
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThreshold catalytic onset of carbon formation on CeO2 during CO2 electrolysis: mechanism and inhibition†
Jiayue
Wang
 a,
Sean R.
Bishop‡
b,
Lixin
Sun
a,
Qiyang
Lu
c,
Gulin
Vardar
a,
Roland
Bliem
a,
Nikolai
Tsvetkov
a,
Ethan J.
Crumlin
d,
Jean-Jacques
Gallet
ef,
Fabrice
Bournel
ef,
Iradwikanari
Waluyo
g and
Bilge
Yildiz
*ac
a,
Sean R.
Bishop‡
b,
Lixin
Sun
a,
Qiyang
Lu
c,
Gulin
Vardar
a,
Roland
Bliem
a,
Nikolai
Tsvetkov
a,
Ethan J.
Crumlin
d,
Jean-Jacques
Gallet
ef,
Fabrice
Bournel
ef,
Iradwikanari
Waluyo
g and
Bilge
Yildiz
*ac
aDepartment of Nuclear Science and Engineering, Massachusetts Institute of Technology (MIT), Cambridge, USA. E-mail: byildiz@mit.edu
bMaterials Processing Center, MIT, Cambridge, MA, USA
cDepartment of Materials Science and Engineering, MIT, Cambridge, MA, USA
dAdvanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, CA, USA
eSorbonne Université, CNRS, Laboratoire de Chimie Physique Matière et Rayonnement – Campus Pierre et Marie Curie, F-75005 Paris, France
fSynchrotron SOLEIL, L'Orme des Merisiers, Saint-Aubin, F-91192 Gif-sur-Yvette, France
gNational Synchrotron Light Source II, Brookhaven National Laboratory, Upton, NY, USA
First published on 22nd May 2019
Abstract
Carbon deposition from CO and other carbon-containing fuels is a major cause of the performance degradation of catalysts and electrocatalysts in many energy conversion devices, including low-temperature solid oxide cells (LT-SOCs). In this work, we present direct observation of carbon deposition on thin-film CeO2 electrodes at LT-SOC operating temperatures (450 °C) in a CO/CO2 atmosphere by in operando X-ray photoelectron spectroscopy. In contrast to the general view that CeO2 is a carbon tolerant material, significant carbon formation was observed on CeO2 during CO2 electrolysis, with no other catalyst present. Moreover, carbon deposition on CeO2 demonstrated an intriguing threshold onset formation against surface Ce3+ concentration. With the aid of Monte Carlo simulations, we propose the neighboring Ce3+–Ce3+ pairs to be a critical catalytic structure that facilitates carbon deposition from CO. Finally, we propose mitigation of carbon deposition on CeO2 by doping CeO2 with non-redox-active cations, and proved this concept using 50% Gd- and 50% Zr-doped CeO2 as an example system. These findings provide an in-depth understanding of the mechanism of carbon deposition on CeO2 during electrochemical reactions and can guide the design of carbon-resistant CeO2-based electrocatalysts.
Introduction
To produce carbon-neutral fuels,1,2 environmentally friendly technologies, including electrolysis3 and thermochemical splitting4 of water and carbon dioxide, have attracted significant attention. In particular, low-temperature solid oxide cells (LT-SOC) that operate from 400 °C to 650 °C are believed to be the next generation technology.5,6 However, carbon poisoning, or “coking”, due to carbon deposition on the catalyst surface during operation, is an important issue for processes involving carbon-containing species.7,8 This coking process can lead to irreversible catalyst deactivation and cause mechanical degradation.9 One major type of carbon poisoning is from CO,10–14 which has been reported as either CO dissociation15,16 (CO → C + O) or the Boudouard disproportionation17–19 (2CO → CO2 + C). At operating temperatures below 700 °C, carbon deposition is thermodynamically more favorable and occurs mainly via the Boudouard reaction.5,20 Consequently, it is important to develop carbon-tolerant electrocatalysts that can operate under LT-SOC operating conditions, with both high catalytic activity and long-term stability.Ceria (CeO2) and ceria-based materials have been widely investigated as catalysts and catalyst supports towards a broad range of reactions.21–23 The catalytic performance of CeO2 is due to its excellent redox activity,24 which can be expressed with the Kröger–Vink notation25 as
 | (1) |
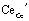 and
and  denote Ce3+ (small polaron26) and doubly positive charged oxygen vacancy, respectively. With this oxygen storage capability, CeO2-based catalysts are also known to be carbon-tolerant as they can oxidize the deposited carbon with lattice oxygen.27
denote Ce3+ (small polaron26) and doubly positive charged oxygen vacancy, respectively. With this oxygen storage capability, CeO2-based catalysts are also known to be carbon-tolerant as they can oxidize the deposited carbon with lattice oxygen.27
LT-SOC fuel electrodes combining ceria-based materials with Ni have yielded high performance but their stability in carbon-containing fuels remains a big challenge.5,28 While carbon deposition has been extensively studied on the Ni surfaces, few studies focus on carbon deposition on CeO2. Despite the high carbon tolerance of CeO2 as a catalyst, carbon deposition may not be negligible on CeO2 in LT-SOCs since the oxygen ion current can greatly affect the carbon tolerance of electrode materials.29 In particular, in electrolysis conditions, O2− are constantly removed from the fuel electrode, making carbon deposition more favorable.30 As an example, recent studies have demonstrated that infiltrating Ni-YSZ with Gd-doped CeO2 did not improve the carbon tolerance during CO2 electrolysis.31,32 Therefore, to obtain a more complete picture of carbon deposition over Ni/CeO2 electrodes, it is important to investigate the onset of carbon formation and carbon deposition mechanism on CeO2 during electrochemical reactions. Moreover, besides Ni/CeO2 electrodes, there has been considerable development of CeO2/perovskite fuel electrodes in LT-SOCs, as a means to improve the electrodes ability to work with carbon-containing fuels without carbon deposition.5,33 Hence a fundamental study of carbon deposition on CeO2 surface would also benefit the design of these conducting-oxide electrodes.
In this work, we systematically studied carbon deposition in CO/CO2 mixture on four types of fluorite-type ceria thin films: undoped ceria (CeO2), 20% Gd-doped ceria (Gd0.2Ce0.8O1.9), 50% Gd-doped and 50% Zr-doped ceria (Gd0.5Ce0.5O1.75, Zr0.5Ce0.5O2). Despite the general opinion that CeO2 is a carbon-tolerant material, we demonstrated for the first time with in operando ambient pressure X-ray photoelectron spectroscopy (APXPS) that carbon is readily deposited on the CeO2 surface within half an hour, under LT-SOC operating conditions (450 °C, CO/CO2 atmosphere, ∼1 V polarization). Moreover, with quantification of both [Ce3+] and the amount of deposited carbon, we demonstrate an intriguing threshold [Ce3+]-carbon relation for the onset of carbon formation on CeO2. Combining APXPS data with Monte Carlo simulation, we propose the most likely catalytic reaction for carbon deposition from CO on CeO2 to be the neighboring Ce3+–Ce3+ pair. Finally, we propose doping CeO2 with non-redox-active cations can effectively mitigate the carbon deposition on CeO2 in the LT-SOCs operating conditions, as exemplified in the case of Gd0.5Ce0.5O1.75 and Zr0.5Ce0.5O2.
