
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFlat-shaped carbon–graphene microcomposites as electrodes for high energy supercapacitors†
Gelines
Moreno-Fernández
a,
Juan L.
Gómez-Urbano
ab,
Marina
Enterría
a,
Teófilo
Rojo
 ab and
Daniel
Carriazo
*ac
ab and
Daniel
Carriazo
*ac
aCIC EnergiGUNE, Parque Tecnológico de Álava, 01510 Miñano, Álava, Spain. E-mail: dcarriazo@cicenergigune.com; Tel: +34 94 529 71 08
bUniversidad Del País Vasco, UPV/EHU, 48080 Bilbao, Spain
cIKERBASQUE, Basque Foundation for Science, 48013 Bilbao, Spain
First published on 3rd June 2019
Abstract
Herein, we report a simple approach for the preparation of phosphate-functionalized carbonaceous graphene-based composites. The homogeneous deposition of a thin layer of phenolic resin on the surface of graphene oxide (GO) sheets was achieved using phosphoric acid as the polymerization catalyst and functionalization agent. Subsequent pyrolysis of the composite yielded homogeneous lamellar-shaped microstructured porous carbon–graphene composites combining enhanced molecular diffusion and high electron transfer. In order to elucidate the effect of GO and porosity on the supercapacitor performance, a graphene free sample and a KOH-activated composite were also prepared and tested in a two-electrode configuration using aqueous and organic electrolytes. It was found that the presence of GO and the KOH activation lead to an increase in the specific surface area combined with a progressive widening of the pores. As a result, the KOH-activated composite reached specific capacitances of 211 and 105 F g−1 when using 1 M H2SO4 and 1.5 M Et4NBF4 electrolytes, respectively. It was also found that phosphorus-functionalization of electrodes leads to an operating voltage of 1.3 V in the aqueous electrolyte, resulting in a considerable increase of the energy density of the cell. Finally, both non-activated and activated graphene-based composites provide excellent capacitance retention, energy and power densities and cycling stability.
1. Introduction
Porous carbons are the most used materials for the fabrication of electrodes for electrochemical double layer capacitors (EDLCs) due to their high specific surface areas, appropriate electronic conductivity and non-toxicity. These porous carbons are generally prepared by the pyrolysis and activation of natural waste due to its abundance and low cost. However, activated carbons derived from natural waste contain a large amount of impurities. Moreover, the porosity and pore size distribution (PSD) are highly dependent on the nature of the waste employed. This can compromise the EDLC performance since a defined porous structure is of paramount importance in supercapacitor electrodes for the EDL formation.1,2 The presence of micropores in these samples is required to ensure large specific surface areas, but the micropore depth and connectivity are also important to enable fast ion transport in the whole electrode material. Hence, the porosity of electrodes should be tailored as a function of the electrolyte used,3–6 which is not possible using highly crosslinked natural polymers as carbon precursors.On the basis of this premise, there is an urgency to explore new carbonaceous materials suitable as electrodes for high energy supercapacitors. In this regard, carbons derived from the pyrolysis of polymers with high purity, well-controlled structure and tunable porosity have been pointed out as promising candidates to replace activated carbon from natural waste, despite their higher cost.7 Among them, phenolic resins have been extensively used as carbon precursors due to their great ability to polymerize and thermoset in diluted aqueous solutions.8
Electrical conductivity, which is another key descriptor in EDLC performance, is sometimes hindered in amorphous porous materials.7,9 Taking this into account, the preparation of homogeneous flat-shaped films of hybrid materials such as graphene10–12 or carbon nanotubes13 with highly conductive behavior and high specific surface area is expected to boost the capacitive performance.
The surface chemistry of electrode materials also plays an important role in their performance. Hence, the incorporation of heteroatoms into the carbon surface/lattice can modify the electronic properties of porous carbon materials, improving their electronic conductivity and at the same time promoting the generation of functional groups that may undergo fast redox reactions, thus providing a pseudocapacitive contribution to the overall capacitance.14 The functionalization of carbon materials with several heteroatoms (oxygen,15 nitrogen,16 boron,17 or sulfur18) has been extensively explored but scarce reports are available on phosphorus-functionalized porous carbon materials, which calls for future research on this heteroatom.19
Phosphorus rich carbons have also been demonstrated to increase the capacitance due to pseudocapacitive processes, even if the mechanisms behind this behavior are not completely understood yet. However, the most interesting feature is the widening of the operating voltage in aqueous supercapacitors to voltages higher than that of water decomposition (1.23 V), which greatly enhances the energy densities, as compared to pure activated carbons.20–23 Previous reports suggest a blocking/protection effect of the oxygen groups from phosphates; for instance, quinones are electrochemically active but unstable due to their reaction with aqueous electrolytes.20
In the present work we have explored the performance of microporous carbons derived from the pyrolysis of graphene oxide–phenolic resin composites as electrodes for high energy supercapacitors. To this end, the materials were tested in a two-electrode configuration using both aqueous and organic electrolytes (1 M H2SO4 and 1.5 M Et4NBF4 in acetonitrile). It was found that the presence of GO sheets during the polymerization of the phenolic resin leads to a dramatic change in the material morphology and to an increase of the accessible microporous surface area. These particular physicochemical properties led to a notable enhancement of the capacitance retention, energy density and cycling stability, specifically, in the organic electrolyte.
