
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA sustainable approach to hierarchically porous carbons from tannic acid and their utilization in supercapacitive energy storage systems†
Noel
Díez
 ,
Guillermo A.
Ferrero
,
Marta
Sevilla
and
Antonio B.
Fuertes
*
,
Guillermo A.
Ferrero
,
Marta
Sevilla
and
Antonio B.
Fuertes
*
Instituto Nacional del Carbón (CSIC), Fco. Pintado Fe 26, Oviedo 33011, Spain. E-mail: abefu@incar.csic.es
First published on 20th May 2019
Abstract
Hierarchically porous carbons with a high surface area have been synthesized by a salt template-assisted chemical activation approach using a biomass-derivative, tannic acid, as the carbon precursor. Tannic acid is pyrolyzed in the presence of KCl, acting as the template and reaction medium, and K2CO3, serving as both the template and activating agent. An important characteristic of our synthesis strategy is that the carbonaceous matter is immersed in a melted mixture constituted by the activating agent and KCl. This reaction medium greatly favours reactivity between the carbon and K2CO3, and yet enhances the carbon yields, which are in the 32–38% range. The porous carbons have large specific surface areas, in the 2250–2750 m2 g−1 range, and their microporosity can be fine-tuned by adjusting the carbonization temperature. When these carbons are tested as the electrode active material for supercapacitors, they exhibit a high capacitance in aqueous and organic electrolytes. The short diffusion pathways provided by their hierarchical architecture allow an excellent electrochemical response in situations of high power demand.
Introduction
Porous carbons have become a key component of energy storage systems by virtue of their high electronic conductivity, large specific surface area and pore size tunability. Porous carbons have already been commercialized as electrode materials for supercapacitors and are considered as a promising conductive host in the cathodes of lithium–sulfur or lithium–selenium batteries.1,2 Given that the demand for porous carbons is expected to continue to grow with the development of these types of technologies, the production of porous carbons under sustainable conditions must be seen as an important challenge.3 Highly activated porous carbons with specific surface areas of over 3000 m2 g−1 have been synthesized by KOH activation of different carbon sources, such as coal, coke, biomass, polymers or nanosized carbons.4–9 However, the use of toxic and corrosive activating agents, such as KOH, incurs operational drawbacks that hinder their implementation on an industrial scale. As an alternative, the use of milder porogenic agents such as KHCO3, K2CO3, potassium oxalate or eutectic salt melts has been explored with some degree of success,10–14 opening the way to more sustainable and environmentally friendly activation processes. On the other hand, biomass and biomass derivatives are the preferred carbon sources owing to their wide availability and low cost.15,16 Generally, the direct carbonization of biomass gives rise to carbons with low or negligible pore development, and chemical or physical activation approaches need to be applied for the generation of porosity within the carbons.15 Traditional activation methods provide carbons with a micropore dominant structure, and consequently mass transport kinetics is restricted.17 In contrast, the presence of meso- and macropores helps to reduce diffusion resistance and improves pore connectivity.17,18 In this regard, carbons with a hierarchical porosity (i.e., carbons with multiple and well-defined porous systems at different length scales) are attracting a great deal of interest in many fields due to the beneficial features of their porous structures with their short diffusion pathways. Hierarchically porous carbons with a large surface area have been prepared by combining different templating methods with physical or chemical activation.19–21 In most cases, these approaches require the use of expensive templates and harmful or corrosive chemicals for the removal of templates or activation products, which hinders their implementation on a large scale. It is therefore desirable to find more simplified synthetic routes that reduce costs, time and environmental impact. Inspired by the bread making process, Deng et al. prepared hierarchically porous carbons by the pyrolysis of a mixture of KHCO3 and different biomass or biomass-derived compounds.12 On the basis of their observations, they concluded that macropores originate due to the gas-generating decomposition of KHCO3, while mesopores and micropores arise due to the reaction of K2CO3 and the carbonized product. Qian et al. also prepared hierarchically porous carbon with a SBET of 1090 m2 g−1 by the co-pyrolysis of a mixture of sodium glutamate and NaCl.22 In this case, NaCl acted as the template for the macropores while micro- and mesopores were formed by the reaction of the carbonized product with CO2 and the intercalation of Na. NaCl has also been used as a salt template for the synthesis of hierarchically porous carbons from lysine, glucose or citric acid, all of which have a specific surface area in the 400–750 m2 g−1 range.23–25 Zhu et al. used various inorganic salts (NaCl, Na2CO3 and Na2SiO3) with different crystal sizes for the synthesis of 3D hierarchically porous carbons using glucose as the carbon source.26 Each ingredient generated cavities in different macro- or meso-pore ranges, and a moderate micropore content (SBET = 1090 m2 g−1) was formed due to the pyrolysis of glucose. Despite their suitable 3D structure, most of these materials possess a modest micropore volume. Since micropores mainly determine the ion storage capacity of these materials, the search for new synthetic routes towards hierarchically porous carbon with an ultra-high surface area and abundant micropores still constitutes an important scientific challenge.In this work, we present a novel sustainable approach for the synthesis of hierarchically porous carbons with a high surface area using a type of tannin as the carbon precursor. Tannins are a family of naturally abundant polyphenols that can be extracted from the bark of trees. They have been proven to be adequate precursors for the synthesis of a variety of carbon materials, such as carbon microspheres27,28 or carbon foam,29 that exhibit a poor or negligible microporosity. Porous tannin-based carbons with a SBET of 1180 m2 g−1 were prepared by Luo et al. by the pre-condensation of a tannin–urea mixture followed by its carbonization in the presence of ZnCl2.30 Szczurek et al. reported the synthesis of tannin–formaldehyde hydrogels activated with KOH, with surface areas of 1800 m2 g−1.31 Tiruye et al. described the preparation of porous carbons (SBET of 1570 m2 g−1) from tannic acid by using a NaCl/ZnCl2 eutectic mixture as the porogenic agent. Herein, we propose a salt template-assisted chemical activation strategy for the production of highly porous carbon networks from tannic acid. Tannic acid, a hydrolysable type of tannin, was selected as the carbon source due to a combination of attractive characteristics. It is a cheap and sustainable carbon precursor, and its carbonization in an inert atmosphere gives rise to char in a much higher yield than that obtained using other biomass precursors (over 25 wt%).32 Moreover, tannic acid melts at a relatively low temperature of ca. 220 °C, which makes it suitable for template carbonization. Our approach is based on the use of KCl which acts as a template at low temperatures (<300 °C) and provides a suitable reaction medium at higher temperatures (>750 °C) and K2CO3 as both the template and activating agent. These inorganic particles are coated with tannic acid during the carbonization/activation treatment, after which they can be easily removed by washing with water. The resultant porous carbons have a specific surface area in the range of 2250–2750 m2 g−1 and a sponge-like structure consisting of thin carbon walls that define interconnected macroscopic voids. The electrochemical performance of the tannic acid-derived porous carbons was analyzed in aqueous (1 M H2SO4) and organic (1 M TEABF4/AN and EMIMTFSI/AN) electrolytes. Due to their high micropore content and hierarchically porous structure, these carbons showed a high capacitance and an excellent electrochemical response at high current densities.
Experimental section
Synthesis of hierarchically porous carbons from tannic acid
Tannic acid was used as the carbon precursor for the synthesis of the carbons. In a typical synthesis, 2.4 g of tannic acid (Aldrich), 2.4 g of K2CO3 (Aldrich) and 16 g of KCl (weight ratio: tannic acid/K2CO3/KCl = 1/1/6.7) were added to 60 mL of distilled water under stirring. Once dissolved, the mixture was frozen in liquid nitrogen and then lyophilized (Telstar Cryodos) at −50 °C and 0.06 mbar. 6 g of this freeze-dried mixture was then pyrolyzed at 750, 800, 850 or 900 °C for 1 h (heating rate of 3 °C min−1) under a nitrogen flow. To remove the inorganic impurities, the carbonized product was simply washed with hot water, and then dried in an oven at 120 °C for 2 h. The carbon samples were denoted as CK-X, where X is the activation temperature in °C. For comparison purposes, a carbon sample was synthesized in the absence of the activating agent at 800 °C using a tannic acid/KCl weight ratio of 1/6.7 (sample code: TK). Another carbon sample was synthesized at 800 °C using only tannic acid and K2CO3 in a weight ratio of 1/1 (sample code: CK).In addition, other activating agents or salt templates were explored. We examined the use of potassium bicarbonate and potassium oxalate as activating agents. In these cases, the activation temperature chosen was 800 °C and the tannic acid/activating agent/KCl weight ratio was 1/1/6.7. These carbon samples were denoted as BK and OK for potassium bicarbonate and potassium oxalate, respectively. Other carbons were synthesized at a temperature of 800 °C by using K2CO3 as the activating agent and NaCl or Na2CO3 as the salt template in the same proportion as before (1/1/6.7). These samples were denoted as CK-N and CK-C, respectively. Additionally, we analyzed the effect that the mixing procedure of the reactants has upon the characteristics of the carbon. In this case, the synthesis procedure involved the freezing of the aqueous mixture in a fridge at −20 °C overnight (instead of with liquid nitrogen) followed by freeze-drying (sample code: CK-F). Another sample was synthesized by mechanically mixing the dry components in a mortar (sample code: CK-M). In both cases, the composition of the reaction mixture was tannic acid/K2CO3/KCl = 1/1/6.7 and the carbonization temperature was 800 °C.