Results
The experimental setup is shown in Fig. 1a and b, which depicts the electrochemical cell consisting of a ceria thin film electrode, an 8% yttria-stabilized zirconia (YSZ) (001) single crystal electrolyte, a Pt grid buried under the ceria electrodes, and a porous Pt counter electrode. The out-of-plane symmetric X-ray diffraction scan for the ceria films on the electrochemical cell is shown in Fig. 1c. The ceria films demonstrated both (001) and (111) orientations: the films grown epitaxially on YSZ are in the (001) direction while the polycrystalline regions grown on Pt grids follow a mixed (001) and (111) texture. Verification of this assignment is shown in ESI Fig. S2.† As explained in ESI Note 1,† all the ceria films have cubic fluorite structure (space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m). To reveal the surface structure, low-energy electron diffraction (LEED) patterns were collected on ceria films epitaxially grown on YSZ (001) without Pt grids at 450 °C, i.e., the same temperature where carbon deposition was studied. As shown in Fig. 1d, the surface of all the ceria thin films on YSZ (001) had a four-fold symmetry, in agreement with the (001) fluorite-type ceria surface structure.24,34 On the other hand, the polycrystalline regions on the Pt grids have yielded no LEED pattern, consistent with previous reports.24
m). To reveal the surface structure, low-energy electron diffraction (LEED) patterns were collected on ceria films epitaxially grown on YSZ (001) without Pt grids at 450 °C, i.e., the same temperature where carbon deposition was studied. As shown in Fig. 1d, the surface of all the ceria thin films on YSZ (001) had a four-fold symmetry, in agreement with the (001) fluorite-type ceria surface structure.24,34 On the other hand, the polycrystalline regions on the Pt grids have yielded no LEED pattern, consistent with previous reports.24
 | ||
| Fig. 1 Schematics of the experimental configuration and sample characterization. (a) The electrochemical cell consists of a thin-film ceria working electrode, a buried Pt grid, an 8% yttria-stabilized zirconia (YSZ) solid electrolyte, and a porous Pt counter electrode. (b) SEM image showing the patterned Pt grid; scale bar, 100 μm. (c) High-resolution X-ray diffraction data of symmetric 2θ-ω scan of the ceria thin-films on the electrochemical cell. (d) Low-energy electron diffraction patterns for the ceria thin films on YSZ (001) without Pt grids, collected at 450 °C in UHV using 110.6 eV electrons (color legends in (c)). A fourfold symmetry is present on all ceria electrodes, highlighted by the dashed square. | ||
Fig. 2 summarizes the surface chemistry evolution during carbon deposition reaction. In the experiment, cathodic (CO2 reduction) and anodic (CO oxidation) biases were applied on the electrochemical cell. Since the width of Pt stripes (∼20 μm) is much smaller than the X-ray spot size (∼1 mm in diameter), the APXPS results reveal an average surface chemistry of the epitaxial and polycrystalline ceria regions. Fig. 2a shows the normalized C 1s, Ce 4d and valence band (VB) photoelectron spectra on Gd0.2Ce0.8O1.9 as a representative case. All the spectra were calibrated by aligning the Ce 4d3/2 peaks at 122 eV35,36 and then normalized to the Ce 4d intensity. Three main features were observed in the C 1s spectra. First, the pronounced rising peak at around 285 eV corresponds to the deposited graphitic carbon, in agreement with previous studies on carbon deposition on ceria.37,38 Secondly, the peak at around 290 eV is an adsorbate peak, which has been commonly attributed to a carbonate species on the ceria surface.36,38,39 Since a detailed investigation of the CO/CO2 adsorption structure on ceria is beyond the scope of this paper, we leave this peak assigned as an adsorbate. Studies on this topic can be found in the ref. 40 and 41. Finally, the two peaks at around 292 eV and 293.5 eV originate from CO and CO2 gas phase.42,43 The assignment of the two gas peaks has been verified by collecting the C 1s spectra while the sample was extracted (ESI Fig. S6†).
 | ||
Fig. 2 Evolution of surface chemistry during carbon deposition. (a) C 1s, Ce 4d, and valence band as a function of applied electrochemical bias at 450 °C in 0.3 Torr 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CO/CO2 on Gd0.2Ce0.8O1.9, with negative values representing cathodic polarization. The spectra taken at 350 °C in 0.02 Torr O2 at OCV are shown as a reference. Both Ce 4d and valence band spectra indicate a more reduced surface under increasing cathodic bias. (b) The relation between carbon deposition and [Ce3+], where the color bands serve as a guide to the eye. Note the carbon intensity does not scale linearly with [Ce3+], instead the onset of significant carbon deposition is only after a threshold [Ce3+] on CeO2 and on Gd0.2Ce0.8O1.9. In addition, carbon deposition was significantly suppressed on both Gd0.5Ce0.5O1.75 and Zr0.5Ce0.5O2. (c) Surface cation composition as a function of [Ce3+]. The open symbols represent the surface composition quantified by APXPS, with the points at [Ce3+] = 0 showing the surface composition in O2. The closed symbols at [Ce3+] = 0 represent the as-prepared surface composition measured by lab-based XPS in UHV (legend in (b)). :1 CO/CO2 on Gd0.2Ce0.8O1.9, with negative values representing cathodic polarization. The spectra taken at 350 °C in 0.02 Torr O2 at OCV are shown as a reference. Both Ce 4d and valence band spectra indicate a more reduced surface under increasing cathodic bias. (b) The relation between carbon deposition and [Ce3+], where the color bands serve as a guide to the eye. Note the carbon intensity does not scale linearly with [Ce3+], instead the onset of significant carbon deposition is only after a threshold [Ce3+] on CeO2 and on Gd0.2Ce0.8O1.9. In addition, carbon deposition was significantly suppressed on both Gd0.5Ce0.5O1.75 and Zr0.5Ce0.5O2. (c) Surface cation composition as a function of [Ce3+]. The open symbols represent the surface composition quantified by APXPS, with the points at [Ce3+] = 0 showing the surface composition in O2. The closed symbols at [Ce3+] = 0 represent the as-prepared surface composition measured by lab-based XPS in UHV (legend in (b)). | ||
The components with the two highest binding energies (shaded region in the plot) in the Ce 4d spectrum arise purely from Ce4+ species.36,44 Note the Ce4+ features decreased under increasing cathodic polarization, indicating the reduction of ceria surface. In the valence band spectra, the feature at about 1.5 eV corresponds to the Ce 4f peak, whose normalized intensity reflects the concentration of Ce3+ at the surface.36,45 Consistent with Ce 4d spectra, the valence band Ce 4f peak intensity increased with increased reduction state at the surface under cathodic polarization. As shown in Fig. 2a and ESI Fig. S7a,† surface [Ce3+] increased monotonically with cathodic polarization for all the ceria samples. At open circuit, both Gd0.2Ce0.8O1.9 and Zr0.5Ce0.5O2 were more reduced than CeO2, indicating a higher reducibility, consistent with previous studies.46–48 On the other hand, 50% Gd-doped CeO2 was less reducible. This could originate from defect–defect interactions in ceria – a high concentration of extrinsic oxygen vacancies and large Gd3+ ions increase the formation energy of additional oxygen vacancy (and Ce3+).49,50
The amount of the deposited carbon under different biases is shown in ESI Fig. S7b.† Due to voltage loss at the electrolyte and the counter electrode, the applied electrical bias was not entirely converted into an electrochemical potential at the ceria electrodes. Therefore, it is not possible to compare the carbon deposition behavior across samples as a function of bias. Instead, we use the surface reduction state, [Ce3+], as a metric in carbon deposition at the ceria surface. This is reasonable, because [Ce3+] depends on the effective oxygen chemical potential determined by the gas composition and the electrochemical potential at the ceria electrodes.51 Carbon deposition as a function of [Ce3+] is plotted in Fig. 2b. There are two important features in the [Ce3+]-carbon relation: firstly, there is a clear threshold of [Ce3+] for the onset of carbon deposition. As shown on CeO2 and Gd0.2Ce0.8O1.9, significant carbon deposition was only observed when [Ce3+] reached a critical value of around 50%. Secondly, the carbon deposition was suppressed on Gd0.5Ce0.5O1.75 and Zr0.5Ce0.5O2. As illustrated, negligible amount of carbon was observed on Zr0.5Ce0.5O2 throughout the entire polarization range even though [Ce3+] exceeded 50%. Although carbon was also observed on Gd0.5Ce0.5O1.75 when polarizing the sample to almost 100% [Ce3+], the deposited carbon intensity was much smaller than that on Gd0.2Ce0.8O1.9.
