
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDynamic covalent urea bonds and their potential for development of self-healing polymer materials†
Zhanhua
Wang
 a,
Satesh
Gangarapu
b,
Jorge
Escorihuela
c,
Guoxia
Fei
a,
Han
Zuilhof
*bd and
Hesheng
Xia
*a
a,
Satesh
Gangarapu
b,
Jorge
Escorihuela
c,
Guoxia
Fei
a,
Han
Zuilhof
*bd and
Hesheng
Xia
*a
aState Key Laboratory of Polymer Materials Engineering, Polymer Research Institute, Sichuan University, Chengdu 610065, P. R. China. E-mail: xiahs@scu.edu.cn
bLaboratory of Organic Chemistry, Wageningen University, Stippeneng 4, 6708 WE Wageningen, The Netherlands. E-mail: Han.Zuilhof@wur.nl
cDepartment of Applied Thermodynamics, Universitat Politècnica de València, Camino de Vera s/n, 46020 Valencia, Spain
dDepartment of Chemical and Materials Engineering, King Abdulaziz University, Jeddah, Saudi Arabia
First published on 18th June 2019
Abstract
Self-healing polymer materials have drawn rapidly increasing interest over the last decade, and have been studied and used in an ever-increasing range of applications. Herein, we successfully make the covalent urea bond – a pinnacle of stability due to strong resonance effects – dynamic in nature through mediation of zinc salts. The dynamic covalent character of urea in the presence of zinc ions is confirmed through dissociation reaction experiments and quantum chemical calculations of small-molecule model urea compounds. In line with our experiments, the modelling results suggest that the presence of zinc ions speeds up the reaction of urea dissociation by two orders of magnitude via the formation of O-bound Zn complexes. Based on such dynamic covalent urea bonds, we then develop a novel class of self-healing polymer materials with excellent healing efficiencies. Different kinds of self-healing and reprocessable polyurea materials were prepared, with polymer properties that can be easily tuned by varying the degree of crosslinking and the molecular weight of the siloxane precursor. Since different kinds of self-healing polyurea materials could easily be prepared due to the commercial availability of a very wide range of amine and isocyanate monomers, this introduction of self-healing properties is expected to have significant potential in a range of applications, such as coatings, paints, and 3D printing. In addition, this introduces polyureas and other urea-containing polymers as a class of highly stable, yet easily reprocessable plastics, which is highly relevant given the globally desired more sustainable use of plastics.
Introduction
The attractiveness of thermoset polymer materials lies in their high stability. However, a resulting challenge therefore typically is that they cannot be reprocessed or repaired upon mechanical damage, which eventually causes environmental pollution, and the lack of repair options limits their lifetime. Endowing polymer materials with self-healing properties can solve this problem and meet sustainable development criteria. Up to now, repairable polymer systems have been realized with reversible1 or irreversible2,3 covalent bonds, supramolecular interactions,4 chemo-mechanical features5–7 and/or shape memory.8–11 In addition, recent studies have also shown that van der Waals forces12 as well as microphase separation13 in thermoplastic polymers can also facilitate self-healing without elaborate chemical modifications. Among these methods, the use of dynamic covalent bonds14–18 to prepare self-healing polymer materials is widely studied, since no external event needs to take place, and the self-healing process can be repeated several times.A lot of dynamic covalent bonds or structures have been successfully utilized to prepare self-healing polymer materials, including alkoxyamine–aldehyde bonds,19 Diels–Alder reaction products,20,21 thiol and selenol radical species amenable to radical association–dissociation,22–27 bond-exchanging vitrimers,28 and so on.29 The most widely used method to prepare self-healing polymer materials based on dynamic covalent chemistry30,31 is the condensation-polymerization of a chain extender, cross-linker and small molecules containing dynamic covalent bonds. However, the introduction of new dynamic bonds increases the complexity of chemical synthesis and may also alter the original polymer properties in undesirable ways. In contrast, if one is able to impart dynamic characteristics to conventional chemical bonds that are used for chain extending and cross-linking, and achieve self-healing functions with those very moieties, then that would be of great importance since no additional external bonds need to be introduced into the polymer networks.
From this point of view, chemical bonds formed from isocyanate chemistry are highly interesting, since the urethane or urea bonds are mildly formed and widely used to prepare many different kinds of functional polymer materials. However, the dynamic isocyanate chemistry is rarely studied, since the dissociative reversion of, for example, carbamate or urea bonds to isocyanates and hydroxyl or amine groups typically occurs at high temperatures (>200 °C), and is associated with deleterious side reactions.32 The dynamic reaction of urethane between isocyanate and hydroxyl groups has indeed been achieved and used for preparing self-healing polymers in the presence of dibutyltin dilaurate (DBTDL),33 zinc acetate,34 or excess hydroxyl moieties,28 by using oxime-based monomers35,36 or vinylogous urethane-based vitrimers37 at relatively high temperature.
Different from the urethane bond, reversibility is harder to achieve for the urea bond, formed by the reaction between amine and isocyanate groups. Cheng et al. reported that self-healing polyurea materials38,39 can be formed by destabilization of urea using a secondary amine group with large steric hindrance, as this will induce weakening of the urea bond and thus increase the dynamic character of the bond.40 However, to the best of our knowledge, dynamic urea bonds formed by primary amine and isocyanate groups have not been reported, and this is primarily attributed to the fast reactions of primary amines and isocyanates that stabilize the reaction products by resonance effects, resulting in highly durable materials.41 This seems understandable, since the reaction of primary amine and isocyanate groups is fast, and the formed urea product is regarded as being highly stabilized by resonance effects.
Yet, in the current study we aimed to do precisely this: turning the urea moiety into a dynamic covalent motif. The reason for such a fundamental approach is that if one is able to develop the field of dynamic urea bond formation and self-healing polyurea materials via dynamic transcarbamoylation processes, it would be feasible to develop entirely novel classes of self-healing polymeric materials. Inspired by the effective catalyzing performance of Zn(OAc)2 on polyurethanes, here we investigated whether we could obtain dynamic covalent urea bonds, formed by primary amines and isocyanate, in the presence of Zn(OAc)2. In other words, we aim to study and thus develop self-healing polymer materials based on the classical covalent urea bond. We studied the reaction kinetics of urea bond formation in the presence and absence of Zn(OAc)2 by nuclear magnetic resonance (NMR), Fourier-transform infrared spectroscopy (FTIR) and gas chromatography-mass spectrometry (GC-MS). The dynamic properties of the urea bond were further confirmed via quantum chemical density-functional theory calculations. Then we set out to unleash the potential of self-healing urea-based polymer materials by preparing two kinds of polydimethylsiloxane–Zn(OAc)2 (PDMS–Zn(OAc)2) cross-linked polymer networks and studied the self-healing properties of these novel materials. Finally, we briefly outline several potential developments based on our findings.