Further KOH activation results in a significant increase of the specific surface area combined with a progressive widening of the micropores, leading to specific capacitances as high as 211 F g−1 and 105 F g−1 when using 1 M H2SO4 and 1.5 M Et4NBF4 electrolytes, respectively.
Interestingly, the removal of the surface-phosphate groups from the composite surface during activation decreases the operating voltage of the electrode material as compared with the functionalized composite, which operates at 1.3 V in the aqueous electrolyte, resulting in a considerable increase of the energy density.
2. Experimental
2.1. Sample preparation
A carbon–graphene composite (hereafter denoted as ResFaGO) was prepared by dissolving 440 mg of resorcinol (Sigma-Aldrich) in 4.0 mL of water and 2.4 mL of ethanol under stirring. Subsequently, 4.0 mL of commercial graphene oxide (GO) (4 mg mL−1, Graphenea) was added and vigorously stirred until complete homogenization. Then, 600 μL of formaldehyde and 100 μL of phosphoric acid were quickly added to the suspension in closed recipients which were transferred to an oven at 85 °C for 70 h to overcome the hydrothermal condensation. ResFaGO carbon was obtained after heating the resins at 800 °C in a tubular oven for 1 h under a dynamic Ar atmosphere. For the preparation of activated carbon–graphene composites, a certain amount of ResFaGO was mixed with KOH in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 in a mortar and subsequently heated at 800 °C under an Ar atmosphere to obtain ResFaGO-A. The as-obtained activated carbon was washed with diluted HCl and hot water several times. A carbon derived from the pyrolysis of the phenolic resin prepared in the absence of graphene oxide (hereafter denoted as ResFa) was prepared for the sake of comparison.
:4 in a mortar and subsequently heated at 800 °C under an Ar atmosphere to obtain ResFaGO-A. The as-obtained activated carbon was washed with diluted HCl and hot water several times. A carbon derived from the pyrolysis of the phenolic resin prepared in the absence of graphene oxide (hereafter denoted as ResFa) was prepared for the sake of comparison.
2.2. Physicochemical characterization
The morphology of the prepared electrodes was studied by Scanning Electron Microscopy (SEM) using a FEI Quanta250 microscope operating at 20 kV. X-ray photoelectron spectroscopy (XPS) measurements were carried out using a UHV spectrometer chamber with base pressure below 10−10 mbar. The chamber features a hemispherical analyser PHOIBOS 150 with a 2D-DLD detector (SPECS) and a monochromatic X-ray source FOCUS 500 (SPECS) with two anodes: Al Kα (hν = 1486.74 eV) and Ag Lα (hν = 2984.3 eV). ASAP2460 and ASAP2020 adsorption analyzers (Micromeritics) were used to measure nitrogen adsorption–desorption isotherms at −196 °C and CO2 adsorption isotherms at 0 °C, respectively. Samples were outgassed at 250 °C for 12 h under vacuum prior to the measurements. The specific surface area (SBET) was calculated according to the Brunauer–Emmett–Teller (BET) method from the nitrogen isotherms in the relative pressure range of 0.05–0.25. The t-plot method was used to calculate the external surface area (SEXT) in the relative pressure range of 0.07–0.25. The total volume of micro–mesopores (VT) was calculated as the amount of N2 adsorbed at a relative pressure of 0.99. The micropore volume (VDR) was calculated by applying the Dubinin–Radushkevich (DR) method in the relative pressure range between 10−6 and 10−1. The mesopore volume was calculated by subtracting the micropore volume from the total pore volume (VT − VDR). The ultramicropore volume (VDR) and the micropore surface area (SDR) were also calculated by applying the Dubinin–Radushkevich (DR) method to the CO2 isotherms in the relative pressure range of 10−5 to 0.03. The pore size distribution (PSD) was determined from both N2 and CO2 isotherms by the 2D-NLDFT heterogeneous surface model using the MicroActive software from Micromeritics. Nitrogen and CO2 were used as probe molecules to assess finite and carbon slit pores, respectively.2.3. Electrode preparation, cell assembling and electrochemical characterization
The electrodes were prepared by mixing 85% of active carbon, 10% of C65 conductive carbon and 5% of polytetrafluoroethylene (PTFE) binder in the presence of ethanol. The mixture was kneaded until plasticity and rolled to obtain a film with typical thickness values of 305 ± 35 μm. Circular shape electrodes of 4–7 mg and 0.95 cm2 were punched from the film and dried at 120 °C under vacuum. The density measured for the electrodes was 0.28, 0.21 and 0.14 g cm−3 for ResFa, ResFaGO and ResFaGO-A, respectively.The electrochemical testing was performed in a two-electrode Swagelok-type cell in which two electrodes with similar masses were separated by a porous glass fiber (Whatman GF/D). The aqueous solution 1 M H2SO4 and the organic one 1.5 M Et4NBF4 in acetonitrile were used as electrolytes. Two stainless steel rods were used as current collectors. In a particular experiment, a two-electrode Swagelok-type cell containing the aqueous solution was equipped with a Hg/HgSO4 reference electrode to follow the potential evolution of individual electrodes using 1 M H2SO4 as the aqueous electrolyte. Cells containing organic electrolytes were assembled in a glovebox. Electrochemical impedance spectroscopy (EIS) (signal amplitude 10 V, frequency range 1 MHz to 10 mHz), cyclic voltammetry (CV) (at the scan rates of 5–100 mV s−1) and galvanostatic charge–discharge (GCD) (at the current densities of 0.2–10 mA g−1) measurements were performed on a multichannel VMP3 generator from Biologic.