Fig. 2c shows the cation fraction of dopants as a function of [Ce3+] during the polarization and carbon deposition process. The surface dopant concentration remained relatively constant before the threshold carbon deposition, but there is an apparent increase after that. For example, the surface Gd concentration in Gd0.2Ce0.8O1.9 and Gd0.5Ce0.5O1.75 increased at high [Ce3+]. The increase of measured cation fraction originates from the fact the XPS signal from Ce gets attenuated more by the deposited carbon, while the dopant are less affected. The observed apparent increase of dopant/Ce ratio thus indicates that more carbon was deposited onto Ce sites than on the dopants (ESI Fig. S12†).
To confirm and better demonstrate the threshold [Ce3+]-carbon relation, we monitored carbon deposition on Gd0.2Ce0.8O1.9 at different biases (i.e., different [Ce3+]) as a function of time. The Ce 4d and C 1s spectra collected during the 1.5 hour measurement at open circuit are shown in Fig. 3a and b respectively. As can be seen, the spectra remained stable throughout the measurement, indicating neither carbon formation nor oxidation state change. Then, a cathodic bias (−0.7 V) was applied to reduce the surface further, but still below the threshold [Ce3+]. The spectra collected immediately after polarization, after 0.5 hours and after 1.5 hours are shown in Fig. 3c and d. A small graphitic carbon peak appeared and the surface Ce became more reduced due to cathodic polarization. Nonetheless, no further change of the surface chemistry took place, with no increase in the carbon peak intensity during the 1.5 hours of measurement time. Upon increasing the applied cathode bias to −1.2 V to reduce the surface beyond the [Ce3+] threshold, significant amount of carbon accumulated already within the first 0.5 hours and the carbon intensity continued to increase during the measurement (Fig. 3f). Meanwhile, a new broad peak appeared at about 125 eV, originating from the carbon KVV Auger peak52 (Fig. 3e). This time-dependent measurement clearly indicates that significant carbon deposition only occurs when the ceria surface reaches a threshold [Ce3+]. After the onset of carbon deposition when [Ce3+] was greater than the threshold, the carbon growth exhibited a self-limiting behavior. The carbon intensity at the surface saturated and stopped growing when the thickness of the deposited carbon reached 1–2 monolayers (ESI Note 7†).
 | ||
| Fig. 3 Time-dependence of carbon deposition on the ceria surface. C 1s and Ce 4d spectra were collected on Gd0.2Ce0.8O1.9 at 450 °C in 9:1 CO/CO2 atmosphere at (a and b) open circuit (OCV), (c and d) −0.7 V, and (e and f) −1.2 V electrical bias in sequence. No significant carbon deposition was observed at OCV and −0.7 V, and the surface remained stable during our measurement. Upon reaching the threshold −1.2 V polarization (i.e., threshold [Ce3+]), the carbon deposition started and increased sharply. Spectra are normalized to the corresponding CO gas peak. | ||
Along with the carbon deposition, the adsorbate peak vanished. If indeed this adsorbate is the carbonate precursor in CO2 electrolysis reaction,36,38 its suppression indicates loss of catalytic activity for CO2 electrolysis because of the surface being covered by deposited carbon. In addition, the gas peak shifted to a lower binding energy, possibly due to a change in the gas adsorption behavior on the carbon-covered surface.53
The change of Ce 4d spectra during carbon deposition is twofold (Fig. 3e): first, the total intensity of the normalized Ce peak decreased as a result of carbon deposition. Second, and more interestingly, the surface became more enriched with Ce3+ during carbon deposition. The increase of surface [Ce3+] can be clearly visualized from the decreasing intensity of Ce4+ peaks in the Ce 4d spectra by comparing the three spectra in Fig. 3e. Possible reasons for this carbon-induced Ce3+ enrichment are discussed in ESI Note 10.†
To summarize, rapid carbon formation from CO only occurred when the surface [Ce3+] exceeded a threshold value of about 50%. This threshold relation is intriguing since the carbon intensity remained negligible prior to the [Ce3+] threshold, despite the considerable amount of Ce3+ and 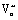 in the near-surface region. In addition, we found the carbon deposition was almost completely suppressed on both Gd0.5Ce0.5O1.75 and Zr0.5Ce0.5O2 throughout the entire polarization range, even after [Ce3+] well exceeded the threshold concentration (50%).
in the near-surface region. In addition, we found the carbon deposition was almost completely suppressed on both Gd0.5Ce0.5O1.75 and Zr0.5Ce0.5O2 throughout the entire polarization range, even after [Ce3+] well exceeded the threshold concentration (50%).
Discussions
We propose that the threshold [Ce3+]-carbon relation can have three origins, which may act individually or synergistically. First, the threshold may arise from the thermodynamics aspects (“thermodynamic threshold”), where the carbon deposition reaction is only thermodynamically favorable beyond a threshold cathodic polarization (i.e., threshold [Ce3+]). Secondly, the threshold can arise from the formation of catalytic sites for carbon deposition (“catalytic threshold”), where the dominant catalytic sites are only formed in significant numbers beyond a threshold [Ce3+]. Thirdly, the threshold may come from the stability of the deposited carbon on the ceria surface (“stability threshold”). For the last case, carbon can be produced at low [Ce3+], but the reaction products stay stably at the ceria surface beyond the threshold of [Ce3+]. In the following paragraphs, we discuss all three possibilities and show the threshold [Ce3+]-carbon relation must be most likely due to a threshold in the formation of the catalytic structure (Ce3+–Ce3+ pair) at the surface (“catalytic threshold”).We first examine the “stability threshold”. Ceria is known for its ability to catalyze the combustion of carbon;54 it may remove (oxidize) the deposited carbon on the surface. As an example, Zr-doped ceria can oxidize the surface carbon with lattice oxygen even at 200 °C.47 Therefore, it is possible that the deposited carbon can only be stabilized on an oxygen-deficient surface, where no O2− species being available to oxidize the deposited carbon. In this scenario, the observed threshold of [Ce3+] should reflect a threshold of 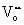 , where a sufficiently oxygen depleted CeO2 surface is unable to inhibit carbon deposition. However, this is in contrast to our observation that carbon deposition was suppressed on Gd0.5Ce0.5O1.75, the most oxygen-deficient ceria surface at a given [Ce3+]. Therefore, we view this scenario to be unlikely.
