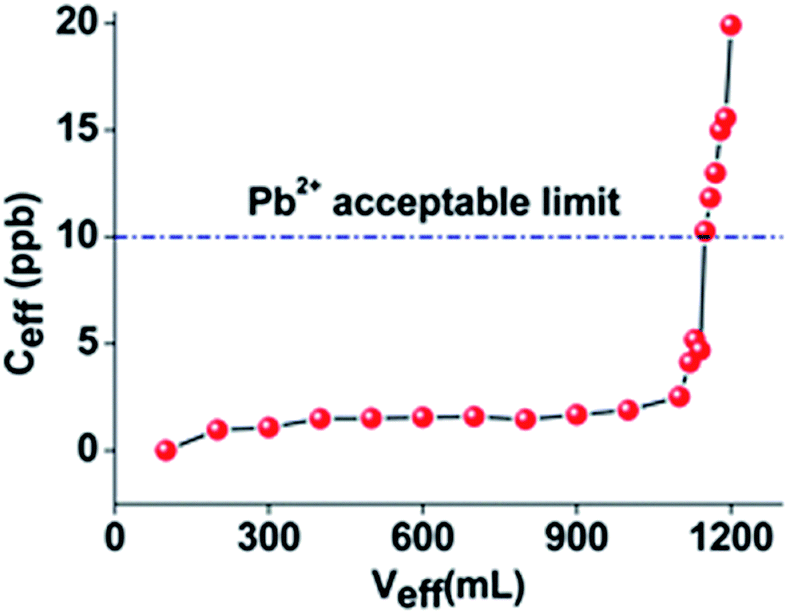 | ||
| Fig. 4 Column sorption data with a wastewater simulant solution (flow rate ∼1.0 mL min−1, one bed volume = 3.5 mL, and stationary phase MOF/sand = 0.05 g:5 g). The initial concentration of Pb2+ was 100 ppb (the concentrations of the other ions are given in the Experimental section). Veff and Ceff are the volume (mL) and Pb2+ concentration (ppb) of the solution passed through the column (i.e. effluent) respectively. | ||
The performance of this column is remarkable considering the fact the stationary phase contained only 50 mg of Ca-MOF. Yet, such a small quantity of the MOF was enough to decontaminate a relatively large volume of the wastewater solution, thus demonstrating its potential for real environmental remediation applications. Of course, regeneration/reuse of the Ca-MOF/sand column is not possible after its use. However, recycling of the most expensive part of the sorbent, i.e. the H6L ligand, can be performed via treatment of the column with a basic solution to decompose the MOF and dissolve the H4L2− ligand followed by acidification of the solution so that the precipitation of the H6L ligand is achieved.
Comparison of Ca-MOF with other heavy metal ion MOF sorbents
At this point, we may compare the performance of Ca-MOF as a heavy metal ion sorbent with that of other MOF sorbent materials. In Table S3,† the important sorption properties of various MOFs tested so far for the capture of heavy metal ions are shown. It can be seen that only a few MOFs display higher Pb2+ sorption capacity than Ca-MOF.5f,g,h In addition, Ca-MOF shows very similar Cd2+ sorption capacity to FJI-H9 MOF (225 vs. 220 mg g−1 for Ca-MOF), which represents the most effective Cd2+ MOF sorbent reported prior to our work.23 Furthermore, Ca-MOF is the only MOF sorbent investigated so far in detail for the sorption of Ni2+ and Zn2+. In addition, we should note that Ca-MOF is the only MOF tested so far for the removal of Pb2+ under flow conditions (i.e. using in an ion exchange column of Ca-MOF), i.e. under conditions simulating the practical wastewater treatment.Finally, it is worth comparing the sorption properties of Ca-MOF with those of other state-of-the-art heavy metal ion sorbents (besides MOFs), including various sulfur-functionalized materials (clays, LDHs, and mesoporous silica),24–27 commercial ion exchange resins28 and metal sulfide ion exchangers (MSIEs).29 From this comparison (Table S4†), it is apparent that the Pb2+ sorption efficiency of Ca-MOF exceeds that of other types of sorbents. In addition, the Cd2+, Ni2+ and Zn2+ uptake capacities of Ca-MOF compare well with those of the leading heavy metal ion sorbents.26,29–31 It is also important to note that Ca-MOF shows not only exceptional maximum sorption capacities but also remarkably fast sorption kinetics and high selectivity for heavy metal ions (especially for Pb2+ and Cd2+), which surpass those of most known sorbents (Tables S3 and S4†).
Characterization of M2+-loaded MOFs – mechanism of ion exchange
In order to characterize the M2+-loaded MOF materials and gain insight into the possible mechanism of the M2+ exchange process, a number of spectroscopic and X-ray diffraction methods were employed. The results obtained are discussed below.:1 while the Pb2+/oxalamide ligand tetra-anion (H2L4−) ratio in complex 2 was found to be 2:1. Both these results were in accordance with the data obtained from the sorption experiments discussed above, where we found that Ca-MOF exchanges its one Ca2+ per formula unit (the Ca2+/H4L2− ratio in Ca-MOF is 1:1) with either one Ni2+ or two Pb2+ ions.