Physicochemical characterization
Scanning electron microscopy (SEM) images were obtained using Quanta FEG650 (FEI) apparatus and transmission electron microscopy (TEM) images were recorded using a JEOL instrument (JEM 2100-F). Thermogravimetric analysis was performed on Q600 TGA apparatus from TA Instruments. X-ray diffraction (XRD) patterns were obtained on a Siemens D5000 instrument operated at 40 kV and 20 mA using a Cu Kα radiation source (λ = 0.15406 nm). Nitrogen adsorption isotherms were recorded at −196 °C using a Micromeritics ASAP 2020 apparatus. Around 0.1 g of the sample, previously degasified at 200 °C for 1 h, was used for each analysis. The apparent surface area was calculated by the BET method. An appropriate relative pressure range was selected to ensure a positive line intersect of multipoint BET fitting (C > 0) and an increase of Vads(1 − p/p0) with p/p0. The total pore volume (VP) was calculated from the amount of nitrogen adsorbed at a relative pressure (p/p0) of 0.90. Pore size distributions were determined by applying the Quenched-Solid Density Functional Theory (QSDFT) method to the nitrogen adsorption data assuming a slit pore model. The pore volume corresponding to micropores (V<2 nm) was determined by the QSDFT method. Elemental analysis of the samples was carried out on a LECO CHNS-932 microanalyzer. To determine the DC electrical conductivity of the porous carbons, a home-made apparatus was used: the carbon sample (∼50 mg) was placed inside a hollow Nylon cylinder with an inner diameter of 8 mm and pressed in between two stainless-steel plungers under a pressure of 7.1 MPa. The thickness of the disc formed by the pressed powdered carbon was determined by measuring the displacement of the upper plunger using a cathetometer. A certain current was passed through the plunger–sample–plunger system by using a DC power source (Promax, DC-405) and then the voltage was measured with a multimeter (Fluke, 45 Dual display). The packing density of the carbon was determined in the same experiment. Temperature programmed desorption (TPD) experiments were carried out on a Micromeritics AutoChem II 2920 coupled to a Pfeiffer-Vacuum OmniStar quadrupole mass spectrometer. Each sample was heated to 900 °C under an argon flow at a heating rate of 3 °C min−1, and the evolution of CO and CO2 (m/z of 28 and 44, respectively) was recorded.Electrochemical characterization
Electrodes were prepared by mixing 85 wt% active material, 10 wt% polytetrafluoroethylene (PTFE) binder (60 wt% suspension in water, Aldrich) and 5 wt% carbon black (Super C65, Timcal) using a small amount of ethanol. The slurries were formed into disc-shaped electrodes with a diameter of 1 cm which were then assembled into two-electrode Swagelok™ type cells. For the determination of the packing density of the electrodes, their thickness was measured with the help of a micrometer. Supercapacitors were built using two electrodes of comparable mass (carbon loading of ca. 10 mg cm−2), electrically isolated by a glassy fibrous separator. Electrochemical measurements were performed using aqueous 1 M H2SO4, 1 M TEABF4 in acetonitrile or EMIMTFSI (1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide, 99%, Ionic Liquids Technologies, Germany) in acetonitrile (wt ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as the electrolyte.
:1) as the electrolyte.
Electrochemical impedance spectroscopy (EIS), cyclic voltammetry (CV) and galvanostatic charge/discharge (CD) experiments were performed at room temperature using a computer-controlled potentiostat (Bio-Logic VMP3 multichannel generator). EIS curves were obtained within a frequency range of 1 mHz to 100 kHz. Cyclic voltammetry experiments were conducted using a cell voltage of 1 V in 1 M H2SO4, 2.7 V in 1 M TEABF4 and 3 V in EMIMTFSI/AN at increasing sweep rates from 1 mV s−1 to 100 mV s−1. Plots of electrode specific capacitance (expressed in farads per gram of active material) vs. cell voltage were calculated using the formula:
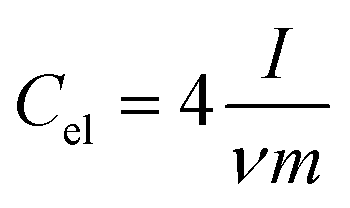 | (1) |
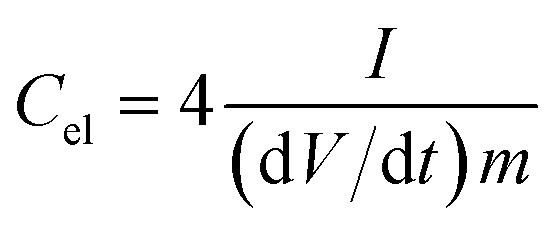 | (2) |
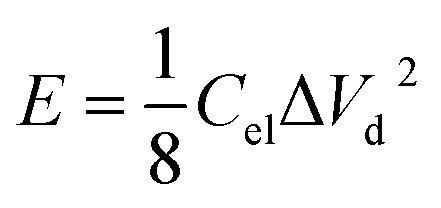 | (3) |
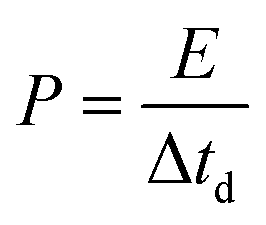 | (4) |
000 cycles. The cycling was performed at a current density of 5 A g−1 for the aqueous electrolyte and 10 A g−1 for 1 M TEABF4 and EMIMTFSI/AN.
