
Controllable assembly of single/double-thin-shell g-C3N4 vesicles via a shape-selective solid-state templating method for efficient photocatalysis†
Lei
Luo
 ab,
Keyan
Li
a,
Anfeng
Zhang
a,
Hainan
Shi
a,
Guanghui
Zhang
a,
Jiani
Ma
b,
Wen
Zhang
b,
Junwang
Tang
*c,
Chunshan
Song
*ad and
Xinwen
Guo
*a
ab,
Keyan
Li
a,
Anfeng
Zhang
a,
Hainan
Shi
a,
Guanghui
Zhang
a,
Jiani
Ma
b,
Wen
Zhang
b,
Junwang
Tang
*c,
Chunshan
Song
*ad and
Xinwen
Guo
*a
aState Key Laboratory of Fine Chemicals, PSU-DUT Joint Center for Energy Research, School of Chemical Engineering, Dalian University of Technology, Dalian 116024, People's Republic of China
bKey Lab of Synthetic and Natural Functional Molecule Chemistry of Ministry of Education, The Energy and Catalysis Hub, College of Chemistry and Materials Science, Northwest University, Xi'an 710127, People's Republic of China
cDepartment of Chemical Engineering, University College London, Torrington Place, London WC1E 7JE, UK
dEMS Energy Institute, PSU-DUT Joint Center for Energy Research, Department of Energy & Mineral Engineering, Pennsylvania State University, University Park, Pennsylvania 16802, USA
First published on 15th April 2019
Abstract
Mimicking natural thylakoid vesicles is an effective approach for facilitating photoabsorption and charge separation over photocatalysts. However, bulk materials cannot function well with a short photocarrier transport distance while thin-layers suffer from preparation difficulty due to the ease of structural collapse. Herein, inspired by natural thylakoid vesicles, a solid-state thermolysis templating method using the shape selectivity of MCM-41 and melamine precursor has been developed to assemble stable thin-shell g-C3N4 vesicles and their heterojunctions. The relatively narrow channel of MCM-41 allows the oligomerization of melamine inside the pore but inhibits its further polymerization into melon. With increasing temperature, oligomers begin to form and migrate out of the channel and polymerize selectively on the open-up outer surface into the vesicle structure. Single- and double-thin-shell g-C3N4 vesicles as well as their heterojunctions have successfully been fabricated through templating by MCM-41, hollow MCM-41 and MOx/MCM-41 (M = Ag, Fe, Co, Cu, and Ni) as evidenced by TEM. A uniform shell thickness can be precisely controlled from 17.5 to 42.1 nm. The tailored g-C3N4 vesicles exhibit enhanced photocatalytic activity and stability for the hydrogen evolution reaction which results from the enhanced photoabsorption and suppressed charge recombination. This new method is versatile for encapsulation of the secondary component including metals and metal oxides in g-C3N4.
Introduction
Considerable attention has been devoted to metal and metal-free photocatalysts for solar energy utilization, which is an attractive approach to advancing sustainable energy and environmental protection.1–4 Recently, graphitic carbon nitride (g-C3N4) has emerged as a metal-free visible-light responsive photocatalyst capable of water splitting,5–14 CO2 photoreduction,15–20 nitrogen fixation,21 organic synthesis22 and pollutant decontamination.23–26 Thermal polymerization of various nitrogen-rich precursors27–35 can be used to fabricate g-C3N4 on a large scale. However, it encounters the problems of a low surface area (SBET < 10 m2 g−1), limited photoabsorption and high charge recombination rate, which greatly suppress the photocatalysis. Nanostructural design and electronic modification are effective means to improve the photocatalytic properties, where the former can not only provide more active sites but also promote mass and charge transfer.36–38 The electronic structural modification can enhance the photoabsorption as well as the charge separation. Hollow nanoparticles, taking advantage of shell selectivity, pH tolerance and stabilized inner components, are promising in versatile applications.39–41 Besides those excellent capacities, hollow photocatalysts are inspired by natural thylakoid vesicles and advantageous because of the shortened charge carrier lengths and light scattering effects inside the cavity to enhance photoabsorption.42–46 The thinner the wall is, the easier it is for the charge carriers to be transported to the outer surface for efficient utilization.The hard-templating method is a widely used route for hollow structure preparation. Hollow g-C3N4 with a tunable wall thickness was first prepared with core/shell silica templates43 through reverse porous replication. Subsequently, a multi-shell g-C3N4 hollow structure was prepared through the same method except for the replacement of the multi-shell silica templates.42 As the traditional hard-templating method relies on reverse porous replication, the precursors should have a low melting point or high solubility to allow precursor pre-casting into the channels. Therefore, the most widely used precursor for morphological control is cyanamide, but it is expensive and virulent. Furthermore, the pre-casting process requires complex hydrothermal treatment and solvent elimination which are time-consuming and energy-intensive. More importantly, the preparation of an ultra-thin-shell structure through the traditional hard-templating method is still difficult because of the easy collapse of the thin shells. Therefore, it is highly desirable to develop a facile, economical and reliable method for thin-shell photocatalyst preparation.