The reaction of Ni(SCN)2 with H6L in H2O/DMF under solvothermal conditions yielded green needles of 1. Complex 1 crystallizes in the monoclinic space group C2/c and the unit cell contains one half of Ni2+, one half of H4L2−, two coordinated H2O molecules and one solvate DMF molecule. The Ni2+ ion resides on an inversion center with 50% site occupancy, adopts an octahedral coordination geometry and is surrounded by six O atoms. The four equatorial O atoms originate from four terminal water molecules while the two apical positions are occupied by two monodentately coordinated carboxylate groups originating from two different ligand moieties (H4L2−) that adopt the η1:η1–μ2 coordination mode leading to a 1D zig-zag chain running parallel to the b axis (Fig. 5). The two pending carboxylic groups (one unique) of the H4L2− ligand form hydrogen bonds with the DMF solvates. There are also several intra- and inter-chain H-bonds involving the terminal H2O molecules and several O donor atoms of the H4L2− ligand that increase the dimensionality of the material to 3D. The purity of complex 1 was verified by PXRD (Fig. S10†).
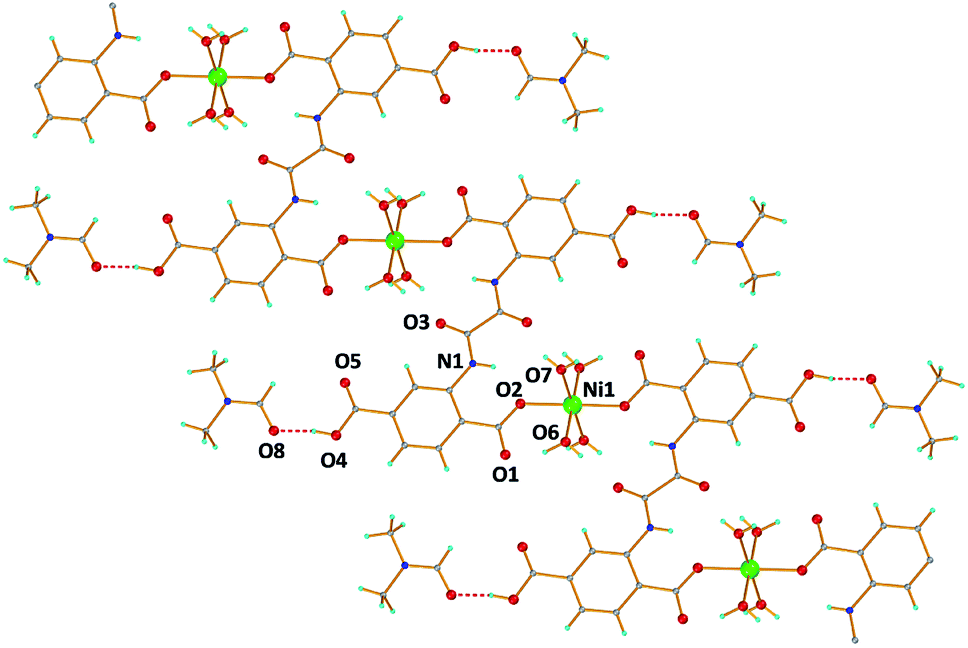 | ||
| Fig. 5 A labeled plot of [Ni(H4L)(H2O)4]·2DMF (1). Color code: Ni green, C grey, O red, N blue, and H cyan. | ||
The reaction of PbCl2 with H6L in H2O/DMF under solvothermal conditions yielded block colorless crystals of 2. Complex 2 crystallizes in the orthorhombic space group Pbcn and the unit cell contains two Pb2+ ions with 50% occupancy, one half of H2L4− and one solvate H2O molecule being disordered over two positions with 50% occupancy. Both Pb2+ ions reside on the 2-fold rotation axis and both are surrounded by six carboxylate O atoms originating from four different H2L4− ligands. The coordination environment around each Pb atom is far from octahedral owing to the significant space occupied by the lone electron pair on each Pb2+. Each of the four carboxylate groups of the tetra-anionic oxalamide ligand (H2L4−) adopts the η1:η2–μ2 coordination mode, bridging either two Pb1 or two Pb2 ions. Therefore, each H2L4− anion serves as a μ8 bridging ligand creating, along with the Pb2+ ions, a double-walled layer laying parallel to the ac plane. The one-dimensional channels formed within the double-walled layer are hosting the solvate H2O molecules (Fig. 6). The purity of complex 2 was verified by PXRD (Fig. S11†). Unfortunately, our efforts to isolate crystalline MOFs of H6L with Zn2+ and Cd2+ have not been fruitful, so far.