Results and discussion
Synthesis and structural properties of the hierarchically porous carbons
Our strategy to produce hierarchically porous carbons from tannic acid involves two key ingredients: (a) a compound (activating agent) that reacts with tannic acid-derived products giving rise to numerous narrow pores, and (b) an unreactive salt (KCl). At low temperatures, KCl acts as a template, generating large voids in the carbonaceous matter, while at higher temperatures it provides a confined reaction medium as a consequence of the formation of a melted phase (the KCl–K2CO3 mixture forms a eutectic liquid mixture above ∼740 °C, vide infra). This synthesis strategy is based on the fact that tannic acid is a substance that melts at a relatively low temperature (∼220 °C). In this state, tannic acid is able to flow and occupy the interparticle voids, which builds up a sponge-like structure due to the templating effect of the inorganic particles (KCl and K2CO3). The synthesis procedure is illustrated in Scheme 1. In the first steps of the thermal treatment (at ca. 220 °C) the tannic acid melts and then fills the interstices in the mixture, covering the KCl and K2CO3 particles. At higher temperatures, the already carbonized product reacts with K2CO3 through redox reactions that generate pores within the carbon framework. Finally, the porous carbon can be recovered from the carbonized product because the inorganic matter is easily removed by merely washing with hot water.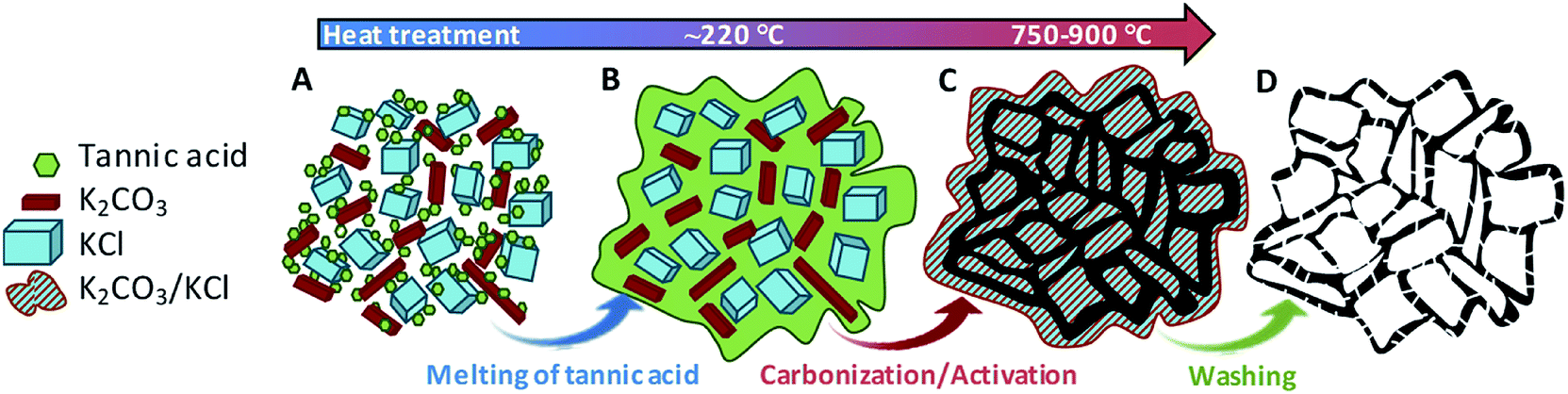 | ||
| Scheme 1 Illustration of the synthesis procedure. (A) Blend of the raw materials; (B) mixture of melted tannic acid and the inorganic salts; (C) carbon–inorganic salt composite; (D) hierarchically porous carbon. | ||
The morphology of the carbon materials was examined by scanning electron microscopy (SEM). As can be seen in Fig. 1a, the carbonized product consists of a continuous carbon matrix embedded within a melted-solidified inorganic phase made of KCl, unreacted K2CO3 and other potassium products. The inorganic phase present in the carbonized sample can be successfully washed out using only water. As shown in Fig. S1,† EDX spectra of the carbonized products before and after washing confirm the complete removal of the potassium salts. The resulting carbon particles exhibit a hierarchical sponge-like structure made up of thin carbon layers surrounding interconnected macroscopic voids (Fig. 1b). This macrostructure was maintained regardless of the temperature of thermal treatment (Fig. S2†). TEM inspection of the porous carbons confirmed the sponge-like structure (Fig. 1c) and revealed that the carbon sheets contain numerous randomly distributed micropores (Fig. 1d), produced by the reaction of carbon with K2CO3. The combination of the chemical activation and the templating effect of the inorganic particles is responsible for the targeted hierarchical micro–macroporous structure defined by large voids and thin carbon layers containing abundant micropores. When the carbon was prepared in the absence of K2CO3 (sample TK) it exhibited a sponge-like structure due to the templating effect of KCl (Fig. S3a and b†) but displayed poor pore development (vide infra). On the other hand, the carbon prepared using only tannic acid and K2CO3 (sample CK) does not have the sponge-like structure (Fig. S3c and d†). These results show that both inorganic ingredients are necessary to obtain a hierarchical microstructure that ensures the development of microporosity inside thin carbon layers, thereby guaranteeing short diffusion pathways (fast kinetics).
 | ||
| Fig. 1 SEM micrographs of a carbonized sample before (a) and after (b) washing. TEM images of a hierarchically porous carbon (c and d). | ||
The porous structure of the hierarchically porous carbons was analyzed by means of N2 physisorption experiments at −196 °C. The N2 adsorption isotherms of the carbons prepared at different temperatures (from 750 to 900 °C) are shown in Fig. 2a. All of the isotherms display a sharp increase in the amount of nitrogen adsorbed at very low relative pressures, indicative of a porosity made up mainly of micropores (pores with a size < 2 nm). The presence of large mesopores or macropores is also evidenced by the pronounced nitrogen uptake at high relative pressures (p/p0 > 0.9). As the carbonization temperature increases from 750 to 900 °C, there is a gradual increase in nitrogen uptake within the 0.1–0.8 relative pressure range, reflecting an enlargement of the pore size from the micropore to the mesopore range. This result is corroborated by the pore size distributions in Fig. 2b, which show that the porosity of these carbons is made up of two pore systems corresponding to: (a) narrow micropores of ∼0.8 nm and (b) wider pores with a size > 1 nm. It can be seen that, as the carbonization temperature increases, there is an enlargement of the pores corresponding to the second system. This is remarkable in the case of carbons CK-850 and CK-900, which have a high number of mesopores. The results clearly show that it is possible to tune the porosity in the micropore and narrow mesopore range by selecting a suitable carbonization temperature. As will be explained later, this finding is of special importance for the performance of the carbons when they are used as electrodes in supercapacitors. The main textural parameters calculated from the N2 adsorption experiments are listed in Table 1. The textural characteristics vary with the carbonization temperature, and carbon CK-900 attained the highest values of specific surface area and total pore and micropore volumes (SBET = 2740 m2 g−1, VP = 1.39 cm3 g−1, V<2 nm = 1.07 cm3 g−1). As mentioned above, all the carbons are highly microporous, especially the sample synthesized at the lowest temperature (CK-750), in which the micropore volume accounts for 87% of the total pore volume. This value decreases slightly to 77% in CK-900 as a consequence of the widening of pores at higher carbonization temperatures.
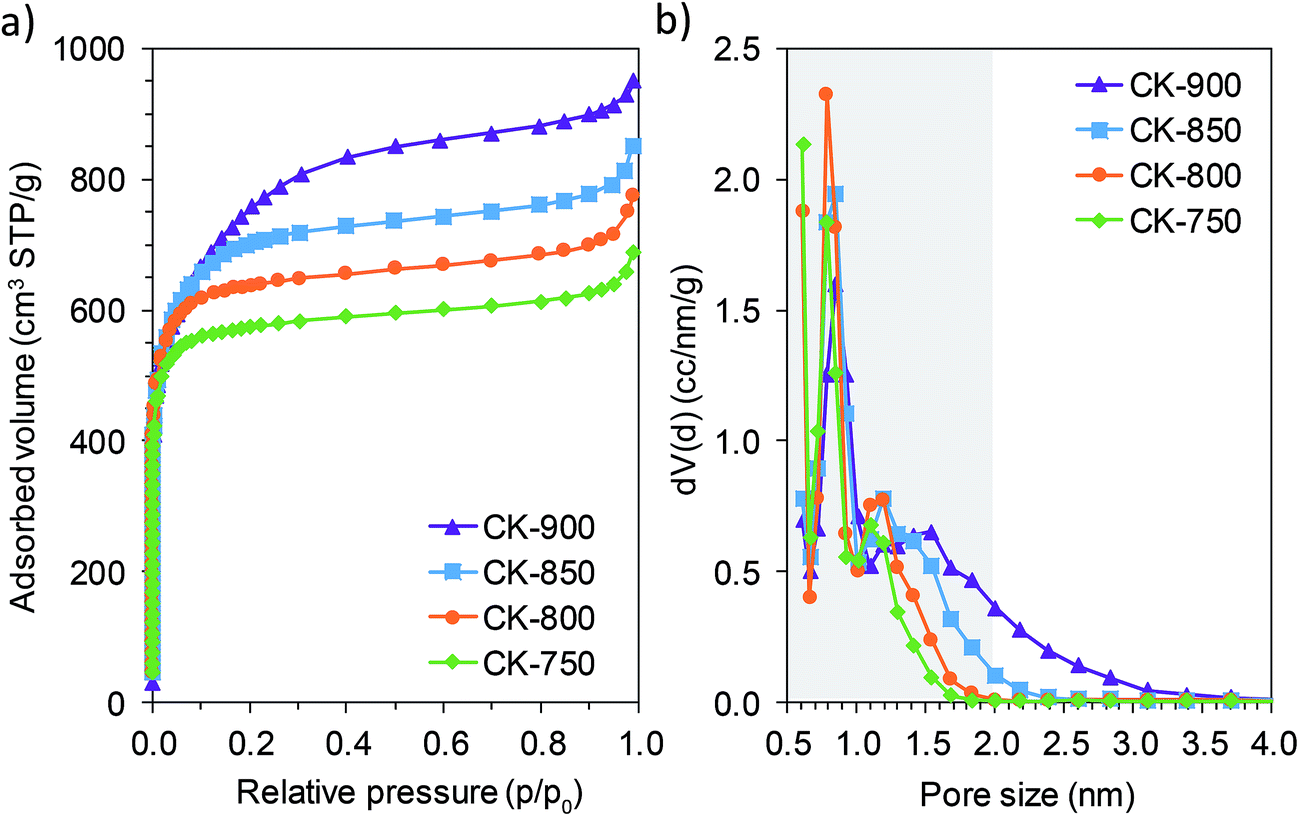 | ||
| Fig. 2 N2 adsorption isotherms (a) and pore size distributions (b) of porous carbons prepared at different temperatures. The shaded area in (b) indicates the micropore range. | ||
| Sample code | Textural properties | Yielda (%) | Conductivity (S cm−1) | Packing density (g cm−3) | ||
|---|---|---|---|---|---|---|
| S BET (m2 g−1) | V p (cm3 g−1) | V <2 nm (cm3 g−1) | ||||
| a Yield calculated by dividing the weight of porous carbon by the weight of tannic acid in the mixture prior to pyrolysis. | ||||||
| CK-750 | 2280 | 0.97 | 0.84 | 38.1 | 1.60 | 0.31 |
| CK-800 | 2510 | 1.08 | 0.93 | 34.4 | 1.65 | 0.34 |
| CK-850 | 2670 | 1.20 | 1.03 | 34.4 | 2.09 | 0.30 |
| CK-900 | 2740 | 1.39 | 1.07 | 32.1 | 3.54 | 0.32 |
The microstructure of the carbon samples was examined by X-ray diffraction. The XRD patterns of CK-800, TK and CK (also synthesized at 800 °C but in the absence of K2CO3 and KCl, respectively) are shown in Fig. S4.† They present low intensity broad bands centred at ca. 22 and 43° typical of turbostratic carbon structures with a very low degree of crystallinity. The large micropore content of CK-800 and CK is evidenced by the intense scattering peak recorded at low values of 2θ.34 The chemical composition of the porous carbons synthesized at different temperatures was evaluated by elemental analysis (see Table S1†). All of them have a low oxygen content ranging from 6.1 to 8.9 wt% (O/C atomic ratio of 0.05–0.07), which decreases with the carbonization temperature. The carbons display a remarkable electrical conductivity that increases from 1.60 S cm−1 in CK-750 to 3.54 S cm−1 in CK-900 as a consequence of the lower oxygen content and the more condensed microstructures expected from using higher carbonization temperatures (see Table 1). The packing density is another parameter that is important for the use of these materials as electrodes and, therefore, the packing density of the carbons in the powdered form was measured. The values obtained ranged from 0.30 to 0.34 g cm−3.