, where a sufficiently oxygen depleted CeO2 surface is unable to inhibit carbon deposition. However, this is in contrast to our observation that carbon deposition was suppressed on Gd0.5Ce0.5O1.75, the most oxygen-deficient ceria surface at a given [Ce3+]. Therefore, we view this scenario to be unlikely.
In addition, the “stability threshold” can originate from a threshold carbon cluster size. Carbon dimers (C2) have been observed to have higher stability than monomers (C1) and are the key intermediates in the growth of graphene on Au surfaces.55 Similarly here, carbon cluster formation may also be a key step in the carbon deposition reaction on ceria. Carbon deposition has been found more favorable near defect sites.18 Therefore, carbon atoms deposited on the surface with a higher defect concentration are more likely to be located closer to each other, promoting the “clustering” process. In this scenario, the observed threshold [Ce3+]-carbon relation should be associated with a threshold surface defect density to ensure that the deposited carbon forms clusters. Clustering of single carbon atoms to form pairs of carbon atoms induces a peak shift in the C 1s spectra as reported by Xu et al.56 We did not find such a peak shift, and therefore we deem this scenario to be unlikely.
We then examine the “thermodynamic threshold”. The reaction environment is already thermodynamically favorable for the Boudouard reaction even without polarization.57 In this case, a thermodynamic threshold can only arise from the CO dissociation reaction. As the surface [Ce3+] depends directly on the overpotential at the electrode,51 the threshold [Ce3+]-carbon relation indicates that a threshold of overpotential is required to split the adsorbed CO. Had this scenario been the case, we would expect the surface overpotential to be a good descriptor for the carbon deposition reaction on ceria. In this case, all the samples reported in Fig. 2c should merge into one common threshold overpotential–carbon relation. However, as illustrated in ESI Fig. S17,† this overpotential–carbon relation cannot explain our observation. In particular, this “thermodynamic threshold” cannot resolve the paradox that at similar surface overpotentials, significant amount of carbon deposition was observed on Gd0.2Ce0.8O1.9 while nearly no carbon was observed on Gd0.5Ce0.5O1.75. Therefore, we assign this scenario to be unlikely as well.
Having excluded the aforementioned scenarios, last but most importantly, we propose that the threshold [Ce3+]-carbon relation should arise from the “catalytic threshold”. This means that the catalytic structure can only be effectively formed on the ceria surface beyond a threshold [Ce3+]. Previous studies have assigned either a single site (i.e., individual Ce3+ site58 or  (ref. 18)) or a neighboring pair of sites (i.e., Ce3+–Ce4+ pair17) as the catalytic structures for carbon deposition from CO on ceria and other oxides. From our observation, we can already exclude the case of single sites. Had the dominant catalytic structure been a single Ce3+ or
(ref. 18)) or a neighboring pair of sites (i.e., Ce3+–Ce4+ pair17) as the catalytic structures for carbon deposition from CO on ceria and other oxides. From our observation, we can already exclude the case of single sites. Had the dominant catalytic structure been a single Ce3+ or  site, the deposited carbon should have increased progressively with [Ce3+] rather than exhibiting a threshold onset. Therefore, we constrain our following discussion to the neighboring defect pairs. As redox-inert Zr and Gd sites don't participate in the reaction,59 five types of surface defect pairs may act as the catalytic structure: Ce3+–Ce4+ pair, Ce3+–Ce3+ pair, Ce3+–O pair, Ce3+–
site, the deposited carbon should have increased progressively with [Ce3+] rather than exhibiting a threshold onset. Therefore, we constrain our following discussion to the neighboring defect pairs. As redox-inert Zr and Gd sites don't participate in the reaction,59 five types of surface defect pairs may act as the catalytic structure: Ce3+–Ce4+ pair, Ce3+–Ce3+ pair, Ce3+–O pair, Ce3+– pair, and Ce4+–
pair, and Ce4+– pair. Meanwhile, despite the difference in the nominal oxygen vacancy concentration at a given [Ce3+], similar [Ce3+]-carbon relations have been observed between CeO2 and Gd0.2Ce0.8O1.9; and between Zr0.5Ce0.5O2 and Gd0.5Ce0.5O1.75, indicating that both the oxygen sites and the
pair. Meanwhile, despite the difference in the nominal oxygen vacancy concentration at a given [Ce3+], similar [Ce3+]-carbon relations have been observed between CeO2 and Gd0.2Ce0.8O1.9; and between Zr0.5Ce0.5O2 and Gd0.5Ce0.5O1.75, indicating that both the oxygen sites and the  play a minor role, if any, in catalyzing carbon deposition. Consequently, only the Ce pairs (Ce3+–Ce4+ or Ce3+–Ce3+) may act as the catalytic structure.
play a minor role, if any, in catalyzing carbon deposition. Consequently, only the Ce pairs (Ce3+–Ce4+ or Ce3+–Ce3+) may act as the catalytic structure.
To resolve the most likely catalytic structure, we conducted a model Metropolis Monte Carlo simulation to quantify Ce pair formation on the ceria surface as a function of [Ce3+]. In principle, one would need to consider all the possible surface terminations to model site-by-site the pairing of reduced ceria cations. Here, we model the Ce3+ pair formation on the (001)-type ceria surface, i.e., the majority surface structure of the electrochemical cell. As will be presented later, the insights drawn from the (001) surface model can be extended to other types of ceria facets as well. Different energies of defect–defect interactions between the nearest-neighbor Ce3+ sites (ECe3–Ce3) were tested, ranging from 0 to 0.2 eV, with positive values representing repulsion. The energies were chosen from a previous calculation, indicating the interaction energy between the nearest-neighbor Ce3+–Ce3+ pair in the bulk of ceria is 0.1 eV.60 As a result, Fig. 4a shows the concentration of Ce3+–Ce4+ and Ce3+–Ce3+ pairs on the equilibrated undoped lattice at different interaction energies. When ECe3–Ce3 = 0 eV (no interaction), the concentration of Ce3+–Ce3+ pairs progressively increased with [Ce3+]. However, when considering the repulsive interactions (ECe3–Ce3 = 0.1, 0.2 eV), a threshold formation of Ce3+–Ce3+ pair appeared. On the other hand, no threshold relation was found in the formation of Ce3+–Ce4+ pairs, as expected.
