
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Anisotropy and enhancement of thermoelectric performance of Sr0.8La0.067Ti0.8Nb0.2O3−δ ceramics by graphene additions†
D.
Srivastava
a,
C.
Norman
a,
F.
Azough
a,
D.
Ekren
a,
K.
Chen
b,
M. J.
Reece
b,
I. A.
Kinloch
a and
R.
Freer
 *a
*a
aSchool of Materials, University of Manchester, M13 9PL, UK. E-mail: Robert.Freer@manchester.ac.uk
bSchool of Engineering and Materials Science, Queen Mary University of London, London E1 4NS, UK
First published on 15th October 2019
Abstract
A-site deficient SrTiO3 ceramics are very promising n-type oxide thermoelectrics but currently limited by their low performance compared to more conventional materials. We show that incorporation of graphene or graphene oxide can significantly improve the transport properties and hence ZT of the ceramic matrix. Powders of Sr0.8La0.067Ti0.8Nb0.2O3−δ were prepared by the mixed oxide route; ceramics and composites with ≤3 wt% graphene or graphene oxide were densified by spark plasma sintering (SPS) at 1473 K for 5 minutes. The microstructures obtained were uniform with an average grain size of 5 μm; the carbon additions were uniformly distributed. Composites employing ‘as-prepared’ powders exhibited three orders of magnitude increase in electrical conductivity, a reduction in thermal conductivity from 4.00 to 2.64 W m−1 K−1, but very modest thermoelectric figure of merit (ZT) values, less than 0.1. Graphene additions yielded superior thermoelectric performance to graphene oxide. Composites prepared with ‘pre-reduced’ oxide powders and 1 wt% graphene were at least 99% dense, with further improvement in electrical conductivity. There was strong anisotropy in their transport properties due to the alignment of the graphene flakes perpendicular to the pressing direction; electrical conductivity was significantly higher perpendicular to the pressing direction; thermal conductivity was lowest parallel to the pressing direction. The highest thermoelectric figure of merit (∼0.25 at 1000 K) was achieved for samples containing graphene measured parallel to the pressing direction. The control of thermoelectric transport properties by additions of carbon species, and the resulting anisotropy in properties could guide the development of processing routes to produce future target materials.
Introduction
Concerns about the use of fossil fuels have initiated research into alternative, renewable sources of energy including thermoelectrics, where power can be generated from waste heat.1,2 Materials with a large thermoelectric figure of merit, (defined as ZT = σS2T/κ, where σ is the electrical conductivity, S is the Seebeck coefficient, κ is the thermal conductivity and T is the absolute temperature) enable efficient conversion of waste heat into useful electrical energy. While many conventional thermoelectric materials (Te based) have high ZT values (1 < ZT < 2),3 the rarity of their starting materials, cost, toxicity and temperature instability limit their widespread exploitation.4,5 As thermoelectrics, oxides have potential disadvantages in terms of modest electrical conductivity6 and high thermal conductivity,4 but are attractive because of their high temperature/chemical stability,7 the high value of S2/κ ratio for layered p-type oxides8,9 and routes to lower thermal conductivity through complex or misfit structures.10 Amongst the n-type oxide materials, SrTiO3 is of major importance due to its high Seebeck coefficient and the relative ease of cationic substitution to modify their thermoelectric properties. The related materials including CaMnO3 (ref. 11 and 12) and ZnO13,14 also show promise as n-type oxide materials along with the CoO2-based layered p-type materials.4,6We have previously shown that controlled doping on Sr/Ti sites of SrTiO3 (STO), in addition to controlled vacancy formation within the system, can introduce significant numbers of charge carriers.15,16 Cationic vacancies in donor doped STO have been shown to increase carrier concentration and generate oxygen vacancies under reducing environment. These point defects play a crucial role in reducing thermal conductivity by scattering of phonons.15,17–19 Various cationic substitution have been reported for Sr and Ti sites to improve the electrical conductivity, with predominately lanthanides on Sr sites20,21 and transition metals (Nb,22 W,23 Ta,7,24 Y25) on Ti sites. These investigations have improved the understanding of the interrelationship between different correlated variables and pushed the ZT values for SrTiO3 based polycrystalline materials up to 0.35 to 0.41 at 1000 K.20,26,27 We recently reported that the introduction of metallic inclusion such as Fe and Cu improved the thermoelectric properties of the base material.28 The metallic inclusions were mainly present at grain boundaries and significantly improved the overall electronic conductivity by reducing the grain boundary resistance. Similarly, we also demonstrated that adjusting the amount of additives can control the distribution of A site ions, and inhibit nano-inclusion formation, thereby significantly increasing the thermoelectric power factor.29 In this work, we seek to understand how the addition of graphene derivatives into the bulk matrix improves the properties of the material. The clear advantages of these additives are their scale and two-dimensional geometry (with lateral dimensions much larger than thickness) which may introduce interesting effects on each of the thermoelectric variables.
Graphene, has the attraction of high electronic mobility30 and has been used to improve the electrical performance of ceramic composites for applications including solar cells, fuel cells and gas sensing.31 Conventional solid state sintering (SSS) and spark plasma sintering (SPS) have also aided the development of ceramic–graphene/reduced-graphene oxide (RGO) composites. Fan et al.32 reported dense GNPs/Al2O3 composites fabricated from ball milled expanded graphite and Al2O3 by SPS. The electrical conductivity reached 5700 S m−1 when the composite contained 15 vol% GNP, which was 170% higher than that of carbon nanotube (CNT)/Al2O3 composites. The percolation threshold for electrical conductivity in the GNPs/Al2O3 composites was reported to be around 3 vol%.32 In general, relatively high Seebeck coefficients can be obtained in graphene-based materials.33,34 The values reported for single layer graphene (SLG) and multi layered graphene (MLG) vary from a few μV K−1 to ∼100 μV K−1 and are dependent on the processing route.35 Theoretical work by Esfarjani et al.36 demonstrated that any complex pathway through granular metals embedded in a semiconducting matrix has the potential to enhance thermoelectric properties (as the semiconductor has a rapidly varying density of states near the chemical potential). It is possible to achieve this interaction effect using graphene based layers at the boundaries in a semiconducting matrix. The thermal conductivity of pristine graphene (∼5000 W m−1 K−1 (ref. 37)) is three orders of magnitude higher than that of SrTiO3, which could be a disadvantage in thermoelectric applications. However, the transport behaviour of graphene and CNT can be tuned by controlling their defect concentration, which could potentially lead to improved ZT. For example, Zhao et al. demonstrated an increase in the Seebeck coefficient and reduction in the thermal conductivity to ∼0.3 W m−1 K−1 by laser treatment of flexible CNT paper.38 Graphene or graphene oxide (GO) inclusions are expected to enhance thermoelectric properties due to their favourable morphology and superior electrical properties. In the last few years there have been several studies of the role and effect of GNPs/GO as fillers. Dey et al.,39 reviewed the use of carbon-based fillers in polymer thermoelectric composites and showed that below the percolation threshold ZT was enhanced because electrical conductivity increases with the content of filler, without significant reduction of the Seebeck coefficient or increase in thermal conductivity; however, the resulting ZT for these polymeric composites was still much less than 1.0.
For inorganic thermoelectrics, graphene has been used as an additive in a range of p-type and n-type materials either in the form of GO or graphene layers such as in doped Bi2Te3,40 CoSb3,41 CuInTe2,42 ZnO,43 Bi2Sb2Te3,44 TiO2,45 La-doped SrTiO3,46 pure SrTiO3,47 and Nb doped SrTiO3.48 The electrical conductivity is increased by the addition of graphene-based phases in all these materials. However, the preparation technique of the composite has a significant effect on the properties. For example, n-type ZnO displayed an 8-fold improvement in ZT for a chemically processed composite including RGO;43 there was simultaneous improvement in electrical conductivity and thermal conductivity, without major change of the Seebeck coefficient. The maximum ZT of 0.28 at 1173 K was for a ZnO based composite containing 1.5 wt% RGO; further additions of RGO degraded ZT. Wu et al.49 have recently demonstrated 180% increase in ZT at 800 K in A-site deficient Nb doped STO (non-stoichiometric) by adding up to 1 wt% GO and SPS processing the reduced mixed powders.