Other synthesis strategies for producing porous carbons from tannic acid were also explored. In this sense, we examined the use of alternative activating agents, salt templates or blending conditions. Thus, as alternatives to K2CO3, other activating agents such as potassium bicarbonate (sample BK) and potassium oxalate (sample OK) were tested. The SEM images of these carbon samples are shown in Fig. S5.† It can be seen that these materials also have a sponge-like macrostructure. The textural characteristics of these carbons are listed in Table S2† and the N2 sorption isotherms and PSDs are plotted in Fig. S6a and b.† The results show that activation with KHCO3 and K2C2O4 also led to hierarchically porous carbons with large specific surface areas of 2180 m2 g−1 and 1990 m2 g−1, and high pore volumes of 0.91 and 0.84 cm3 g−1, respectively. However, the carbon produced with K2CO3 still provided the best textural properties (i.e. a higher BET surface area and total pore and micropore volumes). When the tannic acid was carbonized without using an activating agent (sample TK – mixture of tannic acid and KCl), the carbon obtained exhibited poor textural development: a BET surface area of 510 m2 g−1 and a pore volume of 0.48 cm3 g−1. Interestingly, this material had a small micropore volume (0.16 cm3 g−1) but a large textural porosity, as can be deduced from the large nitrogen uptake at p/p0 > 0.8 (see Fig. S6a†), due to the templating effect of KCl.
The synthesis of carbon samples using other salt templates such as NaCl (code: CK-N) or Na2CO3 (code: CK-C) was also investigated. The use of NaCl or Na2CO3 as the templates gives rise to sponge-like carbons with smaller voids than those of CK-800, as observed by SEM inspection (Fig. S7†). On the other hand, sample CK which was prepared in the absence of an inert salt, had a bulky microstructure (Fig. S3c and d†). The N2 adsorption isotherms and pore size distributions of these carbons are plotted in Fig. S6c and d.† Compared to CK-800, samples CK-N, CK-C and CK exhibit a lower N2 uptake at low relative pressures, as a consequence of their lower micropore content. Indeed, the BET specific surface areas of CK, CK-N and CK-C are 1770, 1890 and 1830 m2 g−1, respectively (Table S2†). These values are significantly lower than that of CK-800 (2510 m2 g−1), which suggests that the presence of KCl as the salt template enhances the porogenic behavior of K2CO3. This may be explained by the fact that the melted phase formed by KCl and K2CO3 at temperatures of ∼740 °C can act as a confining medium that will boost the activation reaction between the potassium carbonate and the carbon (vide infra). The sorption isotherms plotted in Fig. S6† show that, at relative pressures higher than 0.8, the carbons prepared with NaCl, Na2CO3 and no salt template exhibit a pronounced N2 uptake due to their large-mesopore and small-macropore content, which is in agreement with the presence of small voids detected by microscopy.
The way in which the materials are blended prior to pyrolysis is also a decisive step that has a strong influence on the porous structure of the carbons. In this respect, a carbon sample was synthesized in the same way as CK-800 with the exception that the aqueous mixture was frozen in a fridge at −20 °C instead of in liquid N2 (−196 °C). The resulting carbon material (labelled CK-F) has larger, prismatic macroscopic voids connected by thick carbon walls (Fig. S8a†). Another alternative consisting in the dry mixing of the solid reactants with the aid of a mill was also analyzed. The carbon material obtained in this way (sample code: CK-M) has a heterogeneous structure, with areas of bulky carbon and cavities of different sizes (Fig. S8b†). The textural properties of these carbons are listed in Table S2.† They exhibit large BET surface areas (2340 and 2130 m2 g−1 for CK-F and CK-M, respectively) and large pore volumes > 0.85 cm3 g−1, the porosity being made up almost exclusively of micropores (>85%). In spite of their good textural properties, the CK-F and CK-M samples have a poorer microstructure compared to the carbons obtained by freezing in liquid N2, which are characterized by a sponge-like network of thin microporous carbon layers enveloping large voids.
Investigations into the activation mechanism
Two important facts that greatly contribute to the success of the activation process need to be pointed out. The first fact is that tannic acid melts at a relatively low temperature (∼220 °C) in the presence of inorganic particles (i.e., KCl and K2CO3) acting as templates that determine the morphological structure (sponge-like) of the final carbon products. The second fact is that the KCl–K2CO3 mixture forms a eutectic liquid mixture above ∼740 °C.35 It can be envisaged that, having the carbonaceous matter immersed in the liquid inorganic phase containing the activating agent, the redox reactions between the semi-carbonized products and K2CO3 will occur with enhanced reactivity.In order to obtain an insight into the transformations taking place during the thermal treatment, thermogravimetric analysis and temperature-programmed desorption (TPD) experiments were performed. The TGA and DTG curves corresponding to the pure tannic acid and the mixture of tannic acid and K2CO3 (weight ratio: 1/1) are shown in Fig. 3a. In the case of