 | ||
| Fig. 4 Quantification of Ce pair formation on the (001) ceria surface with Monte Carlo simulation. (a) The response of Ce pair formation to the interaction energy between neighboring Ce3+ cations, with positive energies representing repulsion. Note a threshold behavior was observed in the Ce3+–Ce3+ pair formation at repulsive interaction energies, and no threshold is found for the formation of Ce3+–Ce4+ pairs, as expected. (b) Effect of doping on the Ce pair concentration, with the interaction energy fixed at 0.1 eV. Note that doping suppresses Ce pair formation. The error bar for the simulation is smaller than the line width. (c–e) Exemplify three scenario, with schematic lattices showing the effect of repulsive interaction and doping on the Ce3+ pair formation: (c) undoped ceria surface without neighboring Ce3+ pair interaction, where Ce3+ and Ce4+ arrange randomly. (d) Undoped ceria surface with repulsive Ce3+–Ce3+ interaction. Since Ce3+ and Ce4+ arrange alternatingly, no Ce3+–Ce3+ pair is formed up to 50% [Ce3+]. (e) Ceria lattice with 50% of its sites occupied by the dopant. Since Ce sites are isolated from each other by the dopants, the Ce3+–Ce3+ pair formation is suppressed. | ||
The effect of doping on Ce pair formation is shown in Fig. 4b, with the interaction energy between neighboring Ce3+ sites fixed at 0.1 eV.60 As described in more detail in the Methods section, the dopant sites are randomly distributed and are treated as immobile during the simulation. Lattices with three different doping ratios were calculated: 0%, 20%, and 50%, which is in accord with the surface composition measured with APXPS (Fig. 2c). As a result, lattices with higher doping ratio exhibited a smaller concentration of Ce3+–Ce4+ and Ce3+–Ce3+ pairs.
The dependence of Ce pair concentrations on the interaction energy and doping concentration is explained as follows. Due to the repulsive interactions between adjacent Ce3+ sites, it is energetically more favorable for Ce3+ to be surrounded by Ce4+ (Fig. 4d). As a result, the Ce3+–Ce3+ pair is difficult to form at low [Ce3+]. However, once [Ce3+] exceeds 50%, any increase in [Ce3+] results in additional Ce3+–Ce3+ pair formation because of site unavailability, regardless of the interaction strength. Consequently, the Ce3+–Ce3+ pair concentration exhibits a threshold formation against [Ce3+]. Regarding the doping effect: since dopants were treated as immobile sites between Ce atoms in the lattice, they impede Ce3+–Ce3+ pair formation by “isolating” Ce sites from each other (Fig. 4e). Note each lattice structure shown in Fig. 4c–e only represents one possible configuration at a given [Ce3+] and dopant concentration. In the real simulation process, a 50 by 50 2D cubic lattice was employed and the simulation results in Fig. 4a and b reflect the average values obtained from energy minimization of different lattice configurations.
As demonstrated in the Monte Carlo simulations, the catalysis of carbon deposition reaction is very likely linked to the Ce3+–Ce3+ pair formation. To better verify this idea, Fig. 5 presents the comparison of the carbon deposition measured by APXPS and the calculated Ce3+–Ce3+ pair concentration. The simulated Ce3+–Ce3+ pair formation exhibits a very similar behavior as the deposited carbon intensity as a function of [Ce3+]: the model not only captures the absolute value of the onset threshold (∼50% [Ce3+]) but also correctly reflects the relative carbon intensity between ceria surfaces at different doping concentrations. As a result, we conclude that our model qualitatively delivers a strong evidence that the neighboring Ce3+–Ce3+ pair is the corresponding catalytic structure for carbon deposition from CO. Even though we modeled the pairing effect on the (001) surface, we believe that the same argument can be also applicable to other major surface terminations. This is because, regardless of the surface termination, two CO molecules must be dissociated nearby to each other, to enable the C–C bond formation during the growth of graphitic carbon. In other words, it is the formation of adjacent catalytic sites for carbon deposition, i.e., the formation of a nearest neighbor “Ce3+–Ce3+ pair”, that is critical for the coking reaction. Since the distance between the nearest Ce–Ce pairs is the same on the (001), (011), and (111) fluorite-type surfaces, we would expect a similar catalytic behavior for carbon deposition on these low-index surface terminations of ceria. Nevertheless, future studies are needed to confirm this.
 | ||
| Fig. 5 Comparison between the measured carbon deposition and the simulated Ce3+–Ce3+ pair formation, as a function of [Ce3+]. The calculated Ce3+–Ce3+ pair density (line) and the measured carbon intensity (points) are in good overall agreement. Note the model captures the threshold onset and the doping effect, supporting the theory that the neighboring Ce3+–Ce3+ pair is the catalytic structure for carbon deposition from CO. All the simulation curves were replotted from Fig. 4b and divided by a common normalization factor to ease comparison. The solid lines represent the Ce3+–Ce3+ pair density calculated with 0.1 eV interaction energy, whereas the shaded regions indicate the variations when changing the interaction energy from 0 eV to 0.2 eV. | ||
To summarize, in this section we discussed three possible origins of the onset of carbon deposition at a threshold [Ce3+] at ceria surfaces. We deduce that this behavior arises most likely because of the formation of Ce3+–Ce3+ pairs after exceeding a threshold of [Ce3+] at the surface. From this, we also deduce that the Ce3+–Ce3+ pair serves as the catalytic structure for carbon deposition from CO. Consistent with this argument, the enhanced carbon resistance on 50% Zr-doped and 50% Gd-doped ceria originates from a suppressed Ce3+–Ce3+ pair formation. In this work, the proposed catalytic structure does not define a specific reaction pathway, and further studies are needed to reveal the atomistic reaction steps enabled by this catalytic structure.