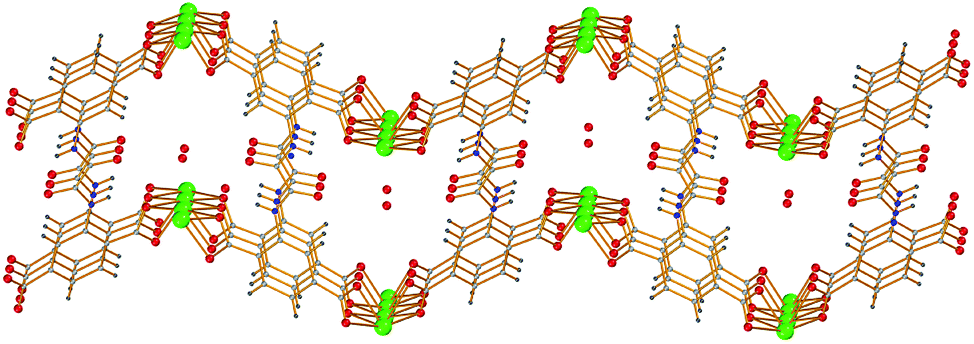 | ||
| Fig. 6 The one-dimensional channels formed within the double-walled layer of [Pb2(H2L)]·2H2O (2) hosting the solvate H2O molecules. Color code: Pb green, C grey, O red, N blue, and H cyan. | ||
This approach has proven very useful in structure studies of materials with limited structural coherence, ranging from crystals with intrinsic disorder to amorphous solids and liquids.32 The experimental atomic PDFs (Fig. S12†) show sharp peaks at low-r distances reflecting the presence of a well-defined short-range atomic order. The PDFs also show subsequent broad oscillations indicating the presence of a medium-range atomic order. The oscillations are very well-defined with the PDFs for Ni-MOF, Zn-MOF and Cu-MOF in comparison to Pb-MOF and Cd-MOF. The observation indicates that the former Ni2+, Zn2+ and Cu2+ loaded MOFs are more ordered locally as compared to the latter (i.e. Pd2+ and Cd2+-loaded materials). As can also be seen in Fig. S12,† the atomic PDFs for Ni-MOF, Zn-MOF and Cu-MOF are similar indicating that the materials share common structural features. In addition, it can be seen that there are some common features in the PDFs of Pb-MOF and Cd-MOF. Selected atomic pair distances for Ni-MOF, Zn-MOF, Cu-MOF, Pb-MOF, Cd-MOF and Ca-MOF (computed) are shown in Fig. 7.
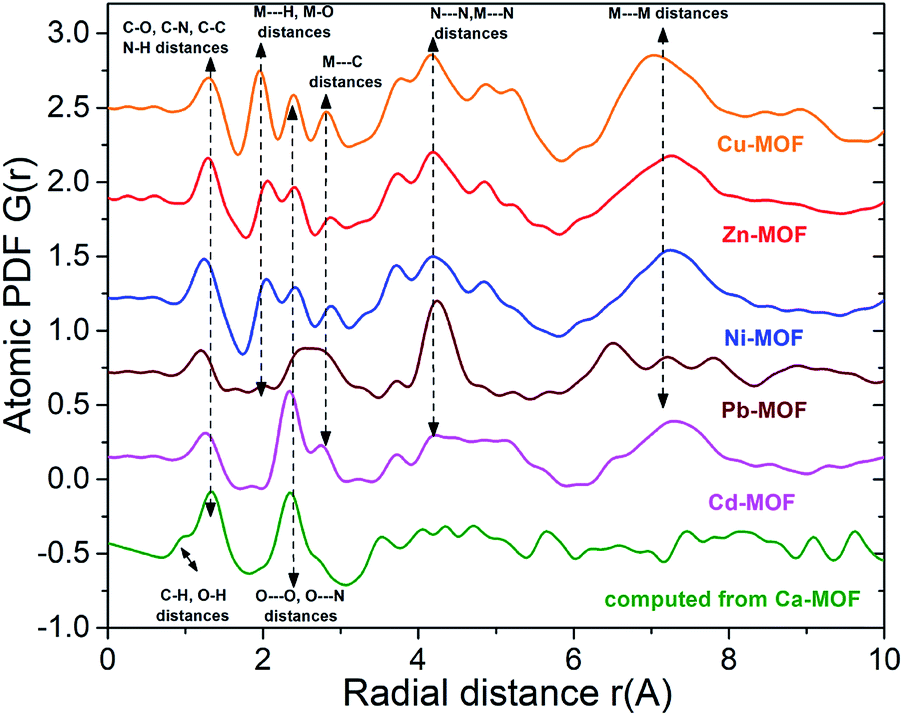 | ||
| Fig. 7 Low-r part of experimental atomic PDFs for Cu-MOF, Zn-MOF, Ni-MOF, Pb-MOF and Cd-MOF. Selected atomic pair distances are marked with vertical broken lines. Computed PDF for Ca-MOF is also shown. | ||
The distances for different metal ion-loaded materials are somewhat different reflecting the different compositions and/or sizes of metal ions. Experimental (for Ni-MOF and Pb-MOF) and computed PDFs derived from crystal structure models of [Ni(H4L)(H2O)4]·2DMF (1) and [Pb2(H2L)]·2H2O (2) are shown in Fig. 8.