the pure tannic acid, it can be seen that a slight weight loss occurs at temperatures below 100 °C due to the loss of moisture. Next, a large weight loss occurs in the 190–385 °C region in a two-step process with maximum peaks at ca. 255 °C and 300 °C. According to the findings of Xia et al.,32 the first degradation peak corresponds to the loss of gallic acid units in the outer layer in the form of benzenetriol and CO2 (see Fig. S9†). At higher temperatures, the benzenediol units still linked to the central glucose ring crosslink through inter- and intra-molecular reactions that give rise to a carbon-rich condensed phase as well as the release of CO2 and different aromatic compounds.32 The slow heating rate employed in the present study (3 °C min−1) favors cross-linking reactions between the semi-degraded products32 and, in consequence, at 800 °C the carbon is obtained in a yield of 24 wt%. When the tannic acid is pyrolyzed in the presence of K2CO3, the weight loss peaks corresponding to thermal degradation shift to lower temperatures (155 °C and 245 °C). This fact can be ascribed to the catalytic effect of K2CO3, which has already been observed by other authors in relation with the carbonization of different carbon precursors.13,14,36,37 At temperatures above 700 °C there is a sharp weight loss, which can be ascribed to the redox reaction between the already carbonized product and K2CO3:
| K2CO3 + 2C → 2K + 3CO | (5) |
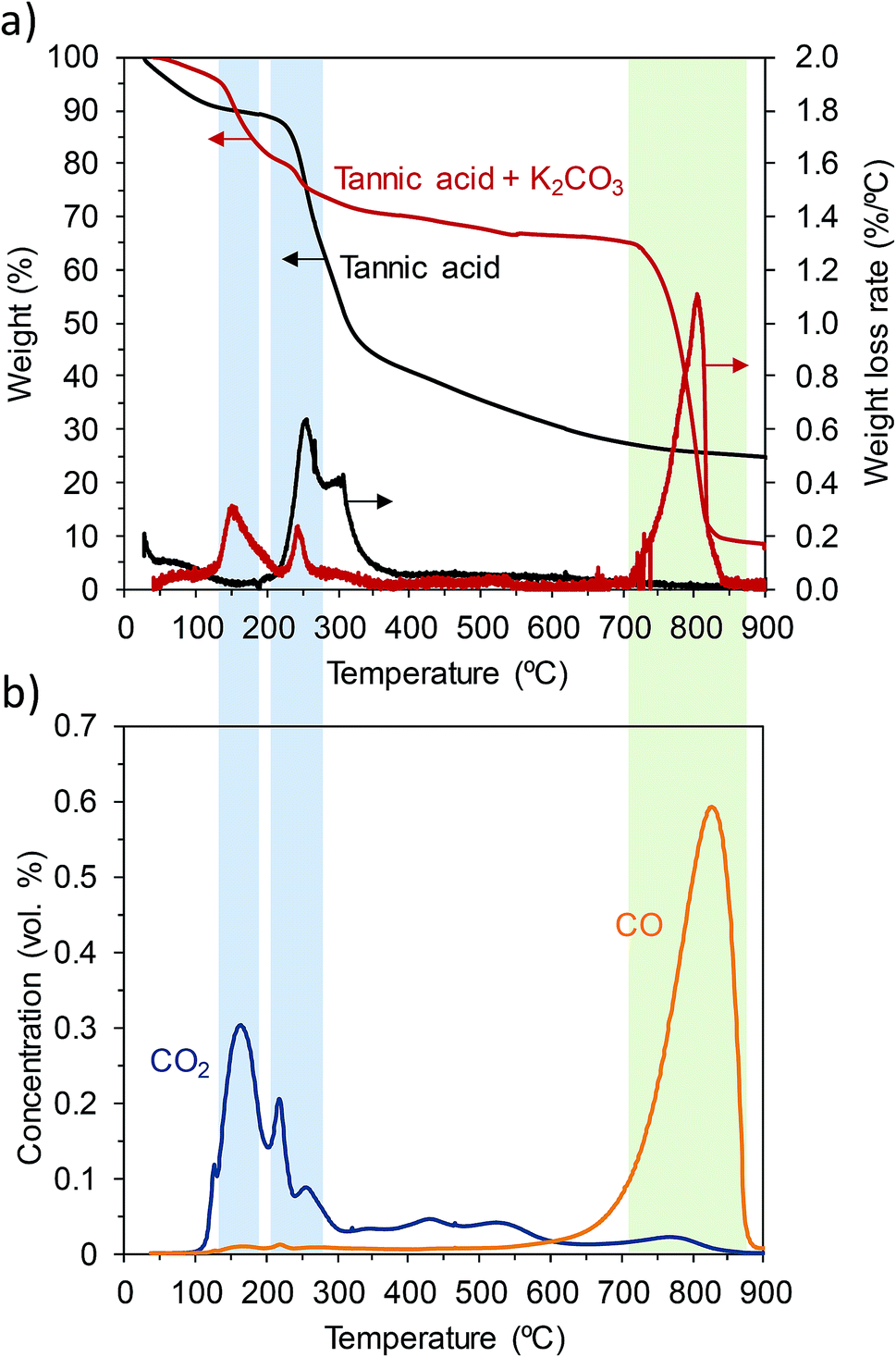 | ||
| Fig. 3 (a) Thermogravimetric (TGA) and differential thermogravimetric (DTG) curves of the pyrolysis of tannic acid and a mixture of tannic acid and K2CO3 and (b) CO2 and CO TPD spectra obtained from a mixture of tannic acid and K2CO3 (heating rate: 3 °C min−1; N2 and Ar atmospheres for TGA and TPD experiments, respectively). | ||
These conclusions have been corroborated by the TPD analysis of a mixture of tannic acid and K2CO3. The results of the analysis displayed in Fig. 3b show the variation in the concentration of evolved gases (CO2 and CO) with temperature. The amounts of CO2 and CO correlated well with the weight loss measured by TGA. Thus, CO2 is mainly released at 150–250 °C, which agrees with the degradation mechanism proposed by Xia et al. (vide supra).32 It can also be seen that, at higher temperatures, small amounts of CO2 are still released due to the condensation and polymerization reactions typical of the carbonization process. CO is mainly released in the 650–900 °C range, due to the redox reaction between the carbonized product and K2CO3 (eqn (5)). Since the amount of CO evolved during the experiment can be calculated by the integration of the TPD curve, we can estimate, on the basis of the stoichiometry of eqn (5), that the degree of conversion of K2CO3 at a temperature of 800 °C was of the order of ca. 30%. The slow thermal decomposition of unreacted K2CO3 at temperatures higher than 800–850 °C (eqn (6)) constitutes a secondary process that also contributes to the generation of porosity within the carbons. CO2 released during the decomposition of K2CO3 results in the gasification of the carbon (eqn (7)), which is responsible for the higher pore development and the larger pore size observed in the carbons activated at 850 °C and, especially, at 900 °C (see Table 1 and Fig. 2b).
| K2CO3 → K2O + CO2 | (6) |
| C + CO2 → 2CO | (7) |
A relevant characteristic of the porous carbons obtained in this way is that they can be produced in high yields. Indeed, the carbon yields (grams of porous carbon per 100 grams of tannic acid) for these carbons are, in all cases, higher than 30% (see Table 1). Surprisingly, the yields of the simultaneous carbonization/activation processes were higher than that of carbonization of solely tannic acid (25%, see Fig. 3a) or the mixture of tannic acid and KCl (26%, Table S2†). In order to explain these high yields, it is important to remember that the carbonization and activation reactions occur within the confined space provided by the melted KCl–K2CO3 mixture. Under these circumstances, the release of volatile compounds is severely restricted and a re-deposition of a fraction of the initially emitted gaseous substances will occur, which explains the observed increase in the carbon yield. In addition, the closer interaction between the carbonized products and the activating agent would favour the activation reactions and, in consequence, will lead to an improvement in pore development, as evidenced by the comparison of the textural properties of carbons produced in the presence of KCl (CK-800) and in its absence (CK). In previous studies, we reported similar results when the activation was performed in the presence of KCl with Na2S2O3 as the activating agent.38,39 Another factor that can contribute to the high carbon yield is the catalytic action of potassium carbonate that reduces the emission of volatiles, thereby favoring the formation of carbonaceous solid products.36 The high carbon yields provided by this synthesis procedure contrasts with the low yields typically associated with the preparation of highly porous carbons from biomass products. Table S3† shows the results of the textural properties and carbon yields for different porous carbons synthesized from biomass products. The table compares the BET surface areas of various K2CO3-activated