The specific capacitance per single electrode Cs (F g−1) was calculated from the discharge galvanostatic plots at different current densities, following the equation:

Energy E (W h kg−1) and power P (W kg−1) densities were calculated according to the following equations:


3. Results and discussion
As described in the experimental section, three different carbon materials were prepared using resorcinol and formaldehyde as polymeric precursors and phosphoric acid as the catalyst of the hydrolysis/condensation as well as the phosphorus precursor. After the polymerization process, homogeneous phenolic resins with a defined porosity and a certain amount of phosphorus were obtained. The as-prepared resins were treated at high temperatures to obtain the final carbons. The reaction processes for ResFa and ResFaGO are summarized in Scheme S1.† The microstructure and morphology of the samples were studied by SEM imaging (Fig. 1). | ||
| Fig. 1 SEM images of ResFA, ResFaGO and ResFaGO-A samples at low (a–c) and high (d–f) magnifications. | ||
As can be observed in Fig. 1a, the ResFa sample presents a sphere-like morphology typical of hydrothermal carbons with uniformly distributed spheres of ca. 2 μm size. The morphology changes drastically for ResFaGO. This carbon composite material shows a flat-shaped morphology in which a layer of carbon of approximately 200 nm thickness is homogeneously coated on the graphene-type sheets (Fig. 1b). The microstructure and particle morphology exhibited by the carbon activated with KOH (ResFaGO-A) are not significantly different compared to the pristine carbon composite, as observed from the SEM images (Fig. 1c). For the sake of comparison pristine GO and rGO were also morphologically evaluated by SEM. Fig. S2† shows the microstructure of these two samples. No major differences are observed between the GO sample and GO pyrolysed at 800 °C, both showing a microporous structure in which very thin graphene sheets are randomly oriented. It can be clearly observed that the deposition of the carbon layer produces a significant increase in the sheet thicknesses within the carbon composites (ResFaGO and ResFaGO-A).
The textural properties of these materials were studied by nitrogen adsorption/desorption at −196 °C and CO2 adsorption at 0 °C (Fig. 2). Both ResFa and ResFaGO present type I isotherms belonging to purely microporous adsorbents.24 The slight increase in nitrogen adsorption observed at high relative pressures, near the saturation point, indicates the presence of interparticle porosity for both samples (Fig. 2a). The porous carbon/reduced graphene oxide composite presents a larger surface area (948 vs. 598 m2 g−1, Table 1) due to an increment in the micropore volume (VDR, Table 1). The increment of SEXT (Table 1) in the latter sample also reveals the generation of large mesopores–narrow macropores by addition of GO sheets during polymerization. It is worth highlighting the significant increase in the specific surface area measured in ResFaGO, 10-fold larger than that of pristine GO and rGO materials (Fig. S3†). The activated sample, ResFaGO-A, presents the largest pore volume (VT, Table 1) and surface area (1961 m2 g−1, Table 1). The isotherm of the activated material also presents a marked micropore contribution but, as evidenced by Vmeso and SEXT values (Table 1) some narrow mesoporosity is generated during activation.