Results and discussion
Dynamic covalent chemistry of simple urea compounds
First, we would like to test our hypothesis whether the urea bond can turn into a dynamic covalent bond in the presence of zinc ions at different temperatures (Fig. 1A). To this end, as a proof of principle, we investigated the potentially dynamic properties of the urea bond by studying the dynamics of diethyl urea in the presence of benzylamine and 2-(p-tolyl)ethylamine, respectively, and zinc acetate (2 wt%), in DMSO at 90 °C, by GC-MS and 1H NMR. We hypothesized that under these conditions either dissociation of the urea bond will produce the original isocyanate, which can then be captured by another amine, or the urea will undergo a Lewis acid-catalyzed nucleophilic addition of an amine onto the urea bond, and exchange one of the original amines.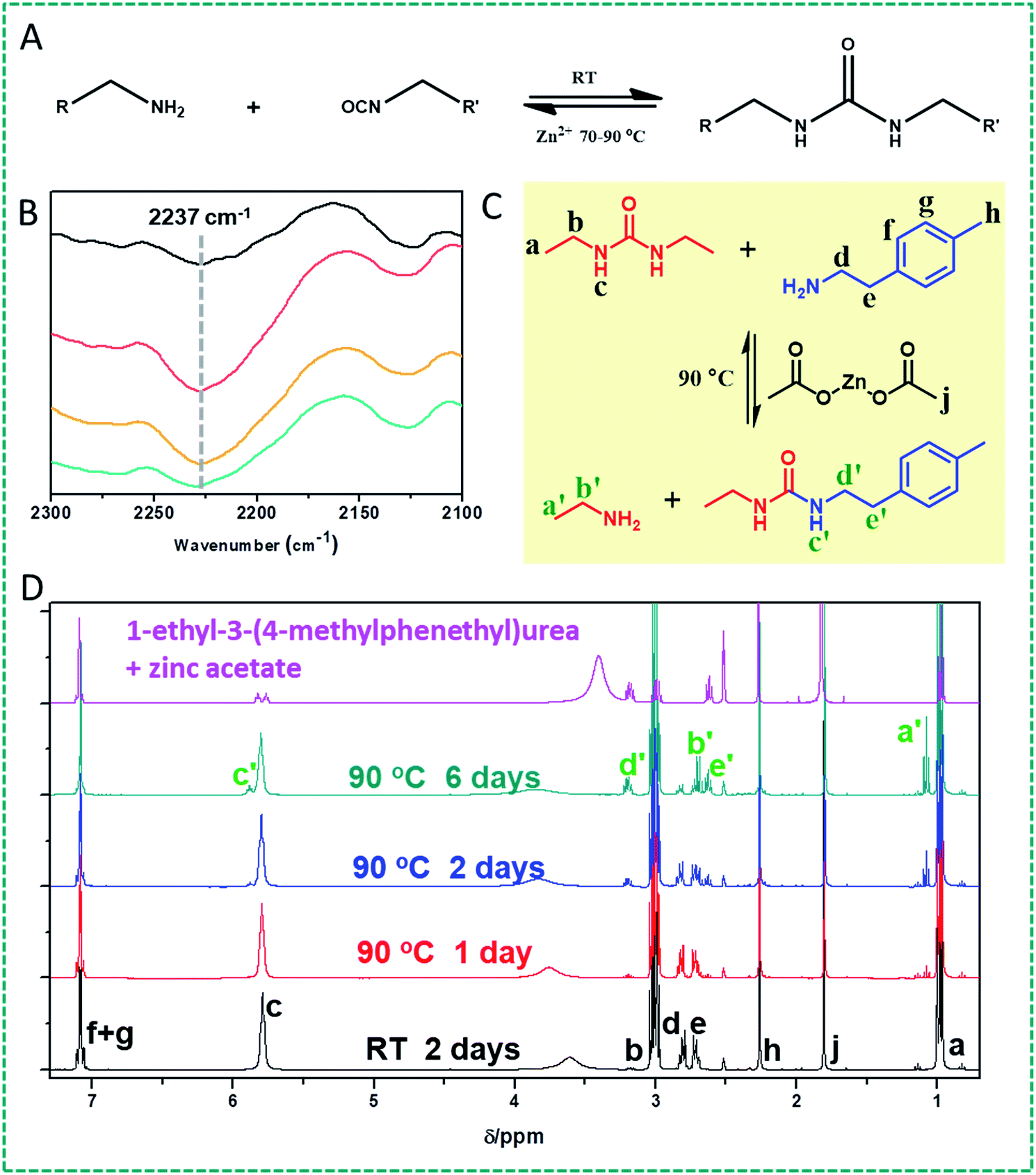 | ||
| Fig. 1 (A) Fundamental question of this study: can the urea bond be made reversible by Lewis acid catalysis in the presence of zinc ions at different temperatures, and thus open up the way for intrinsically self-healing polyureas? (B) FTIR spectra of a linear PDMS–Zn(OAc)2: a freshly prepared sample (black curve, top); the fresh sample was heated at 90 °C for 16 h (red curve), and then cooled down to room temperature for 24 h (yellow curve), and finally kept at room temperature for 72 h (dark cyan curve, bottom). (C) Reaction between 1,3-diethylurea and 2-(p-tolyl)ethylamine in the presence of Zn(OAc)2. (D) 1H NMR spectra of a 1,3-diethylurea, 2-(p-tolyl)ethylamine and Zn(OAc)2 mixture (reaction shown in (C)) reacted in DMSO at room temperature for 2 days (black curve), and at 90 °C for different time periods (red, blue and dark cyan curves); the pink spectrum (top) is that of a mixture of independently prepared 1-ethyl-3-(4-methylphenethyl)urea and zinc acetate. | ||
First of all, indeed the transamination products 1-ethyl-3-benzylurea and 1-ethyl-3-(4-methylphenethyl)urea are formed. These products show up in GC-MS after about 1 h reaction time. In addition, upon continued reaction (typically 24 h and longer), even the follow-up product [1,3-dibenzyl urea or di(4-methylphenethyl)urea] could be observed (Fig. S1†). The asymmetric urea compound can dissociate into two amine and isocyanate compounds. The newly formed benzyl isocyanate and 4-methylphenethyl isocyanate will react with an amine to either re-form or produce 1,3-dibenzyl urea or di(4-methylphenethyl)urea compounds. The yield of these formed products depends on the specific reaction conditions. In a control experiment, no transamination products were observed by GC-MS under identical reaction conditions in the absence of Zn salts, showing that Zn(OAc)2 plays a vital role in the dynamic urea bond formation (Fig. S2†).
In order to further investigate whether the mechanism behind this dynamic character is based on dissociation into the corresponding amine and isocyanate or a catalyst-assisted exchange of amines, FTIR was used to characterize the structural change. A urea-terminated linear PDMS was prepared by reacting amine-terminated polydimethylsiloxane (DMS-A12, Mn = 1000 g mol−1) with phenethyl isocyanate. This reaction was chosen because the product was a very viscous liquid which can stick onto a KBr plate for IR measurement. After heating this urea-terminated PDMS in the presence of 5 wt% Zn(OAc)2 at 90 °C for 16 h, an obvious isocyanate peak at 2237 cm−1 was detected, which confirmed that some urea bonds were dissociated into the constituent amine and isocyanate groups. After cooling the liquid down to room temperature for 72 h, the FTIR curve almost recovers to the original one (Fig. 1B). These results suggest that the dynamic character of urea bonds is achieved by dissociation into the corresponding amine and isocyanate. During this process the concomitant changes of the amine and urea signals in the IR spectra are not obvious and hard to differentiate because of hydrogen bonding formation and the negligible decomposition of the urea group to amine and isocyanate groups (Fig. S3†).
The dynamic character of the urea bond at elevated temperatures under Zn ion catalysis was further confirmed by 1H NMR studies. We choose 1,3-diethyl urea as a model molecule to study the dynamic properties of urea in the presence of zinc ions. We first conducted the NMR experiment by heating a mixture of 1,3-diethyl urea, 2-(p-tolyl)ethylamine and Zn(OAc)2 in DMSO-d6 at 90 °C. If the urea bond would exhibit reversibility, then the re-formed ethyl isocyanate will react with 2-(p-tolyl)ethylamine to form the new compound 1-ethyl-3-(4-methylphenethyl)urea (Fig. 1C). Fig. 1D shows the 1H NMR spectra of the compounds obtained after treatment for different time periods. The new peaks a′ and b′, and c′, d′ and e′ belonging to ethylamine and 1-ethyl-3-(4-methylphenethyl)urea are confirmed by comparison with the spectrum observed for independently prepared 1-ethyl-3-(4-methylphenylethyl)urea. Especially, a new peak (c′) at 5.87 ppm attributed to the newly formed urea bond confirms the formation of 1-ethyl-3-(4-methylphenethyl)urea. Similar NMR results were also observed upon heating a mixture of 1,3-diethyl urea and Zn(OAc)2 in DMSO-d6 at 90 °C. If the urea bond is reversible under these conditions, then it can be expected that ethylamine and ethyl isocyanate will be formed. Indeed, NMR peaks corresponding to ethylamine were observed (see a′ and b′). No signal for ethyl isocyanate was found, which we attribute to reaction of the isocyanate with the water in the DMSO-d6 (Fig. S4†). Fig. S5† shows the NMR spectra of samples with this initial composition when reacted for 16 h at different temperatures, from which we can see that the peak attributed to the new urea bond appears when the reaction temperature is more than 70 °C. This indicates that the urea bond becomes evidently dynamic at this temperature.
Quantum chemical calculations
With these results in hand, we aimed to further delineate the mechanism by B3LYP/6-311+G(d,p) quantum chemical methods (with a Lan2LDZ pseudopotential to model the Zn cation). To this end, we studied the dissociation of diethyl urea to ethylamine and ethyl isocyanate. All calculations were run with the Gaussian 16 suite of programs, and used the SMD solvent model implemented therein to mimic the effects of DMSO.The decomposition of diethyl urea (RC) into CH3–CH2–NCO and CH3–CH2–NH2 can be described as starting with C–N bond lengthening, or as starting with a proton shift, or as a reaction in which both processes take place simultaneously. The C–N bond lengthening route does not lead to a viable minimum even if the C⋯N distance is extended to >4 Å, at which point this route was abandoned. When starting this H-transfer, it became quickly clear that upon transfer of a proton from one N atom to the other, none of the C–N bonds changed drastically. In other words, the dissociation reaction takes place sequentially, starting with H+ transfer followed by C–N cleavage. The reaction profile proceeds through a first transition state (TS1) in which one of the N–H bonds of an amide group lengthens and eventually breaks, and the transferred proton forms a N–H bond with the N atom of the other amino group (Fig. 2A: the proton is transferred from N1 to N2). In this TS, the N1–H bond is elongated to 1.33 Å and the proton starts to bind with the N2 amide atom with a distance of 1.30 Å between them. Because of the H+ migration, the electron density in the amide bond changes, leading to slight bond length changes of the N1–C and N2–C bonds: the N1–C bond shortens from 1.38 to 1.33 Å, while the N2–C bond increases from 1.38 Å to 1.51 Å. The (zero-point energy corrected) energy of TS1 is ca. 44 kcal mol−1 higher than that of the urea reactant, clearly in line with the high stability of urea at room temperature, making this the rate-determining step.