 | ||
| Fig. 8 Comparison between experimental (Ni-MOF and Pb-MOF) and model (1 and 2) PDFs. | ||
As can be seen in Fig. 8, the local atomic structure of Ni-MOF is close to that of the respective crystal structure of 1. The comparison of the PDF of Pb-MOF with that computed from the structure of 2 also revealed several similarities between them. First three PDF peaks for Pb-MOF are well-defined and positioned at about 1.2, 2.6 and 4.2 Å. Similarly to the PDF of Pb-MOF, the computed PDF of 2 shows peaks at about 1.2 and 4.2 Å. Contrary to the PDF of Pb-MOF, the calculated PDF of 2 does not show a well-defined peak but only a spike in the vicinity of 2.4 A. The spike can be attributed to Pb–O distances and it also appears as a second neighbour peak in the computed PDF of Ca-MOF. Evidently, the Pb-MOF and compound 2 show common structural features.
| [Ca(H4L)(DMA)2]·2DMA + 2Pb2+ + xH2O → [Pb2(H2L)]·xH2O + Ca2+ + 2H+ + 4DMA | (1) |
| [Ca(H4L)(DMA)2]·2DMA + 1.5Cd2+ + xH2O → [Cd1.5(H4L)0.5(H2L)0.5]·xH2O + Ca2+ + H+ + 4DMA | (2) |
| [Ca(H4L)(DMA)2]·2DMA + M2+ + (x + 4)H2O →[M(H4L)(H2O)4]·xH2O + Ca2+ + 4DMA (M2+ = Ni2+, Zn2+) | (3) |
Based on the results from spectroscopic, thermal and X-ray diffraction methods, some suggestions for the possible structures of metal ion-loaded MOFs can be provided. Thus, PDF, IR and TGA revealed that Ni-MOF and Zn-MOF present identical structures which are closely related to the structure of [Ni(H4L) (H2O)4]·2DMF (1) reported in this work. In addition, PDF data indicated that Pb-MOF has some common structural features with [Pb2(H2L)]·2H2O (2) also described in this work. These results are consistent with the chemical composition (metal to ligand molar ratio) of Ni-MOF, Zn-MOF and Pb-MOF determined from the sorption data. Finally, PDF and IR data showed that there are some similarities between the structures of Pb-MOF and Cd-MOF, but they are not identical. This is also in accordance with the sorption data which revealed somewhat different metal to ligand molar ratios for Pb-MOF and Cd-MOF which are 2:1 and 1.5:1, respectively.
Determination of Pb2+, Cu2+, Cd2+, and Zn2+ by ASV at a modified GPE with Ca-MOF
From the above discussed sorption data and our previous work,12 it became apparent that Ca-MOF is an excellent and selective sorbent for various heavy metal ions. Such a highly efficient sorbent as Ca-MOF is suitable to be used in electrodes for preconcentration of heavy metal ions and their determination via anodic stripping voltammetry (ASV). We have thus fabricated a stand-alone and ready-to-use MOF-based syringe electrode. The analytical performance of the modified GPE with Ca-MOF was investigated by conducting preliminary quantitative ASV analyses at various concentrations (10–60 μg L−1) of Pb2+, Cu2+, Zn2+ and Cd2+. These metal cations were selected as model analytes in order to show the performance of the Ca-MOF/sensor in a wide range of potentials. Besides, the Ca-MOF/sensor was capable of detecting more than one heavy metal ion in a single run, such as Pb2+ and Cd2+ (Fig. S13†).The sensor exhibited linear concentration dependence in the examined concentration ranges for all metal ions with correlation coefficients (R2) higher than 0.99 in all cases, as depicted in Fig. 9 (panels A–C) and Fig. S14.† The limits of detection (LODs) (calculated as three times the standard deviation of the y residuals of the calibration points divided by the slope of the calibration plot) were 0.64 μg L−1 for Pb2+, 1.4 μg L−1 for Cu2+, 1.1 μg L−1 for Zn2+ and 1.3 μg L−1 for Cd2+, which were comparable to, or lower than, those of other electrochemical sensors.33–35 The within-sensor reproducibility (expressed as the % relative standard deviation (% RSD) of repetitive measurements (n = 5) of 30 μg L−1 of each cation at the same sensor) was lower than 4.2%, while the between-sensor reproducibility (% RSD at four different sensors) was lower than 5.8% at the 30 μg L−1 level. Besides, the shelf-life of the modified GPE with Ca-MOF was tested over a period of 2 months and a t-test demonstrated that the ASV response remained statistically stable. These results verify the suitability of the proposed sensor for monitoring selected trace heavy metal ions. Besides, the ASV detection sensitivity of Pb2+, Cu2+, Cd2+, and Zn2+ at the modified GPE with Ca-MOF was about 5 times higher than that of the unmodified GPE (Fig. 9D), and also better-shaped stripping peaks with a lower background were obtained, indicating the enhanced electrochemical properties of Ca-MOF.
