
Boosting inverted perovskite solar cell performance by using 9,9-bis(4-diphenylaminophenyl)fluorene functionalized with triphenylamine as a dopant-free hole transporting material†
Hong Duc
Pham‡
 a,
Lidón
Gil-Escrig‡
b,
Krishna
Feron
cd,
Sergei
Manzhos
e,
Steve
Albrecht
bg,
Henk J.
Bolink
*f and
Prashant
Sonar
*a
a,
Lidón
Gil-Escrig‡
b,
Krishna
Feron
cd,
Sergei
Manzhos
e,
Steve
Albrecht
bg,
Henk J.
Bolink
*f and
Prashant
Sonar
*a
aInstitute of Future Environment, School of Chemistry, Physics and Mechanical Engineering, Queensland University of Technology (QUT), 2 George Street, Brisbane, QLD-4001, Australia. E-mail: sonar.prashant@qut.edu.au
bYoung Investigator Group Perovskite Tandem Solar Cells, Helmholtz-Zentrum Berlin für Materialien und Energie GmbH, Kekuléstraße 5, 12489 Berlin, Germany
cCSIRO Energy Centre, NSW-2304, Australia
dCentre for Organic Electronics, University of Newcastle, Callaghan, NSW 2308, Australia
eCentre Énergie Matériaux Télécommunications, Institut National de la Recherche Scientifique, 1650, boulevard Lionel-Boulet, Varennes, QC J3X1S2, Canada
fInstituto de Ciencia Molecular Universidad de Valencia, Valencia 46022, Spain
gTechnical University Berlin, Faculty IV – Electrical Engineering and Computer Science, Marchstr. 23, 10587 Berlin, Germany
First published on 15th April 2019
Abstract
In this study, two newly developed small molecules based on 9,9-bis(4-diphenylaminophenyl)fluorene functionalized with triphenylamine moieties, namely TPA-2,7-FLTPA-TPA and TPA-3,6-FLTPA-TPA, are designed, synthesized and characterized. The electrochemical, optical and thermal properties of both materials are investigated using various techniques. Afterwards, these materials are employed as dopant-free hole transporting materials (HTMs) in planar inverted perovskite solar cell devices with the aim of determining the device performance and studying their stability in comparison with reference N4,N4,N4′′,N4′′-tetra([1,10-biphenyl]-4-yl)-[1,1′:4′,1′′-terphenyl]-4,4′′-diamine (TaTm)-based devices. Under 1 sun conditions, TPA-3,6-FLTPA-TPA-based devices achieve a power conversion efficiency (PCE) of 13.9% whereas TPA-2,7-FLTPA-TPA-based devices exhibit the highest PCE of 17.1% mainly due to an improvement in the fill factor (FF). Meanwhile, the devices prepared using TaTm as the reference HTM exhibit an overall efficiency of 15.9%. In addition to the higher efficiency, our newly developed HTM TPA-2,7-FLTPA-TPA-based devices demonstrate good stability which is comparable to those with TaTm under similar aging test conditions.
Introduction
Compared to inorganic semiconductors, organic counterparts have been developed and employed intensively either as the active layer or charge transporting layer for various optoelectronic devices including organic light emitting diodes (OLEDs), organic solar cells (OSCs), organic thin-film transistors (OTFTs), perovskite solar cells (PSCs) and other types of devices. Organic semiconducting materials offer several benefits such as versatility of synthesis, which provides great scope for tuning optoelectronic properties, lowering the production cost, compatibility with several substrates (transparent glass, flexible plastic, paper and clothes), light weight and low-temperature fabrication and large scale processing.1–4 In this regard, Li's research group has provided a great overview of OSC materials potentially suitable for commercialization based on high performance, simple device fabrication processes, high stability, low cost and green processing.5 In addition to OSCs, recently, PSCs have attracted much attention due to their promising power conversion efficiency (PCE) which has recently reached 23.7%. This device efficiency is now close to that of commercially used silicon solar cells.6 This high efficiency can only be achieved with the use of solid-state organic hole transporting layers (HTLs) in such PSC devices where the organic semiconducting material plays a extremely important critical role. The efficiency and stability of PSC devices are major device parameters and they are being continuously improved. This would have not been possible without the exploration of new charge transporting layers with high hole mobility incorporated into PSC devices. The performance can be enhanced with the use of an HTL because this layer assists in the collection of holes upon dissociation of excitons created after light absorption. In addition to their role of extraction and transport of holes, which is necessary to boost the open circuit voltages (Voc) and achieve high-performance devices,7–10 these HTLs also protect the perovskite layer from penetration of harmful elements such as moisture and ion ingress from the contact electrodes. In addition to its dual role, the HTL also plays a key role in achieving fabrication of perovskite solar cell devices on a large scale, first in the form of prototypes, and ultimately to push this technology to the next level – commercial use.Basically, perovskite devices can be categorized into the conventional/regular (n–i–p) structure and the planar inverted (p–i–n) structure, based on their configuration.11 In the first layout, a compact or mesoporous metal oxide electron transporting layer (ETL, n-type) is deposited first followed by an active perovskite layer (i) and then a p-type HTL layer (p). Meanwhile, the inverted (p–i–n) structure consists of a hole transporting layer (HTL, p-type) which is deposited first, followed by a perovskite active layer (i) and then an electron transporting layer (ETL, n-type). For n–i–p devices, usually high performance n-type mesoporous TiO2 needs to be deposited at a high temperature (around 400 °C) which restricts the use of flexible plastics as the substrate. But there are some exceptional oxides such as SnO2 which do not require high temperature annealing and exhibit relatively high solar cell performance.12 For the p–i–n architecture, on the other hand, all layers can be simply fabricated via low-temperature solution-processing techniques, which is beneficial to fabricate large-scale devices using various substrates.13–15 Moreover, this kind of device can cope with some limitations of the regular ones, including (a) the increase in the production cost due to the requirement of high temperature processing and (b) high instability and large hysteresis.16
Generally, organic hole transporting materials (HTMs) for PSCs can be divided into two main groups: polydispersed polymers and monodispersed small molecules. The latter offer various advantages, including a defined molecular structure, tunable energetics, and good batch-to-batch reproducibility in comparison with polymeric ones.16,17 Among various reported small molecule-based HTMs, one based on a molecular engineered fluorene–dithiophene (FDT) core substituted with N,N-di-p-methoxyphenylamine donor groups with a simple dissymmetric structure has been successfully used with state-of-the-art devices and achieves power conversion efficiencies of 20.2% which is one of the higher performance devices reported in the literature.18 Though a higher efficiency was achieved, a dopant was required. Though commercial polymeric HTM based p–i–n perovskite devices, such as poly(3,4-ethylenedioxythiophene:polystyrene sulfonate) (PEDOT:PSS) and poly[bis(4-phenyl)(2,4,6-trimethylphenyl)amine] (PTAA), displayed high performance,19,20 they suffer from limitations which impact the practical production of PSCs. While the hydrophilic and strong acidic nature of PEDOT:PSS has a negative impact on the long-term stability of the device, the use of PTAA is limited due to its extremely expensive price which is about 50 times the price of gold.16 Thus the exploration of novel small molecular HTMs for inverted p–i–n PSCs has begun.
To date, even though small molecular HTM based planar inverted devices have achieved some promising PCEs [such as Trux-OMeTAD (18.05%),21 TPA-TVT-TPA (16.32%),16 TPASB (16.2%),22 and TPAC3M (16.58%)23], the stability of the devices is still low or not understood. Recent studies have shown that the combination of molybdenum trioxide (MoO3) and suitable organic semiconductors may enhance the stability and efficiency of PSCs.24 Indeed, Bolink's research group reported that the combination of MoO3 and a derivative of an arylamine (N4,N4,N4′′,N4′′-tetra([1,10-biphenyl]-4-yl)-[1,1′:4′,1′′-terphenyl]-4,4′′-diamine or TaTm) results in good performance and good stability, avoiding the use of doped HTMs in inverted PSCs.25 MoO3 which has a deep work-function was shown to induce a free hole density at the interface of organic semiconductors and was found to improve the current density and fill factor (FF) in perovskite cells by minimizing the sheet resistance.26,27 The use of TaTm in a doped charge transport layer has recently been found to be beneficial for the performance of vacuum deposited perovskite solar cells.28 In that case, in order to avoid recombination at the interface between the perovskite and the doped layer, a thin layer of pure (undoped) organic charge transport molecules was inserted between the perovskite and the doped layer. However, the stability of solar cells using doped transport layers has been shown to be worse than that of those employing non-doped layers.29
Fluorene is one of the most promising building blocks for synthesizing light-emitting conjugated materials due to its high photoluminescence quantum yield, good thermal and chemical stability, high solubility in common organic solvents, and good film-forming properties.30–32 It has been shown that substitution of a bulky aromatic group such as triphenylamine at the central position of fluorine inhibits keto defect formation, which further gives stable blue emission in OLED devices. Additionally, such molecular design also enhances the thermal stability of both the material and the device.33 Apart from OLED applications, various fluorene derivative based HTMs for perovskite solar cells have been developed.34–46 However, they rely on additives which damage the long-term stability of the devices and are mainly employed in conventional/regular (n–i–p) structures. There are hardly any studies regarding fluorene-based HTMs fabricated for inverted devices.