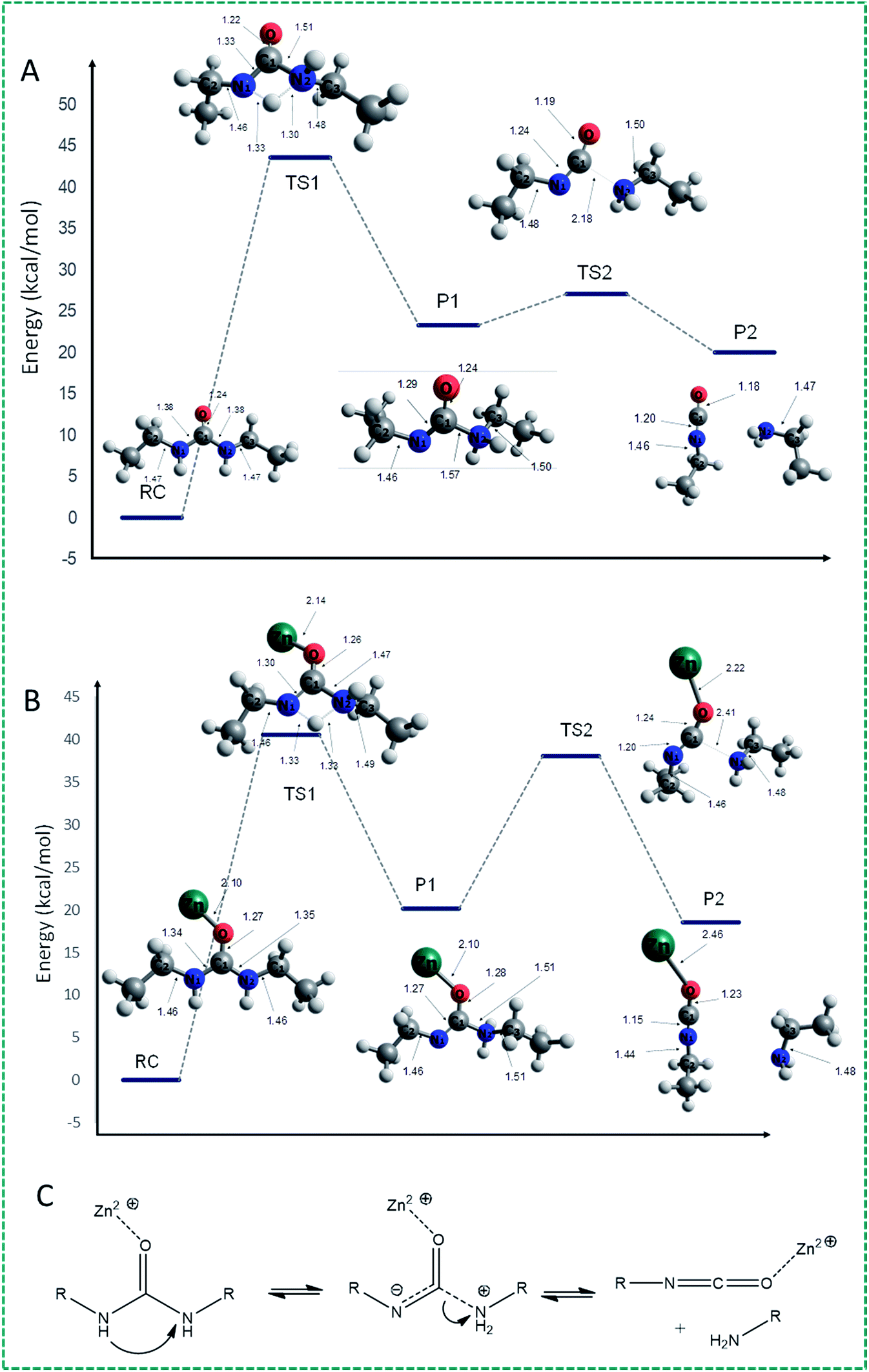 | ||
| Fig. 2 (A) Energy profile of the decomposition reaction of diethyl urea into ethyl isocyanate and ethyl amine. (B) Energy profile of the Zn(II) catalyzed-decomposition of diethylurea into ethyl isocyanate and ethyl amine. The Zn atom is coordinated to the carbonyl O atom. (C) Schematic illustration of the mechanism of the Zn-catalyzed reversible dissociation and re-formation of ureas. | ||
Subsequently, a zwitterionic intermediate is formed, in which the N1–C and N2–C bond distances are 1.29 and 1.57 Å, respectively. Starting from diethyl urea as the reactant, the overall reaction to this intermediate is endothermic by 23.3 kcal mol−1. Next, the N2–C bond is elongated, reaching 2.18 Å in the second TS (TS2), while the N1–C bond is shortened to 1.24 Å and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond decreases from 1.24 Å (in the initial diethyl urea) to 1.19 Å. This second transition state has an energy barrier of only 3.7 kcal mol−1, to yield ethyl isocyanate and ethyl amine as decomposition products (P2) by an exothermic process of −2.8 kcal mol−1 from the zwitterionic intermediate. The overall decomposition reaction from urea to isocyanate and amine is therefore endothermic by more than 20 kcal mol−1, in line with the high stability of urea-based compounds.
O bond decreases from 1.24 Å (in the initial diethyl urea) to 1.19 Å. This second transition state has an energy barrier of only 3.7 kcal mol−1, to yield ethyl isocyanate and ethyl amine as decomposition products (P2) by an exothermic process of −2.8 kcal mol−1 from the zwitterionic intermediate. The overall decomposition reaction from urea to isocyanate and amine is therefore endothermic by more than 20 kcal mol−1, in line with the high stability of urea-based compounds.
When studying the urea decomposition reaction in the presence of Zn(II), there are multiple possible pathways, as the Zn atom can bind to the carbonyl O atom, or to any of the N atoms in the amino group, i.e. to the N atom from which the proton is transferred (N1), or to the N atom to which the proton is transferred (N2). In the first pathway (see Fig. 2B), in which the Zn atom is bound to the carbonyl O atom, the first TS (TS1) is characterized by a proton transfer energy barrier of just over 40 kcal mol−1. In this transition state, the N1–H and N2–H bonds are – analogous to the uncatalyzed route – elongated to 1.33 Å and shortened to 1.30 Å, respectively, with the N1–C bond shortened from 1.34 to 1.30 Å, and the N2–C bond increased from 1.35 Å to 1.47 Å. The full proton transfer yields the intermediate (P1) with an endothermic energy step of 19.6 kcal mol−1. Therefore, the role of the O-bound Zn is to reduce the energy of both the TS and the zwitterionic intermediate by ca. 3–4 kcal mol−1, suggesting a significant catalytic effect of Zn cations. This yields an expected rate acceleration at 90 °C of roughly 2 orders of magnitude, in line with the absence of observed exchange reactions in the absence of Zn salts. In contrast, the second transition (TS2) state now involves a greatly increased activation energy of 18.8 kcal mol−1. This TS structure is only slightly lower in energy than the first one, suggesting complex and likely condition-dependent kinetics of this reaction, which will finally yield product P2.
On the other hand, there is an alternative pathway which involves coordination of Zn2+ to the N atom (the Zn–N1 distance is 2.16 Å) from which the proton migrates to the other N atom (N2). In this structure, the N1–C distance is somewhat increased (1.38 to 1.45 Å) while Zn⋯N coordination shortens the N2–C bond distance slightly (1.38 to 1.35 Å) (Fig. S6†). The proton transfer TS – with similar N⋯H distances as before – now has an energy higher than 48 kcal mol−1, i.e. this reaction will be appreciably slower than the uncatalyzed one. Also the following C–N lengthening TS has to pass an energy barrier of 20.0 kcal mol−1, but this is evidently not the rate-limiting step. Finally, coordination of Zn2+ to the N2 atom does not lead to a viable reaction progress: the proton transfer TS is at least 55 kcal mol−1 above the reactants, which makes this an uncompetitive route.
We interpret these data as indicating that the decomposition reaction of urea is nearly absent at room temperature, either in the presence or absence of Zn2+ due to significant energy barriers. However, at elevated temperatures, crossing these barriers can become viable, even though the overall reaction (from urea to isocyanate + amine) is endothermic. Here we thus calculate a significant effect of Zn2+ cations, with catalytic rate differences of typically two orders of magnitude. In reality, these kinetics will likely be more complicated, as acid/base catalysis will likely also play a role. In addition, the coordination of Zn might change during the reaction, and possibly more Zn ions might be involved. We aim to address these facets in future work, but it suffices to state that quantum chemistry calculations confirm that Zn ions catalyze the decomposition of urea, and that the overall Zn-catalyzed dissociation process can be viewed as sketched in Fig. 2C.