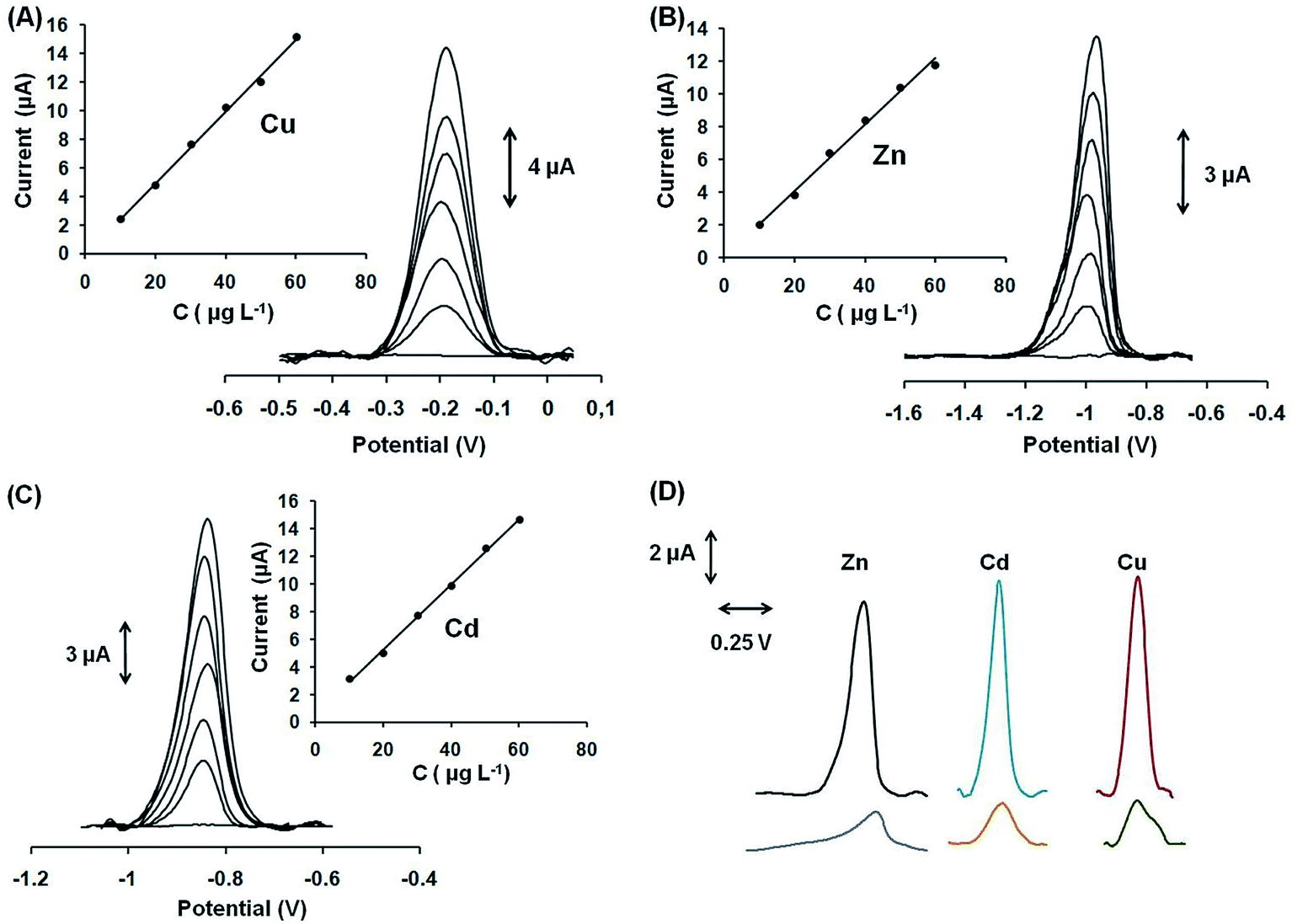 | ||
| Fig. 9 SWASV voltammograms and respective calibrations plots in the range of 10–60 μg L−1 (step 10 μg L−1) of (A) Cu2+ in 0.1 mol L−1 acetate buffer, (B) Zn2+ in 1 × 10−3 mol L−1 KCl, and (C) Cd2+ in 0.1 mol L−1 acetate buffer (pH 4.5). (D) SWASV voltammograms of 30 μg L−1 of Zn2+, Cd2+, and Cu2+ (black, blue, and red lines respectively) obtained with the modified GPE with Ca-MOF and SWASV voltammograms of 30 μg L−1 of Zn2+, Cd2+, and Cu2+ (gray, orange, and green lines, respectively) obtained with the bare GPE. Other voltammetric parameters are noted in the text. | ||
Compared to conventional, bismuth- and antimony-film electrodes, which are the most commonly used mercury-free transducers in ASV determination of heavy metals, the presented modified GPE with Ca-MOF is also able to determine Cu+2. At bismuth or antimony film electrodes, Cu determination is normally not possible since the oxidation peak of Cu appears outside the useful potential window of these electrodes.36–38 On the other hand, the existing MOF-based electrodes have shown single metal ion ASV detection properties (as they can determine only one cation), while the modified GPE with Ca-MOF presented multi-detection capabilities.7–10 Only two reports appear in the pertinent literature that use MOF-based electrodes for the ASV detection of four metal ions, but in these applications the MOFs were drop-cast on bare glassy carbon electrodes (GCEs).11 According to this protocol, each time and before each electrochemical measurement the bare GCE is abraded with appropriate paper, polished with alumina slurries, sonicated in suitable solutions for cleaning and then the MOF is dripped onto the GCE and air-dried. Therefore, these MOF-based electrodes cannot be considered as ready-to-use sensors, since they require a multistep reconstruction before each electrochemical measurement. Last but not least, so far, no MOF-based electrodes have been reported for ASV trace determination of Zn(II).
Conclusions
In conclusion, the MOF [Ca(H4L)(DMA)2]·2DMA (Ca-MOF) was shown to be a highly selective and efficient sorbent for heavy metal ions, especially for Pb2+ and Cd2+ being some of the most dangerous pollutants in various types of wastewater. It is also important that a low cost column based on Ca-MOF and silica sand was found particularly effective to decontaminate relatively large quantities of simulated wastewater containing traces of Pb2+ (100 ppb) and a series of competitive ions in high concentrations. The latter result indicated that Ca-MOF might find