carbons37,40–45 and of other recently reported highly porous carbons synthesized using other activating agents.5–7,22,31,46,47 In general, the porous carbons that exhibit high pore development (SBET > 2000 m2 g−1) are obtained at the expense of extensive carbon etching that results in a poor carbon yield (5–15%). In contrast, high carbon yields usually imply poor textural development. When tannic acid is used as the carbon source and K2CO3 as the activating agent it is possible to combine high carbon yields (>30%) with good textural development (SBET > 2000 m2 g−1). In support of this, Fig. 4 shows the textural parameters of different porous carbons expressed per gram of raw material. It can be seen that the porous carbon produced from tannic acid is situated amongst the highest values, and is only exceeded by the gulfweed-based carbon reported by Li et al.5 However, the synthesis approach they employed consisted in a two-heating step procedure based on the use of a corrosive activating agent such as KOH (KOH/gulfweed weight ratio = 4). Another point that is important to highlight regarding our synthesis strategy is that it requires the use of a relatively small amount of the activating agent (tannic acid/K2CO3 wt ratio of 1/1), unlike the KOH-activated carbons that need high weight ratios, normally in the 2–5 range.4
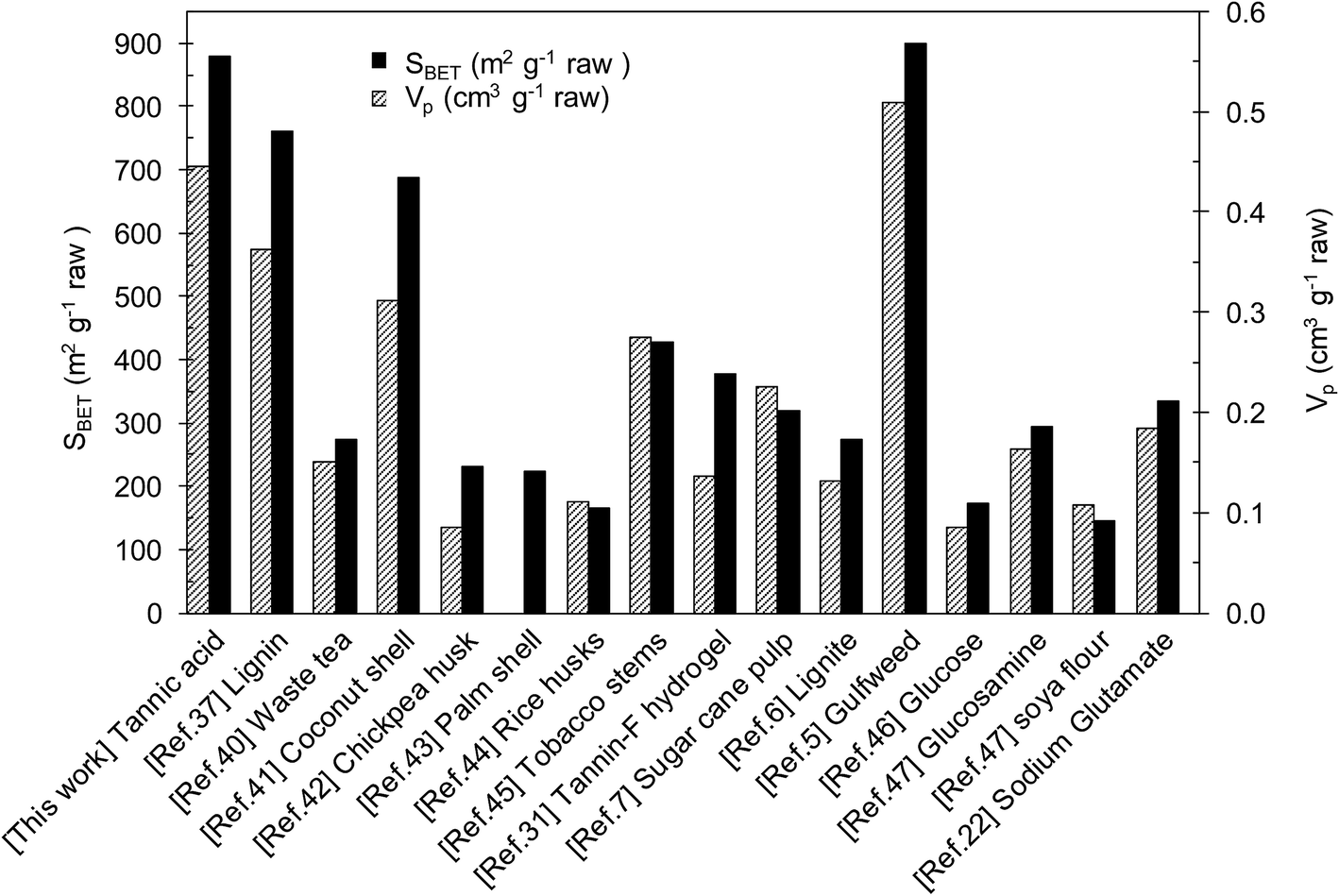 | ||
| Fig. 4 Textural characteristics, expressed per gram of raw material, of activated carbons prepared by using different precursors and activating agents. | ||
Electrochemical performance of the hierarchically porous carbons
The materials developed in this work possess a combination of properties which makes them attractive for their implementation in capacitive energy storage devices, either by themselves or as supports for electroactive species. They have good electronic conductivity (ca. 2–4 S cm−1) that promotes fast electron conduction/transport, a 3D architecture composed of thin carbon walls which ensure short diffusion pathways for fast ion transport, and a high surface area due mainly to micropores which are the most efficient ion stores. Consequently, high specific capacitances as well as a high capacitance retention at high rates can be expected from these materials. In view of their characteristics, CK-800 (micro–macroporous) and CK-900 (micro–meso–macroporous) carbons were selected and processed into electrodes with a carbon loading analogous to that of commercial devices (ca. 10 mg cm−2) and assembled in symmetric two-electrode cells. The electrodes made with CK-800 and CK-900 had a thickness of 300–350 μm and a packing density of 0.37 and 0.35 g cm−3, respectively. The electrochemical performance of these carbons was studied in different electrolytes (1 M H2SO4, 1 M TEABF4 in AN and EMIMTFSI in AN, wt/wt = 1/1), which allowed stable cycling of the cells at different voltages (1, 2.7 and 3 V, respectively).Their capacitance was calculated from the discharge branch of the galvanostatic profiles. The evolution of capacitance with increasing current density in the different electrolytes is shown in Fig. 5a–c. In 1 M H2SO4, the carbons exhibit capacitances up to ca. 260 F g−1 at low current densities and still retain capacitances as high as 98–116 F g−1 at an ultra-high current density of 80 A g−1. These values are superior to those of commercially available activated carbons (see Fig. 5a) and lie within the state-of-the-art for biomass-derived carbons even though we have used a higher electrode mass loading.15,48,49 The good rate capability of these materials was further corroborated by cyclic voltammetry. As can be seen in Fig. S10,† the voltammograms still preserve a rectangular shape at a scan rate of 100 mV s−1 with only a small reduction in the enclosed area, which is indicative of a small decrease in capacitance. It must be pointed out that this excellent electrochemical performance at high current loads was achieved despite the use of thick electrodes (340–350 μm) that approximate to carbon loadings typical of commercial devices. CK-800 shows higher values of capacitance over the whole range of current densities studied, probably due to its higher content of micropores in the range of ca. 0.7 nm (see Fig. 2b), which are the most efficient for EDL formation in aqueous electrolytes.50Fig. 6a shows the charge–discharge galvanostatic plots of both carbons in 1 M H2SO4 at a high current density of 20 A g−1. It can be seen that the discharge branches present low voltage drops and only a small equivalent distributed resistance (EDR). The low resistance to ion diffusion is also evidenced by the small Warburg region in the Nyquist plots, shown in Fig. S11a.† The rapid response of the cells built with these carbons is borne out by the low time constants (of 1.6 s in both materials) calculated from the characteristic frequency f0 at a phase angle of −45° (Fig. S11d†). All these results confirm the above assumptions, i.e. fast electron and ion transport is facilitated by the highly conductive 3D architecture and the large surface area and micropore content provide a high ion storage capacity. Finally, the stable cycling performance of the supercapacitors at 1 V was confirmed over 10000 cycles of charge–discharge at 5 A g−1, recording a capacitance retention higher than 90% in both systems (Fig. S12a†).