Herein, we report two new fluorene-based small molecules, 4,4′-(2,7-bis(4-(bis(4-methoxyphenyl)amino)phenyl)-9H-fluorene-9,9-diyl)bis(N,N-diphenylaniline) (denoted as TPA-2,7-FLTPA-TPA) and 4,4′-(3,6-bis(4-(bis(4-methoxyphenyl)amino)phenyl)-9H-fluorene-9,9-diyl)bis(N,N-diphenylaniline) (denoted as TPA-3,6-FLTPA-TPA). These molecules were designed and synthesized successfully by varying the substitution position of triphenylamine (TPA) groups via bromine atoms at the 2 and 7 positions or at the 3 and 6 positions on the 9,9-bis(4-diphenylaminophenyl)fluorene core (FLTPA). After characterizing the materials’ properties, they were implemented as dopant-free hole transporting materials in inverted CH3NH3PbI3 devices. A thin layer of MoO3 was deposited in order to ensure good ohmic contact between the ITO and the HTM layers. For the fabrication of p–i–n architectures in this work, the perovskite film was deposited by vacuum techniques. Thus, poly(3,4-ethylenedioxythiophene:polystyrene sulfonate) (PEDOT:PSS) was not selected as the reference HTM because it does not block electrons and may cause losses in built-in potential of the devices.47 Instead of using PEDOT:PSS, TaTm was chosen as the reference HTM for comparison with the two new materials. The aim of this work is to investigate the effect of the different substitutions (2,7 vs. 3,6-positions) in the FLTPA core on its electrochemical, optical and thermal properties. Moreover, the comparison of device performance and stability of these new pristine HTMs with TaTm based devices under similar conditions was carried out.
Results and discussion
Molecular design and synthesis
In this work, the rational design of these new HTMs is based on symmetrical donor–π linker–donor (D–π–D) structures. In this geometry, a 9,9-bis(4-diphenylaminophenyl)fluorene (FLTPA) unit is chosen as the π linker because it is a well-known bulky skeleton which is beneficial for hole injection ability and high thermal stability on account of the presence of electron-rich TPA and electron-deficient fluorenone (FL) groups.33,48–51 Meanwhile, the other TPA units are selected to be donors. In consequence of their outstanding features (such as strong electron-donating and high hole transporting ability, amorphous structure forming ability, and a 3D propeller structure),52,53 they have been widely employed in solar cells54–56 and organic light emitting diodes (OLEDs).17,52,53 These TPA groups were introduced at the 2 and 7 positions or at the 3 and 6 positions on the FLTPA core. While the 2,7-substitution makes the linear structure of the HTM, the twisted one is created by the 3,6-substitution. Although there are several reports related to the substitution at the 2,7-position in the FLTPA group, the 3,6-derivatives remain unexplored. Furthermore, there are few earlier studies reporting the effect of substitution at different positions of small molecular HTMs on PSCs' performance.57,58The synthesis strategy of TPA-2,7-FLTPA-TPA and TPA-3,6-FLTPA-TPA is illustrated in Scheme 1, and a detailed synthesis procedure can be found in the Experimental section. Briefly, 4,4′-(2,7-dibromo-9H-fluorene-9,9-diyl)bis(N,N-diphenylaniline) (compound 2) was made from commercial 2,7-dibromo-9H-fluoren-9-one (compound 1) with excessive triphenylamine in methanesulfonic acid at 140 °C for 48 h. Meanwhile, in order to synthesize 4,4′-(3,6-dibromo-9H-fluorene-9,9-diyl)bis(N,N-diphenylaniline) (compound 5), 3,6-dibromo-9H-fluoren-9-one (compound 4) was prepared by using commercially available 3,6-dibromophenanthrene-9,10-dione (compound 3) in the presence of potassium hydroxide (KOH) and potassium permanganate (KMnO4) at 130 °C for 72 h. Afterwards, compound 4 was used as the starting material to synthesize compound 5 under similar reaction conditions mentioned above. The use of a large excess of triphenylamine in two cases, producing compounds 2 and 5, aims to prevent the reaction of free para-positions with more fluorenone to give a bridged species.33 Simultaneously, the precursor 4-methoxy-N-(4-methoxyphenyl)-N-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)aniline (TPA-BE) was prepared as described previously.7,16 Finally, the synthesis of the new target HTMs TPA-2,7-FLTPA-TPA and TPA-3,6-FLTPA-TPA was conducted by using the classical Suzuki coupling reaction between compound 2 with TPA-BE or compound 5 with TPA-BE, respectively. In the Suzuki reaction, while 2 M potassium carbonate (K2CO3) was used as a base, tetrakis(triphenylphosphine)palladium(0) played the role of a catalyst. The reaction was stirred in an anhydrous toluene solvent at 120 °C for 48 h. Upon purification, the yields of these reactions were found to be 31% for TPA-2,7-FLTPA-TPA and 26% for TPA-3,6-FLTPA-TPA. The purity and chemical structures of both compounds were confirmed by proton and C13 nuclear magnetic resonance (NMR) spectroscopy, which are shown in Fig. S1 and S2 (ESI†). These materials exhibit good solubility in most common organic solvents, including chloroform, dichloromethane and chlorobenzene.