We have previously reported that the incorporation of graphene in La doped SrTiO3 ceramic (sintered conventionally in a reducing atmosphere of Ar/5% H2) resulted in significant improvement in ZT values.46 The highest ZT of 0.12 at 1000 K for the base ceramic was improved to values of ≥0.26 (approximately temperature-independent) from ambient to 1000 K for a graphene loading of 0.6 wt%. More recently Feng et al.47 reported a ZT value of 0.09 at 760 K for pure SrTiO3 containing RGO, and Okhay et al.48 reported a ZT value of 0.29 at 1160 K for Nb-doped SrTiO3 containing RGO.
From this background we sought to understand whether there were significant differences between the use of graphene and reduced graphene oxide as additives at different loading levels. Furthermore, in view of the two dimensional nature of graphene and GO we also wanted to evaluate whether there was any significant anisotropy in the thermoelectric properties. For the base ceramic matrix, an A-site deficient, La, Nb doped SrTiO3, 80% SrTiO3 – 20% La1/3NbO3 (hereafter referred to as L2 = Sr0.8La0.067Ti0.8Nb0.2O3−δ)15 was chosen. Whilst this has moderate thermoelectric properties, it has the advantage of optimised aliovalent doping giving ∼13.5% of A-site vacancies.15 This also provides a direct comparison with our earlier work on La doped SrTiO3 ceramic composites.46 It is well established that SPS sintering of ceramics offers much shorter processing times (minutes compared to hours), minimal grain growth during processing and a reducing atmosphere during the heat treatment at elevated temperature. The final objective was therefore to assess whether high quality thermoelectric SrTiO3-based composites could be prepared by SPS processing alone (in minutes) without first having to subject the powders to heat treatment in a reducing atmosphere.
Material and methods
Ceramics of L2 composition (Sr0.8La0.067Ti0.8Nb0.2O3−δ) were prepared using the mixed oxide route. The starting materials were TiO2 (Sachtleben Chemie GmbH); SrCO3 (Sigma Aldrich, UK); La2O3 (Molycorp Inc, USA); Nb2O5 (Solvay, UK); all were at least 99.9% purity; La2O3 was heated to 1200 K for 10 hours prior to use. The powders were combined in the required proportions to give 80% SrTiO3 – 20% La1/3NbO3, and mixed for 24 h with yttria-stabilised zirconia (YSZ) media (5 mm diameter) and propan-2-ol. The resulting slurry was dried and calcined in air at 1573 K for 12 hours. A single step calcination was sufficient to obtain single phase L2 material. The homogenous calcined powders were mixed with graphene or GO.Graphene was prepared by solvent exfoliation of GNP flakes (Grade M5, XG sciences) using a process previously reported.46,50 In brief, 5 g of GNP-M5 was placed a solution of 250 ml phenol and methanol in a ratio of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 0.05 g of cetyl-triethyl ammonium bromide (CTAB). The mixture was sonicated using an ultrasonic probe for 1 hour, followed by centrifugation at 2000 rpm to remove any unexfoliated material. GO was prepared by a modified Hummer's method,51,52 where graphite (grade 2369, Angstrom Sciences) was oxidised using KMnO4 in concentrated H2SO4. Characterisation details of the graphene and GO flakes in terms of X-ray diffraction, microstructure and thermogravimetric analysis are given in ESI, Fig. SI-1 to SI-4.†
:1 and 0.05 g of cetyl-triethyl ammonium bromide (CTAB). The mixture was sonicated using an ultrasonic probe for 1 hour, followed by centrifugation at 2000 rpm to remove any unexfoliated material. GO was prepared by a modified Hummer's method,51,52 where graphite (grade 2369, Angstrom Sciences) was oxidised using KMnO4 in concentrated H2SO4. Characterisation details of the graphene and GO flakes in terms of X-ray diffraction, microstructure and thermogravimetric analysis are given in ESI, Fig. SI-1 to SI-4.†
The L2 ceramic powder was mixed with either a dispersion of graphene or suspension of GO in concentrations of 0.1, 1.0 wt% and 1.0, 3.0 wt% respectively. The concentrations of GO were deliberately higher than those used for graphene in order to allow for the loss of carbon with functional groups attached to GO. The mixtures were ultra-sonicated for 30 minutes to achieve uniform dispersions, then dried on a hot plate at 350 K with mechanical stirring at 100 rpm (to minimize segregation of the two phases) and dry mixed in Super Mixer at 2000 rpm with 1:2 ratio of powder to yttria-stabilized zirconia balls. The resulting mixture was sieved through 300 mesh and sintered using SPS (HPD25, FCT System GmbH). Powders were packed into a graphite die set and pressed uniaxially at 30 MPa. The samples were sintered by SPS at 1473 K and 50 MPa for 5 minutes with a heating rate of ±100 K min−1.
The density of the sintered samples was determined using the Archimedes method. X-ray diffraction (XRD) was carried out using a Philips X'pert diffractometer with Cu source; the 2θ scan angle was 10° to 85° with step of size 0.030° and a dwell time of 10 s at each step. The XRD spectra were refined using the Rietveld program Topas.53
The thermal diffusivity of the samples was determined from 300 K to 1100 K using an in-house laser flash facility.54 The specific heat capacity was determined by differential scanning calorimetry (Netzsch STA 449 C)55 using a sapphire reference. An STA 449 C was used to determine the thermogravimetric behaviour of composites and dried graphene derivatives in different atmospheres.
The electrical conductivity (σ) and Seebeck coefficient (S) of the bulk samples were determined from 300 K to 1050 K in a helium atmosphere using a ZEM-III (Ulvac-Riko). Samples were typically 10 mm × 3 mm × 3 mm in size. In order to assess the loss of graphene or GO from the sintered samples, fracture surface microstructures were examined using a Quanta 650-FEGSEM equipped with an Oxford Instruments EDX system (energy dispersive spectroscopy). For grain size and compositional analysis of the composites, polished samples were examined using the same FEGSEM.
Raman spectroscopy (Renishaw InVia, 633 nm) was performed on the fracture surfaces of SPS sintered samples to assess the degree of graphene dispersion. The alignment of the graphene flakes in the ceramic matrix was determined by polarised Raman spectroscopy. The Raman spectra were recorded under a backscattering geometry and a vertical/vertical (VV) combination of incident and scattered polarization (the polarization for incident light was parallel to that of the scattered light). Samples were rotated on a 360° stage, and the change in intensity of the G-band peaks with the rotation angle was recorded.
Results and discussion
Samples directly prepared by SPS
The sample compositions, abbreviation codes and densities are listed in Table 1. After SPS processing all of the samples were dense (94–99% theoretical, Table 1), and uniformly light grey in colour, much lighter than the L2 samples prepared under reducing conditions;15 the lighter colour suggests that they were not reduced as effectively as the L2 sample sintered in Ar/5% H2.15| Composition | Sample code | Density, (% theoretical) |
|---|---|---|
| L2 = Sr0.8La0.067Ti0.8Nb0.2O3−δ | L2-SPS | 99.5% |
| L2 + 0.1 wt% G | L201G-SPS | 99.5% |
| L2 + 1.0 wt% G | L210G-SPS | 98.4%, |
| L2 + 1.0 wt% GO | L210GO-SPS | 98.1% |
| L2 + 3.0 wt% GO | L230GO-SPS | 94.8% |
The XRD spectra (Fig. 1) exhibited strong peaks of the cubic perovskite L2 material and a few minor peaks including TiO2, carbon (graphite) and possibly a phase related to Sr5LaTi3Nb7O30.56 The lattice parameters of the L2-SPS and the graphene-bearing samples (3.912 ± 0.001 Å) are comparable to those of the L2 samples sintered in air (3.913 Å).16 This suggests that the unit cell is not enlarged significantly by oxygen vacancies in the heavily reduced L2 samples sintered in Ar/5% H2 (ref. 15) in previous work. The main lattice parameter for the L2-GO samples (3.914 ± 0.001 Å) was only marginally larger than that of the pure L2 but without any systematic trend with G/GO concentration.