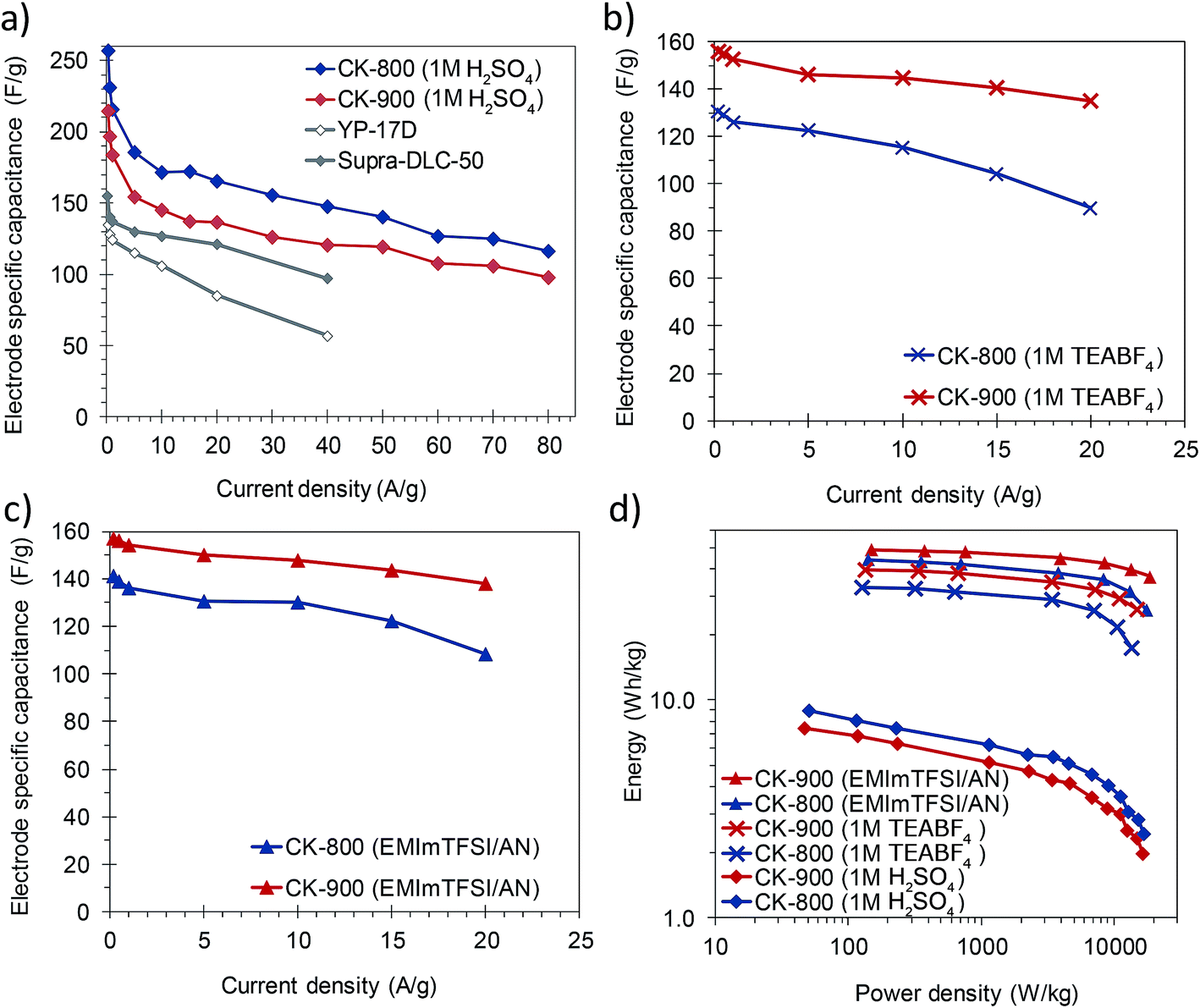 | ||
| Fig. 5 Rate capability of CK-800 and CK-900 in (a) 1 M H2SO4, (b) 1 M TEABF4 and (c) EMIMTFSI/AN, calculated by galvanostatic charge–discharge cycling. (d) Ragone plot of the symmetric cells. | ||
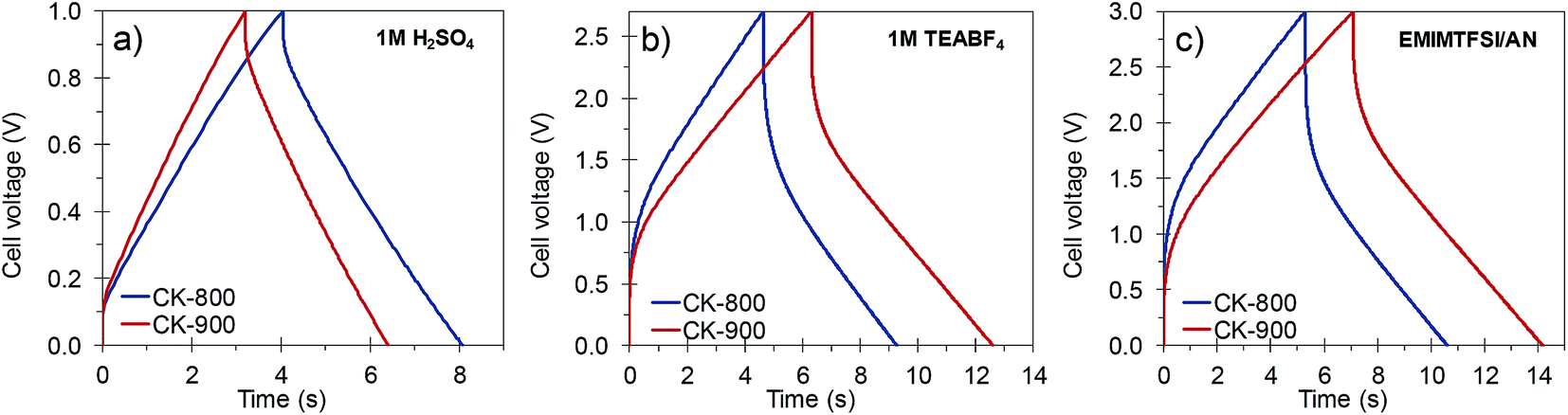 | ||
| Fig. 6 Galvanostatic charge–discharge profiles of the porous carbons in (a) 1 M H2SO4, (b) 1 M TEABF4 and (c) EMIMTFSI/AN at a current density of 20 A g−1. | ||
In order to increase the cell voltage and, therefore, the amount of energy stored in the device, the electrochemical performance of the carbons was also tested in the organic electrolyte 1 M TEABF4, as well as in the ionic liquid EMIMTFSI dissolved in AN. The use of these electrolytes allowed the cell voltage to be increased to 2.7 and 3 V, respectively. The stability at these high voltages was demonstrated over 10000 cycles of charge–discharge at 10 A g−1, with a capacitance retention higher than 85% in all the systems (see Fig. S12b and c†). The rate capability of these electrolytes – as observed in a series of galvanostatic charge/discharge experiments – is plotted in Fig. 5b and c. In both organic electrolytes the capacitance and rate capability of CK-900 are better than those of CK-800 over the whole range of current densities. Because of the favorable porous structure of CK-900 it was possible to achieve capacitance values of ca. 160 F g−1, as well as a capacitance retention of ca. 88% at a current density of 20 A g−1, in both organic electrolytes. Such values of capacitance and rate capability compare favorably with those of the state-of-the-art carbons.8,10,51 The values of surface area normalized capacitance of CK-900 are higher than those of CK-800 (CK-900: 5.7 μF cm−2 in both electrolytes; CK-800: 5.2 and 5.6 μF cm−2 in 1 M TEABF4 and EMIMTFSI/AN, respectively). From these results, it can be inferred that the larger size of the micropores in CK-900, as well as the presence of some small mesopores, makes it possible to take full advantage of these electrolytes. The higher capacitance values and rate capability of CK-900 in the organic electrolytes were also confirmed by cyclic voltammetry (Fig. S10b and c†). A slightly higher resistance to ion diffusion in the micropores of CK-800 is evidenced by the larger EDR observed in the galvanostatic charge–discharge curves (Fig. 6b and c) and the larger Warburg region in the Nyquist plots (Fig. S11b and c†). Thus, the values of the EDR deduced from the Nyquist plots for CK-800 in 1 M TEABF4 and EMIMTFSI/AN are 2.1 ohm and 2.5 ohm, respectively, and they drop to 1.6 ohm and 1.7 ohm, respectively, for the CK-900 carbon. As a result, the time constants also decrease from 16.7 s in 1 M TEABF4 and 12.5 s in EMIMTFSI/AN for CK-800 to 9.1 s and 8.3 s, respectively, for CK-900. In the case of CK-900, the increase in the cell voltage when using the ionic liquid-containing electrolyte led to energy density values of 49 W h kg−1 at 151 W kg−1 and 37 W h kg−1 at a high power of 18.8 kW kg−1 (Fig. 5d).
Conclusions
Hierarchically porous carbons have been successfully synthesized by the salt template-assisted chemical activation of tannic acid. The thermal treatment of a mixture of tannic acid, KCl (acting as the template and confining reaction medium) and K2CO3 (acting as the template and activating agent) gave rise to a sponge-like carbon structure consisting of interconnected microporous thin carbon layers. No toxic or corrosive chemicals were used, thereby ensuring the sustainability and environmental friendliness of the process. The carbons produced in this way have a large specific surface area in the 2250–2750 m2 g−1 range and pore volumes of ∼1.0–1.4 cm3 g−1. Despite their extensive pore development and surface area, the carbons were produced in a yield of >30%, which contrasts with the low yields typically obtained when using other biomass or biomass-derived precursors. The porous carbons were tested as supercapacitor electrodes with a carbon loading of ca. 10 mg cm−2, and exhibited a high capacitance in aqueous (260 F g−1 in 1 M H2SO4) and organic (160 F g−1 in 1 M TEABF4 and EMIMTFSI/AN) electrolytes. Moreover, the hierarchical architecture of the carbons presented little resistance to ion diffusion in the three electrolytes, which allowed a fast response at high current densities.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research work was supported by the Spanish MINECO-FEDER (CTQ2015-63552-R) and regional GRUPIN2018 (IDI/2018/000148). We also acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).References
- J.-G. Wang, K. Xie and B. Wei, Nano Energy, 2015, 15, 413–444 CrossRef CAS.