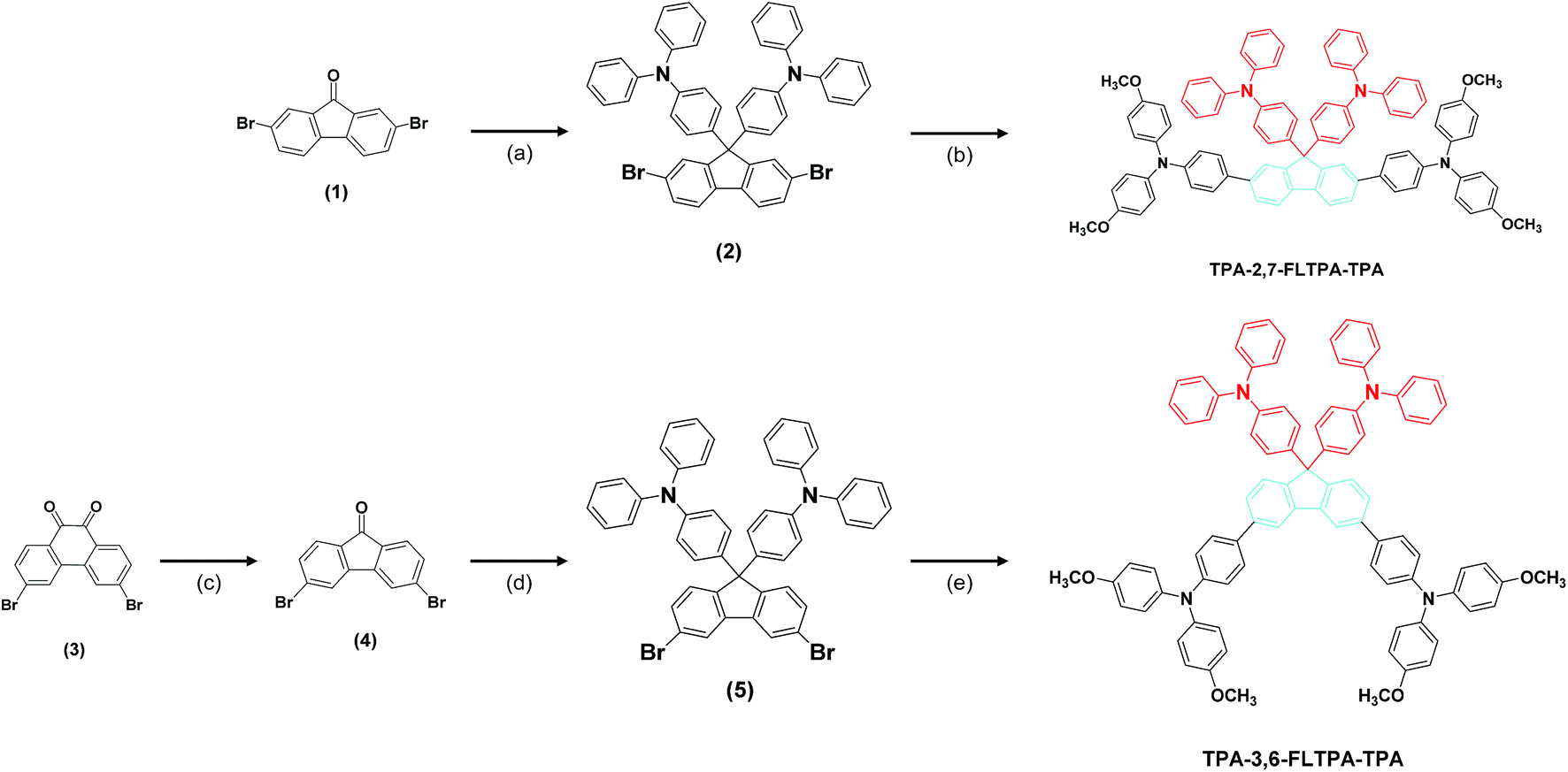 | ||
| Scheme 1 Synthesis routes of TPA-2,7-FLTPA-TPA and TPA-3,6-FLTPA-TPA. Reagents and conditions: (a) triphenylamine, MeSO3H, 140 °C, 48 h; (b) 2 M K2CO3, toluene, Pd(PPh3)4, 120 °C, 48 h; (c) KOH, KMnO4, 130 °C, 72 h; (d) triphenylamine, MeSO3H, 140 °C, 48 h; (e) 2 M K2CO3, toluene, Pd(PPh3)4, 120 °C, 48 h. | ||
Ab initio calculations
To understand the frontier orbital electron density distributions of TPA-2,7-FLTPA-TPA and TPA-3,6-FLTPA-TPA, density functional theory (DFT) calculations were performed at the B3LYP/LANL2DZ level with a polarizable continuum model (PCM) of the chloroform solvent.59–61 Optical absorption spectra were computed with TD-DFT.62 The resulting lowest unoccupied molecular orbital (LUMO) and highest occupied molecular orbital (HOMO) are illustrated in Fig. 1. The relevant data are summarized in Table S1 (ESI†). Generally, the electron distribution of the LUMO and HOMO of both materials exhibits the same trend. In the LUMO, the electron density is primarily localized on the fluorene core and its adjacent phenyl groups, while that of the HOMO is mainly delocalized on the fluorene core and TPA end-capping units. The HOMO values of TPA-2,7-FLTPA-TPA and TPA-3,6-FLTPA-TPA are calculated to be −4.85 and −4.92 eV, respectively; the LUMO values are −1.56 and −1.29 eV, respectively. As the result, the band gap of each material is estimated to be 3.29 eV for TPA-2,7-FLTPA-TPA and 3.63 eV for TPA-3,6-FLTPA-TPA. For comparison, the HOMO and LUMO values of TaTm computed at the same level of theory are −5.02 and −1.57 eV, respectively. The obtained theoretical HOMO–LUMO values at the single-molecule level and in an approximate solvent model are useful to compare the electronic properties of the molecules. For quantitative analysis, the frontier energy levels are measured in a relevant aggregate state using a photoelectron spectroscopy measurement technique, as described below.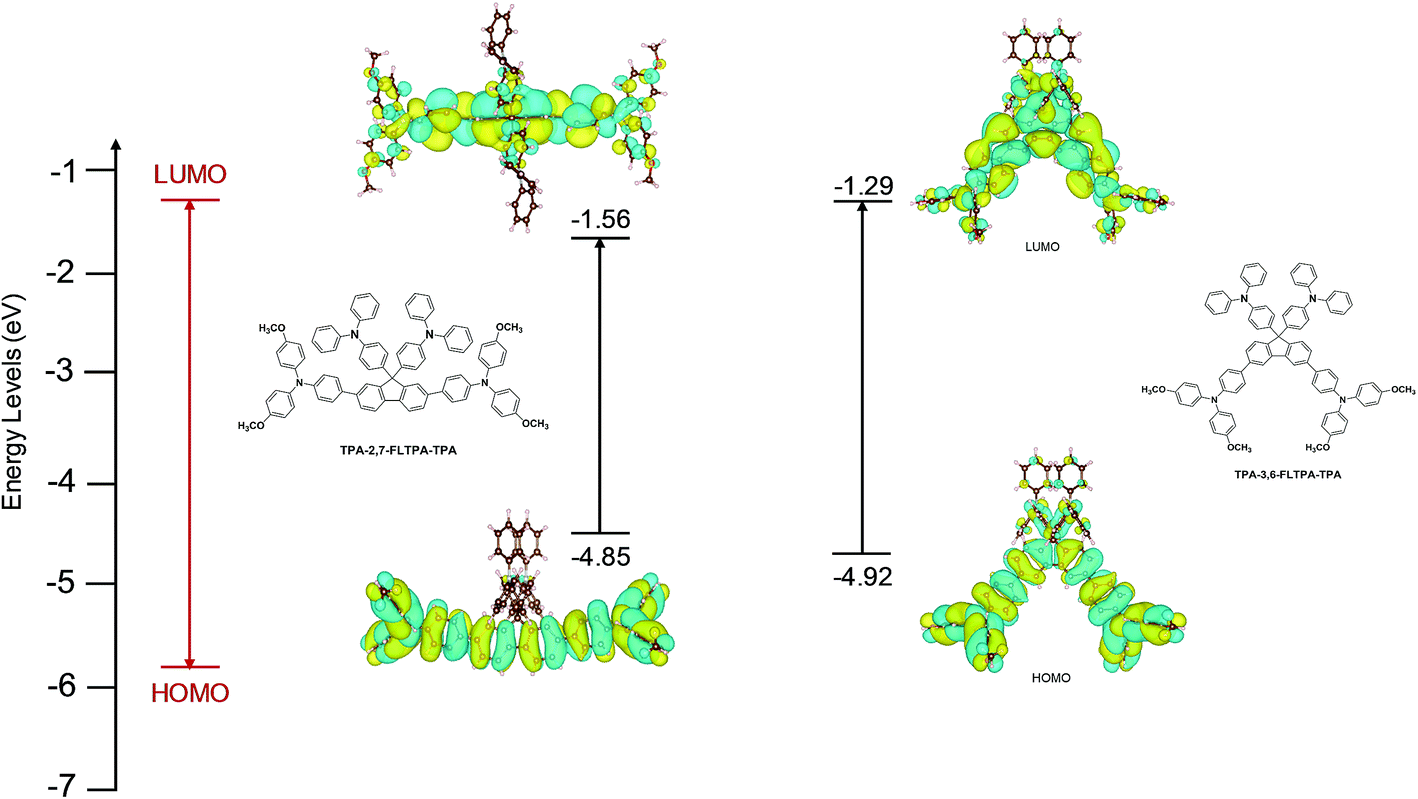 | ||
| Fig. 1 Calculated energy levels and orbital isosurfaces of the HOMO and LUMO of TPA-2,7-FLTPA-TPA (left) and TPA-3,6-FLTPA-TPA (right). | ||
The high LUMO positions of both molecules are beneficial for their use as HTMs, as are the relatively deep HOMO energies comparable to the valence band maximum of the perovskite. The computed lowest-energy absorption peaks are at 431 nm for TPA-2,7-FLTPA-TPA and 384 nm for TPA-3,6-FLTPA-TPA and are due to HOMO-to-LUMO transitions (Table S1, ESI†). The redshift of TPA-2,7-FLTPA-TPAvs.TPA-3,6-FLTPA-TPA observed experimentally is thus confirmed (the redshift of the absolute positions of the computed peaks vs. experimental ones is expected for transitions with some charge transfer character).