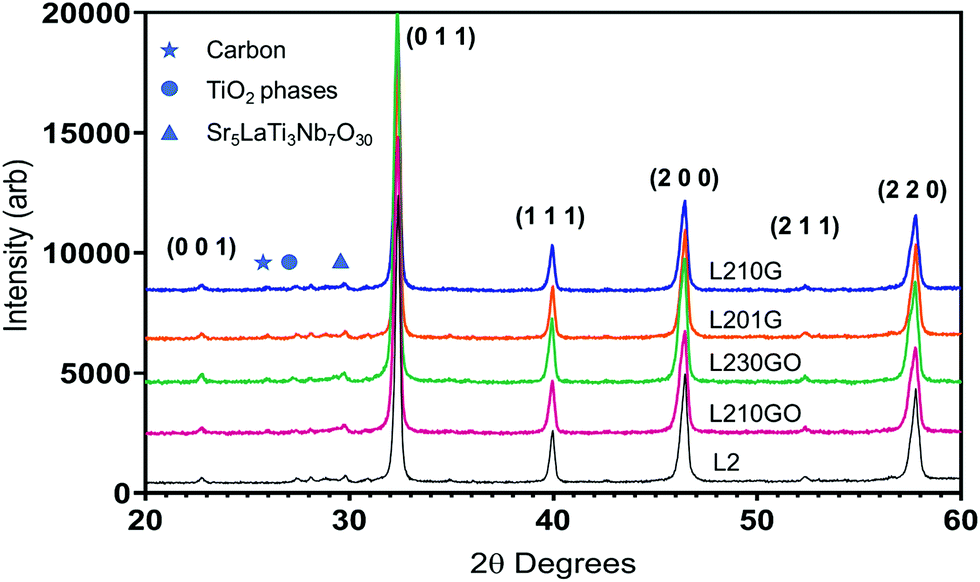 | ||
| Fig. 1 XRD spectra of SPS sintered samples. | ||
Fig. 2 shows micrographs of the fracture surfaces of L210GO-SPS samples (Fig. 2a–c) and L210G-SPS samples (Fig. 2d–f). The grain sizes of all of the samples sintered by SPS were approximately 5 μm, which is significantly smaller than for the L2 samples processed under Ar/H2 atmosphere at one bar total pressure, which was around 10 μm.15 The reduction in grain size for the SPS samples reflects the application of pressure and reduction in sintering time. In the only two other closely related investigations of SrTiO3 composites containing RGO, Feng et al.47 used pure, commercially prepared SrTiO3 powder and SPS processing; the grain sizes were in the range 14–18 μm for the ceramics and ∼1 μm for composites containing RGO. Okhay et al.48 used Nb doped SrTiO3 and sintered under one bar total pressure; ceramic grain sizes were 10–41 μm, and marginally smaller at 7–27 μm for the RGO containing composites.
 | ||
| Fig. 2 SEM micrographs of fractured surfaces of: L210GO-SPS (a–c); L210G-SPS (d–f). | ||
In the present investigation the GO flakes in the SPS processed samples were typically 10 μm in size (Fig. 2a–c); their distribution varied and there is some evidence of aggregation into smaller round or crumpled regions. The latter can produce rounded pores when the samples are fractured or polished (for example Fig. 2b and c) and flakes are pulled out. This is in strong contrast to the elongated pores generated by ‘fibre’ pull-out in the L210G-SPS samples (Fig. 2d–f); the elongated pores appear to be aligned in the direction normal to pressing direction. The distribution of graphene flakes in the matrix of the L210G-SPS samples was much more uniform than for the distribution of GO in the L210GO-SPS samples. Fig. 2e shows thin graphene flakes, which are partially transparent. The inset shows a graphene flake bridging the gap between the grains through a void. This should improve the electrical properties and provide reinforcement to improve the mechanical fracture strength as suggested by Porwal et al.57Fig. 2f shows graphene flakes wedged between the grains, with no gap around the flakes or loosely held flakes.
All of the composite samples showed semiconducting behaviour with electrical conductivity increasing with temperature until approximately 700 K; at higher temperatures it decreased (Fig. 3a). The improvement in the electrical conductivity by the addition of graphene or GO is highlighted in Fig. 3b. Irrespective of measurement temperature, the electrical conductivity increases with the loading of graphene/GO. Compared to pure L2-SPS, the increase with graphene/GO is typically three orders of magnitude, with the greatest increase achieved for samples containing 1 wt% graphene, followed by the samples containing 3 wt% GO. The highest electrical conductivity was obtained for the L210G-SPS samples, with σ = 3.0 × 103 S m−1 at 700 K, which was 3 times higher than for L201G-SPS. These values are comparable with the highest reported by Feng et al.47 for the pure SrTiO3–RGO composites, but lower than the conductivities reported by Okhay et al. for Nb-SrTiO3–RGO composites.48 It would appear that the graphene/reduced GO flakes located between the grains lead to increased mobility of the charge carriers and a change in the local defect concentrations. The limited evidence available47,48 supports this interpretation. Fig. 3b indicates that samples containing reduced GO flakes underperform compared to samples containing graphene flakes, primarily due to lower electrical conductivity of GO due to its highly functionalised nature.58 It should be noted that the sp3 bonding will be partially reduced to sp2 by the sintering process. There is a suggestion that the higher loadings of RGO required for equivalent levels of carbon addition (in the matrix) have a detrimental effect on the sintering and final density of the samples sintered both conventionally46 and by SPS (Table 1). Okhay et al.48 also found that increased levels of RGO significantly reduced the final sintered density. However, Table 1 shows that for equivalent loadings of G and GO, the densities are almost the same.
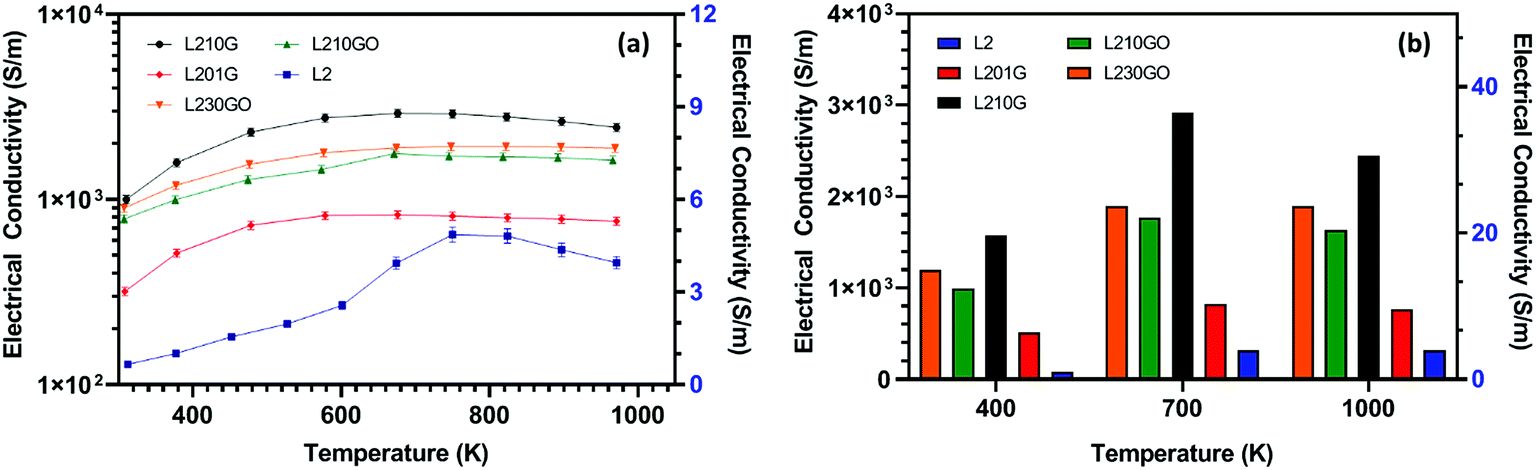 | ||
| Fig. 3 (a) Temperature dependence of electrical conductivity of composite samples sintered in SPS conditions (right hand conductivity axis is only for L2 samples); (b) bar chart demonstrating the difference in electrical conductivity of composite samples prepared by addition of graphene/GO compared to base L2 samples at 400 K, 700 K, 1000 K. | ||
The Seebeck coefficients of the samples sintered by SPS are presented in Fig. 4a; there is a sharp reduction in S values as the graphene/GO content in the matrix increased, consistent with the findings of Okhay et al.48 Data for the baseline reference (L2-SPS) was relatively constant around −380 μV K−1, with minimal change across the temperature range. All of the samples demonstrate a small uniform increase in the absolute Seebeck coefficient at temperatures above 700 K and different gradients at lower temperatures; the changes are related to the semiconductor-metallic conduction transition at 600–700 K. However, this also confirms that the Seebeck coefficients of the samples are affected by the presence of increasing amounts of carbon species. This is exemplified by the data for the L210G-SPS and L230GO-SPS samples; the former sample exhibits the highest electrical conductivity but has significantly higher absolute Seebeck coefficient than that for L230GO-SPS samples (by a factor of at least 2) which is counter-intuitive for thermoelectric materials. This could, potentially, be related to a change in polarity of the Seebeck coefficients in multilayer graphene.59 It can be speculated that the increased GO content increased the contribution from p-type behaviour of the multilayer graphene is working against the n-type matrix; however, the overall conductivity is not increased as the charge carriers are controlled primarily by matrix whose density is reduced.