Self-healing polymers
Given the wide-ranging use of polyureas for high-stability polymers, and these results obtained for model compounds with dynamic covalent urea bonds, we aimed to develop self-healing urea-based polymers. Our first strategy to design self-healing polymer materials based on this zinc ion-catalyzed reversible urea bond is shown in Fig. 3A: a primary amine-terminated polydimethylsiloxane (DMS-A21, Mn = 5000 g mol−1, DMS-A15 or DMS-A12) was crosslinked using 1,6-diisocyanate (HDI) and (20–25% aminopropylmethylsiloxane)-dimethylsiloxane copolymer (AMS-1203) with Zn(OAc)2 as a catalyst. The obtained polymer materials (P1–P6, ESI†) were named PDMS–Zn(OAc)2-I, which was characterized by IR (Fig. S7†), and the mechanical properties are summarized in Table S1.† Upon being subjected to a mechanical scratch, the PDMS–Zn(OAc)2-I (P1) networks can be repaired by thermal treatment at 90 °C for 30 min (Fig. 3B and C). The self-healing properties of the PDMS–Zn(OAc)2-I (P1) network were further verified by tensile strength experiments. The materials were first cut into two pieces, and then put together followed by heating the polymer at 90 °C for 24 h. Remarkably, the stress strength and the maximum elongation before breaking (elongation at break point) of this cut-off sample both fully recovered to the original value (Fig. 3D), which indicates that the as-prepared PDMS–Zn(OAc)2-I films possess excellent self-healing properties. The degree of elongation before breaking of the PDMS–Zn(OAc)2 networks decreases with increasing the cross-linker AMS-1203 content due to the enhanced cross-linking density, but all of these three PDMS–Zn(OAc)2 samples displayed excellent self-healing properties, with a healing efficiency of more than 90% as deduced from both tensile stress and elongation before breaking experiments (Table S1,† P2–P4). Upon decreasing the molecular weight of amine-terminated polydimethylsiloxane from 5000 to 3000 (Table S1,† P5), the elongation at break and tensile strength decreased, again due to the effectively enhanced cross-linking density. However, this polymer still showed a healing efficiency of 93%. Further decreasing the molecular weight of amine-terminated polydimethylsiloxane to 1000 (Table S1,† P6) makes the self-healing properties disappear, because the roughness of the cut-off sample prevents the two parts from being placed intimately together. In order to outline the role of zinc ions in the self-healing process, several control experiments were conducted. First of all, we tested the self-healing behavior of the several PDMS films with different catalysts (Fig. 4A). In the absence of zinc ions, the tensile stress only recovers to 45%. In the presence of Zn(OAc)2 and Zn(CF3SO3)2, PDMS networks exhibit excellent self-healing properties. However, in the presence of magnesium acetate no self-healing was observed. Therefore the nature of the metal ion is crucial. All of the above results demonstrate that the zinc ion plays a very important role during the self-healing process, rather than the anion, which does not seem to be catalytically critical. We also examined the self-healing behavior at room temperature in the presence of Zn(OAc)2; no self-repairing was observed, indicating that the self-healing process is a thermally activated process, in line with the computations. Finally, we studied the influence of zinc concentration on the self-healing behavior of the PDMS–Zn(OAc)2. A clearly increased self-healing is observed with an increased Zn concentration, up to a saturation limit: when the zinc concentration is more than 0.5 wt%, we achieve more than 90% healing efficiency, while further increasing the Zn concentration to >1 wt% does not seem to add any further benefits (Fig. 4B). We propose that the self-healing properties of the PDMS–Zn(OAc)2 – under these conditions – can be attributed to the dynamic nature of urea bonds catalyzed by zinc ions, which is schematically shown in Fig. 4C.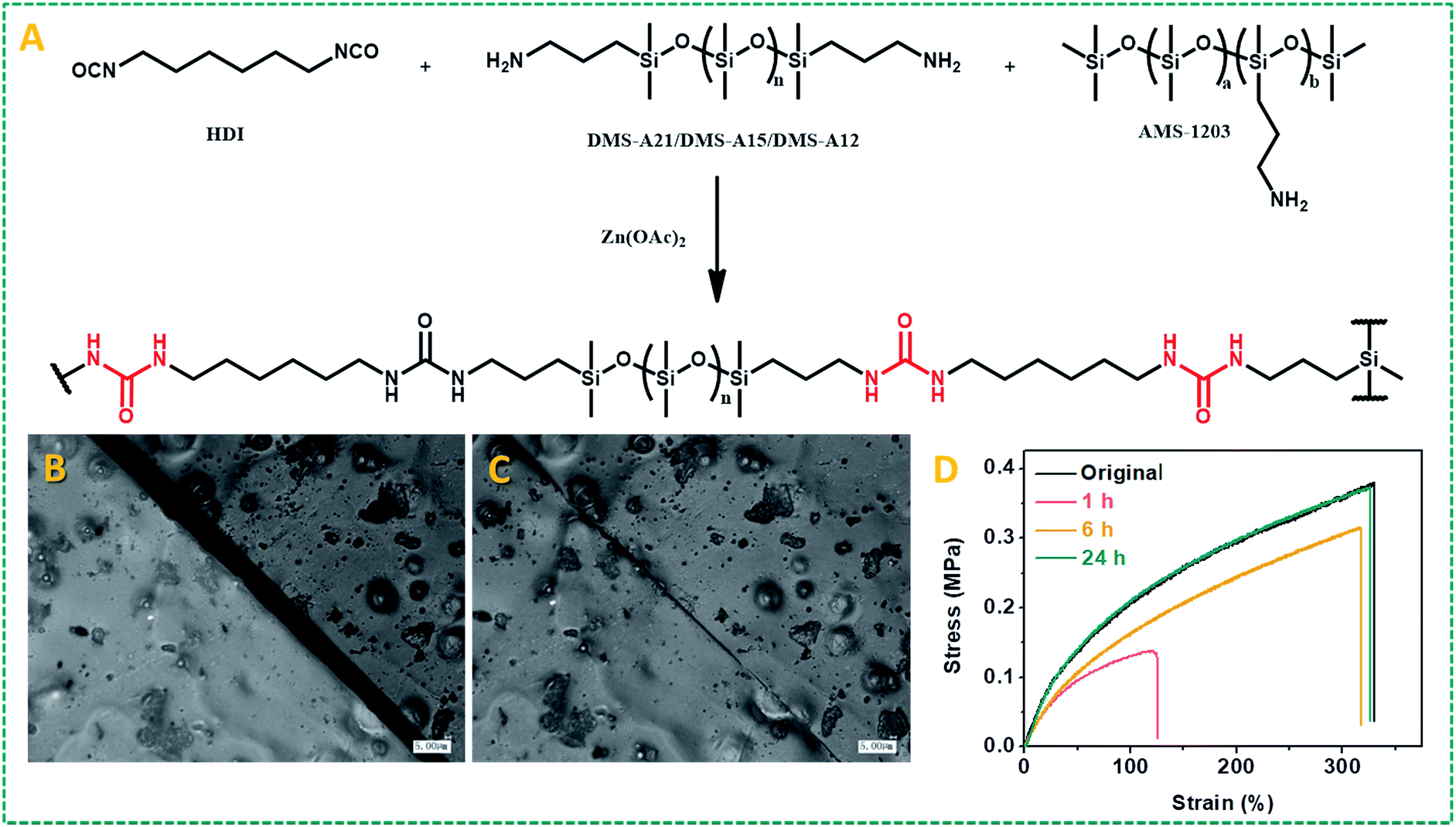 | ||