 | ||
| Fig. 2 (a) N2 adsorption/desorption isotherms at −196 °C and (b) CO2 adsorption isotherms at 0 °C for the studied samples and their corresponding (c–e) PSDs calculated from both N2 and CO2 adsorption isotherms by 2D-NLDFT, N2-carbon finite pores and DFT, CO2-carbon slit pores, respectively. | ||
| N2a | CO2b | |||||||
|---|---|---|---|---|---|---|---|---|
| V T (cm3 g−1) | V meso (cm3 g−1) | V DR (cm3 g−1) | S BET (m2 g−1) | S EXT (m2 g−1) | S DR (m2 g−1) | V DR (cm3 g−1) | S DR (m2 g−1) | |
| a Data obtained from N2 adsorption–desorption isotherms at −196 °C. b Data obtained from CO2 adsorption isotherms at 0 °C. c Data obtained from Dubinin–Radusckevich. d Data obtained from the t-plot. e Data obtained from Dubinin–Radusckevich. | ||||||||
| ResFa | 0.36 | 0.13 | 0.24 | 598 | 109 | 670 | 0.18 | 449 |
| ResFaGO | 0.48 | 0.11 | 0.37 | 948 | 138 | 1054 | 0.26 | 666 |
| ResFaGO-A | 1.07 | 0.21 | 0.87 | 1961 | 901 | 2434 | 0.44 | 1110 |
On the other hand, the ultramicroporosity of the studied samples was further analyzed by CO2 adsorption (Fig. 2b).25,26 The CO2 uptake increases from the bulk material (ResFa) to the carbon/graphene composite (ResFaGO) and further increases for the activated composite (ResFaGO-A). Hence, both the presence of graphene sheets and chemical activation increase the micropore volume and the available microporous area (VDRCO2 and SDRCO2, Table 1). Regarding the shape of the CO2 isotherms, the convex curves observed for ResFa and ResFaGO indicate the presence of narrow micropores while the sloping isotherm of ResFaGO-A reveals the presence of wider micropores. The PSDs calculated for both CO2 and N2 isotherms are displayed together in Fig. 2c–e for a general overview of the materials porosity. ResFa and ResFaGO samples present a similar pore size distribution with negligible mesopore contribution but bearing micropores smaller than 0.6 nm. The composite material presents a larger micropore volume (Fig. 2c and d, Table 1). The activated sample presents a greater contribution of wider micropores (from 0.6 to 2 nm) and no contribution of narrow mesopores. It can be concluded that the presence of GO sheets acting as the support during the polymerization process not only causes significant changes in the material morphology at the microscale (Fig. 1b), leading to an increase in its specific surface area, but it also favors ion accessibility to the micropores as compared to bulk materials (ResFa).27 Further activation of the composite material causes an increment of the micropore volume as well as widening of the micropore size. This could be ascribed to the collapse of adjacent micropores during the activation process.28
The surface elemental composition of the three different materials was evaluated by EDX mapping (Table S1, Fig. S1†). As compared with the carbon/graphene composite (ResFaGO), the bulk material (ResFa) presents relatively large amounts of oxygen (5.6 vs. 2.4 wt%) and phosphorus (10.3 vs. 2.0 wt%) on its surface. The XPS spectrum registered for sample ResFA and the deconvolution of the high-resolution core level spectra of P2p (Fig. S4†) confirm that phosphorus is present on the surface of the carbon composites as phosphate, diphosphate or metaphosphate functional groups.
The activation of the composite material (ResFaGO-A) not only increases the oxygen content up to 6.3 wt% but also seems to remove all the phosphorus functional groups from the material surface. Hence, KOH reacts preferentially with the Lewis acid sites (phosphorus in phosphates) during activation generating potassium phosphates, which are further removed from the material surface during the washing step.
The differences verified in morphological, porous and surface chemistry properties of the prepared materials enable a suitable evaluation of the effect of these critical parameters on the supercapacitor performance.
The electrochemical performance of these materials was first evaluated by electrochemical impedance spectroscopy. Fig. 3 includes the Nyquist plots registered for ResFa, ResFaGO and ResFaGO-A in both aqueous (1 M H2SO4) and organic (1.5 M Et4NBF4) electrolytes. As can be seen in Fig. 3a, the curves acquired for the three electrode materials in 1 M H2SO4 show a vertical line in the low-medium frequency regime, which is characteristic of their capacitive behavior, combined with a semicircle registered at high frequencies that can be ascribed to charge transfer processes. The first cut with the X-axis is ascribed to the resistance of the electrolyte and the amplitude of the semicircle is ascribed to the contact and internal resistances.29 The sum of all resistances corresponds to the equivalent series resistance (ESR) and their values are summarized in Table 2. ESR follows the trend ResFaGO-A < ResFaGO < ResFa, which points out the positive influence of graphene and chemical activation on enhancing the electrode electronic conductivity. Hence, previous studies demonstrated that KOH activation is selective towards carbon atoms in disordered environments leaving behind the most graphitic domains which increase the overall structural order and, consequently, the electronic conductivity of the material.10
 | ||
| Fig. 3 Electrochemical impedance spectroscopy plots for ResFa, ResFaGO and ResFaGO-A: Nyquist plots and variation of C′′ with frequency for the aqueous (a and c) and the organic electrolytes (b and d). | ||
| Electrolyte | Sample | ESR (Ω) | τ (s) | ΔV (V) | C (F g−1) | C retention (%) | E (W h kg−1) |
|---|---|---|---|---|---|---|---|
| 1 M H2SO4 | ResFa | 1.00 | 2.94 | 1.3 | 140 | 62 | 8.2 |
| ResFaGO | 0.25 | 0.61 | 1.3 | 147 | 69 | 8.6 | |
| ResFaGO-A | 0.17 | 0.07 | 1.1 | 211 | 77 | 9.1 | |
| 1.5 M Et4NBF4/AN | ResFa | 1.84 | — | 2.5 | 18 | 38 | 3.9 |
| ResFaGO | 1.73 | 1.98 | 2.7 | 61 | 55 | 15.4 | |
| ResFaGO-A | 1.73 | 0.69 | 2.6 | 105 | 72 | 25.5 |
In contrast to the aqueous electrolyte, the Nyquist plots registered in 1.5 M Et4NBF4/AN (Fig. 3b) show a larger ESR resistance and similar semicircle amplitude at high frequency regardless of the material tested. The increase in the ESR values when using 1.5 M Et4NBF4 instead of 1 M H2SO4 should be ascribed to the lower ionic conductivity of the electrolyte combined with the larger ion size in the organic electrolyte compared with the aqueous ones.