 | ||
| Fig. 4 Temperature dependence of: (a) Seebeck coefficient; (b) thermal conductivity; and (c) ZT values for composite samples (right hand ZT scale only applies to L2 samples). | ||
The thermal conductivity of the composites (Fig. 4b) indicates an inverse relationship with temperature for all of the samples; the electronic contribution to the total thermal conductivity is minimal. For L2-SPS, the thermal conductivity decreases from 4.3 W m−1 K−1 at room temperature to 3.0 W m−1 K−1 at 1000 K. These values are comparable to data obtained for the L2 samples pressureless sintered in Ar/H2.15 Thus the reduction in grain size from ∼10 μm (pressureless sintering) to ∼5 μm for SPS processing does not have any significant impact on thermal conductivity. However, the incorporation of carbon species (graphene or GO) does have a marked effect, reducing thermal conductivity above 400 K (Fig. 4b). The lowest thermal conductivity of 2.64 W m−1 K−1 was obtained for L210G-SPS at 975 K, lower than the thermal conductivity reported by Popuri et al.17 for similar SrTiO3 containing A-site vacancies, by Feng et al.47 for SrTiO3–RGO composites and by Okhay et al.48 for Nb-SrTiO3–RGO composites. The presence of the graphene or RGO clearly reduces thermal conductivity, but there are no clear differences between the data in terms of the type or the amount of the additive for modest levels of carbon addition.
The thermoelectric figure of merit for the composite samples (Fig. 4c), shows a marked improvement upon addition of graphene/GO; this is directly linked to the increase in electrical conductivity. While the L2-SPS sample had a maximum ZT of 0.0002 at 950 K (similar to that for L2 samples sintered in air16), the composite samples demonstrated an increase of at least two orders of magnitude. The composite L210G-SPS exhibited the highest ZT of 0.065 at 950 K. This is only marginally lower than the value reported by Feng et al.47 for pure SrTiO3–RGO composites (0.09 at 750 K). However, these values are at least a factor of 5 lower than ZT for heavily reduced L2 samples prepared by conventional sintering in Ar/5% H2.15 It is clear that while SPS enables rapid processing, the low PO2 atmosphere within the SPS facility does not reduce the strontium titanate-based samples sufficiently for optimum thermoelectric performance. To overcome these limitations, batches of L2 powders were first reduced in Ar/5% H2 at 1600 K for 24 hours. The reduced L2 powder (denoted as L2R) was milled and mixed with 1.0 wt% graphene (the best performing sample from the first set of experiments). The L2R base powder samples and composites were then sintered by SPS.
Samples prepared from pre-reduced powders
The samples prepared from reduced L2 powders (i.e. L2R as explained above) were of high density, greater than 99% theoretical and were dark grey in colour (compared to those listed in Table 1 which were light grey), which suggests the presence of additional Ti3+ ions. X-ray diffraction spectra for the reduced samples are shown in Fig. 5. Topas refinement suggests a minor peak for graphite at 26.5 degrees for L2R10G-SPS, a residue from SPS processing in graphite. The lattice parameters for both types of samples are very similar to those for the L2 samples prepared in Ar/H2 (3.923 ± 0.001 Å)15 and significantly higher than those for the composites prepared with non-reduced L2 powders (3.913 ± 0.001 Å). The strong reducing atmosphere and the creation of oxygen vacancies led to significantly larger lattice parameters in the final SPS samples. A rutile-based minor phase (TixNb1−xO2−y) was detected in both L2R-SPS and L2R10G-SPS samples (∼1 mass%).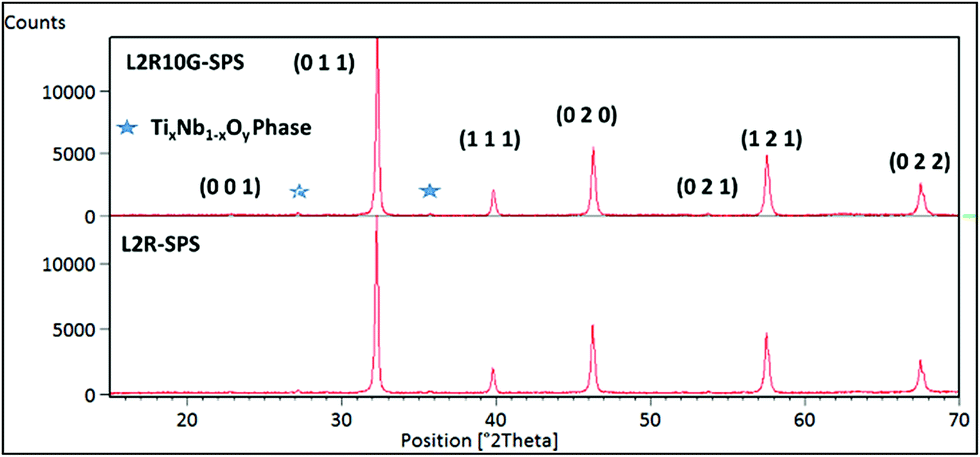 | ||
| Fig. 5 XRD spectra for samples prepared by SPS using reduced L2 powders: ceramic of L2R-SPS and composite based on L2R powder and 1.0 wt% graphene (L2R10G-SPS). | ||
SEM images of polished and fractured surfaces of L2R10G-SPS are shown in Fig. 6a–c. The L2R-SPS samples were dense, homogeneous with minimal voids and pores (Fig. SI-5 in ESI†). At high resolution there was evidence of the rutile-based (Ti1−xNbxOy) minor phase. The L2R10G-SPS samples (Fig. 6a–c) contained voids, characteristic of graphene pull-out, which were also seen in Fig. 2. The voids frequently exhibit ‘zig-zag’ morphology, related to the original geometry of the graphene flakes during sintering. In addition, the voids and flakes in the fracture surface images (Fig. 6b and c) provide good evidence of orientation as a result of the pressing process. The low magnification images suggest orientation of the graphene flakes normal to pressing direction. There is no macroscopic evidence of interactions between the graphene flakes and the matrix phase, and no indication of carbide phases being formed. The micrographs of the polished surfaces of L2R10G-SPS samples suggest the presence of core–shell structures (e.g. Fig. SI-5†), which have also been observed in reduced and conventionally sintered samples.60 In contrast, core–shell structures were not visible in SPS samples prepared from non-reduced L2 powder, indicating the effect of strong reduction of the L2 powder prior to SPS processing.
 | ||
| Fig. 6 SEM micrographs: polished surface of: (a) L2R10G-SPS; and fracture surfaces (b and c) of L2R10G-SPS. | ||
Since processing under reducing environments leads to oxygen non-stoichiometry of the base L2 material Sr0.8La0.067Ti0.8Nb0.2O3−δ, thermogravimetric (TGA) analysis was undertaken to determine the value of δ. TGA plots for L2-SPS, L2R-SPS and L2R10G-SPS samples heated under oxidising conditions are presented in Fig. SI-6 in the ESI.† The heating curve for the L2R-SPS sample suggests absorption of oxygen, with the onset close to 800 K. The amount of oxygen uptake in this system is directly related to oxygen vacancies denoted by δ. For L2R-SPS, the δ value was calculated to be 0.055, which is significantly lower than for the L2 sample sintered in Ar/5% H2 for 24 hours (δ = 0.084).15 This difference is in part due to lower temperature of 1600 K used for reduction of the L2 powders, but also the much less reducing atmosphere provided by SPS. This interpretation is supported by the TGA curve for the L2-SPS sample (Fig. SI-6†); oxygen absorption with temperature is negligible, suggesting minimal oxygen vacancies were created by SPS processing alone. The more complex TGA curve for L2R10G-SPS sample follows the same trend as for other samples up to 850 K, but then shows weight loss with increasing temperature as the graphene flakes in the matrix started to oxidise. The weight loss of approximately 1 mass% corresponds to the original graphene addition in the matrix. It is anticipated that the graphene helps to remove oxygen from the perovskite during heat treatment and this may explain the compensation of oxygen uptake in the control sample L2R-SPS.