| Fig. 3 (A) Schematic representation of the synthetic route of the PDMS–Zn(OAc)2-I network. (B) Optical image of the as-prepared PDMS–Zn(OAc)2-I film (P1) with a scratch (scar bar: 5 μm). (C) Optical image of the damaged PDMS–Zn(OAc)2-I film after thermal treatment at 90 °C for 30 min (scar bar: 5 μm). (D) Stress–strain curves of the original PDMS–Zn(OAc)2-I (P1) films and the cut-off films after thermal treatment at 90 °C for different time periods. | ||
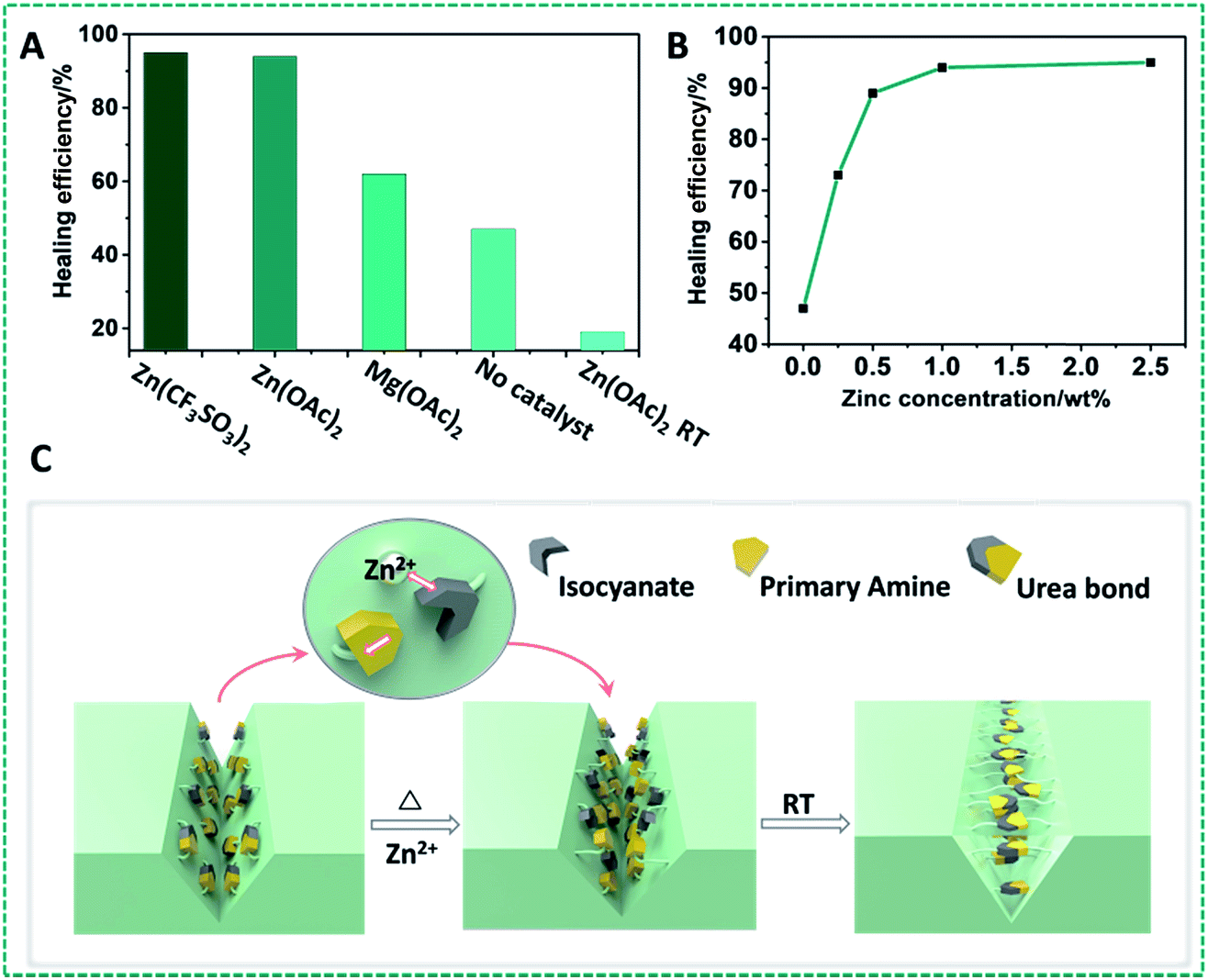 | ||
| Fig. 4 (A) Healing efficiency of PDMS materials with different catalysts at 90 °C. (B) Healing efficiency of the PDMS–Zn(OAc)2-I network (P4) as a function of Zn(OAc)2 concentration. (C) Schematic illustration of the self-healing process of the PDMS–Zn(OAc)2 networks: in the presence of Zn ions, urea moieties can open up and subsequently restore the materials by re-forming in novel geometries. | ||
Many self-healing PDMS materials relying on dynamic covalent or non-covalent bonds have been developed. The main challenge in this field is to achieve repairable PDMS materials with high mechanical strength. Such strong and repairable materials may be achieved by varying the monomer composition and molecular weight, using a cross-linking network structure and/or introducing supramolecular interactions. To realize this goal, another kind of PDMS–Zn(OAc)2-II (P7: PDMS–IPDI–triHDI–Zn(OAc)2, a detailed preparation procedure can be found in the ESI†) material was prepared by reacting a DMS-A12 (primary amine-terminated polydimethylsiloxane, Mn = 1000 g mol−1) monomer with isophorone diisocyanate (IPDI), followed by cross-linking with a tri-functional homopolymer of hexamethylene diisocyanate (tri-HDI) (Fig. 5A). In this system, the hydrogen bonding density was raised by decreasing the molecular weight of the siloxane homopolymer, resulting in an obvious increase of the mechanical strength. In addition, the degree of crystallinity was reduced by replacing HDI with IPDI as the chain extender, which will increase the elastic properties of the materials. The result of these changes is that these self-healing urea-linked PDMS–Zn(OAc)2-II materials had a high tensile stress (7.0 MPa) and elongation at break (more than 600%), much higher than those of P2–P5 (Table S1†). Due to the catalyzing role of zinc ions, this material still showed excellent self-healing properties as shown in Fig. 5B and C. Two PDMS–Zn(OAc)2-II (P7) films with different widths (1 cm and 0.6 cm) were cut into two pieces, and two of them (one is 1 cm, the other is 0.6 cm) were put together and then heated at 110 °C for 16 h, the healed samples can hold a weight of at least 500 g (Fig. 5B). Tensile experiment results also demonstrated that both the tensile stress and elongation at break can recover to about 90% after healing at 110 °C for 16 h (Fig. 5C). We summarized the mechanical properties of reported self-healing PDMS materials and the results are presented in Fig. 5D. We can see that most of the as-developed self-healing PDMS materials possess weak mechanical strength. Only one example reported had a mechanical strength 9.46 MPa higher than our PDMS materials, but that material displayed a low elongation at break.52 This comparison illustrates that our self-healing PDMS materials not only display excellent healing efficiency, but also possess superior mechanical properties. In this particular system, 110 °C was used to achieve the self-healing, resulting from the presence of many hydrogen bonds. Extensive H-bonding results in frozen segments at lower temperatures, and effects that repair can only take place upon sufficient heating to break the hydrogen bonds, so as to allow polymer segments to be able to move. This elevated temperature is not essential for Zn-catalyzed repair of polyureas. To demonstrate that the Zn-catalyzed self-healing of polyureas also takes place at much lower temperatures, we changed the PDMS monomer from DMS-A12 to DMS-A21 (molecular weight is changed from 1000 to 5000) and prepared another kind of PDMS–Zn(OAc)2-II (P8; see the ESI† for details of the preparation). This polymer is much softer than P7 due to the reduced hydrogen bonding, and displays a relatively low degree of cross-linking. As Fig. S8† shows, this material can be repaired at 70 °C within 30 min, with more than 90% recovery of both the tensile stress and elongation at break. In other words, the repair is smooth even upon gentle heating.