practical application in heavy metal ion remediation of polluted water. Pair distribution function (PDF) data provided us insight into the structure of M2+-exchanged materials. Thus, these data revealed close similarities of the structures of the Ni2+, Zn2+ and Cu2+-loaded materials with that of the crystalline [Ni(H4L)(H2O)4]·2DMF (1) compound reported in this work. In addition, PDF studies indicated that the Pb2+-loaded material shares common structural features with the [Pb2(H2L)]·2H2O (2) compound also described here. Motivated by the excellent heavy metal ion sorption properties of Ca-MOF, we have developed a simple ready-to-use electrochemical sensor based on modified graphite paste with Ca-MOF. This sensor was capable of analysing Pb2+, Cu2+, Zn2+ and Cd2+via anodic stripping voltammetry (ASV) and particularly low detection limits (0.64–1.4 μg L−1) were achieved for these metal ions. Actually, this is the first time that a MOF shows such a dual function, i.e. it is suitable for both sorption and electrochemical determination of heavy metal ions. Thus, an avenue is opened for the development of a new family of materials that would be employed in both environmental remediation and monitoring. Research in our groups is currently focused towards this direction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AP would like to thank the State Scholarships foundation (IKY) for a post-doctoral research scholarship, funded by the Action “Supporting Post-Academic Researchers” from the resources of the NF “Human Resources Development, Education and Lifelong Learning” with Priority Axes 6, 8, and 9 and co-funded by the European Social Fund – ESF and the Greek State. This work was partially supported by the Special Account for Research Grants (SARG) of the National and Kapodistrian University of Athens (UoA). The work was also supported in part by DOE grant DE-SC0006877. The Bodossaki Foundation is gratefully acknowledged for donating the TGA to UoA.Notes and references
- (a) B. Sarkar, Heavy metals in the environment, CRC Press, 2002 CrossRef; (b) G. M. Naja and B. Volesky, Toxicity and sources of Pb, Cd, Hg, Cr, As, and radionuclides in the environment, in Heavy Metals in the Environment, ed. L. K. Wang, J. P. Chen, Y. Hung and N. K. Shammas, CRC Press, 2009 Search PubMed; (c) L. M. Plum, L. Rink and H. Haase, Int. J. Environ. Res. Public Health, 2010, 7, 1342–1365 CrossRef CAS.
- A. Diamantis, A. Margariti, A. D. Pournara, G. S. Papaefstathiou, M. J. Manos and T. Lazarides, Inorg. Chem. Front., 2018, 5, 1493–1511 RSC and references cited therein.