To complement the SEM images of the fracture surfaces of the samples (Fig. 6), Raman spectroscopy was used to confirm the presence of graphene flakes after sintering. The Raman map (Fig. SI-7†) for L2R10G-SPS confirms that the graphene was well distributed with some degree of aggregation, but more importantly that graphene had survived the SPS processing. Raman mapping was also used to determine the orientation of the graphene flakes in the matrix. Under VV polarisation configuration,61 the intensity from the graphene flake is dependent on cos4ϕ, where ϕ is the angle between the monitored graphene flake and axis of laser polarization. The decrease in intensity of the Raman G-band with respect to ϕ is an indication of spatial orientation.61 The intensity of the G-band at approximately 1580 cm−1 for different ϕ angles has been normalised relative to the intensity at 0° and is presented for two sites in Fig. 7. The absolute intensity at 90° for site-1 was non-zero and may be attributed to some contribution from background-misoriented edges,62 but I90° ≪ I0°. A line for cos4ϕ is plotted in the figure and shows the expected trend in intensity variation with ϕ for a perfectly oriented flake in a matrix, as approximated in site 1. The signal from site 2 reduced initially but then increased beyond 50 degrees. This suggests that the graphene flake may be located between matrix grains and be partly under the surface. However, it is possible that flake at site 2 may not be oriented in the same way as at site 1, and might be folded or crumpled, or may contain multiple flakes.
 | ||
| Fig. 7 (a) SEM image of graphene flake on fracture surface at site 1; (b) SEM image of graphene flake in elongated crack on fracture surface at site 2; (c) Plots for L2R10G-SPS of the normalized intensity of Raman G band at 1580 cm−1 using VV polarization. The dotted line represents IG ∝ cos4ϕ. | ||
The SEM images in Fig. 6 and the Raman data presented in Fig. 7 confirm that the graphene flakes are partially oriented relative to the pressing direction. Consequently, the thermoelectric properties need to be determined both along the plane and across the plane of graphene flakes relative to the pressing direction, to determine whether there is anisotropy (Fig. 8). A pilot study of samples prepared from powders which had not been pre-reacted (L2-SPS type powders) indicated the thermoelectric properties were very modest; for this reason, a detailed investigation with respect to pressing direction was not undertaken.
 | ||
| Fig. 8 Schematic diagram showing the cross-plane and in-plane directions for measurement of thermoelectric properties; cross-plane is parallel to the pressing direction; in-plane is perpendicular to the pressing direction. | ||
The electrical conductivity of L2R-SPS and L2R10G-SPS measured in orthogonal directions is shown in Fig. 9a. The L2R-SPS samples show similar conductivity when measured in different orthogonal directions, confirming that the properties in these samples are isotropic. The electrical conductivity of L2R-SPS is significantly higher than for the L2-SPS samples (Fig. 3a). Comparison of data for the L2R-SPS samples with that of the L2R10G-SPS samples for the in-plane direction shows an increase in electrical conductivity due to the presence of graphene by typically a factor of two. At temperatures below 650 K the electrical conductivity for L2R10G-SPS in the cross-plane direction is comparable with that for the L2R-SPS samples, highlighting the anisotropy between the two directions for L2R10G-SPS (Fig. 9a). Thus, for the in-plane direction a significant fraction of graphene flakes are aligned in the same direction as the current flow, facilitating current paths for the charge carriers. With higher carrier mobility in the graphene than in the ceramic, the overall electrical conductivity is significantly increased in an in-plane direction.
 | ||
| Fig. 9 Temperature dependence of: (a) electrical conductivity; and (b) Seebeck coefficients of L2R-SPS and L2R10G-SPS measured in orthogonal directions. | ||
With increasing temperature above 650 K the difference in conductivity for the two orthogonal directions for the graphene-containing samples decreases steadily (Fig. 9a). This may reflect a change in conduction mechanism with temperature in the cross-plane direction; potentially at lower temperatures the current passes through bulk ceramic material alone, and bypasses the graphene flakes that are oriented perpendicular to the current direction. At higher temperatures, electrical conductivity in the cross-plane direction increases above that for the L2R-SPS samples, perhaps because of an increasing contribution from the graphene to conduction through thermally activated carriers and processes that are more effective at higher temperatures. The highest electrical conductivities here for L2R10G in-plane are comparable with our earlier data for La-SrTiO3–G composites,46 and marginally lower, at the lowest temperatures, than the conductivities reported for the Nb-rich SrTiO3–RGO composites of Okhay et al.48
In contrast to the electrical conductivity data, the Seebeck coefficients of the composite samples L2R10G-SPS (Fig. 9b) do not exhibit any significant anisotropy with respect to the measurement direction. However, as anticipated, the Seebeck coefficients of the composite samples are approximately 15–20% lower than those for the L2R-SPS samples at all temperatures, with the usual inverse relationship with electrical conductivity. The fact that the Seebeck coefficients of the L2R10G-SPS samples have been reduced by the presence of the graphene (here and in earlier studies47,48) and are also isotropic may be related, in part, to the effective p-type behaviour of graphene flakes as a result of doping;59,63 the presence of both electrons and holes as charge carriers will lower the overall Seebeck coefficients. In samples containing true monolayers of graphene, ballistic carrier transport (low effective mass, m*) and low carrier concentrations in the graphene flakes would also encourage very low absolute Seebeck coefficients.