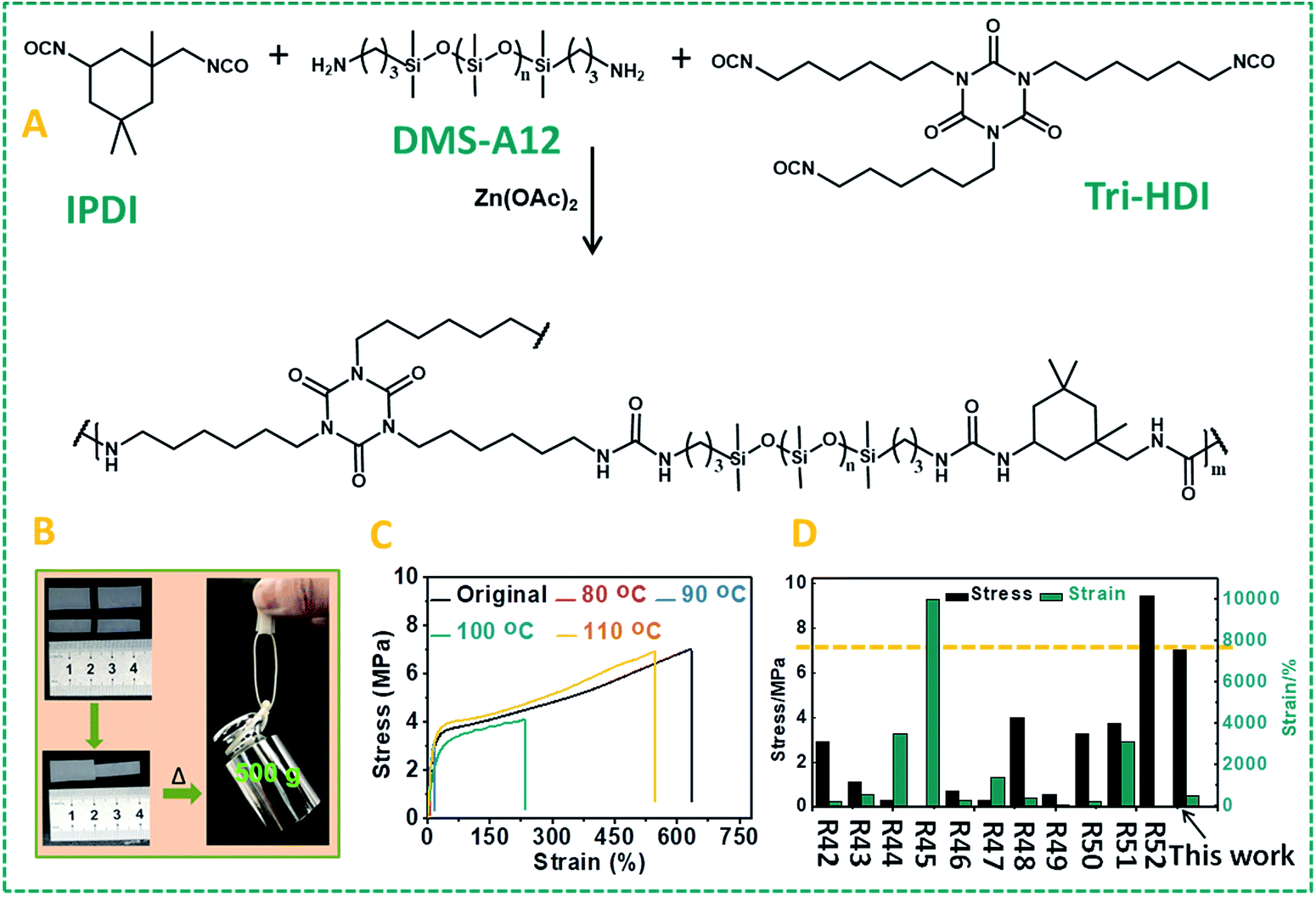 | ||
| Fig. 5 (A) Schematic representation of the synthetic route of the PDMS–Zn(OAc)2-II (P6) network. (B) Picture showing the cut-off and healed PDMS–Zn(OAc)2-II (P7) films, and the ability of even thin repaired pieces to hold 0.5 kg. (C) Stress–strain curves of the original PDMS–Zn(OAc)2-II (P7) films and the self-healed films after thermal treatment at different temperatures. (D) Comparison of our PDMS–Zn(OAc)2-II (P7) materials with previously reported self-healing PDMS in the literature42–52 in terms of breaking strength and elongation at break. | ||
In order to study the influence of the zinc salt on the thermal stability of these materials, we conducted thermogravimetric analysis (TGA) experiments of PDMS–Zn(OAc)2-II with different concentrations of Zn(OAc)2, and the results are shown in Fig. S9.† All of these materials exhibit relatively good thermal stability up to 250 °C. The PDMS–Zn(OAc)2-II with concentrations of 1 wt% and 2 wt% start to decompose at around 255 °C, while this degradation behavior happens at around 280 °C for PDMS–Zn(OAc)2-II without zinc. This means that addition of the zinc salt at these concentrations will reduce the thermal stability of the materials only slightly.
In addition, in order to study the influence of the zinc salt on the microscopic structure of materials, ultrathin films of PDMS–Zn(OAc)2-II (P7) were examined by transmission electron microscopy (TEM). The results showed that no microphase separation was observed for the polymer networks, but only aggregates formed with zinc salts were found (Fig. S10†), which we tentatively attribute to the low solubility of Zn(OAc)2 in tetrahydrofuran. Although there are some (catalytically likely non-active) aggregates at these higher wt%, overall, zinc ions can still catalyze the dynamic chemistry of urea bonds effectively, as also indicated by the observation that only 0.5 wt% zinc catalyst is needed to obtain excellent healing efficiency.
Mechanism of self-healing in polyurea polymers
In order to verify the proposed self-healing mechanism and dynamic properties of the urea bond, we first tested the solubility of the cross-linked PDMS–Zn(OAc)2 materials (P4) in toluene. As shown in Fig. 6A, the PDMS–Zn(OAc)2 materials can be dissolved in toluene within 4 h by heating at 90 °C, while only swelling behavior (Fig. 6B) was observed after immersion in toluene at room temperature for 24 h, and also upon immersion at 90 °C for 24 h without the zinc catalyst (Fig. 6C). In the absence of reversible dynamic bonds, it is expected that materials can only be swelled by solvent, but cannot be dissolved. This experiment shows the dynamic character of the urea bond upon heating with Zn salts, and the absence of dynamicity when either zinc or an elevated temperature is absent.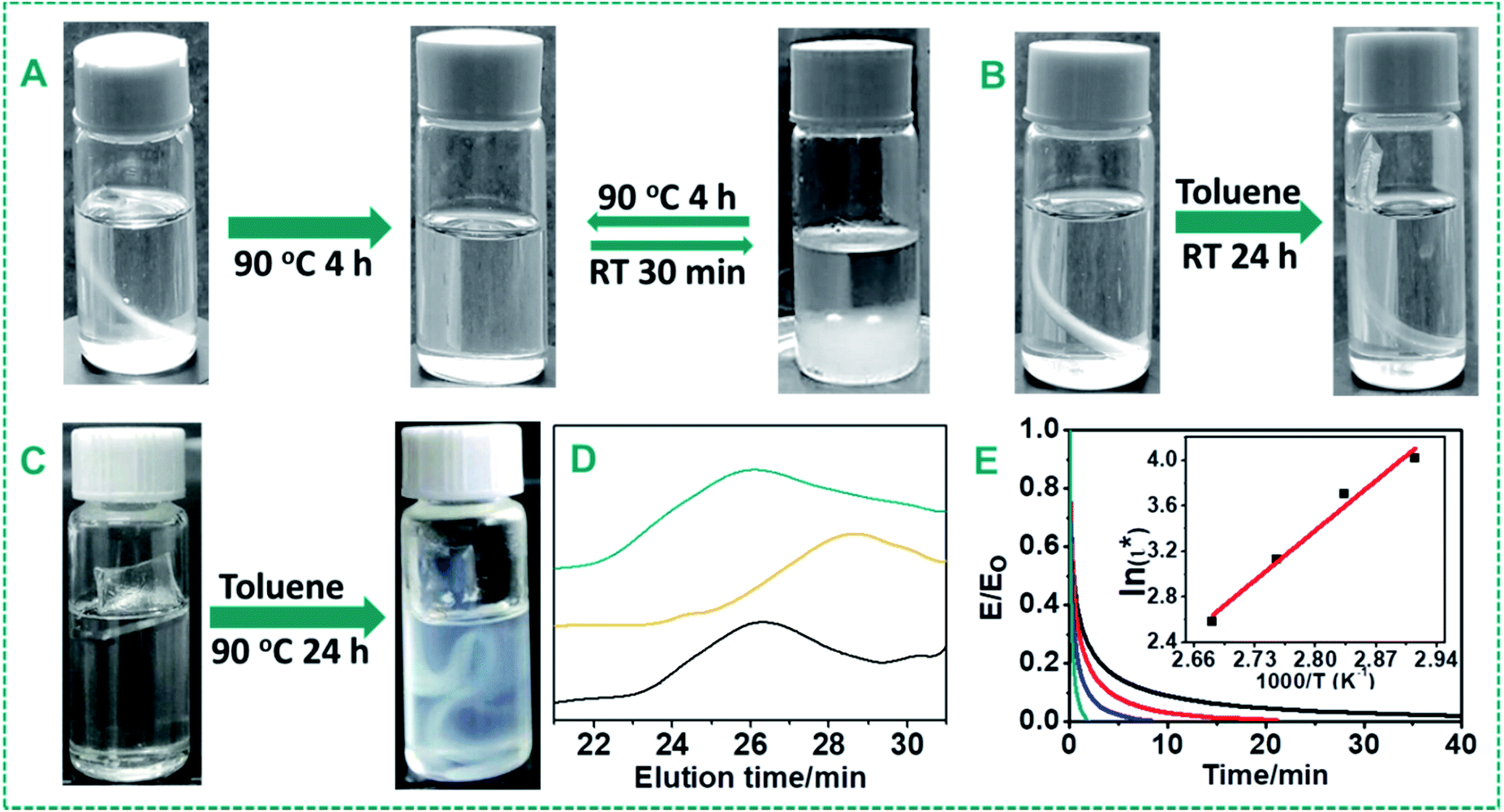 | ||
Fig. 6 (A) A PDMS–Zn(OAc)2-I film (P4) immersed into toluene at 90 °C for 4 h and then cooled down to r.t. for 30 min. (B) Freshly prepared PDMS–Zn(OAc)2-I (P4) films immersed into toluene at r.t. for 24 h. (C) Freshly prepared PDMS films (no zinc) immersed into toluene at 90 °C for 24 h. (D) GPC curve of a linear PU–PDMS. Black curve: a linear PDMS–Zn(OAc)2 formed by IPDI and DMS-A12 with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio; yellow curve: the linear PDMS–Zn(OAc)2 was dissolved in toluene and then 1 equivalent DMS-A12 and 2.5 wt% Zn(OAc)2 were added and the resulting mixture incubated at 90 °C for 12 h; dark cyan curve: another equivalent IPDI was added into the solution and stirred for 20 min, and then the solvent was evaporated at r.t. for 12 h. (E) Normalized stress–relaxation curves of PDMS–Zn(OAc)2-II (P7) at various temperatures (black: 70 °C, red: 80 °C, blue: 90 °C, dark cyan: 100 °C). Inset: fitting line of the relaxation times according to the Arrhenius equation, and the relaxation activation energy obtained from the slope. :1 ratio; yellow curve: the linear PDMS–Zn(OAc)2 was dissolved in toluene and then 1 equivalent DMS-A12 and 2.5 wt% Zn(OAc)2 were added and the resulting mixture incubated at 90 °C for 12 h; dark cyan curve: another equivalent IPDI was added into the solution and stirred for 20 min, and then the solvent was evaporated at r.t. for 12 h. (E) Normalized stress–relaxation curves of PDMS–Zn(OAc)2-II (P7) at various temperatures (black: 70 °C, red: 80 °C, blue: 90 °C, dark cyan: 100 °C). Inset: fitting line of the relaxation times according to the Arrhenius equation, and the relaxation activation energy obtained from the slope. | ||
Secondly, a linear PDMS was synthesized by reacting IPDI with amine-terminated polydimethylsiloxane (DMS-A12, Mn = 1000 g mol−1) in the presence of 2.5 wt% Zn(OAc)2. This system was chosen because of the better solution of the forming polymer in THF for further gel permeation chromatography (GPC) measurements. The fully finished reaction between DMS-A12 and IPDI formed a linear polyurea with a Mn of 12.3 × 103 g mol−1 as analyzed by GPC. The addition of another equivalent of DMS-A12 resulted in degradation of the molecular weight to 4.5 × 103 g mol−1 after 12 h of incubation at 90 °C in the presence of Zn(OAc)2. Surprisingly, addition of another equivalent of IPDI led to the recovery of the molecular weight to 12.3 × 103 g mol−1 again (Fig. 6D). These results demonstrate that the urea bonds are dissociated into isocyanate and amine groups after heat treatment with the help of zinc ions, and can re-form into the urea bond upon cooling.