It is important to note the evolution of a 45° slope region at medium frequencies for ResFaGO and ResFaGO-A electrodes, indicating hindered molecular diffusion in the EDL formation.30
In order to gain more insight into the molecular transfer properties, the frequency dependence of the imaginary part of the capacitance is presented in Fig. 3c and d for aqueous and non-aqueous electrolytes, respectively. The maximum is related to the transition from resistive to capacitive behavior. The inverse of the maximum frequency is called the time constant (τ) and is the minimum time needed to charge and discharge a supercapacitor. ResFa presents a large EDL relaxation time in 1 M H2SO4 (Fig. 3c) while it does not clearly show a maximum within the low-frequency region in 1.5 M Et4NBF4 (Fig. 3d, Table 2). This fact evidences the hindered mobility of ions, particularly large ions existing in the organic electrolyte, within the small/deep pores present in this bulky sample (Fig. 1). Indeed, it was previously reported that micropores between 0.65 and 0.83 nm in width are the most effective in forming the electric double-layer.20 The hindered formation of the double layer observed for both ResFa and ResFaGO can be therefore explained by the CO2 adsorption analysis (Fig. 2), where both electrode materials presented micropores smaller than 0.6 nm. In contrast, the activated sample (ResFaGO-A) presents the lowest value of τ, i.e., the fastest EDL formation and the fastest charge–discharge behavior, in both aqueous and organic electrolytes (Fig. 3, Table 2). This is in agreement with the larger contribution of wider micropores (from 0.6 to 2 nm) observed in N2 adsorption measurements (Fig. 2e).
From these results, it can be claimed that the charge–discharge performance is improved in ResFaGO electrodes compared to ResFa due to the presence of smaller depth micropores in the former sample, induced by the presence of graphene sheets as the support (Fig. 1), thus favoring the accessibility of the ions to the micropores. The activation of ResFaGO further enhances the ionic transport due to the presence of wider micropores which promotes the formation of the EDL (Fig. 2).
The cyclic voltammetry (CV) curves registered for the studied electrode materials are displayed in Fig. 4. Voltage windows were rigorously determined by the experiments to avoid electrolyte decomposition. As compared with the activated carbon/graphene composite, ResFa and ResFaGO electrodes present a clear enlargement in the operating voltage window when using 1 M H2SO4 as the electrolyte (Fig. 4a and c). This enlargement of the voltage window (1.3 V) to values above the theoretical decomposition of water (1.23 V) seems to be related to the presence of phosphorus on the ResFa and ResFaGO electrode surfaces (Table S1, Fig. S1 and S4†), which blocks the active redox sites and thus prevents water decomposition.21,22 As shown in Fig. S5† a similar contribution to the enlarged voltage window (1.3 V) is recorded for the negative (0.65 V) and positive (0.65 V) electrodes, resulting in a symmetric device. CV-curve shapes are quasi-rectangular with the presence of redox peaks at low scan rates evidencing both pseudocapacitive and EDLC energy storage mechanisms.
 | ||
| Fig. 4 Cyclic voltammograms of ResFa, ResFaGO and ResFaGO-A samples recorded at 5 and 100 mV s−1 for the aqueous (a and c) and organic (b and d) electrolytes. | ||
The substitution of the aqueous electrolyte with the organic electrolyte 1.5 M Et4NBF4 allows an increase in the operating voltage window up to 2.5–2.7 V, as expected. Fig. 4b and c show a rectangular-shaped CV curve characteristic of the EDLC energy storage mechanism.
The specific capacitances in both aqueous and organic electrolytes were calculated from GCD curves presented in Fig. 5 and listed in Table 2. For the 1 M H2SO4 electrolyte, ResFa and ResFaGO electrodes present similar capacitance (∼140 F g−1) while ResFaGO-A delivers much higher capacitance (211 F g−1, Fig. 5a). When the current density is increased (Fig. 5c) the lack of conductivity/molecular diffusion of the ResFa sample becomes evident because of the dramatic decrease of the specific capacitance and the deviation from the linear shape of the GCD curve. The sluggish electron conduction and the low surface area accessible to large ions of the electrolyte are further verified by observation of the GCD curves in the organic electrolyte (Fig. 5b and d). The low specific capacitance (18 F g−1, Table 2) observed for the ResFa sample as compared with the composite (61 F g−1) and the activated composite material (105 F g−1) is in good agreement with the progressive widening of the pore size distribution and the enlargement of the surface area of ResFaGO and ResFaGO-A (Fig. 2).The absence of pores larger than 0.60 nm, which are required for the double-layer formation when using Et4NBF4 salt,31,32 prevents the ResFa electrode from suitable charge–discharge, as previously observed in the EIS analysis (Fig. 3d). It is worth noting the relatively large specific capacitance measured in the organic electrolyte (61 F g−1) for the non-activated carbon–graphene composite (ResFaGO), which is further enhanced by the activation process giving rise to comparable specific capacitance values reported for other representative nanostructured carbons (Table S2†).
 | ||
| Fig. 5 Galvanostatic charge–discharge profiles of ResFa, ResFaGO and ResFaGO-A samples recorded at 0.2 A g−1 and 10 A g−1 for the aqueous (a and c) and organic (b and d) electrolytes. | ||
Regardless of the electrolyte used, the ResFaGO-A electrode displays the best performance at high scan rates and high current densities (Fig. 4c, d, 5c and d). This fact supports the positive effect that both graphene presence and the activation process have on the ionic diffusion and electronic conductivity (Fig. 3).