Optical properties
UV absorption spectroscopy of TPA-2,7-FLTPA-TPA and TPA-3,6-FLTPA-TPA in chloroform solution (SL) and solid state thin films (TF) was carried out and the normalized spectra are illustrated in Fig. 2a. The corresponding data are displayed in Table 1. Overall, both compounds exhibit an absorption band at a low wavelength (below 400 nm). Also, the UV-vis spectra in SL and TF have similar peaks, indicating that there is no significant aggregation or crystallization in solid films63 and there is also a poor π–π intermolecular interaction occurring in the solid state.64 In the case of TPA-3,6-FLTPA-TPA one maximum absorption peak was observed within the measurement range whereas for TPA-2,7-FLTPA-TPA, there are two absorption peaks.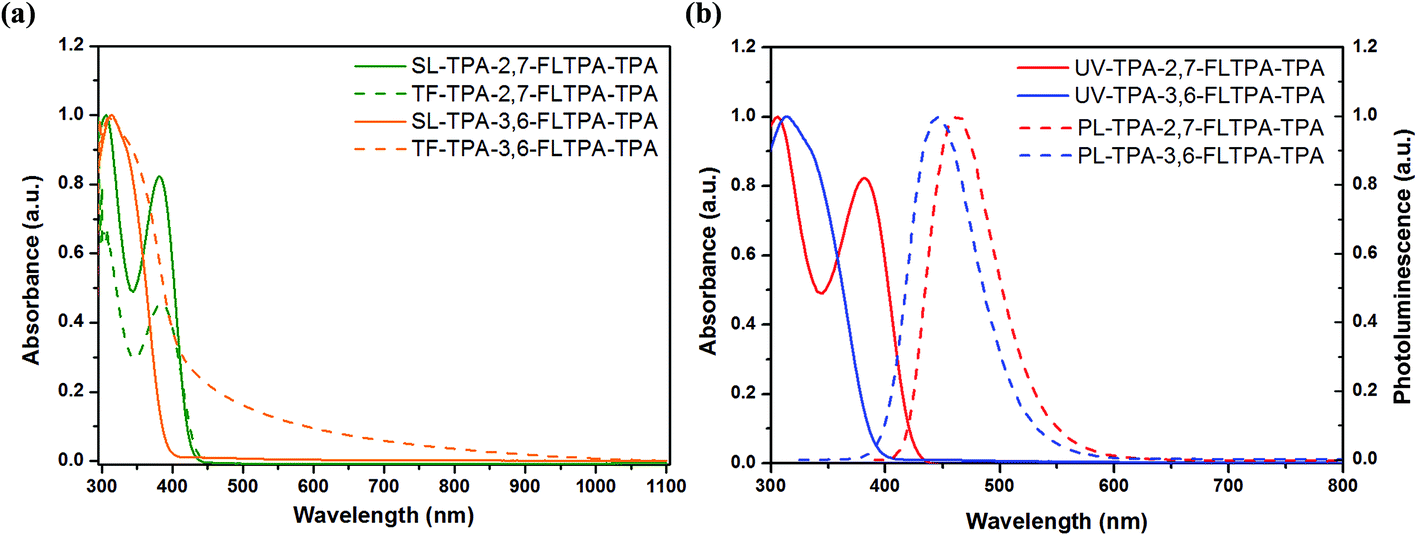 | ||
| Fig. 2 (a) UV-Vis absorption spectra in CF solution (SL, solid line) and films (TF, dashed line) and (b) normalised absorption (solid line) and emission spectra (dashed line) in CF solutions of TPA-2,7-FLTPA-TPA and TPA-3,6-FLTPA-TPA. | ||
| HTMs | λ max [nm] | λ PL [nm] | Stokes shift [nm] | E optg [eV] | E HOMO [eV] | E LUMO [eV] | T d [°C] | T g [°C] | |
|---|---|---|---|---|---|---|---|---|---|
| Solutiona | Filmb | ||||||||
| a Absorption spectrum was measured in a chloroform (CF) solution. b Film was prepared by spin-coating the CF solution containing the sample onto a glass substrate at a spin speed of 1000 rpm at room temperature. c Emission spectrum was analysed in a CF solution. d Optical bandgap was calculated from the formula 1240/λonset. e The oxidation potential was also measured by photoelectron spectroscopy in air (PESA). f E PESALUMO = EPESAHOMO + Eoptg. | |||||||||
| TPA-2,7-FLTPA-TPA | 305, 382 | 298, 384 | 463 | 157 | 2.95 | −5.45 | −2.50 | 434 | 162 |
| TPA-3,6-FLTPA-TPA | 313 | 315 | 446 | 133 | 2.88 | −5.57 | −2.69 | 456 | 167 |
In the case of TPA-2,7-FLTPA-TPA, the absorption spectra in SL show one strong peak at 305 nm which is attributed to the π–π* local electron transition of the TPA end-capping units, and another, weaker peak at 382 nm attributed to the π–π* electron transition between the fluorene core and two adjacent phenyl conjugated moieties. Meanwhile, TPA-2,7-FLTPA-TPA thin films exhibited strong absorption at 298 nm and a weaker feature at 384 nm. For TPA-3,6-FLTPA-TPA, the absorption maxima are observed at 313 nm in SL and 315 nm in TF. The slightly red-shifted and broad absorption in TF compared to that in SL indicates very weak intermolecular interactions in the solid-state in TPA-2,7-FLTPA-TPA while in TPA-3,6-FLTPA-TPA, the broad spectral feature at low wavelength appearing in TF but not in SL is indicative of intermolecular transitions.65 The optical band gap of TPA-2,7-FLTPA-TPA and TPA-3,6-FLTPA-TPA in the thin film is estimated to be 2.95 and 2.88 eV, respectively. The wide band gap nature of these molecules is obvious due to the non-planar conjugated backbone and weak electron donating nature of the phenylene moiety. As the peaks are due to HOMO-to-LUMO transitions (as confirmed by TD-DFT calculations), the high optical band gap is an indicator of a high LUMO energy (in agreement with ab initio calculations) which is beneficial for an HTM.
Furthermore, photoluminescence (PL) in chloroform solutions was characterized and is illustrated in Fig. 2b. The response data are listed in Table 1. The maximum peak of TPA-2,7-FLTPA-TPA is located at 463 nm, whereas for TPA-3,6-FLTPA-TPA, the maximum peak is observed at 446 nm. The Stokes shift, which is estimated from the gap between the maximum peaks of absorption and emission, of two compounds is found to be 157 nm for TPA-2,7-FLTPA-TPA and 133 nm for TPA-3,6-FLTPA-TPA. The relatively large Stokes shift of both compounds can be beneficial for high pore filling capability in materials with a good hole-transporting ability.63,66–68
Thermal properties
The thermal behavior of TPA-2,7-FLTPA-TPA and TPA-3,6-FLTPA-TPA was characterized by using thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) measurements. As shown in Fig. S3 (ESI†), a 5% weight loss is recorded at 434 °C for TPA-2,7-FLTPA-TPA and 456 °C for TPA-3,6-FLTPA-TPA in TGA experiments. The observed decomposition temperature is one of the highest values reported for HTMs and this clearly demonstrates the outstanding thermal stability of these compounds. The outstanding thermal stability of both compounds is attributed to their rigid aromatic backbone and higher Stokes shift. During the second scan of DSC investigations (shown in Fig. S4, ESI†), TPA-2,7-FLTPA-TPA and TPA-3,6-FLTPA-TPA show amorphous phases with higher glass transition temperatures (Tg) of 162 and 167 °C, respectively. This indicates a more stable amorphous state of two new materials, being quite indispensable to optoelectronic devices since their operating temperature is higher than room temperature. The measured glass transition temperature is higher than that of the conventional classical HTM Spiro-OMeTAD (124 °C). From an industrial perspective, higher Tg is an absolutely critical measure for the commercialization of perovskite solar cells using any HTM material. Overall, the introduction of TPA terminal substituents at different positions (2,7 vs. 3,6) of the 9,9-bis(4-diphenylaminophenyl)fluorene core has little impact on the thermal stability of both compounds and this is expected since most of the molecular building blocks are the same and only their substitution patterns are altered.Electrochemical properties