Conclusions
In this work, we systematically studied the carbon deposition reaction during CO2 electrolysis on four fluorite-type ceria thin films: CeO2, Gd0.2Ce0.8O1.9, Gd0.5Ce0.5O1.75, and Zr0.5Ce0.5O2. By performing in operando APXPS measurements, we revealed that, despite the general opinion that CeO2 is a carbon-tolerant catalyst, carbon deposition can readily occur on CeO2 surfaces in LT-SOCs conditions. This observation clearly highlights the importance of investigating carbon deposition on CeO2 surfaces to mitigate carbon poisoning in LT-SOCs. Moreover, carbon deposition exhibits an onset at a threshold value of 50% Ce3+ concentration when dopant concentration is relatively low or zero. This intriguing threshold [Ce3+]-carbon relation deepens the understanding of the carbon deposition reaction mechanism: combining APXPS data with Monte Carlo simulation, we propose the most likely catalytic structure for carbon deposition to be the neighboring Ce3+–Ce3+ pair. Inspired by this mechanism, we propose that doping CeO2 with non-redox-active cations can effectively mitigate the carbon deposition on CeO2 under LT-SOC operating conditions. This idea has been verified with in operando APXPS on 50% Gd and 50% Zr-doped CeO2, where carbon formation has been effectively suppressed.Since Ce3+ is a key enabler in a wide range of electrocatalytic reactions,24,36 suppressing carbon deposition while maintaining a high [Ce3+] is required for developing carbon-tolerant ceria-based electrocatalysts. Our results indicate that suppressing Ce3+–Ce3+ pair formation at high [Ce3+] can be the key to alleviate carbon deposition from CO. This includes ways to isolate Ce3+ cations from each other, for example by doping ceria with non-redox-active cations (such as 50% Zr- and 50% Gd-doped CeO2 in this work). When used as fuel cell electrodes, it has been suggested that higher Gd concentrations in Gd-doped ceira can improve electrode's electrical conductivity as well as dimensional stability.61 Previous studies have also shown that heavily Zr-doped ceria benefits oxygen surface exchange62 as well as bulk diffusivity.63 Therefore, both enhanced carbon resistance and electrocatalytic performance could be realized simultaneously on heavily doped ceria surfaces. In addition, as the number of Ce3+ sites per unit surface area of doped ceria is lower than the undoped case, one may also consider increasing the total surface area of the doped ceria by employing nanoparticles.64,65
Methods
Film fabrication
The electrochemical cells were fabricated on 10 mm × 10 mm × 0.5 mm 8% yttria-stabilized zirconia (YSZ) (001) single crystal substrates (MTI Corporation), which served as the solid electrolyte. Pt paste (SPI Supplies) was applied on the back of the YSZ substrate and then sintered in air at 800 °C for 1 hour as the counter electrode (CE). Buried, micro-patterned Pt grids were fabricated through photolithography on the front side of the substrate as the current collector (ESI Fig. S1†). The thin film working electrode (WE) was deposited on top of YSZ and Pt grids using pulsed laser deposition (PLD), where a KrF excimer laser with 248 nm wavelength was used. During deposition, the substrate temperature was kept at 750 °C in an oxygen pressure of 10 mTorr.In operando APXPS
The APXPS measurements were carried out at Beamline 9.3.2 of the Advanced Light Source, Lawrence Berkeley National Laboratory (ALS); the NAP-XPS end station of the Pierre and Marie Curie University set on TEMPO beamline at Synchrotron SOLEIL; and the IOS beamline 23-ID-2 of the National Synchrotron Light Source II (NSLS-II) at Brookhaven National Lab. The electrochemical cells described above were placed on a ceramic heater, with thermocouples mounted directly onto the surface for temperature measurements. The thermocouple along with a Pt wire was also used to apply electrochemical bias on the sample, where the Pt wire was welded to a Pt foil beneath the CE. During the experiment, cathodic (CO2 reduction) and anodic (CO oxidation) biases were applied on the electrochemical cell to drive surface redox reaction as well as to change the oxidation state of surface Ce cations.Samples were preconditioned at 350 °C in 20 mTorr O2 at the beginning of the measurement to remove adventitious carbon. The operando environment studied in this work was 450 °C in 0.15 Torr (SOLEIL, NSLS-II) and 0.3 Torr (ALS) 9:1 CO/CO2 atmosphere, under cell bias ranging from +0.7 to −1.5 V. The APXPS spectra were collected at an incident photon energy of 370 eV (SOLEIL) and 400 eV (ALS, NSLS-II). CeO2, Gd0.5Ce0.5O1.75, and Zr0.5Ce0.5O2 were tested in ALS while Gd0.2Ce0.8O1.9 were tested in all of the three beamlines.
All the XPS spectra were quantified with CasaXPS software. Carbon deposition intensity was calculated by integrating the graphitic carbon area in the C 1s spectra and then normalized to the CO gas peak intensity (ESI Note 4†). [Ce3+] was calculated as the peak area ratio of Ce3+ features to the total area of Ce 4d (ESI Note 5†).
Monte Carlo simulation
A 50 × 50 2D square lattice with periodic boundary conditions was employed to simulate the ceria (001) surface. Since the aim of this simulation is to quantify Ce pair formation, the simulated lattice consists purely of Ce and dopant sites, and no oxygen sites were considered. In the simulated square 2D lattice, each atom has four nearest neighboring sites and represents either a Ce (Ce3+ or Ce4+) or a dopant. Only the interaction between two nearest Ce3+ sites was considered, while all other interactions were ignored. Therefore, the system Hamiltonian can be expressed as H = NCe3–Ce3·ECe3–Ce3, where NCe3–Ce3 and ECe3–Ce3 denote the Ce3+–Ce3+ pair number and the interaction energy between them, respectively. In this model, Ce3+ and Ce4+ sites were set free to switch while dopant sites were considered immobile. This assumption is in agreement with the significant lower mobility of cations compared to that of polarons in ceria.66 The Metropolis Monte Carlo simulation was conducted as follows:(1) Randomly pick two Ce sites on the lattice and calculate the energy difference ΔH of switching Ce3+ and Ce4+.
(2) If the switch results in a reduced system energy (ΔH < 0), accept it with a probability of 1. Otherwise, accept the switch with a probability of exp (ΔH/kBT), where kB and T denote the Boltzmann constant and temperature respectively.
In accord with the experimental environment, the simulation temperature was set to be 450 °C. 100 random configurations were generated as initial guesses at each [Ce3+] and doping ratio. Then each of the 100 initial lattices was evolved by switching sites according to the Metropolis scheme. Each Monte Carlo step (MCS) consisted of 2500 switching trials, and the ensemble average was estimated by averaging over the last 200 MCS after equilibrium. Finally, the concentration of Ce pairs (both Ce3+–Ce3+ and Ce3+–Ce4+) were calculated as the mean value of the 100 thermodynamically equilibrated lattices (ESI Note 11†).
Author contributions
J. W., S. R. B., and B. Y. conceived the experiment design. J. W. and Q. L. prepared the samples and J. W. conducted the sample characterization. All authors carried out the APXPS experiments. J. W. performed the Monte Carlo simulations. All authors contributed to writing the manuscript. B. Y. supervised the project.Conflicts of interest
The authors declare that they have no competing financial interests.Acknowledgements
The authors are grateful for the funding support from the National Aeronautics and Space Administration in support of the Mars Oxygen ISRU Experiment (award number NNH17CH01C), and from Exelon Corp (award number 025610-00007). The authors acknowledge the facility support from the Microsystems Technology Laboratories and the Center for Materials Science and Engineering (NSF under award number DMR-1419807) at MIT. This research used the synchrotron radiation facilities at the National Synchrotron Light Source II (23-ID-2 beamline), a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704; the Advanced Light Source which is supported by the DOE Office of Basic Energy Sciences under Contract No. DE-AC02-05CH11231; and the TEMPO beamline at the Synchrotron SOLEIL. The authors thank Peter A. Crozier (ASU), William Chueh (Stanford), Ulrike Diebold, Alexander K. Opitz (TU Wien) and Franziska Hess (MIT) for the helpful discussions on the carbon deposition reaction mechanisms. The authors also thank Dongha Kim (MIT) and Maximilian Jansen (ETH) for the assistance of APXPS measurement. J. W. and B. Y. thank Dr Rachael Rothman (University of Sheffield) for her helpful advice and input on the preparation of this manuscript. J. W. thanks Hongzhou Ye and Charles Settens (MIT) for the helpful discussions on the Monte Carlo simulation and XRD measurements.References
- A. Schoedel, Z. Ji and O. M. Yaghi, The role of metal–organic frameworks in a carbon-neutral energy cycle, Nat. Energy, 2016, 1, 16034 CrossRef CAS.
- C. Graves, S. D. Ebbesen, M. Mogensen and K. S. Lackner, Sustainable hydrocarbon fuels by recycling CO2 and H2O with renewable or nuclear energy, Renewable Sustainable Energy Rev., 2011, 15, 1–23 CrossRef CAS.
- K. Chen and S. P. Jiang, Reviewmaterials degradation of solid oxide electrolysis cells, J. Electrochem. Soc., 2016, 163, F3070–F3083 CrossRef CAS.
- J. R. Scheffe and A. Steinfeld, Oxygen exchange materials for solar thermochemical splitting of H2O and CO2: a review, Mater. Today, 2014, 17, 341–348 CrossRef CAS.