The evaluation of the dependence of specific capacitance on the applied current density was performed by charging–discharging the studied electrodes from 0.1 to 10 A g−1 (Fig. 6a and b). For all the tested electrodes, the capacitance retention (Cretention) in the aqueous electrolyte is greater than that in the organic one. Regardless of the electrolyte used, the capacitance fading follows the trend ResFaGO-A > ResFaGO > ResFa where retention ratios as high as 77% and 72% are achieved for ResFaGO-A in 1 M H2SO4 and 1.5 M Et4NBF4, respectively.
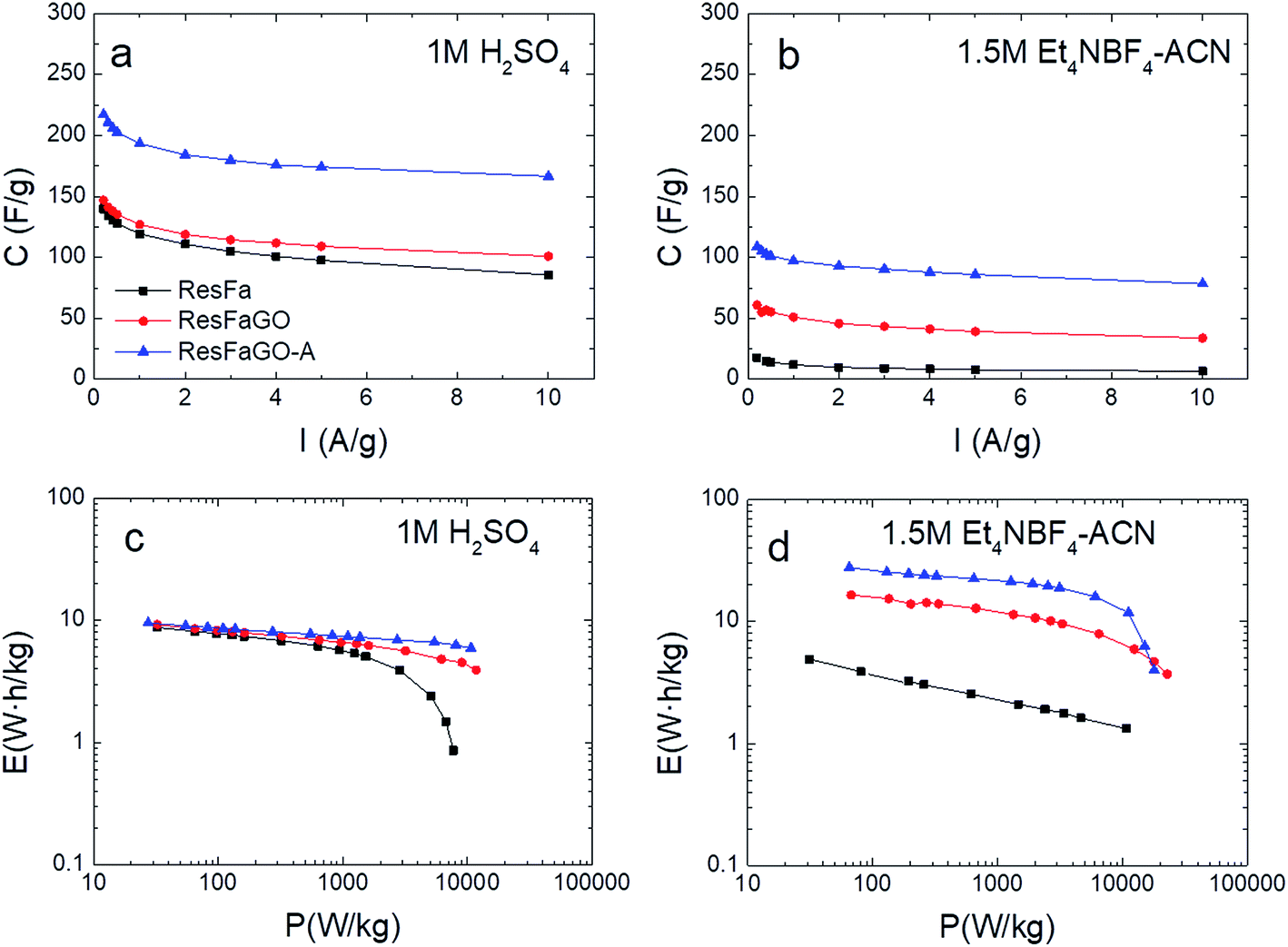 | ||
| Fig. 6 Capacitance retention vs. current density and the Ragone plot of ResFa, ResFaGO and ResFaGO-A samples in the aqueous (a and c) and organic (b and d) electrolytes. | ||
The energy and the power densities calculated for all the samples in the two different electrolytes are presented in the Ragone plots in Fig. 6c and d. Regarding the 1 M H2SO4 electrolyte, ResFaGO-A yields 1.5 times higher capacitance than that registered for ResFa and ResFaGO samples (210 F g−1vs. 140 and 147 F g−1, Fig. 5). Nevertheless, the three studied materials show similar energy values at low power densities (Fig. 6c and Table 2). The widening in the working voltage window to 1.3 V of the latter devices results in comparable energy values around ∼8.5 W h kg−1. Consistent with poor capacitance retention (Fig. 6a), the ResFa electrode displays a strong drop in the energy delivered at high power densities (Fig. 6c and Table 2) due to the narrow pore size distribution and hard-to-access deep micropores which slow down the supercapacitor charge–discharge and thus the power delivery.