To achieve high-performance PSCs, good alignment of energy levels between the HTL and the perovskite layer is very crucial. In this work, the HOMO values of both materials are measured using a photoelectron spectroscopy in air (PESA) (Fig. 3a and b) technique. Meanwhile, the LUMO values of the materials are estimated using the equation EPESALUMO = EPESAHOMO + Eoptg. The corresponding HOMO and LUMO data are presented in Table 1. The energy values calculated using the PESA technique are used further to evaluate new HTMs in the actual device design because PESA gives more appropriate and realistic values since the measurements are performed on the thin film. The HOMO value calculated by PESA is found to be −5.57 eV and −5.45 eV for TPA-3,6-FLTPA-TPA and TPA-2,7-FLTPA-TPA HTMs respectively and the obtained values are quite close to the VBM value of the main light absorbing perovskite active layer (−5.46 eV). The deeper (than VBM) HOMO of TPA-3,6-FLTPA-TPA than TPA-2,7-FLTPA-TPA is expected to provide lower performance.63,69 Furthermore, the LUMO of both materials is higher than the CBM of the perovskite layer (−3.93 eV), which confirms the electron blocking ability of the two new HTMs. The HOMO of TaTm (−5.4 eV)29 is similar to the HOMO value of TPA-2,7-FLTPA-TPA; hence we expect to achieve at least as good efficiencies as with the TaTm devices.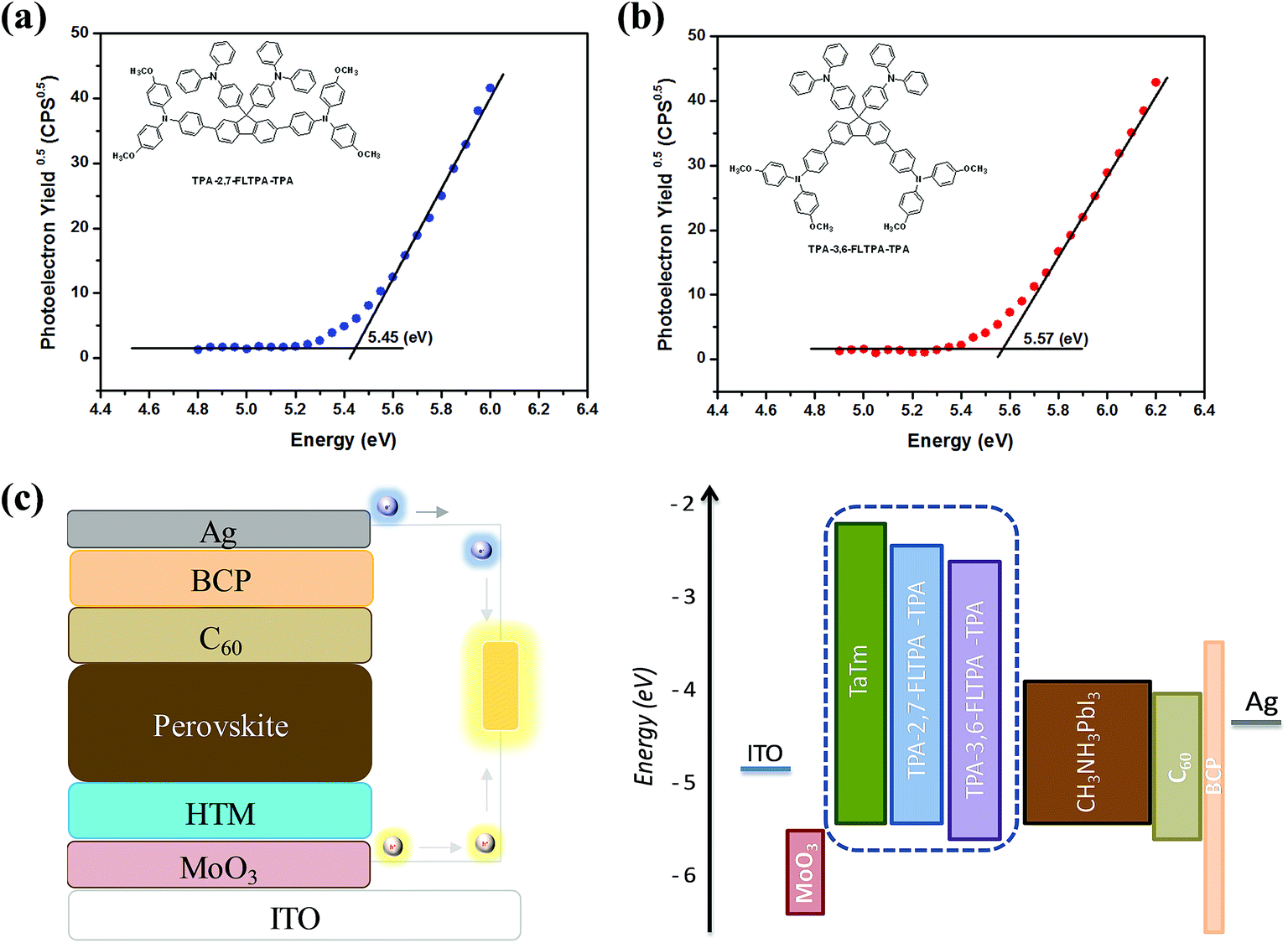 | ||
| Fig. 3 Photoelectron spectroscopy in air (PESA) spectra of (a) TPA-2,7-FLTPA-TPA and (b) TPA-3,6-FLTPA-TPA; and (c) diagram of the device structure and energy levels. | ||
Perovskite solar cells
The photovoltaic performances of PSCs based on these newly reported HTMs are evaluated using ITO/MoO3/HTM/CH3NH3PbI3/C60/BCP/Ag device configurations. In these layouts, indium tin oxide (ITO) on glass is used as a transparent conducting cathode whereas silver (Ag) acts as a transparent conducting anode. MoO3 plays as an assisting hole transporting layer, whereas commercially available TaTm and our novel organic materials are employed as HTMs. Subsequently, a CH3NH3PbI3 perovskite layer is deposited as the active layer followed by fullerene C60 as an electron transporting compact layer. 2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline (BCP) is used as an electron extracting and hole blocking layer as per our previous study.29 The chemical structures of all these charge transporting layers are presented in Fig. S5 (ESI†). The energy diagram of each layer in the device architecture is shown in Fig. 3c, whereas the device fabrication and measurement can be found in the Experimental section.Current–voltage (J–V) performance of PSCs with different HTMs (Fig. 4a) was measured under simulated sun irradiation (AM 1.5G, 100 mW cm−2). The respective parameters are summarized in Table 2. Devices with the TPA-2,7-FLTPA-TPA HTM yielded the highest efficiency of up to 17.1%, while the PCE of TPA-3,6-FLTPA-TPA based devices is found to be around 13.9% and the reference ones with the TaTm HTM displayed a PCE of 15.9%. From the J–V curves and the forward reverse scan, it is clear that there is no hysteresis observed in any of the fabricated devices (Fig. S6, ESI†) and this is a good indication of the lack of charge trapping. The outstanding PCE of TPA-2,7-FLTPA-TPA can be attributed to the higher Voc, Jsc and FF values compared to those of other HTM-based devices. The obtained Voc of FLTPA derivative-based devices is measured to be close to 1.05 V which is close to the devices based on commercial TaTm (1.037 V). Generally, a higher Voc is ascribed to deeper HOMO energy levels of HTLs, leading to a better PCE. In this work, the Voc of TPA-2,7-FLTPA-TPA-based devices is higher than that of TaTm-based ones. The variation of Voc might be attributed to the band bending at the interlayer interfaces. The slightly higher HOMO of TPA-2,7-FLTPA-TPA might be helpful for better hole extraction vs. TaTm (which is supported by a better FF). In addition to Voc, the short circuit current (Jsc) for TPA-2,7-FLTPA-TPA is close to 21 mA cm−2 whereas those of TPA-3,6-FLTPA-TPA and TaTm are 19.85 and 20.42 mA cm−2, respectively. The lower Jsc of the TPA-3,6-FLTPA-TPA HTM in comparison with other HTMs may be attributed to the mismatched energy level between the HOMO of the HTM and the valence band of the perovskite, resulting in a negative impact on the efficient hole-transport ability between the hole transporting layer and the perovskite one. Furthermore, the FF values of these HTMs are in the order: TPA-2,7-FLTPA-TPA (78%) > TaTm (75%) > TPA-3,6-FLTPA-TPA (67%). The worse FF of TPA-3,6-FLTPA-TPA-based devices could be ascribed to their higher series resistance (Rs).16,17 From all the above observations, it is clear that TPA-2,7-FLTPA-TPA exhibits better performance than the twisted molecular structure of TPA-3,6-FLTPA-TPA and this might be attributed to a more linear conjugated structure arising from the attachment of the TPA group at the 2,7 position. The devices were characterized by measuring the EQE as a function of the incident light wavelength (Fig. 4b). The perovskite solar cells with different HTMs show a similar photocurrent response of about 75–85% over the whole visible spectrum. To ensure sufficient statistics, at least two different devices each containing four cells were evaluated for both HTMs tested (Fig. S7, ESI†).
 | ||
| Fig. 4 (a) J–V characteristics under 100 mW cm−2 illumination, (b) EQE spectra, and (c) maximum power point tracking over 40 min for an unencapsulated device, measured in an inert atmosphere. | ||
| HTLs | Additives | V oc [V] | J sc [mA cm−2] | FF | PCEb [%] |
|---|---|---|---|---|---|
| a Cell size (active area): 1 mm2. Photovoltaic performance at 1000 W m−2 (AM 1.5G) at a constant scan speed of 30 mV s−1 for inverted CH3NH3PbI3 devices. b An average device efficiency of a total of 8 cells. | |||||
| TaTm | None | 1.037 | 20.42 | 75 | 15.9 |
| TPA-2,7-FLTPA-TPA | None | 1.052 | 20.82 | 78 | 17.1 |
| TPA-3,6-FLTPA-TPA | None | 1.051 | 19.85 | 67 | 13.9 |
A forward bias was applied to the perovskite solar cells in the dark (Fig. S8, ESI†). In all the HTM based devices, we could observe a typical diode behaviour. The leakage current is low, indicating the high quality of the diodes independent of the HTM employed. A low leakage current density is beneficial for maximum open circuit voltages as the recombination due to leakage is diminished.