- (a) M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS PubMed; (b) S. Horike, S. Shimomura and S. Kitagawa, Nat. Chem., 2009, 1, 695–704 CrossRef CAS PubMed; (c) P. Kumar, A. Pournara, K. H. Kim, V. Bansai, S. Rapti and M. J. Manos, Prog. Mater. Sci., 2017, 86, 25–74 CrossRef CAS; (d) J. M. Taylor, T. Komatsu, S. Dekura, K. Otsubo, M. Takata and H. Kitagawa, J. Am. Chem. Soc., 2015, 137, 11498–11506 CrossRef CAS PubMed.
- (a) C. K. Brozek and M. Dinca, Chem. Soc. Rev., 2014, 43, 5456–5467 RSC; (b) D. T. Genna, A. G. Wong-Foy, A. J. Matzger and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 10586–10589 CrossRef CAS; (c) J. Yu, Y. Cui, C. Wu, Y. Yang, Z. Wang, M. O'Keeffe, B. Chen and G. Qian, Angew. Chem., Int. Ed., 2012, 51, 10542–10545 CrossRef CAS PubMed; (d) Z. Wang, J. H. Zhang, J. J. Jiang, H. P. Wang, Z. W. Wei, X. Zhu, M. Pan and C. Y. Su, J. Mater. Chem. A, 2018, 6, 17698–17705 RSC; (e) Y. X. Tan, Y. P. He and J. Zhang, Chem. Commun., 2011, 47, 10647–10649 RSC.
- (a) A. V. Desai, B. Manna, A. Karmakar, A. Sahu and S. K. Ghosh, Angew. Chem., Int. Ed., 2016, 55, 7811–7815 CrossRef CAS PubMed; (b) L. Carlucci, G. Ciani, S. Maggini, D. M. Proserpio and M. Visconti, Chem.–Eur. J., 2010, 16, 12328–12341 CrossRef CAS PubMed; (c) H. R. Fu, Z. X. Xu and J. Zhang, Chem. Mater., 2015, 27, 205–210 CrossRef CAS; (d) H. Fei, C. H. Pham and S. R. J. Oliver, J. Am. Chem. Soc., 2012, 134, 10729–10732 CrossRef CAS PubMed; (e) Y. Feng, Y. Bi, W. Zhao and T. Zhang, J. Mater. Chem. A, 2016, 4, 7596–7600 RSC; (f) J. Li, X. Wang, G. Zhao, C. Chen, Z. Chai, A. Alsaedi, T. Hayat and X. Wang, Chem. Soc. Rev., 2018, 47, 2322–2356 RSC; (g) M. Mon, R. Bruno, J. Ferrando-Soria, D. Armentano and E. Pardo, J. Mater. Chem. A, 2018, 6, 4912–4947 Search PubMed; (h) Q. Gao, J. Xu and X.-H. Bu, Coord. Chem. Rev., 2019, 378, 17–31 Search PubMed; (i) S. Rapti, S. A. Diamantis, A. Dafnomili, A. Pournara, E. Skliri, G. S. Armatas, A. C. Tsipis, I. Spanopoulos, C. D. Malliakas, M. G. Kanatzidis, J. C. Plakatouras, F. Noli, T. Lazarides and M. J. Manos, J. Mater. Chem. A, 2018, 6, 20813–20821 Search PubMed; (j) R. J. Drout, K. Otake, A. J. Howarth, T. Islamoglu, L. Zhu, C. Xiao, S. Wang and O. K. Farha, Chem. Mater., 2018, 30, 1277–1284 Search PubMed.
- A. Economou and C. Kokkinos, Advances in Stripping Analysis of Metals, in Electrochemical Strategies in Detection Science, ed. D. Arrigan and M. Thomson, RSC, 2016 Search PubMed.
- F. Cai, Q. Wang, X. Chen, W. Qiu, F. Zhan, F. Gao and Q. Wang, Biosens. Bioelectron., 2017, 98, 310–316 Search PubMed.
- M. Roushani, A. Valipour and Z. Saedi, Sens. Actuators, B, 2016, 233, 419–425 Search PubMed.
- H. Guo, Z. Zheng, Y. Zhang, H. Lin and Q. Xu, Sens. Actuators, B, 2017, 248, 430–436 Search PubMed.
- H. Guo, D. Wang, J. Chen, W. Weng, M. Huang and Z. Zheng, Chem. Eng. J., 2016, 289, 479–485 Search PubMed.
- (a) D. Wang, Y. Ke, D. Guo, H. Guo, J. Chen and W. Weng, Sens. Actuators, B, 2015, 216, 504–510 Search PubMed; (b) M. Lu, Y. Deng, Y. Luo, J. Lv, T. Li, J. Xu, S.-W. Chen and J. Wang, Anal. Chem., 2018, 91, 888–895 Search PubMed.