- Z. Gao, L. V. Mogni, E. C. Miller, J. G. Railsback and S. A. Barnett, A perspective on low-temperature solid oxide fuel cells, Energy Environ. Sci., 2016, 9, 1602–1644 RSC.
- L. Fan, B. Zhu, P.-C. Su and C. He, Nanomaterials and technologies for low temperature solid oxide fuel cells: Recent advances, challenges and opportunities, Nano Energy, 2018, 45, 148–176 CrossRef CAS.
- D. Neagu, et al., Nano-socketed nickel particles with enhanced coking resistance grown in situ by redox exsolution, Nat. Commun., 2015, 6, 8120 CrossRef PubMed.
- B. Liu, et al., Microwaves effectively examine the extent and type of coking over acid zeolite catalysts, Nat. Commun., 2017, 8, 514 CrossRef CAS PubMed.
- P. Boldrin, et al., Strategies for carbon and sulfur tolerant solid oxide fuel cell materials, incorporating lessons from heterogeneous catalysis, Chem. Rev., 2016, 116, 13633–13684 CrossRef CAS PubMed.
- S. R. Bishop, et al., Oxygen generation from carbon dioxide for advanced life support, ECS Trans., 2008, 11, 173–179 CAS.
- S. D. Ebbesen and M. Mogensen, Electrolysis of carbon dioxide in solid oxide electrolysis cells, J. Power Sources, 2009, 193, 349–358 CrossRef CAS.
- M. P. Kohn, M. J. Castaldi and R. J. Farrauto, Auto-thermal and dry reforming of landfill gas over a Rh/AL2O3 monolith catalyst, Appl. Catal., B, 2010, 94, 125–133 CrossRef CAS.
- J. Harmsen, W. Jansen, J. Hoebink, J. Schouten and H. Brongersma, Coke deposition on automotive three-way catalysts studied with leis, Catal. Lett., 2001, 74, 133–137 CrossRef CAS.
- A. Goguet, et al., Study of the origin of the deactivation of a Pt/CeO2 catalyst during reverse water gas shift (rwgs) reaction, J. Catal., 2004, 226, 382–392 CrossRef CAS.
- J.-X. Liu, B.-Y. Zhang, P.-P. Chen, H.-Y. Su and W.-X. Li, CO dissociation on face-centered cubic and hexagonal close-packed nickel catalysts: A first-principles study, J. Phys. Chem. C, 2016, 120, 24895–24903 CrossRef CAS.
- E. Putna, R. Gorte, J. Vohs and G. Graham, Evidence for enhanced dissociation of CO on Rh/ceria, J. Catal., 1998, 178, 598–603 CrossRef CAS.
- Y. Liu, et al., Mechanism of CO disproportionation on reduced ceria, ChemCatChem, 2010, 2, 336–341 CrossRef CAS.
- M. Kogler, et al., High-temperature carbon deposition on oxide surfaces by CO disproportionation, J. Phys. Chem. C, 2016, 120, 1795–1807 CrossRef CAS PubMed.
- G. M. Bremmer, et al., In situ tem observation of the boudouard reaction: multi-layered graphene formation from co on cobalt nanoparticles at atmospheric pressure, Faraday Discuss., 2017, 197, 337–351 RSC.
- K. Girona, J. Laurencin, J. Fouletier and F. Lefebvre-Joud, Carbon deposition in CH4/CO2 operated sofc: Simulation and experimentation studies, J. Power Sources, 2012, 210, 381–391 CrossRef CAS.
- J. A. Rodriguez, D. C. Grinter, Z. Liu, R. M. Palomino and S. D. Senanayake, Ceria-based model catalysts: fundamental studies on the importance of the metal-ceria interface in CO oxidation, the water-gas shift, CO2 hydrogenation, and methane and alcohol reforming, Chem. Soc. Rev., 2017, 46, 1824–1841 RSC.
- T. Montini, M. Melchionna, M. Monai and P. Fornasiero, Fundamentals and catalytic applications of CeO2-based materials, Chem. Rev., 2016, 116, 5987–6041 CrossRef CAS PubMed.
- W. C. Chueh, Y. Hao, W. Jung and S. M. Haile, High electrochemical activity of the oxide phase in model ceria–Pt and ceria–Ni composite anodes, Nat. Mater., 2012, 11, 155–161 CrossRef CAS PubMed.
- W. C. Chueh, et al., Highly enhanced concentration and stability of reactive Ce3+ on doped CeO2 surface revealed in operando, Chem. Mater., 2012, 24, 1876–1882 CrossRef CAS.
- H. L. Tuller and S. R. Bishop, Point defects in oxides: Tailoring materials through defect engineering, Annu. Rev. Mater. Res., 2011, 41, 369–398 CrossRef CAS.
- H. Tuller and A. Nowick, Small polaron electron transport in reduced CeO2 single crystals, J. Phys. Chem. Solids, 1977, 38, 859–867 CrossRef CAS.
- E. L. Lawrence and P. A. Crozier, Oxygen transfer at metal-reducible oxide nanocatalyst interfaces: Contrasting carbon growth from ethane and ethylene, ACS Appl. Nano Mater., 2018, 1, 1360–1369 CrossRef CAS.
- V. Duboviks, et al., A raman spectroscopic study of the carbon deposition mechanism on Ni/CGO electrodes during CO/CO2 electrolysis, Phys. Chem. Chem. Phys., 2014, 16, 13063–13068 RSC.
- Y. Lin, Z. Zhan, J. Liu and S. A. Barnett, Direct operation of solid oxide fuel cells with methane fuel, Solid State Ionics, 2005, 176, 1827–1835 CrossRef CAS.
- W. Li, Y. Shi, Y. Luo, Y. Wang and N. Cai, Carbon deposition on patterned nickel/yttria stabilized zirconia electrodes for solid oxide fuel cell/solid oxide electrolysis cell modes, J. Power Sources, 2015, 276, 26–31 CrossRef CAS.
- T. L. Skafte, P. Blennow, J. Hjelm and C. Graves, Carbon deposition and sulfur poisoning during CO2 electrolysis in nickel-based solid oxide cell electrodes, J. Power Sources, 2018, 373, 54–60 CrossRef CAS.
- T. L. Skafte, C. Graves, P. Blennow and J. Hjelm, Carbon deposition during CO2 electrolysis in ni-based solid-oxide-cell electrodes, ECS Trans., 2015, 68, 3429–3437 CrossRef CAS.
- H. Kan and H. Lee, Enhanced stability of NiFe/GDC solid oxide fuel cell anodes for dry methane fuel, Catal. Commun., 2010, 12, 36–39 CrossRef CAS.
- V. F. Solovyov, et al., Highly efficient solid state catalysis by reconstructed (001) ceria surface, Sci. Rep., 2014, 4, 4627 CrossRef PubMed.
- D. Mullins, S. Overbury and D. Huntley, Electron spectroscopy of single crystal and polycrystalline cerium oxide surfaces, Surf. Sci., 1998, 409, 307–319 CrossRef CAS.
- Z. A. Feng, M. L. Machala and W. C. Chueh, Surface electrochemistry of CO2 reduction and CO oxidation on Sm-doped CeO2-x: coupling between Ce3+ and carbonate adsorbates, Phys. Chem. Chem. Phys., 2015, 17, 12273–12281 RSC.