In the case of 1.5 M Et4NBF4 (Fig. 6d), important differences are verified in the whole range of power densities for the devices assembled using the three different electrode materials. Hence, the restricted molecular diffusion in ResFa severely limits the energy density in the organic electrolyte compared with that of ResFaGO and ResFaGO-A (3.9 W h kg−1vs. 15.4 and 25.5 W h kg−1, Fig. 6d, Table 2).
In view of the results, ResFaGO and ResFaGO-A, which exhibit the best performance in both electrolytes, were subjected to a cycling test at 10 A g−1. Fig. 7 shows that almost 90% of the capacitance is still retained after 10000 cycles, which evidences the high stability of both materials in both aqueous and organic electrolytes.
 | ||
| Fig. 7 Cycling performance at 10 mA g−1 for ResFaGO and ResFaGO-A samples in the aqueous electrolyte 1 M H2SO4 (a) and the organic electrolyte 1.5 M Et4NBF4 in acetonitrile (b). | ||
4. Conclusions
In this work, we have introduced a novel synthesis of a carbon composite derived from the pyrolysis of a thin layer of phenolic resin precursor deposited on the surface of GO sheets, using phosphoric acid as the polymerization catalyst and functionalization agent. According to the characterization, the carbon–graphene composite shows a lamellar structure, pores in the micropore scale and phosphate group functionalization. Furthermore, a graphene-free microporous carbon was prepared for the sake of comparison, following the same synthesis route.The composite was evaluated as the electrode for supercapacitors in both aqueous and organic electrolytes. It was found that the performance of the composites that integrate graphene in the structure was much better in terms of capacitance and capacitance retention than the graphene-free microporous carbons, especially when the organic electrolyte is used. This behavior should be ascribed to the template effect that graphene oxide has on the final microstructure of the composites, which leads to an easy access to the smaller depth micropores promoting the electric double layer formation and enhancing the ion diffusion into the porous structure. Moreover, the presence of phosphorus on the electrode surface seems to have a positive impact on the supercapacitor performance when using 1 M H2SO4: (i) broadening the working voltage window to 1.3 V and (ii) increasing the capacitance through fast redox reactions (pseudocapacitance).
Further activation of the graphene composites maintains the morphology and microstructure of the composites but produces an enlargement of the specific surface area combined with a progressive widening of the pore sizes. Hence, further improvement in the electrochemical performance of these samples is observed, especially in the organic electrolyte.
Summarizing, carbon–graphene composites are promising materials to be used as electrodes for supercapacitors since their great physicochemical properties lead to interesting values of energy and power densities, as well as long cycling stability in both aqueous and organic electrolytes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the European Union (Graphene Flagship, Core 2, Grant number 785219), the Spanish Ministry of Science and Innovation (MICINN/FEDER) (MAT2015-64617-C2-2-R and RTI2018-096199-B-I00) and the Basque Government (Elkartek 2018) for the financial support of this work. J. L. G. U. is very thankful to the “Ministerio de ciencia, innovación y universidades” for the FPU grant (16/03498). We also want to acknowledge the company GRAPHENEA for supplying the graphene oxide used in this work and Dr M. A. Muñoz for the acquisition of the XPS spectrum.References
- C. Lei, N. Amini, F. Markoulidis, P. Wilson, S. Tennison and C. Lekakou, J. Mater. Chem. A, 2013, 1, 6037 RSC.
- J. Liu, N. P. Wickramaratne, S. Z. Qiao and M. Jaroniec, Nat. Mater., 2015, 14, 763–774 CrossRef CAS PubMed.
- L. Borchardt, M. Oschatz and S. Kaskel, Mater. Horiz., 2014, 1, 157–168 RSC.
- B. Song, C. Sizemore, L. Li, X. Huang, Z. Lin, K. Moon and C.-P. Wong, J. Mater. Chem. A, 2015, 3, 21789–21796 RSC.
- C. Zheng, X. Zhou, H. Cao, G. Wang and Z. Liu, J. Power Sources, 2014, 258, 290–296 CrossRef CAS.