Finally, the stability was measured for the TPA-2,7-FLTPA-TPA and TaTm HTM based devices. The maximum power point tracking of a representative cell with a TPA-2,7-FLTPA-TPA HTL at 25 °C and without a UV filter is reported in Fig. 4c, showing no signs of degradation after 40 min under continuous illumination. We can claim that the stability of this new HTM-based device is comparable to that of devices using TaTm as an HTL.
A list of recently reported small molecular fluorene based hole transporting materials is summarized in Table S2† and their chemical structures are revealed in Fig. S9 (ESI†). While there are several fluorene derivatives developed for perovskite solar cells, almost all these materials are fabricated in conventional/regular (n–i–p) layouts. Particularly, they required additives to enhance the device performance, which impede the long-term stability of the devices and increase the production cost of PSCs. Meanwhile, our new materials do not rely on additives and are employed in inverted devices.
A cost evaluation of our two newly developed materials and the commercially used TaTm standard material is provided in Table 3. The synthesis cost of the two new hole transporting compounds is estimated by following the same procedure reported in earlier published papers63,70,71 by taking Australian chemical suppliers’ cost into account. Based on reported HTMs with a low cost, a negligible cost-per-peak-watt contribution of 0.004$ W-1 has been calculated for the EDOT-OMeTPA HTM.71 Additionally, the high synthetic accessibility of this compound greatly facilitates reduction of toxic chemical wastes and this is a great green synthesis methodology with lower cost HTMs for generating environment friendly materials. The price of the reference materials used in this study (TaTm) was provided by TCI America Research Chemicals, a commercial chemical supplier company. The flowchart demonstrates the synthesis of 1 g of TPA-3,6-FLTPA-TPA and 1 g of TPA-2,7-FLTPA-TPA and both are illustrated in Fig. S10 and S11, respectively (ESI†). The cost of our newly developed high-performance TPA-2,7-FLTPA-TPA HTM (taking both efficiency and stability data into consideration) is half that of commercial TaTm. We believe that the synthesis cost of our new materials will be reduced when the synthesis is carried out on a large-scale.
| HTLs | Synthesis cost [$ g−1] | Commercial price [$ g−1] |
|---|---|---|
| TaTm | — | 264 |
| TPA-2,7-FLTPA-TPA | 92.89 | — |
| TPA-3,6-FLTPA-TPA | 105.56 | — |
Conclusions
In summary, we have reported two triphenylamine substituted fluorene based newly developed organic hole transporting materials, namely TPA-2,7-FLTPA-TPA and TPA-3,6-FLTPA-TPA, using low cost starting precursors via an elegant synthetic method. The newly synthesised materials exhibit outstanding thermal stability with a high glass transition temperature compared to conventional Spiro-OMeTAD thus satisfying one of the important requirements for commercialization. To the best of our knowledge, for the first time, dopant free fluorene based HTMs were integrated into inverted perovskite solar cell devices. The devices fabricated with the TPA-2,7-FLTPA-TPA HTL exhibit an outstanding power conversion efficiency of 17.1% with an impressive fill factor reaching 78%. The obtained fill factor is one of the highest values among reported TPA derivative-based HTMs using CH3NH3PbI3 as an active layer in inverted devices. Interestingly, the synthesis cost of our new materials is quite low. Our elegant molecular design clearly indicates the a good potential of these newly developed cost efficient HTMs for fabricating stable high-performance perovskite devices using the most appropriate inverted device geometry required for industrial applications. The obtained device performance suggests that the linear molecular structure is better than a twisted one. Our obtained results therefore suggest further research avenues using FLTPA based low-cost and stable HTMs for the development of PSCs.Experimental section
Materials and instruments
All chemicals and reagents were purchased from commercial vendors and used directly without any further purification. Synthesized compounds were characterized by 1H-NMR and 13C-NMR, performed on a Varian-400 spectrometer or a Bruker 600 MHz spectrometer. High-resolution mass spectra were recorded on an LTQ Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) equipped with an electrospray ionisation (ESI) source, operating in the positive ion mode at a resolution of 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 (at m/z 400). Reserpine ([M + H]+, m/z 609.28066) was used as a lock mass calibrant to increase the measurement accuracy. Thermal analysis was performed using a Pegasus Q500TGA thermogravimetric analyzer in a nitrogen atmosphere at a heating rate of 10 °C min−1. Differential scanning calorimetry (DSC) was conducted under nitrogen using a Chimaera instrument Q100 DSC. The sample was heated at 10 °C min−1 from 30 °C to 300 °C. Absorption spectra were recorded on a Shimadzu UV-1800 spectrometer. Photoelectron spectroscopy in air (PESA) measurements were conducted on an AC-2 photoelectron spectrometer (Riken-Keiki Co.).
000 (at m/z 400). Reserpine ([M + H]+, m/z 609.28066) was used as a lock mass calibrant to increase the measurement accuracy. Thermal analysis was performed using a Pegasus Q500TGA thermogravimetric analyzer in a nitrogen atmosphere at a heating rate of 10 °C min−1. Differential scanning calorimetry (DSC) was conducted under nitrogen using a Chimaera instrument Q100 DSC. The sample was heated at 10 °C min−1 from 30 °C to 300 °C. Absorption spectra were recorded on a Shimadzu UV-1800 spectrometer. Photoelectron spectroscopy in air (PESA) measurements were conducted on an AC-2 photoelectron spectrometer (Riken-Keiki Co.).
Synthesis
The synthesis of TPA precursors was carried out following an earlier published synthetic procedure.7,16Perovskite device fabrication
Materials: photo-lithographically patterned ITO covered glass substrates were purchased from Naranjo Substrates (www.naranjosubstrates.com). MoO3, PbI2 and C60 were purchased from Aldrich and CH3NH3I and BCP from Lumtec. TaTm was provided by Novaled.Devices were prepared on pre-cleaned ITO substrates subsequently cleaned with soap, water and isopropanol in an ultrasonic bath, followed by UV-ozone treatment. The substrates were then transferred to a vacuum chamber integrated into a nitrogen-filled glovebox (MBraun, H2O and O2 < 0.1 ppm) and evacuated to a pressure of 3 × 10−6 mbar. For thickness calibration, we individually sublimed MoO3 and TaTm materials and a calibration factor was obtained by comparing the thickness inferred from quartz crystal microbalance (QCM) sensors with that measured with a mechanical profilometer (Ambios XP1). Once the materials were calibrated, a 5 nm thick MoO3 film was deposited at 0.1 Å s−1 deposition rate. Following the film deposition, the chamber was vented and the substrates were transferred to another vacuum chamber integrated in the same glovebox evacuated to 1 × 10−6 mbar. The vacuum chamber is equipped with six temperature-controlled evaporation sources (Creaphys) fitted with ceramic crucibles. The thermal sources were directed upwards with an angle of approximately 90° with respect to the bottom of the evaporator. The substrate holder-to-evaporation source distance is approximately 20 cm. Three QCM sensors were used, two for monitoring the deposition rate of each evaporation source while the third one close to the substrate holder for monitoring the total deposition rate. Then, 10 nm thick films of TaTm were deposited by heating the crucible at 250 °C with a deposition rate of 0.5 Å s−1. The HTMs tested were deposited by a solution process using a 5 mg mL−1 toluene solution (3000 rpm, 30 s). After deposition, the films were annealed at 100 °C for 10 min. Once the hole transport layers were deposited, the perovskite was deposited by co-evaporation of CH3NH3I and PbI2. The use of clean QCM sensors for perovskite evaporation is important to avoid false readings due to perovskite formation on the sensor. For a more accurate deposition the density of PbI2 (6.16 g cm−3) is set in the equipment. For CH3NH3I the density is assumed to be 1 g cm−3, and the z factor for both materials is set to 1. The calibration of the deposition rate for CH3NH3I was found to be difficult due to non-uniform layers and the soft nature of the material impeded accurate thickness measurements. Hence, CH3NH3I was evaporated by keeping the temperature constant at 70 °C, while the CH3NH3I:PbI2 ratio was controlled offline using grazing incident X-ray diffraction by adjusting the PbI2 deposition temperature. The optimum deposition temperatures were found to be 250 °C for PbI2 and 70 °C for CH3NH3I. This was followed by deposition of electron selective contacts by thermal evaporation, C60 (25 nm) and BCP (8 nm). The devices were finished with the thermal evaporation of the metal electrode under a base pressure of 2 × 10−6 mbar to a thickness of 100 nm of Ag.