- A. Margariti, S. Rapti, A. D. Katsenis, T. Friščić, Y. Georgiou, M. J. Manos and G. S. Papaefstathiou, Inorg. Chem. Front., 2017, 4, 773–781 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found Crystallogr., 2008, 64, 112–122 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 Search PubMed.
- F. Pinakidou, E. Kaprara, M. Katsikini, E. C. Paloura, K. Simeonidis and M. Mitrakas, Sci. Total Environ., 2016, 551–552, 246–253 Search PubMed.
- A. Margariti, A. D. Pournara, M. J. Manos, T. Lazarides and G. S. Papaefstathiou, Polyhedron, 2018, 153, 24–30 Search PubMed.
- Handbook on the Toxicology of Metals, ed. G. F. Nordberg, B. A. Fowler, M. Nordberg and L. T. Friberg, Elsevier, 3rd edn, 2007 Search PubMed.
- V. Coman, B. Robotin and P. Ilea, Resour., Conserv. Recycl., 2013, 73, 229–238 Search PubMed.
- K. Tanong, J.-F. Blais and G. Mercier, Recent Pat. Eng., 2014, 8, 13–23 Search PubMed.
- A. Benhammou, A. Yaacoubi, L. Nibou and B. Tanouti, J. Colloid Interface Sci., 2005, 282, 320–326 Search PubMed.
- (a) M. J. Manos and M. G. Kanatzidis, Chem.–Eur. J., 2009, 15, 4779–4784 Search PubMed; (b) M. J. Manos, N. Ding and M. G. Kanatzidis, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3696–3699 Search PubMed.
- G. P. Jeppu and T. P. Clement, J. Contam. Hydrol., 2012, 129–130, 46–53 Search PubMed.
- H. Xue, Q. Chen, F. Jiang, D. Yuan, G. Lv, L. Liang, L. Liu and M. Hong, Chem. Sci., 2016, 7, 5983–5988 Search PubMed.
- I. L. Lagadic, M. K. Mitchell and B. D. Payne, Environ. Sci. Technol., 2001, 35, 984–990 Search PubMed.
- L. Ma, Q. Wang, S. M. Islam, Y. Liu, S. Ma and M. G. Kanatzidis, J. Am. Chem. Soc., 2016, 138, 2858–2866 Search PubMed.
- S. Ma, Q. Chen, H. Li, P. Wang, S. M. Islam, Q. Gu, X. Yang and M. G. Kanatzidis, J. Mater. Chem. A, 2014, 2, 10280–10289 Search PubMed.
- J. S. Lee, S. Gomez-Salazar and L. L. Tavlarides, React. Funct. Polym., 2001, 49, 159–172 Search PubMed.
- S. Chamarthy, C. W. Seo and W. E. Marshall, J. Chem. Technol. Biotechnol., 2001, 76, 593–597 Search PubMed.
- M. J. Manos and M. G. Kanatzidis, Chem. Sci., 2016, 7, 4804–4824 Search PubMed.
- D. Sarma, S. M. Islam, K. S. Subrahmanyam and M. G. Kanatzidis, J. Mater. Chem. A, 2016, 4, 16597–16605 Search PubMed.
- J.-R. Li, L. Xu, M.-L. Fu, Y.-X. Wang and H. Xiao, Inorg. Chem. Front., 2018, 5, 403–412 Search PubMed.
- (a) H. P. Klug and L. E. Alexander, X-Ray Diffraction Procedures for Polycrystalline and Amorphous Materials, Wiley, 1974 Search PubMed; (b) T. Egami and S. Billinge, Underneath the Bragg Peaks: Structural Analysis of Complex Materials, Pergamon Press, 2012 Search PubMed.
- L. Xiao, H. Xu, S. Zhou, T. Song, H. Wang, S. Li, W. Gan and Q. Yuan, Electrochim. Acta, 2014, 143, 143–151 Search PubMed.
- N. Serrano, J. M. Díaz-Cruz, C. Ariño and M. Esteban, Anal. Bioanal. Chem., 2009, 396, 1365–1369 Search PubMed.
- P. S. Adarakatti, V. Gangaiah and A. Siddaramanna, Mater. Sci. Semicond. Process., 2018, 84, 157–166 Search PubMed.
- A. Economou and P. R. Fielden, Analyst, 2003, 128, 205–213 Search PubMed.
- C. Kokkinos and A. Economou, Curr. Anal. Chem., 2008, 4, 183–190 Search PubMed.
- N. Serrano, J. M. Díaz-Cruz, C. Ariño and M. Esteban, TrAC, Trends Anal. Chem., 2016, 77, 203–213 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: EDS, PXRD, IR, NMR, TGA, PDFs and crystallographic data in CIF electronic format. CCDC 1905924 and 1905925. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ta03337h |
| This journal is © The Royal Society of Chemistry 2019 |