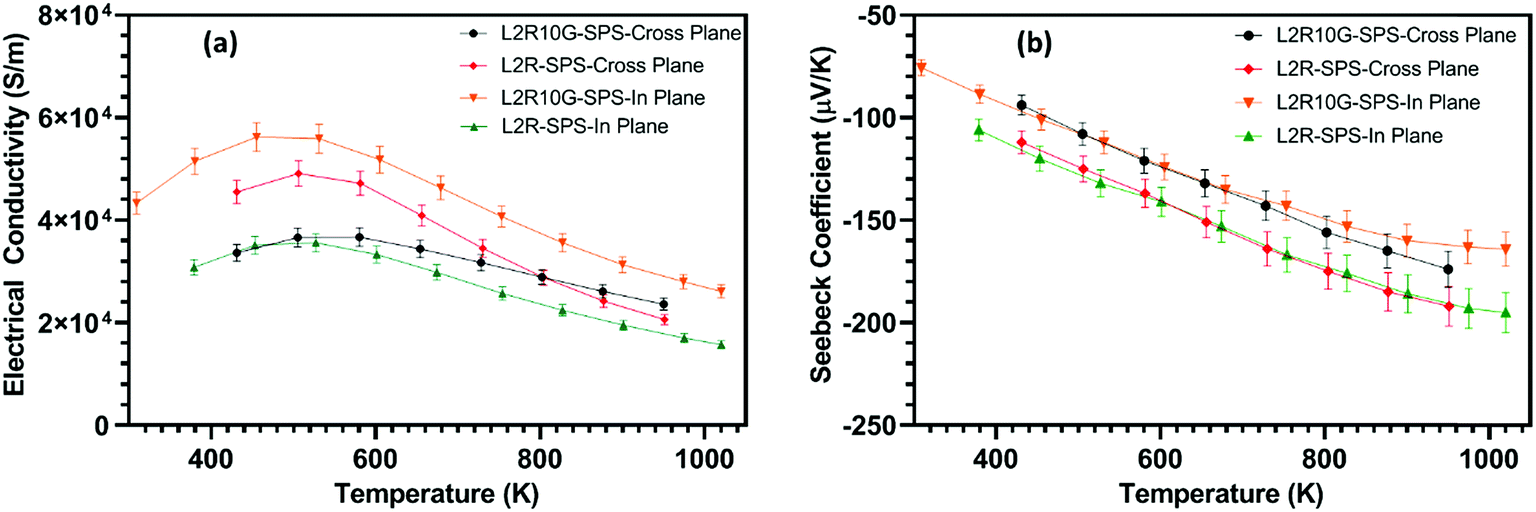
The standard approach for measuring thermal conductivity of ceramic samples is along the pressing direction. This implies that the composite L2R10G-SPS samples will normally be measured in a cross-plane direction with respect to the orientation of the graphene flakes (Fig. 8a). In order to measure thermal diffusivity (and thereby determine thermal conductivity) disc shaped samples were cut for each of the relevant directions from the original SPS samples. The resulting thermal conductivity data for the two directions are presented in Fig. 10a. The cross-plane thermal conductivity for L2R10G-SPS was consistently 15–20% lower than that for the L2R-SPS samples as the heat travelled across the plane of the graphene flakes. The lowest value of thermal conductivity (2.7 W m−1 K−1 at 1050 K) was obtained for the L2R10G-SPS-cross-plane sample. This value is lower than the minimum value of 3.0 W m−1 K−1 recorded for both La-SrTiO3–G composites (at 1000 K),46 and Nb-rich SrTiO3–RGO composites at 1200 K.48 In contrast, the thermal conductivity of the L2R10G-SPS-in plane samples was almost 20% higher than for the L2R-SPS samples (Fig. 10a). While, the thermal conductivity of the L2R-SPS samples was isotropic, it decreased from 4.75 W m−1 K−1 at 400 K to 3.1 W m−1 K−1 at 1050 K. Hence, the thermal conductivity of the composite L2R10G samples showed almost 35% anisotropy; the values were higher when measured along the direction in which the graphene flakes were predominantly oriented in the ceramic matrix. This is also consistent with the anisotropic behaviour of our composite samples prepared from GNP flakes (instead of graphene) as shown in Fig. SI-8;† the thermal diffusivity of the GNP composites, measured in a cross-plane direction, is lower by a factor of 15–20 compared to diffusivity measured in an in-plane direction (note different scales used for thermal diffusivity for each set of data in Fig. SI-8†). Thus, the two dimensional nature of the graphene and GNP flakes gives rise to distinct anisotropy in both the thermal and electrical transport properties of the composites. It is noted that the thermal conductivity of graphene can be tuned by one order of magnitude by defect engineering, whilst having much less impact on electrical conductivity.64
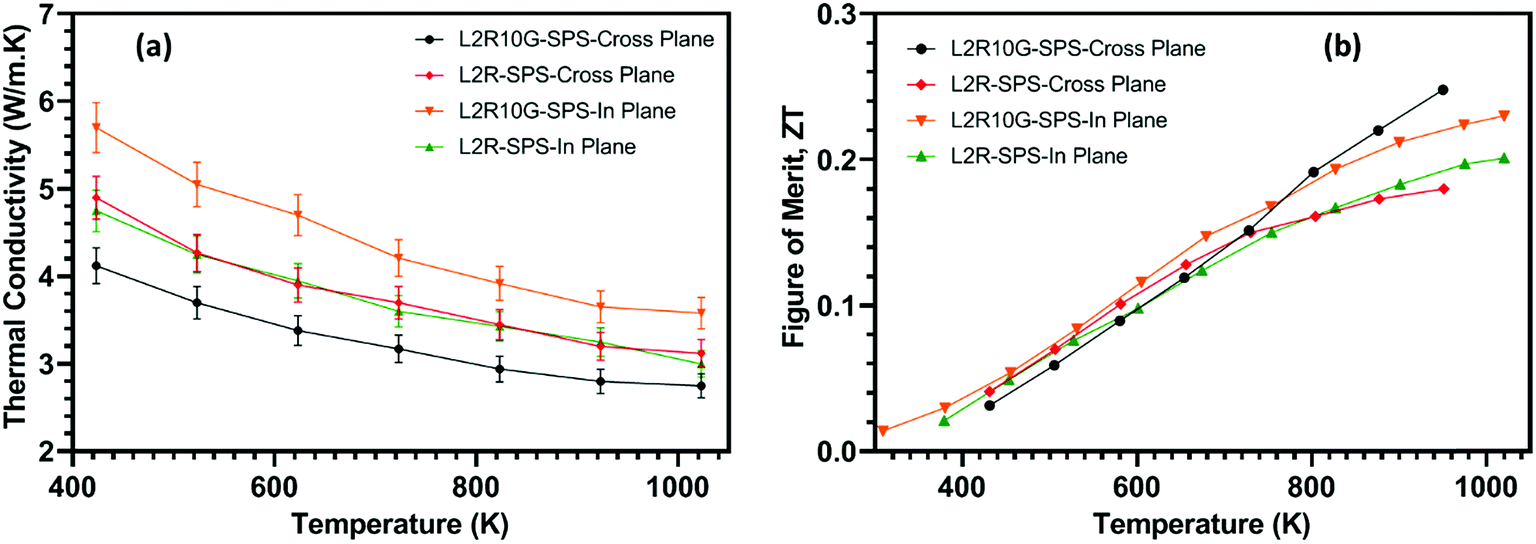 | ||
| Fig. 10 Temperature dependence of: (a) thermal conductivity of L2R-SPS and L2R10G-SPS measured in cross plane and in-plane directions; (b) direction-dependent thermoelectric figure of merit data (ZT) for L2R-SPS and L2R10G-SPS samples. The uncertainty bars (±10%) have been omitted for clarity. | ||
The resulting thermoelectric figure of merit (ZT) of the composite samples L2R10G-SPS and the L2R-SPS samples were determined for the cross-plane and in-plane directions and are presented in Fig. 10b. For the L2R-SPS samples, the ZT values did not show any anisotropy within the measurement uncertainty. The maximum ZT of ∼0.2 for the L2R-SPS samples was obtained at 1050 K. For the composite L2R10G-SPS samples measured in the in-plane direction, the higher electrical conductivity (Fig. 9a) was offset by the higher thermal conductivity of the samples (Fig. 10a) compared to that of the L2R-SPS samples. The maximum ZT for L2R10G-SPS-in-plane sample was 0.225 at 1000 K, which is a ∼12% improvement compared to the L2R-SPS samples. Above 700 K the difference in ZT between the graphene-containing and graphene-free samples becomes more obvious and values for the L2R10G-SPS samples measured in the cross-plane direction steadily increase above those for all other samples. The consistently lower thermal conductivity (Fig. 10a) and enhanced electrical conductivity above 700 K (Fig. 9a) leads to the highest value of ∼0.25 at 1000 K for L2R10G-SPS samples in the cross-plane direction (Fig. 10b). This 20% increase in the ZT values compared to the graphene-free samples demonstrates the improvement in thermoelectric properties afforded by the inclusion of graphene in the ceramic matrix. To aid comparisons between the thermoelectric data we have included a tabulated summary of thermal conductivity, Seebeck coefficients, electrical conductivity and figure of merit as a function of temperature in Table SI-1 in ESI.† Data from other related investigations are also included in the table.
It is interesting to note that the maximum ZT of 0.25 at 1000 K obtained here for the Sr0.8La0.067Ti0.8Nb0.2O3−δ samples prepared with 1 wt% graphene and processed by SPS is virtually the same as for the pressureless sintered La-SrTiO3–G composites with the same graphene loading,46 and also for the Nb-SrTiO3–RGO composites at 1000 K.48 Our SPS processed samples showed the same temperature dependence of ZT, increasing with measurement temperature (Fig. 10b) as the Nb-SrTiO3–RGO composites processed under 1 bar total pressure; our earlier work on La-SrTiO3–G composites exhibited higher ZT values at lower temperatures and thus a much wider effective ‘thermal window’ for the composites.46
To provide further insight into the transport parameters the modified Heikes formula60,65 (eqn (1)) was used to calculate carrier concentration (n) for the different samples. With this data the usual conduction relationship (eqn (2)) was utilised to calculate carrier mobility (μ).
 | (1) |
| σ = neμ | (2) |
 | (3) |
The resulting data for carrier concentration, carrier mobility and carrier effective mass are presented in Table 2.
| Calculated carrier concentration (cm−3) | Carrier mobility μ (cm2 V−1 s−1) | Carrier effective mass, m* | |
|---|---|---|---|
| L2R-SPS | 3.6 × 1021 | 0.55 | 6.4m0 |
| L2R10G-SPS-in plane | 4.3 × 1021 | 0.77 | 6.0m0 |
| L2R10G-SPS-cross plane | 4.4 × 1021 | 0.46 | 5.9m0 |
It is evident from Table 2 that carrier concentration is higher in the samples containing graphene, and that improved electrical performance in samples containing graphene flakes aligned along the direction of measurement arises, in part, from the higher carrier mobility. In contrast, for the samples where properties were measured in a direction normal to the orientation of graphene flakes the carrier mobility was significantly lower than that for the control samples (L2R-SPS). These results support the concepts that the presence of the graphene flakes will increase the concentration of carriers, and will aid carrier mobility along the plane of graphene flakes, because of the imposed anisotropy and the high carrier mobility of graphene itself.