Device characterization
The external quantum efficiency (EQE) was estimated using the cell response at different wavelengths (measured with a white light halogen lamp in combination with band-pass filters), where the spectrum mismatch was corrected using a calibrated Si reference cell (MiniSun simulator by ECN, Netherlands). The un-encapsulated devices were measured in a N2-filled glovebox. The J–V characteristic curves were collected in the dark and under illumination using a Keithley 2400 source measure unit. The device active area was 0.04 cm−2 (defined as the overlap between the ITO and the top metal contact) and measured through a shadow mask with 0.01 cm2 aperture to prohibit lateral current collection from outside the active area.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. D. M. is thankful to QUT for a QUTPRA scholarship for conducting his doctoral research. We are very thankful to the Central Analytical Research Facility (CARF), Institute of Future Environments (IFE) and the Queensland University of Technology (QUT) for providing facility and equipment support. P. S. is thankful to QUT for the financial support from the Australian Research Council (ARC) for the Future Fellowship (FT130101337) and QUT core funding (QUT/322120-0301/07). Financial support from the Spanish Ministry of Economy and Competitiveness (MINECO) via the Unidad de Excelencia María de Maeztu MDM-2015-0538, MAT2017-88821-R and the Generalitat Valenciana (Prometeo/2016/135) is acknowledged. L. G. E. is funded by the Helmholtz Innovation Lab HySPRINT, which is financially supported by the Helmholtz Association.References
- Nat. Mater., 2013, 12, 591 Search PubMed.
- A. Facchetti, Nat. Mater., 2013, 12, 598–600 CrossRef CAS PubMed.
- A. Mishra and P. Bauerle, Angew. Chem., Int. Ed. Engl., 2012, 51, 2020–2067 CrossRef CAS PubMed.
- O. Ostroverkhova, Chem. Rev., 2016, 116, 13279–13412 CrossRef CAS PubMed.
- R. Xue, J. Zhang, Y. Li and Y. Li, Small, 2018, 14, 1801793 CrossRef PubMed.
- NREL, Best Research-Cell Efficiencies, http://www.nrel.gov/ncpv/images/efficiency_chart.jpg, accessed April 2019.
- H. D. Pham, Z. Wu, L. K. Ono, S. Manzhos, K. Feron, N. Motta, Y. Qi and P. Sonar, Adv. Electron. Mater., 2017, 3, 1700139 CrossRef.
- N. H. Tiep, Z. Ku and H. J. Fan, Adv. Energy Mater., 2016, 6, 1501420 CrossRef.
- T. H. Le, Q. D. Dao, M. P. Nghiem, S. Peralta, R. Guillot, Q. N. Pham, A. Fujii, M. Ozaki, F. Goubard and T. T. Bui, Chem.–Asian J., 2018, 13, 1302–1311 CrossRef CAS PubMed.
- H. D. Pham, K. Hayasake, J. Kim, T. T. Do, H. Matsui, S. Manzhos, K. Feron, S. Tokito, T. Watson, W. C. Tsoi, N. Motta, J. R. Durrant, S. M. Jain and P. Sonar, J. Mater. Chem. C, 2018, 6, 3699–3708 RSC.
- H. D. Pham, X. Li, W. Li, S. Manzhos, A. K. K. Kyaw and P. M. Sonar, Energy Environ. Sci., 2019, 12, 1177–1209 RSC.
- Q. Jiang, L. Zhang, H. Wang, X. Yang, J. Meng, H. Liu, Z. Yin, J. Wu, X. Zhang and J. You, Nat. Energy, 2016, 2, 16177 CrossRef.
- L. Meng, J. You, T. F. Guo and Y. Yang, Acc. Chem. Res., 2016, 49, 155–165 CrossRef CAS PubMed.
- T. Liu, K. Chen, Q. Hu, R. Zhu and Q. Gong, Adv. Energy Mater., 2016, 6, 1600457 CrossRef.
- W. Yan, S. Ye, Y. Li, W. Sun, H. Rao, Z. Liu, Z. Bian and C. Huang, Adv. Energy Mater., 2016, 6, 1600474 CrossRef.
- H. D. Pham, H. Hu, K. Feron, S. Manzhos, H. Wang, Y. M. Lam and P. Sonar, Sol. RRL, 2017, 1, 1700105 CrossRef.
- H. D. Pham, H. Hu, F.-L. Wong, C.-S. Lee, W.-C. Chen, K. Feron, S. Manzhos, H. Wang, N. Motta, Y. M. Lam and P. Sonar, J. Mater. Chem. C, 2018, 6, 9017–9029 RSC.
- M. Saliba, S. Orlandi, T. Matsui, S. Aghazada, M. Cavazzini, J.-P. Correa-Baena, P. Gao, R. Scopelliti, E. Mosconi, K.-H. Dahmen, F. De Angelis, A. Abate, A. Hagfeldt, G. Pozzi, M. Graetzel and M. K. Nazeeruddin, Nat. Energy, 2016, 1, 15017–15024 CrossRef CAS.
- J. H. Heo, H. J. Han, D. Kim, T. K. Ahn and S. H. Im, Energy Environ. Sci., 2015, 8, 1602–1608 RSC.
- Y. Shao, Y. Yuan and J. Huang, Nat. Energy, 2016, 1, 15001 CrossRef CAS.
- C. Huang, W. Fu, C. Z. Li, Z. Zhang, W. Qiu, M. Shi, P. Heremans, A. K. Jen and H. Chen, J. Am. Chem. Soc., 2016, 138, 2528–2531 CrossRef CAS PubMed.
- Y. Li, Z. Xu, S. Zhao, B. Qiao, D. Huang, L. Zhao, J. Zhao, P. Wang, Y. Zhu, X. Li, X. Liu and X. Xu, Small, 2016, 12, 4902–4908 CrossRef CAS PubMed.
- S. J. Park, S. Jeon, I. K. Lee, J. Zhang, H. Jeong, J.-Y. Park, J. Bang, T. K. Ahn, H.-W. Shin, B.-G. Kim and H. J. Park, J. Mater. Chem. A, 2017, 5, 13220–13227 RSC.
- S. Wang, T. Sakurai, W. Wen and Y. Qi, Adv. Mater. Interfaces, 2018, 5, 1800260 CrossRef.
- B. Dänekamp, N. Droseros, D. Tsokkou, V. Brehm, P. P. Boix, M. Sessolo, N. Banerji and H. J. Bolink, J. Mater. Chem. C, 2019, 7, 523–527 RSC.
- M. Kröger, S. Hamwi, J. Meyer, T. Riedl, W. Kowalsky and A. Kahn, Appl. Phys. Lett., 2009, 95, 123301 CrossRef.
- J. Xu, O. Voznyy, R. Comin, X. Gong, G. Walters, M. Liu, P. Kanjanaboos, X. Lan and E. H. Sargent, Adv. Mater., 2016, 28, 2807–2815 CrossRef CAS PubMed.
- C. Momblona, L. Gil-Escrig, E. Bandiello, E. M. Hutter, M. Sessolo, K. Lederer, J. Blochwitz-Nimoth and H. J. Bolink, Energy Environ. Sci., 2016, 9, 3456–3463 RSC.
- J. Avila, L. Gil-Escrig, P. P. Boix, M. Sessolo, S. Albrecht and H. J. Bolink, Sustainable Energy Fuels, 2018, 2, 2429–2434 RSC.