The dynamic properties of the PDMS–Zn(OAc)2 materials were further investigated by tensile stress relaxation analysis (Fig. 6E). The modulus dropped sharply within the initial tens of seconds, mainly due to the dissociation of hydrogen bonds, disentanglement of the polymer chains and molecular relaxation, and then decreased gradually because of the thermal dynamic exchange of the urea bonds in the networks. Relaxation activation energy (Ea,r = 52 kcal mol−1) was calculated from the slope of the fitting line of the relaxation times according to the Arrhenius equation, shown in the inset diagram of Fig. 6E. Such relatively low relaxation activation energy imparts excellent healing properties to the materials.
From dynamic thermomechanical analysis (DMA) data we can see that compared with the pure PDMS film the modulus of the PDMS–Zn(OAc)2-I (P4) films drops sharply at ∼90 °C (Fig. S11†), indicating a thermal transition here. This result cannot confirm the dynamic mechanism at a molecular level, but considering the GPC and solubility experiment results, we believe that this transition comes from the dissociation of the urea bond into isocyanate and amine groups. In this case the cross-linked polymer network is open to some extent, which reduces the cross-linking density. The result of this is that we can see a sharp decrease in the storage modulus of the PDMS–Zn(OAc)2 film. In addition, due to the sharp decrease in the degree of cross-linking, the PDMS–Zn(OAc)2 film becomes too soft to be stretched, which hampers obtaining modulus data when that temperature is higher than the dynamic temperature. All of the results from GPC, solubility experiments and DMA confirmed that the self-healing mechanism is based on the dissociation of the urea bond at elevated temperatures in the presence of zinc ions, and that the newly formed isocyanate and amine groups will re-form into a urea bond after cooling down to room temperature.
Conclusions
Despite the high resonance-induced stability of urea moieties, we show here for the first time the dynamic character of ureas in the presence of zinc ions via well-controlled dissociation and exchange experiments and via quantum chemical calculations of small-molecule model urea compounds. We estimate that the presence of zinc ions as a catalyst speeds up the dissociation of ureas by roughly two orders of magnitude, via the formation of O-bound Zn complexes. Relying on this dynamic urea bond, we developed self-healing Zn(OAc)2-containing polymeric materials that, upon mechanical scratching, can be fully repaired via a simple thermal treatment. Due to the wide range of commercially available primary amines and isocyanate monomers, this method can be used to prepare many kinds of novel self-healing and reprocessable polyurea materials. As a result, dynamic polyurea materials are expected to have significant potential in a range of applications, such as coatings, paints, and 3D printing.53,54 Studies on the influence of different catalysts on the dynamic character of urea bonds are currently ongoing in our labs.Experimental section
Materials
Amine-terminated polysiloxane (DMS-A12: Mn = 900–1000 g mol−1, DMS-A15: Mn = 3000 g mol; DMS-A21: Mn = 5000 g mol−1) and (20–25% aminopropylmethylsiloxane)-dimethylsiloxane copolymer (AMS-1203: Mn = 25000 g mol−1) were purchased from Gelest Inc. 1,6-Diisocyanate (HDI), isophorone diisocyanate (IPDI), HDI trimer, phenethyl isocyanate, zinc acetate, magnesium acetate, zinc trifluoromethanesulfonate, 2-(p-tolyl)ethylamine, N,N′-diethylurea and anhydrous tetrahydrofuran were obtained from the best reagent companies.
Experimental methods
Characterization
The weight-average molecular weight of the synthesized PU-PDMS was determined by gel permeation chromatography (GPC; HLC-8320) with THF as an eluent phase and PMMA as a standard sample. PDMS–PUR–Zn(OAc)2 was characterized by IR spectroscopy on a Fourier transform infrared spectrometer (FTIR; Nicolet 560) in the frequency range of 400–4000 cm−1, and further analyzed by 1H NMR (Bruker ARX-400 at 400 MHz). Dynamic Mechanical Thermal Analysis (DMTA) experiments were performed using a TA Instruments Q800 machine in the dual cantilever mode. The test temperatures ranged from −30 to 150 °C and the heating rate was 3 °C min−1. The strain amplitude used was 50 μm, and the stretching frequency was 1 Hz. Mechanical tensile-stress experiments were conducted on an Instron 5567 machine (USA) at room temperature with a strain rate of 50 mm min−1. The dimension of specimens was 35 × 8 × 0.7 mm3. At least four samples of each loading fraction were tested. GC and GC-MS analyses were performed by taking 50 μL aliquots of the reaction studied in DMSO-d6 and injecting them in a vial filled with 500 μL water and 500 μL 1,2-dichloroethane. The sample was gently mixed by shaking, and injections into the GC and GC-MS were from the 1,2-dichloroethane layer. Thermogravimetric analysis (TGA) experiments were performed on a NETZSCH TG 209 instrument at a linear heating rate of 10 °C min−1 from 30 to 800 °C under a nitrogen atmosphere. TEM measurements were conducted with a Tecnai G2 F20 S-TWIN transmission electron microscope (FEI).Computational methods
The geometries of all the reactants, products and transition states (TS) were optimized at the B3LYP/6-311+G(d,p) level of theory. Frequency analysis was carried out on all stationary points to confirm their nature (minimum, TS) on the potential energy surface. The universal solvation model SMD was used to mimic the solvent dimethyl sulfoxide (DMSO) for the calculation of the free energy of solvation. All calculations were carried out with the Gaussian16 software package, and Gaussview 6.0 was used as visualization software.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Key R&D Program of China (2017YFE0111500), the National Natural Science Foundation of China (51703143), the EU Horizon 2020 research and innovation programme (No. 734164), the Young Talent Team Science and Technology Innovation Project of Sichuan Province (2016TD0010), and a grant from the State Key Laboratory of Polymer Materials Engineering of Sichuan University (Grant No. sklpme2017-3-04).Notes and references
- N. Kuhl, S. Bode, M. D. Hager and U. S. Schubert, in Self-Healing Materials, ed. M. D. Hager, S. VanDerZwaag and U. S. Schubert, Springer-Verlag Berlin, Berlin, 2016, vol. 273, pp. 1–58 Search PubMed.
- B. Ghosh and M. W. Urban, Science, 2009, 323, 1458–1460 CrossRef CAS PubMed.
- Y. Yang and M. W. Urban, Angew. Chem., Int. Ed., 2014, 53, 12142–12147 CrossRef CAS PubMed.
- F. Herbst, D. Dohler, P. Michael and W. H. Binder, Macromol. Rapid Commun., 2013, 34, 203–220 CrossRef CAS PubMed.