The present work has shown marked differences between graphene and RGO, with the former being superior for La–Nb-SrTiO3 composites. It has also confirmed that there is significant anisotropy in the thermoelectric transport properties in composites containing two dimensional flakes of graphene. In terms of future studies, there is scope to adjust the composition of the A-site species to maximise cation vacancies,15,48 but also set against the background that for the composites, the graphene or RGO is almost certainly affecting the oxygen vacancy content of the SrTiO3-grains in contact with the graphene.47 To achieve the very highest performance in SrTiO3–graphene/RGO composites almost certainly requires the grain boundary resistance to be minimised.66
Conclusions
This work presents important new information on the role and effects of carbon additions on a leading n-type thermoelectric oxide and the enhancement of transport properties. High density, high quality Sr0.8La0.067Ti0.8Nb0.2O3−δ ceramics and composites including ≤3 wt% graphene or graphene oxide were synthesised by SPS processing; grain sizes were typically 5 μm. Composites prepared from mixed oxide powders that were not subjected to a reducing atmosphere heat treatment before SPS exhibited three orders of magnitude increase in electrical conductivity over the ceramic samples, a reduction in thermal conductivity to 2.64 W m−1 K−1 at 975 K, but very modest ZT values, less than 0.1. In all cases, the incorporation of graphene yielded superior thermoelectric performance to the use of graphene oxide. However, the use of SPS processing alone for 5 minutes at 1473 K was insufficient to reduce the strontium titanate grains adequately for optimum thermoelectric performance.Samples prepared from powders which were first reduced in Ar/5% H2 before SPS processing were of very high density (greater than 99% theoretical), dark grey in colour, with evidence of core–shell structures. The oxygen vacancy deficiency, denoted by δ, was determined to be 0.055. Raman mapping confirmed that the distribution of graphene grains in the composites was relatively uniform, with partial orientation compared to the pressing direction. Composite samples including 1 wt% graphene exhibited strong anisotropy in transport properties; electrical conductivity was significantly higher for the in-plane direction, although they were closer at the highest temperatures. Thermal conductivity was lowest for the cross-plane direction (2.7 W m−1 K−1 at 1000 K). The consistently lower thermal conductivity and enhanced electrical conductivity above 700 K leads to the highest ZT value of ∼0.25 at 1000 K for composite samples in the cross-plane direction. This 20% increase in the ZT values compared to the graphene-free samples demonstrates the benefit gained by inclusion of graphene in the ceramic matrix. The control of thermoelectric transport properties by additions carbon species, and the resulting anisotropy in properties could provide guidance in the optimum routes to synthesise future target materials.
Conflicts of interest
There are no conflicts of interest.Acknowledgements
The authors are grateful to the EPSRC for the provision of funding for this work (EP/H043462, EP/I036230/1, EP/L014068/1, EP/L017695/1 acknowledged by RF). All research data supporting this publication are directly available within the publication. D. S. acknowledges support through the provision of a President's Scholarship from the University of Manchester. We are grateful for the constructive comments of Dr Jianyun Cao on earlier versions of the manuscript.References
- G. J. Snyder and E. S. Toberer, Nat. Mater., 2008, 7, 105 CrossRef CAS PubMed.
- K. Biswas, J. He, I. D. Blum, C.-I. Wu, T. P. Hogan, D. N. Seidman, V. P. Dravid and M. G. Kanatzidis, Nature, 2012, 489, 414 CrossRef CAS PubMed.
- X. Zhang and L.-D. Zhao, J. Materiomics, 2015, 1, 92–105 CrossRef.
- K. Koumoto, Y. F. Wang, R. Z. Zhang, A. Kosuga and R. Funahashi, Annu. Rev. Mater. Res., 2010, 40, 363–394 CrossRef CAS.
- T. M. Tritt, Annu. Rev. Mater. Res., 2011, 41, 433–448 CrossRef CAS.
- J. He, Y. F. Liu and R. Funahashi, J. Mater. Res., 2011, 26, 1762–1772 CrossRef CAS.
- J. W. Fergus, J. Eur. Ceram. Soc., 2012, 32, 525–540 CrossRef CAS.
- W. Koshibae and S. Maekawa, Phys. Rev. Lett., 2001, 87, 236603 CrossRef CAS PubMed.
- I. Terasaki, Y. Sasago and K. Uchinokura, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 12685–12687 CrossRef.
- H. Ohta, K. Sugiura and K. Koumoto, Inorg. Chem., 2008, 47, 8429–8436 CrossRef CAS PubMed.
- L. Bocher, M. H. Aguirre, D. Logvinovich, A. Shkabko, R. Robert, M. Trottmann and A. Weidenkaff, Inorg. Chem., 2008, 47, 8077–8085 CrossRef CAS PubMed.
- M. Molinari, D. A. Tompsett, S. C. Parker, F. Azough and R. Freer, J. Mater. Chem. A, 2014, 2, 14109–14117 RSC.
- G. K. Ren, J. L. Lan, C. C. Zeng, Y. C. Liu, B. Zhan, S. Butt, Y. H. Lin and C. W. Nan, JOM, 2014, 67, 211–221 CrossRef.
- M. Ohtaki, J. Ceram. Soc. Jpn., 2011, 119, 770–775 CrossRef CAS.
- D. Srivastava, C. Norman, F. Azough, M. C. Schäfer, E. Guilmeau, D. Kepaptsoglou, Q. M. Ramasse, G. Nicotra and R. Freer, Phys. Chem. Chem. Phys., 2016, 18, 26475–26486 RSC.
- D. Srivastava, F. Azough, M. Molinari, S. C. Parker and R. Freer, J. Electron. Mater., 2015, 44, 1803–1808 CrossRef CAS.
- S. R. Popuri, A. J. M. Scott, R. A. Downie, M. A. Hall, E. Suard, R. Decourt, M. Pollet and J. W. G. Bos, RSC Adv., 2014, 4, 33720–33723 RSC.
- A. V. Kovalevsky, A. A. Yaremchenko, S. Populoh, A. Weidenkaff and J. R. Frade, J. Phys. Chem. C, 2014, 118, 4596–4606 CrossRef CAS.
- M. C. Verbraeken, T. Ramos, K. Agersted, Q. Ma, C. D. Savaniu, B. R. Sudireddy, J. T. S. Irvine, P. Holtappels and F. Tietz, RSC Adv., 2015, 5, 1168–1180 RSC.
- A. V. Kovalevsky, A. A. Yaremchenko, S. Populoh, P. Thiel, D. P. Fagg, A. Weidenkaff and J. R. Frade, Phys. Chem. Chem. Phys., 2014, 16, 26946–26954 RSC.
- H. Muta, K. Kurosaki and S. Yamanaka, J. Alloys Compd., 2003, 350, 292–295 CrossRef CAS.
- S. Ohta, T. Nomura, H. Ohta and K. Koumoto, J. Appl. Phys., 2005, 97, 34106 CrossRef.
- A. V. Kovalevsky, S. Populoh, S. G. Patricio, P. Thiel, M. C. Ferro, D. P. Fagg, J. R. Frade and A. Weidenkaff, J. Phys. Chem. C, 2015, 119, 4466–4478 CrossRef CAS.