- P. Sonar, J. Zhang, A. C. Grimsdale, K. Mullen, M. Surin, R. Lazzaroni, P. Leclere, S. Tierney, M. Heeney and I. McCulloch, Macromolecules, 2004, 37, 709–715 CrossRef CAS.
- R. Grisorio, C. Piliego, P. Fini, P. Cosma, P. Mastrorilli, G. Gigli, G. P. Suranna and C. F. Nobile, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6051–6063 CrossRef CAS.
- I. H. Jung, Y. K. Jung, J. Lee, J.-H. Park, H. Y. Woo, J.-I. Lee, H. Y. Chu and H.-K. Shim, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7148–7161 CrossRef CAS.
- C. Ego, A. C. Grimsdale, F. Uckert, G. Yu, G. Srdanov and K. Müllen, Adv. Mater., 2002, 14, 809–811 CrossRef CAS.
- S. Daskeviciu Te, N. Sakai, M. Franckevicius, M. Daskeviciene, A. Magomedov, V. Jankauskas, H. J. Snaith and V. Getautis, Adv. Sci., 2018, 5, 1700811 CrossRef PubMed.
- Y. C. Chen, S. K. Huang, S. S. Li, Y. Y. Tsai, C. P. Chen, C. W. Chen and Y. J. Chang, ChemSusChem, 2018, 11, 3225–3233 CrossRef CAS PubMed.
- B. Xu, D. Bi, Y. Hua, P. Liu, M. Cheng, M. Grätzel, L. Kloo, A. Hagfeldt and L. Sun, Energy Environ. Sci., 2016, 9, 873–877 RSC.
- A. Krishna, D. Sabba, J. Yin, A. Bruno, L. J. Antila, C. Soci, S. Mhaisalkar and A. C. Grimsdale, J. Mater. Chem. A, 2016, 4, 8750–8754 RSC.
- K. Liu, Y. Yao, J. Wang, L. Zhu, M. Sun, B. Ren, L. Xie, Y. Luo, Q. Meng and X. Zhan, Mater. Chem. Front., 2017, 1, 100–110 RSC.
- L. Bai, Z. Wang, Y. Han, Z. Zuo, B. Liu, M. Yu, H. Zhang, J. Lin, Y. Xia, C. Yin, L. Xie, Y. Chen, Z. Lin, J. Wang and W. Huang, Nano Energy, 2018, 46, 241–248 CrossRef CAS.
- Y. Hua, J. Zhang, B. Xu, P. Liu, M. Cheng, L. Kloo, E. M. J. Johansson, K. Sveinbjörnsson, K. Aitola, G. Boschloo and L. Sun, Nano Energy, 2016, 26, 108–113 CrossRef CAS.
- N. J. Jeon, H. Na, E. H. Jung, T.-Y. Yang, Y. G. Lee, G. Kim, H.-W. Shin, S. Il Seok, J. Lee and J. Seo, Nat. Energy, 2018, 3, 682–689 CrossRef CAS.
- Z. Li, J. Chen, H. Li, Q. Zhang, Z. Chen, X. Zheng, G. Fang, H. wang and Y. Hao, RSC Adv., 2017, 7, 41903–41908 RSC.
- R. Tiazkis, S. Paek, M. Daskeviciene, T. Malinauskas, M. Saliba, J. Nekrasovas, V. Jankauskas, S. Ahmad, V. Getautis and M. Khaja Nazeeruddin, Sci. Rep., 2017, 7, 150 CrossRef PubMed.
- Y. Wang, T. S. Su, H. Y. Tsai, T. C. Wei and Y. Chi, Sci. Rep., 2017, 7, 7859 CrossRef PubMed.
- G. Wu, Y. Zhang, R. Kaneko, Y. Kojima, K. Sugawa, T. H. Chowdhury, A. Islam, Q. Shen, M. Akhtaruzzaman, T. Noda and J. Otsuki, Sol. RRL, 2017, 1, 1770134 CrossRef.
- W. Yu, J. Zhang, X. Wang, X. Liu, D. Tu, J. Zhang, X. Guo and C. Li, Sol. RRL, 2018, 2, 1800048 CrossRef.
- L. Gil-Escrig, C. Momblona, D. Forgács, S. Pla, F. Fernández-Lázaro, M. Sessolo, Á. Sastre-Santos and H. J. Bolink, Org. Electron., 2016, 37, 396–401 CrossRef CAS.
- A. L. Capodilupo, V. Vergaro, E. Fabiano, M. De Giorgi, F. Baldassarre, A. Cardone, A. Maggiore, V. Maiorano, D. Sanvitto, G. Gigli and G. Ciccarella, J. Mater. Chem. B, 2015, 3, 3315–3323 RSC.
- Q. Fang, B. Xu, B. Jiang, H. Fu, W. Zhu, X. Jiang and Z. Zhang, Synth. Met., 2005, 155, 206–210 CrossRef CAS.
- A. m. Thangthong, N. Prachumrak, R. Tarsang, T. Keawin, S. Jungsuttiwong, T. Sudyoadsuk and V. Promarak, J. Mater. Chem., 2012, 22, 6869–6877 RSC.
- S. Jiao, Y. Liao, X. Xu, L. Wang, G. Yu, L. Wang, Z. Su, S. Ye and Y. Liu, Adv. Funct. Mater., 2008, 18, 2335–2347 CrossRef CAS.
- Y. Shirota, J. Mater. Chem., 2005, 15, 75–93 RSC.
- Y. Shirota, J. Mater. Chem., 2000, 10, 1–25 RSC.
- J. Wang, K. Liu, L. Ma and X. Zhan, Chem. Rev., 2016, 116, 14675–14725 CrossRef CAS PubMed.
- Z. Ning and H. Tian, Chem. Commun., 2009, 5483–5495, 10.1039/b908802d.
- P. Agarwala and D. Kabra, J. Mater. Chem. A, 2017, 5, 1348–1373 RSC.
- X. Zong, W. Qiao, Y. Chen, H. Wang, X. Liu, Z. Sun and S. Xue, ChemistrySelect, 2017, 2, 4392–4397 CrossRef CAS.
- X. Liu, F. Kong, R. Ghadari, S. Jin, W. Chen, T. Yu, T. Hayat, A. Alsaedi, F. Guo, Z. a. Tan, J. Chen and S. Dai, Energy Technol., 2017, 5, 1788–1794 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- W. Kohn and L. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- C. Adamo and D. Jacquemin, Chem. Soc. Rev., 2013, 42, 845–856 RSC.
- H. D. Pham, T. T. Do, J. Kim, C. Charbonneau, S. Manzhos, K. Feron, W. C. Tsoi, J. R. Durrant, S. M. Jain and P. Sonar, Adv. Energy Mater., 2018, 8, 1703007 CrossRef.
- S. Ma, H. Zhang, N. Zhao, Y. Cheng, M. Wang, Y. Shen and G. Tu, J. Mater. Chem. A, 2015, 3, 12139–12144 RSC.
- S. T. Ang, A. Pal and S. Manzhos, J. Chem. Phys., 2018, 149, 044114 CrossRef PubMed.
- D. Alberga, G. F. Mangiatordi, F. Labat, I. Ciofini, O. Nicolotti, G. Lattanzi and C. Adamo, J. Phys. Chem. C, 2015, 119, 23890–23898 CrossRef CAS.
- H. Li, K. Fu, A. Hagfeldt, M. Gratzel, S. G. Mhaisalkar and A. C. Grimsdale, Angew. Chem., Int. Ed., 2014, 53, 4085–4088 CrossRef CAS PubMed.
- W. J. Chi, P. P. Sun and Z. S. Li, Nanoscale, 2016, 8, 17752–17756 RSC.
- W.-J. Yin, J.-H. Yang, J. Kang, Y. Yan and S.-H. Wei, J. Mater. Chem. A, 2015, 3, 8926–8942 RSC.
- T. P. Osedach, T. L. Andrew and V. Bulović, Energy Environ. Sci., 2013, 6, 711–718 RSC.
- M. L. Petrus, T. Bein, T. J. Dingemans and P. Docampo, J. Mater. Chem. A, 2015, 3, 12159–12162 RSC.
- L. A. Estrada and D. C. Neckers, J. Org. Chem., 2009, 74, 8484–8487 CrossRef CAS PubMed.
- N. Fomina and T. E. Hogen-Esch, Macromolecules, 2008, 41, 3765–3768 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta01681c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2019 |