- Y. Yuanyuan, L. Ruiyi, L. Zaijun, L. Junkang, G. Zhiguo and W. Guangli, Electrochim. Acta, 2014, 125, 330–337 CrossRef.
- C. Young, T. Park, J. W. Yi, J. Kim, M. S. A. Hossain, Y. V. Kaneti and Y. Yamauchi, ChemSusChem, 2018, 11, 3546–3558 CrossRef CAS PubMed.
- J. Zhou, Z. Qiu, J. Zhou, W. Si, H. Cui and S. Zhuo, Electrochim. Acta, 2015, 180, 1007–1013 CrossRef CAS.
- Y. Meng, D. Gu, F. Zhang, Y. Shi, L. Cheng, D. Feng, Z. Wu, Z. Chen, Y. Wan, A. Stein and D. Zhao, Chem. Mater., 2006, 18, 4447–4464 CrossRef CAS.
- M. Enterría, F. J. Martín-Jimeno, F. Suárez-García, J. I. Paredes, M. F. R. Pereira, J. I. Martins, A. Martínez-Alonso, J. M. D. Tascón and J. L. Figueiredo, Carbon, 2016, 105, 474–483 CrossRef.
- K. Zhang, B. T. Ang, L. L. Zhang, X. S. Zhao and J. Wu, J. Mater. Chem., 2011, 21, 2663 RSC.
- Y. Qian, I. M. Ismail and A. Stein, Carbon, 2014, 68, 221–231 CrossRef CAS.
- M. C. Gutiérrez, D. Carriazo, A. Tamayo, R. Jiménez, F. Picó, J. M. Rojo, M. L. Ferrer and F. del Monte, Chem. - Eur. J., 2011, 17, 10533–10537 CrossRef PubMed.
- J. P. Paraknowitsch and A. Thomas, Energy Environ. Sci., 2013, 6, 2839 RSC.
- K. Sun, S. Yu, Z. Hu, Z. Li, G. Lei, Q. Xiao and Y. Ding, Electrochim. Acta, 2017, 231, 417–428 CrossRef CAS.
- M. J. Mostazo-López, R. Ruiz-Rosas, A. Castro-Muñiz, H. Nishihara, T. Kyotani, E. Morallón and D. Cazorla-Amorós, Carbon, 2018, 129, 510–519 CrossRef.
- V. Thirumal, A. Pandurangan, R. Jayavel and R. Ilangovan, Synth. Met., 2016, 220, 524–532 CrossRef CAS.
- A. Elmouwahidi, J. Castelo-Quibén, J. F. Vivo-Vilches, A. F. Pérez-Cadenas, F. J. Maldonado-Hódar and F. Carrasco-Marín, Chem. Eng. J., 2018, 334, 1835–1841 CrossRef CAS.
- M. Enterría and J. L. Figueiredo, Carbon, 2016, 108, 79–102 CrossRef.
- D. Hulicova-Jurcakova, A. M. Puziy, O. I. Poddubnaya, F. Suárez-García, J. M. D. Tascón and G. Q. Lu, J. Am. Chem. Soc., 2009, 131, 5026–5027 CrossRef CAS.
- D. Carriazo, M. C. Gutiérrez, F. Picó, J. M. Rojo, J. L. G. Fierro, M. L. Ferrer and F. del Monte, ChemSusChem, 2012, 5, 1405–1409 CrossRef CAS.
- C. Huang, A. M. Puziy, O. I. Poddubnaya, D. Hulicova-Jurcakova, M. Sobiesiak and B. Gawdzik, Electrochim. Acta, 2018, 270, 339–351 CrossRef CAS.
- N. Díez, C. Botas, E. Goikolea and D. Carriazo, J. Mater. Sci., 2017, 52, 11191–11200 CrossRef.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2008 Search PubMed.
- D. Cazorla-Amorós, J. Alcañiz-Monge and A. Linares-Solano, Langmuir, 1996, 12, 2820–2824 CrossRef.
- D. Lozano-Castelló, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2004, 42, 1233–1242 CrossRef.
- J. Liu, F. Mirri, M. Notarianni, M. Pasquali and N. Motta, J. Power Sources, 2015, 274, 823–830 CrossRef CAS.
- P. J. M. Carrott and J. J. Freeman, Carbon, 1991, 29, 499–506 CrossRef CAS.
- P. L. Taberna, P. Simon and J. F. Fauvarque, J. Electrochem. Soc., 2003, 150, A292 CrossRef CAS.
- J. Chmiola, G. Yushin, R. Dash and Y. Gogotsi, J. Power Sources, 2006, 158, 765–772 CrossRef CAS.
- A. García-Gómez, G. Moreno-Fernández, B. Lobato and T. A. Centeno, Phys. Chem. Chem. Phys., 2015, 17, 15687–15690 RSC.
- G. Moreno-Fernandez, S. Perez-Ferreras, L. Pascual, I. Llorente, J. Ibañez and J. M. Rojo, Electrochim. Acta, 2018, 268, 121–130 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta03295a |
| This journal is © The Royal Society of Chemistry 2019 |