- A. A. Yaremchenko, S. Populoh, S. G. Patricio, J. Macias, P. Thiel, D. P. Fagg, A. Weidenkaff, J. R. Frade and A. V. Kovalevsky, Chem. Mater., 2015, 27, 4995–5006 CrossRef CAS.
- C. Chen, T. S. Zhang, R. Donelson, T. T. Tan and S. Li, J. Alloys Compd., 2015, 629, 49–54 CrossRef CAS.
- Z. L. Lu, H. R. Zhang, W. Lei, D. C. Sinclair and I. M. Reaney, Chem. Mater., 2016, 28, 925–935 CrossRef CAS.
- S. Ohta, H. Ohta and K. Koumoto, J. Ceram. Soc. Jpn., 2006, 114, 102–105 CrossRef CAS.
- D. Srivastava, C. Norman, F. Azough, M. C. Schäfer, E. Guilmeau and R. Freer, J. Alloys Compd., 2018, 731, 723–730 CrossRef CAS.
- D. Ekren, F. Azough, A. Gholinia, S. J. Day, D. Hernandez-Maldonado, D. M. Kepaptsoglou, Q. M. Ramasse and R. Freer, J. Mater. Chem. A, 2018, 6, 24928–24939 RSC.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- S. Bai and X. P. Shen, RSC Adv., 2012, 2, 64–98 RSC.
- Y. C. Fan, L. J. Wang, J. L. Li, J. Q. Li, S. K. Sun, F. Chen, L. D. Chen and W. Jiang, Carbon, 2010, 48, 1743–1749 CrossRef CAS.
- W. S. Bao, S. Y. Liu and X. L. Lei, J. Phys.: Condens. Matter, 2010, 22, 315502 CrossRef CAS PubMed.
- Y. M. Zuev, W. Chang and P. Kim, Phys. Rev. Lett., 2009, 102, 096807 CrossRef PubMed.
- N. S. Sankeshwar, S. S. Kubakaddi and B. G. Mulimani, in Advances in Graphene Science, InTech, Rijeka, 2013, ch. 9, pp. 219–271 Search PubMed.
- K. Esfarjani, M. Zebarjadi and Y. Kawazoe, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 085406 CrossRef.
- M. S. Fuhrer, C. N. Lau and A. H. MacDonald, MRS Bull., 2010, 35, 289–295 CrossRef CAS.
- W. Y. Zhao, S. F. Fan, N. Xiao, D. Y. Liu, Y. Y. Tay, C. Yu, D. H. Sim, H. H. Hng, Q. C. Zhang, F. Boey, J. Ma, X. B. Zhao, H. Zhang and Q. Y. Yan, Energy Environ. Sci., 2012, 5, 5364–5369 RSC.
- A. Dey, O. P. Bajpai, A. K. Sikder, S. Chattopadhyay and M. A. S. Khan, Renewable Sustainable Energy Rev., 2016, 53, 653–671 CrossRef CAS.
- B. B. Liang, Z. J. Song, M. H. Wang, L. J. Wang and W. Jiang, J. Nanomater., 2013, 210767 Search PubMed.
- B. Feng, J. Xie, G. S. Cao, T. J. Zhu and X. B. Zhao, J. Mater. Chem. A, 2013, 1, 13111–13119 RSC.
- H. J. Chen, C. Y. Yang, H. L. Liu, G. H. Zhang, D. Y. Wan and F. Q. Huang, CrystEngComm, 2013, 15, 6648–6651 RSC.
- D. S. Chen, Y. Zhao, Y. N. Chen, B. A. Wang, H. Y. Chen, J. Zhou and Z. Q. Liang, ACS Appl. Mater. Interfaces, 2015, 7, 3224–3230 CrossRef CAS PubMed.
- C. Li, X. Y. Qin, Y. Y. Li, D. Li, J. Zhang, H. F. Guo, H. X. Xin and C. J. Song, J. Alloys Compd., 2016, 661, 389–395 CrossRef CAS.
- W. H. Nam, Y. S. Lim, W. Kim, H. K. Seo, K. S. Dae, S. Lee, W.-S. Seo and J. Y. Lee, Nanoscale, 2017, 9, 7830–7838 RSC.
- Y. Lin, C. Norman, D. Srivastava, F. Azough, L. Wang, M. Robbins, K. Simpson, R. Freer and I. A. Kinloch, ACS Appl. Mater. Interfaces, 2015, 7, 15898–15908 CrossRef CAS PubMed.
- X. Feng, Y. Fan, N. Nomura, K. Kikuchi, L. Wang, W. Jiang and A. Kawasaki, Carbon, 2017, 112, 169–176 CrossRef CAS.
- O. Okhay, S. Zlotnik, W. Xie, K. Orlinski, M. J. Hortiguela Gallo, G. Otero-Irurueta, A. J. S. Fernandes, D. A. Pawlak, A. Weidenkaff and A. Tkach, Carbon, 2019, 143, 215–222 CrossRef CAS.
- C. Wu, J. Li, Y. Fan, J. Xing, H. Gu, Z. Zhou, X. Lu, Q. Zhang, L. Wang and W. Jiang, J. Alloys Compd., 2019, 786, 884–893 CrossRef CAS.
- Y. Lin, J. Jin, O. Kusmartsevab and M. Song, J. Phys. Chem. C, 2013, 117, 17237–17244 CrossRef CAS.
- M. Hirata, T. Gotou, S. Horiuchi, M. Fujiwara and M. Ohba, Carbon, 2004, 42, 2929–2937 CAS.
- J. P. Rourke, P. A. Pandey, J. J. Moore, M. Bates, I. A. Kinloch, R. J. Young and N. R. Wilson, Angew. Chem., 2011, 123, 3231–3235 CrossRef.
- H. M. Rietveld, Acta Crystallogr., 1967, 22, 151–158 CrossRef CAS.
- R. Taylor, J. Phys. E: Sci. Instrum., 1980, 13, 1193–1199 CrossRef CAS.
- M. J. O'Neill, Anal. Chem., 1966, 38, 1331–1336 CrossRef.
- X. L. Zhu, X. Q. Liu and X. M. Chen, J. Am. Ceram. Soc., 2011, 94, 1829–1836 CrossRef CAS.
- H. Porwal, P. Tatarko, S. Grasso, J. Khaliq, I. Dlouhy and M. J. Reece, Carbon, 2013, 64, 359–369 CrossRef CAS.
- C. Punckt, F. Muckel, S. Wolff, I. A. Aksay, C. A. Chavarin, G. Bacher and W. Mertin, Appl. Phys. Lett., 2013, 102, 023114 CrossRef.
- J. H. Warner, F. Schäffel, A. Bachmatiuk and M. H. Rümmeli, in Graphene, ed. J. H. Warner, F. Schäffel, A. Bachmatiuk and M. H. Rümmeli, Elsevier, 2013, pp. 61–127 Search PubMed.
- D. Srivastava, C. Norman, F. Azough, M. C. Schäfer, E. Guilmeau, D. Kepaptsoglou, Q. M. Ramasse, G. Nicotra and R. Freer, Phys. Chem. Chem. Phys., 2016, 18, 26475–26486 RSC.
- Z. Li, R. J. Young and I. A. Kinloch, ACS Appl. Mater. Interfaces, 2013, 5, 456–463 CrossRef CAS PubMed.
- A. C. Ferrari and D. M. Basko, Nat. Nanotechnol., 2013, 8, 235–246 CrossRef CAS PubMed.
- T. A. Amollo, G. T. Mola, M. S. K. Kirui and V. O. Nyamori, Crit. Rev. Solid State Mater. Sci., 2018, 43, 133–157 CrossRef CAS.
- J. Oh, H. Yoo, J. Choi, J. Y. Kim, D. S. Lee, M. J. Kim, J.-C. Lee, W. N. Kim, J. C. Grossman, J. H. Park, S.-S. Lee, H. Kim and J. G. Son, Nano Energy, 2017, 35, 26–35 CrossRef CAS.
- H. Taguchi, M. Sonoda and M. Nagao, J. Solid State Chem., 1998, 137, 82–86 CrossRef CAS.
- R. Z. Zhang, C. L. Wang, J. C. Li and K. Koumoto, J. Am. Ceram. Soc., 2010, 93, 1677–1681 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta02883h |
| This journal is © The Royal Society of Chemistry 2019 |