- M. Jia, S. Lu, Y. Chen, T. Liu, J. Han, B. Shen, X. Wu, S.-J. Bao, J. Jiang and M. Xu, J. Power Sources, 2017, 367, 17–23 CrossRef CAS.
- L. Wei and G. Yushin, Nano Energy, 2012, 1, 552–565 CrossRef CAS.
- J. Wang and S. Kaskel, J. Mater. Chem., 2012, 22, 23710–23725 RSC.
- S. Li, K. Han, J. Li, M. Li and C. Lu, Microporous Mesoporous Mater., 2017, 243, 291–300 CrossRef CAS.
- Y. Wu, J.-P. Cao, X.-Y. Zhao, Z.-Q. Hao, Q.-Q. Zhuang, J.-S. Zhu, X.-Y. Wang and X.-Y. Wei, Electrochim. Acta, 2017, 252, 397–407 CrossRef CAS.
- K. Zou, Y. Deng, J. Chen, Y. Qian, Y. Yang, Y. Li and G. Chen, J. Power Sources, 2018, 378, 579–588 CrossRef CAS.
- Y. Zhu, S. Murali, M. D. Stoller, K. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz and M. Thommes, Science, 2011, 332, 1537–1541 CrossRef CAS.
- N. Díez, R. Mysyk, W. Zhang, E. Goikolea and D. Carriazo, J. Mater. Chem. A, 2017, 5, 14619–14629 RSC.
- M. Sevilla, G. A. Ferrero and A. B. Fuertes, Carbon, 2017, 114, 50–58 CrossRef CAS.
- N. Fechler, T. P. Fellinger and M. Antonietti, Adv. Mater., 2013, 25, 75–79 CrossRef CAS.
- J. Deng, T. Xiong, F. Xu, M. Li, C. Han, Y. Gong, H. Wang and Y. Wang, Green Chem., 2015, 17, 4053–4060 RSC.
- J. i. Hayashi, M. Uchibayashi, T. Horikawa, K. Muroyama and V. G. Gomes, Carbon, 2002, 40, 2747–2752 CrossRef CAS.
- J. i. Hayashi, T. Horikawa, I. Takeda, K. Muroyama and F. N. Ani, Carbon, 2002, 40, 2381–2386 CrossRef CAS.
- J. Deng, M. Li and Y. Wang, Green Chem., 2016, 18, 4824–4854 RSC.
- H. Lu and X. S. Zhao, Sustainable Energy Fuels, 2017, 1, 1265–1281 RSC.
- H. Wang, Z. Xu, A. Kohandehghan, Z. Li, K. Cui, X. Tan, T. J. Stephenson, C. K. King'ondu, C. M. Holt and B. C. Olsen, ACS Nano, 2013, 7, 5131–5141 CrossRef CAS PubMed.
- B. Conway, in Electrochemical Supercapacitors, Similarities and differences between supercapacitors and batteries for storing electrical energy, Springer, Boston, MA, 1999, pp. 11–31 Search PubMed.
- S. Dutta, A. Bhaumik and K. C.-W. Wu, Energy Environ. Sci., 2014, 7, 3574–3592 RSC.
- D. W. Wang, F. Li, M. Liu, G. Q. Lu and H. M. Cheng, Angew. Chem., 2008, 120, 379–382 CrossRef.
- A. H. Lu, G. P. Hao, Q. Sun, X. Q. Zhang and W. C. Li, Macromol. Chem. Phys., 2012, 213, 1107–1131 CrossRef CAS.
- W. Qian, J. Zhu, Y. Zhang, X. Wu and F. Yan, Small, 2015, 11, 4959–4969 CrossRef CAS.
- F. Pan, Y. Duan, A. Liang, J. Zhang and Y. Li, Electrochim. Acta, 2017, 238, 375–383 CrossRef CAS.
- S. Zhu, J. Li, Q. Li, C. He, E. Liu, F. He, C. Shi and N. Zhao, Electrochim. Acta, 2016, 212, 621–629 CrossRef CAS.
- Z. Wang, S. Luo, F. Chen, D. Wang, Y. Liu, X. Qi, C. Shi and N. Zhao, RSC Adv., 2016, 6, 54718–54726 RSC.
- S. Zhu, J. Li, C. He, N. Zhao, E. Liu, C. Shi and M. Zhang, J. Mater. Chem. A, 2015, 3, 22266–22273 RSC.
- F. L. Braghiroli, V. Fierro, M. T. Izquierdo, J. Parmentier, A. Pizzi and A. Celzard, Bioresour. Technol., 2014, 151, 271–277 CrossRef CAS PubMed.
- F. L. Braghiroli, V. Fierro, M. T. Izquierdo, J. Parmentier, A. Pizzi and A. Celzard, Carbon, 2012, 50, 5411–5420 CrossRef CAS.
- G. Tondi, V. Fierro, A. Pizzi and A. Celzard, Carbon, 2009, 47, 1480–1492 CrossRef CAS.
- R. Luo, C. Liu, J. Li, C. Wang, X. Sun, J. Shen, W. Han and L. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 32737–32744 CrossRef CAS PubMed.
- A. Szczurek, G. Amaral-Labat, V. Fierro, A. Pizzi and A. Celzard, Microporous Mesoporous Mater., 2014, 196, 8–17 CrossRef CAS.
- Z. Xia, A. Singh, W. Kiratitanavit, R. Mosurkal, J. Kumar and R. Nagarajan, Thermochim. Acta, 2015, 605, 77–85 CrossRef CAS.
- M. D. Stoller and R. S. Ruoff, Energy Environ. Sci., 2010, 3, 1294–1301 RSC.
- Y. S. Yun, S. Y. Cho, J. Shim, B. H. Kim, S. J. Chang, S. J. Baek, Y. S. Huh, Y. Tak, Y. W. Park and S. Park, Adv. Mater., 2013, 25, 1993–1998 CrossRef CAS PubMed.
- J. Yaokawa, K. Oikawa and K. Anzai, Calphad, 2007, 31, 155–163 CrossRef CAS.
- J. Kopyscinski, M. Rahman, R. Gupta, C. A. Mims and J. M. Hill, Fuel, 2014, 117, 1181–1189 CrossRef CAS.
- J. i. Hayashi, A. Kazehaya, K. Muroyama and A. P. Watkinson, Carbon, 2000, 38, 1873–1878 CrossRef CAS.
- A. B. Fuertes, G. A. Ferrero, N. Diez and M. Sevilla, ACS Sustainable Chem. Eng., 2018, 6, 16323–16331 CrossRef CAS.
- M. Sevilla, N. Diez, G. A. Ferrero and A. B. Fuertes, Energy Storage Materials, 2019, 18, 356–365 CrossRef.
- I. I. Gurten, M. Ozmak, E. Yagmur and Z. Aktas, Biomass Bioenergy, 2012, 37, 73–81 CrossRef CAS.
- L. Yue, Q. Xia, L. Wang, L. Wang, H. DaCosta, J. Yang and X. Hu, J. Colloid Interface Sci., 2018, 511, 259–267 CrossRef CAS PubMed.
- J. i. Hayashi, T. Horikawa, K. Muroyama and V. G. Gomes, Microporous Mesoporous Mater., 2002, 55, 63–68 CrossRef CAS.
- D. Adinata, W. M. A. W. Daud and M. K. Aroua, Bioresour. Technol., 2007, 98, 145–149 CrossRef CAS PubMed.
- K. Foo and B. Hameed, Bioresour. Technol., 2011, 102, 9814–9817 CrossRef CAS PubMed.
- W. Li, L.-B. Zhang, J.-H. Peng, N. Li and X.-Y. Zhu, Ind. Crops Prod., 2008, 27, 341–347 CrossRef CAS.
- X. Liu and M. Antonietti, Carbon, 2014, 69, 460–466 CrossRef CAS.
- M. Sevilla, G. A. Ferrero and A. B. Fuertes, Chem. Mater., 2017, 29, 6900–6907 CrossRef CAS.
- H. Lu and X. Zhao, Sustainable Energy Fuels, 2017, 1, 1265–1281 RSC.
- S. Zhu, J. Li, C. He, N. Zhao, E. Liu, C. Shi and M. Zhang, J. Mater. Chem. A, 2015, 3, 22266–22273 RSC.
- F. Béguin and E. Frackowiak, Carbons for electrochemical energy storage and conversion systems, CRC Press, 2009 Search PubMed.
- Q.-L. Zhu, P. Pachfule, P. Strubel, Z. Li, R. Zou, Z. Liu, S. Kaskel and Q. Xu, Energy Storage Materials, 2018, 13, 72–79 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta01712g |
| This journal is © The Royal Society of Chemistry 2019 |