- Z. Liu, et al., Ambient pressure XPS and IRRAS investigation of ethanol steam reforming on Ni-CeO2(111) catalysts: an in situ study of C–C and O–H bond scission, Phys. Chem. Chem. Phys., 2016, 18, 16621–16628 RSC.
- Y. Yu, et al., CO2 activation and carbonate intermediates: an operando AP-XPS study of CO2 electrolysis reactions on solid oxide electrochemical cells, Phys. Chem. Chem. Phys., 2014, 16, 11633–11639 RSC.
- T. Staudt, et al., Electronic structure of magnesia ceria model catalysts, CO2 adsorption, and CO2 activation: A synchrotron radiation photoelectron spectroscopy study, J. Phys. Chem. C, 2011, 115, 8716–8724 CrossRef CAS.
- M. Favaro, et al., Subsurface oxide plays a critical role in CO2 activation by Cu(111) surfaces to form chemisorbed CO2, the first step in reduction of CO2, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6706–6711 CAS.
- S. Chen, et al., Probing surface structures of CeO2, TiO2, and Cu2O nanocrystals with CO and CO2 chemisorption, J. Phys. Chem. C, 2016, 120, 21472–21485 CrossRef CAS.
- S. Blomberg, et al. , Phys. Rev. Lett., 2013, 110, 117601 CrossRef CAS PubMed.
- B. Eren, C. Heine, H. Bluhm, G. A. Somorjai and M. Salmeron, Catalyst chemical state during CO oxidation reaction on Cu(111) studied with ambient-pressure X-ray photoelectron spectroscopy and near edge X-ray adsorption fine structure spectroscopy, J. Am. Chem. Soc., 2015, 137, 11186–11190 CrossRef CAS PubMed.
- C. Zhang, et al., Multielement activity mapping and potentiacs catalysisal mapping in solid oxide electrochemical cells through the use of operando XPS, ACS Catal., 2012, 2, 2297–2304 CrossRef CAS.
- Z. A. Feng, F. El Gabaly, X. Ye, Z.-X. Shen and W. C. Chueh, Fast vacancy-mediated oxygen ion incorporation across the ceria–gas electrochemical interface, Nat. Commun., 2014, 5, 4374 CrossRef CAS PubMed.
- S. Grieshammer, M. Nakayama and M. Martin, Association of defects in doped non-stoichiometric ceria from first principles, Phys. Chem. Chem. Phys., 2016, 18, 3804–3811 RSC.
- E. Aneggi, et al., Ceriazirconia particles wrapped in a 2D carbon envelope: Improved low temperature oxygen transfer and oxidation activity, Angew. Chem., Int. Ed., 2015, 54, 14040–14043 CrossRef CAS PubMed.
- S. Bishop, K. Duncan and E. Wachsman, Defect equilibria and chemical expansion in non-stoichiometric undoped and gadolinium-doped cerium oxide, Electrochim. Acta, 2009, 54, 1436–1443 CrossRef CAS.
- S. R. Bishop, T. S. Stefanik and H. L. Tuller, Electrical conductivity and defect equilibria of Pr0.1Ce0.9O2-δ, Phys. Chem. Chem. Phys., 2011, 13, 10165–10173 RSC.
- C. Chatzichristodoulou and P. V. Hendriksen, Oxygen nonstoichiometry and defect chemistry modeling of Ce0.8Pr0.2O2, J. Electrochem. Soc., 2010, 157, B481–B489 CrossRef CAS.
- D. Chen and H. L. Tuller, Voltagecontrolled nonstoichiometry in oxide thin films: Pr0.1Ce0.9O2 case study, Adv. Funct. Mater., 2014, 24, 7638–7644 CrossRef CAS.
- J. S. Murday, B. I. Dunlap, F. L. Hutson and P. Oelhafen, Carbon KVV auger line shapes of graphite and stage-one cesium and lithium intercalated graphite, Phys. Rev. B, 1981, 24, 4764–4770 CrossRef CAS.
- S. Porsgaard, et al., Charge state of gold nanoparticles supported on titania under oxygen pressure, Angew. Chem., Int. Ed., 2011, 50, 2266–2269 CrossRef CAS PubMed.
- E. Ramírez-Cabrera, A. Atkinson and D. Chadwick, The influence of point defects on the resistance of ceria to carbon deposition in hydrocarbon catalysis, Solid State Ionics, 2000, 136–137, 825–831 CrossRef.
- S. Riikonen, A. V. Krasheninnikov, L. Halonen and R. M. Nieminen, The role of stable and mobile carbon adspecies in copper-promoted graphene growth, J. Phys. Chem. C, 2012, 116, 5802–5809 CrossRef CAS.
- L. Xu, et al., Transformation of carbon monomers and dimers to graphene islands on CO(0001): Thermodynamics and kinetics, J. Phys. Chem. C, 2013, 117, 2952–2958 CrossRef CAS.
- K. Sasaki and Y. Teraoka, Equilibria in fuel cell gases: Ii. the C–H–O ternary diagrams, J. Electrochem. Soc., 2003, 150, A885–A888 CrossRef CAS.
- M. Swanson, V. V. Pushkarev, V. I. Kovalchuk and J. L. d'Itri, The dynamic surface chemistry during the interaction of CO with ceria captured by raman spectroscopy, Catal. Lett., 2007, 116, 41–45 CrossRef CAS.
- J. Paier, C. Penschke and J. Sauer, Oxygen defects and surface chemistry of ceria: Quantum chemical studies compared to experiment, Chem. Rev., 2013, 113, 3949–3985 CrossRef CAS PubMed.
- S. Grieshammer and M. Martin, Influence of defect interactions on the free energy of reduction in pure and doped ceria, J. Mater. Chem. A, 2017, 5, 9241–9249 RSC.
- R. O’hayre, S.-W. Cha, F. B. Prinz and W. Colella, Fuel Cell Fundamentals, John Wiley & Sons, 2016 Search PubMed.
- N. Yang, et al., Role of associated defects in oxygen ion conduction and surface exchange reaction for epitaxial samaria-doped ceria thin films as catalytic coatings, ACS Appl. Mater. Interfaces, 2016, 8, 14613–14621 CrossRef CAS PubMed.
- T. Wakita and M. Yashima, Structural disorder in the cubic Ce0.5Zr0.5O2 catalyst: A possible factor of the high catalytic activity, Appl. Phys. Lett., 2008, 92, 101921 CrossRef.
- J.-h. Myung, D. Neagu, D. N. Miller and J. T. Irvine, Switching on electrocatalytic activity in solid oxide cells, Nature, 2016, 537, 528 CrossRef CAS PubMed.
- Y. Chen, et al., A robust and active hybrid catalyst for facile oxygen reduction in solid oxide fuel cells, Energy Environ. Sci., 2017, 10, 964–971 RSC.
- S. Beschnitt, T. Zacherle and R. A. De Souza, Computational study of cation diffusion in ceria, J. Phys. Chem. C, 2015, 119, 27307–27315 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta03265g |
| ‡ Present Address: Redox Power Systems, LLC, College Park, MD, USA. |
| This journal is © The Royal Society of Chemistry 2019 |