- C. E. Diesendruck, N. R. Sottos, J. S. Moore and S. R. White, Angew. Chem., Int. Ed., 2015, 54, 10428–10447 CrossRef CAS PubMed.
- J. F. Patrick, M. J. Robb, N. R. Sottos, J. S. Moore and S. R. White, Nature, 2016, 540, 363–370 CrossRef CAS PubMed.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794–797 CrossRef CAS PubMed.
- Y. Heo and H. A. Sodano, Adv. Funct. Mater., 2014, 24, 5261–5268 CrossRef CAS.
- X. F. Luo and P. T. Mather, ACS Macro Lett., 2013, 2, 152–156 CrossRef CAS.
- D. Habault, H. J. Zhang and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7244–7256 RSC.
- Y. H. Kim and R. P. Wool, Macromolecules, 1983, 16, 1115–1120 CrossRef CAS.
- M. W. Urban, D. Davydovich, Y. Yang, T. Demir, Y. Z. Zhang and L. Casabianca, Science, 2018, 362, 220–225 CrossRef CAS PubMed.
- Y. Yang, D. Davydovich, C. C. Hornat, X. L. Liu and M. W. Urban, Chem, 2018, 4, 1928–1936 CAS.
- W. K. Zou, J. T. Dong, Y. W. Luo, Q. Zhao and T. Xie, Adv. Mater., 2017, 29, 18 Search PubMed.
- F. Garcia and M. M. J. Smulders, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3551–3577 CrossRef CAS PubMed.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- C. N. Bowman and C. J. Kloxin, Angew. Chem., Int. Ed., 2012, 51, 4272–4274 CrossRef CAS PubMed.
- H. Otsuka, K. Aotani, Y. Higaki and A. Takahara, J. Am. Chem. Soc., 2003, 125, 4064–4065 CrossRef CAS PubMed.
- S. Yu, R. C. Zhang, Q. Wu, T. H. Chen and P. C. Sun, Adv. Mater., 2013, 25, 4912–4917 CrossRef CAS PubMed.
- X. L. Lu, G. X. Fei, H. S. Xia and Y. Zhao, J. Mater. Chem. A, 2014, 2, 16051–16060 RSC.
- Y. Amamoto, H. Otsuka, A. Takahara and K. Matyjaszewski, Adv. Mater., 2012, 24, 3975–3980 CrossRef CAS PubMed.
- M. Hernandez, A. M. Grande, W. Dierkes, J. Bijleveld, S. van der Zwaag and S. J. Garcia, ACS Sustainable Chem. Eng., 2016, 4, 5776–5784 CrossRef CAS.
- S. B. Ji, W. Cao, Y. Yu and H. P. Xu, Adv. Mater., 2015, 27, 7740–7745 CrossRef CAS PubMed.
- S. M. Kim, H. Jeon, S. H. Shin, S. A. Park, J. Jegal, S. Y. Hwang, D. X. Oh and J. Park, Adv. Mater., 2018, 30, 1705145 CrossRef PubMed.
- A. Rekondo, R. Martin, A. R. de Luzuriaga, G. Cabanero, H. J. Grande and I. Odriozola, Mater. Horiz., 2014, 1, 237–240 RSC.
- Z. Q. Lei, H. P. Xiang, Y. J. Yuan, M. Z. Rong and M. Q. Zhang, Chem. Mater., 2014, 26, 2038–2046 CrossRef CAS.
- D. J. Fortman, J. P. Brutman, C. J. Cramer, M. A. Hillmyer and W. R. Dichtel, J. Am. Chem. Soc., 2015, 137, 14019–14022 CrossRef CAS PubMed.
- K. Imato, M. Nishihara, T. Kanehara, Y. Amamoto, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2012, 51, 1138–1142 CrossRef CAS PubMed.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898–952 CrossRef PubMed.
- Y. H. Jin, C. Yu, R. J. Denman and W. Zhang, Chem. Soc. Rev., 2013, 42, 6634–6654 RSC.
- E. Delebecq, J. P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS PubMed.
- N. Zheng, Z. Z. Fang, W. K. Zou, Q. Zhao and T. Xie, Angew. Chem., Int. Ed., 2016, 55, 11421–11425 CrossRef CAS PubMed.
- Y. Yang and M. W. Urban, Polym. Chem., 2017, 8, 303–309 RSC.
- W. X. Liu, C. Zhang, H. Zhang, N. Zhao, Z. X. Yu and J. Xu, J. Am. Chem. Soc., 2017, 139, 8678–8684 CrossRef CAS PubMed.
- D. Fu, W. Pu, Z. Wang, X. Lu, S. Sun, C. Yu and H. Xia, J. Mater. Chem. A, 2018, 6, 18154–18164 RSC.
- W. Denissen, M. Droesbeke, R. Nicolay, L. Leibler, J. M. Winne and F. E. Du Prez, Nat. Commun., 2017, 8, 7 CrossRef PubMed.
- H. Z. Ying, Y. F. Zhang and J. J. Cheng, Nat. Commun., 2014, 5, 9 Search PubMed.
- Y. F. Zhang, H. Z. Ying, K. R. Hart, Y. X. Wu, A. J. Hsu, A. M. Coppola, T. A. Kim, K. Yang, N. R. Sottos, S. R. White and J. J. Cheng, Adv. Mater., 2016, 28, 7646–7651 CrossRef CAS PubMed.
- S. Zechel, R. Geitner, M. Abend, M. Siegmann, M. Enke, N. Kuhl, M. Klein, J. Vitz, S. Grafe, B. Dietzek, M. Schmitt, J. Popp, U. S. Schubert and M. D. Hager, NPG Asia Mater., 2017, 9, e420 CrossRef CAS.
- H. Kim and M. W. Urban, Langmuir, 2000, 16, 5382–5390 CrossRef CAS.
- L. Yang, Y. L. Lin, L. S. Wang and A. Q. Zhang, Polym. Chem., 2014, 5, 153–160 RSC.
- X. Y. Jia, J. F. Mei, J. C. Lai, C. H. Li and X. Z. You, Chem. Commun., 2015, 51, 8928–8930 RSC.
- X. Y. Jia, J. F. Mei, J. C. Lai, C. H. Li and X. Z. You, Macromol. Rapid Commun., 2016, 37, 952–956 CrossRef CAS PubMed.
- C. H. Li, C. Wang, C. Keplinger, J. L. Zuo, L. Jin, Y. Sun, P. Zheng, Y. Cao, F. Lissel, C. Linder, X. Z. You and Z. A. Bao, Nat. Chem., 2016, 8, 619–625 Search PubMed.
- Y. L. Rao, A. Chortos, R. Pfattner, F. Lissel, Y. C. Chiu, V. Feig, J. Xu, T. Kurosawa, X. D. Gu, C. Wang, M. Q. He, J. W. Chung and Z. N. Bao, J. Am. Chem. Soc., 2016, 138, 6020–6027 CrossRef CAS PubMed.
- J. F. Mei, X. Y. Jia, J. C. Lai, Y. Sun, C. H. Li, J. H. Wu, Y. Cao, X. Z. You and Z. N. Bao, Macromol. Rapid Commun., 2016, 37, 1667–1675 CrossRef CAS PubMed.
- F. B. Madsen, L. Y. Yu and A. L. Skov, ACS Macro Lett., 2016, 5, 1196–1200 CrossRef CAS.
- J. Zhao, R. Xu, G. X. Luo, J. Wu and H. S. Xia, J. Mater. Chem. B, 2016, 4, 982–989 RSC.
- J. Zhao, R. Xu, G. X. Luo, J. Wu and H. S. Xia, Polym. Chem., 2016, 7, 7278–7286 RSC.
- X. Z. Yan, Z. Y. Liu, Q. H. Zhang, J. Lopez, H. Wang, H. C. Wu, S. M. Niu, H. P. Yan, S. H. Wang, T. Lei, J. H. Li, D. P. Qi, P. G. Huang, J. P. Huang, Y. Zhang, Y. Y. Wang, G. L. Li, J. B. H. Tok, X. D. Chen and Z. A. Bao, J. Am. Chem. Soc., 2018, 140, 5280–5289 CrossRef CAS PubMed.
- J. C. Lai, J. F. Mei, X. Y. Jia, C. H. Li, X. Z. You and Z. A. Bao, Adv. Mater., 2016, 28, 8277–8282 CrossRef CAS PubMed.
- Y. Yang, E. M. Terentjev, Y. Wei and Y. Ji, Nat. Commun., 2018, 9, 2291 CrossRef PubMed.
- J. C. Lai, L. Li, D. P. Wang, M. H. Zhang, S. R. Mo, X. Wang, K. Y. Zeng, C. H. Li, Q. Jiang, X. Z. You and J. L. Zuo, Nat. Commun., 2018, 9, 2725 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta02054c |
| This journal is © The Royal Society of Chemistry 2019 |