The encapsulated structure is important to improve the stability of the inner layer via the chemical tolerance of the exterior39 and meanwhile realize a cascade reaction in organic synthesis.47 In the case of artificial synthesis, precise regulation of the component distribution is of great significance in dividing the redox sites48–50 and promoting charge separation. Modifying g-C3N4 with noble metal nanoparticles including platinum, gold and silver can efficiently enhance the photocatalysis through the surface plasmon resonance (SPR) effect. The resonance could induce the formation of an intensive electromagnetic field to promote the charge separation and trap the resonant photons for efficient higher photoabsorption. In addition, the noble metal nanoparticles could also act as electron sinks, which trap the free electrons and thereafter promote the charge separation. On the other hand, the preparation of vesicles with functional interiors has been a challenge so far.
In the present work, a shape-selective solid-state (solvent-free) templating method based on the shape selectivity of MCM-41![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 51 sieves using melamine as the precursor was developed to fabricate thin-shell g-C3N4 vesicles. The proper pore size of MCM-41 allows melamine molecules to selectively deposit and polymerize on the opened-up surface but not in the relatively narrow channels. On the premise of retaining the pore structure, single- and double-shell g-C3N4 vesicles were successfully prepared by adjusting the amount of opened-up surface. Through this versatile method, the encapsulation of metal and metal oxides in the vesicle structure is successfully achieved. The photocatalytic activity of the tailored g-C3N4 was evaluated by water splitting under visible light irradiation, and a substantially enhanced H2 evolution rate (27.5 μmol h−1) was achieved compared to the bulk.
51 sieves using melamine as the precursor was developed to fabricate thin-shell g-C3N4 vesicles. The proper pore size of MCM-41 allows melamine molecules to selectively deposit and polymerize on the opened-up surface but not in the relatively narrow channels. On the premise of retaining the pore structure, single- and double-shell g-C3N4 vesicles were successfully prepared by adjusting the amount of opened-up surface. Through this versatile method, the encapsulation of metal and metal oxides in the vesicle structure is successfully achieved. The photocatalytic activity of the tailored g-C3N4 was evaluated by water splitting under visible light irradiation, and a substantially enhanced H2 evolution rate (27.5 μmol h−1) was achieved compared to the bulk.
Results and discussion
Shape-selective templating method to fabricate single-shell g-C3N4
Molecular sieves exhibit shape selectivity52 and are widely used in the petrochemical industry to highly selectively produce various value-added chemicals. Depending on the size of the channel in the sieves, only molecules of a certain size or a certain shape can pass through or be produced. Introducing those types of molecular sieves into the area of morphology control will greatly broaden the range of materials. In the case of g-C3N4 preparation through thermolysis, various chemicals can be used as the precursor. Among them, melamine is one of the most readily available precursors. Since melamine sublimates above 300 °C, if it can migrate or deposit into the channel before thermolysis, the method will be solvent-free in the solid-state and the traditional hydrothermal operation will be avoided.Spherical MCM-4153 with a 2.0 nm pore size distribution (Fig. S1†) and a single opened-up surface was used as the sacrificial template. Readily available melamine was used as the precursor. The synthetic process is illustrated in Fig. 1a. Through calcination of the manually ground mixture of melamine and MCM-41, followed by thermolysis and desilication, single-shell g-C3N4 vesicles were prepared and denoted as SSCN. Different from the traditional templating method, the thermal sublimation and migration properties of melamine are fully utilized to avoid the pre-casting operation. The solid-state synthesis can simplify the operation while realizing morphological control. Fig. S2† shows the transmission electron microscope (TEM) images of MCM-41@g-C3N4 nanoparticles at different electron beam radiation times. With the prolonged irradiation time, the internal structure gradually shrinks, and voids appear in the particles and gradually increase to 74 nm. The yolk–shell structure not only proves the initial core–shell structure of g-C3N4@MCM-41 composite but also shows the excellent structural stability of g-C3N4. The morphology of SSCN is shown in Fig. 1b and c. SSCN has a uniform vesicular structure with a shell thickness of ca. 30.0 ± 4.5 nm, and the formation of the bulk (via self-template polymerization of melamine) is significantly inhibited as suggested by the uniform morphology. Before polymerizing into g-C3N4, melamine undergoes a migration into the channel or attaches on the outer surface, which is necessary to realize morphology control without the pre-casting operation. Furthermore, no impurity particles were observed in the cavity, suggesting that the polymerization of melamine in the MCM-41 channels was significantly inhibited; otherwise, the final sample would appear as a porous three-dimensional spherical structure. It is clear that the spherical MCM-41 plays three roles in the formation of the single-shell g-C3N4: (a) the silica prevents melamine from undergoing self-templating polymerization, (b) the external surface templates the formation of a shell structure, and (c) the narrow channel avoids polymerization to form the conventional ordered mesoporous materials. As the spherical MCM-41 has a hydroxyl rich surface, the hydrogen bonding between the hydroxyl and the amino groups of melamine would play a role in enriching the melamine molecules and enhancing the polymerization. The surface of the spherical MCM-41 is divided into two parts, including the channel and the external opened-up surface. As the calculated kinetic diameter of melamine molecules is smaller than the average pore size of the spherical MCM-41 (0.66 vs. 2.00 nm), the vaporized melamine molecule can first migrate through the channel. However, restricted by the relatively narrow channel that only accommodates a few molecules, the formation of oligomers is dominant inside the channel and the formation of a planar-stacking structure of g-C3N4 is significantly difficult. Moreover, it is quite difficult to polymerize such an ultralow concentration of melamine. In contrast, the external opened-up surface shows no confinement for this transition. The key point of this strategy lies in the appropriate size of the template channel, the precursor and the product, which is well-known as shape selectivity that is widely applied in the selective catalysis of zeolites.52 The above experimental results suggest that the polymerization of melamine selectively occurs on the opened-up outer surface of MCM-41.
 | ||
| Fig. 1 (a) Schematic illustration of the solid-state shape-selective templating method for SSCN preparation. (b, c) TEM images of the SSCN. | ||
The phase of the tailored g-C3N4 was identified as the typical pristine g-C3N4 as evidenced by X-ray diffraction (XRD) patterns, Fourier transform-infrared spectroscopy (FT-IR) and X-ray photoelectron spectroscopy (XPS) as displayed in Fig. S3.† Bulk g-C3N4 (BCN) was prepared for comparison under parallel experimental conditions without any templates. For the bulk g-C3N4, the typical diffraction peaks at about 27.50° and 13.04° correspond to the inter-layer structural stacking units (200) and the in-plane repeated heptazine units (100). In contrast, the single-shell g-C3N4 exhibits a much weaker (200) diffraction, which suggests decreased stacking layers, consistent with the morphological evolution from the bulk to vesicles. Meanwhile, the (100) diffraction was absent for SSCN, attributed to the minor fragmental g-C3N4 composition which greatly increases the structural disorder of the shells as evidenced by the scanning electron microscope (SEM) images (Fig. S4†). FT-IR and XPS spectra are also consistent with the structure as shown in the ESI.†
To delineate the thermal migration behavior of melamine during the solid-state synthesis, the products calcined with or without MCM-41 at different temperatures were compared. Fig. S5† shows the wide-angle and small-angle XRD patterns of the products calcined at different temperatures, with their phase shown in Table S2.† With increasing calcination temperature, melamine gradually polymerized into g-C3N4. When the temperature increases to 350 °C, melamine starts to oligomerize. Upon rising to 400–500 °C, the oligomers further transform to melem, which further polymerizes into g-C3N4 (melon) at 500 °C and above. On the other hand, the products show a significant difference in the absence of MCM-41. Although melamine could not polymerize below 300 °C, it forms melem when the temperature is above 350 °C without showing the formation of oligomers. This phenomenon suggests that MCM-41 promotes the transition of melamine, which may be attributed to the enrichment of melamine caused by capillary condensation of MCM-41 pores. As can be seen from Fig. S5c,† with increasing the temperature, the small angle diffraction peak increases first, then decreases, and reaches the maximum at 450 °C. According to Bragg's law (2dsinθ = nλ), the higher angle shift of the diffraction peak corresponds to the narrower pore channel, suggesting melamine migration into the channel. Consequently, when the angle of the diffraction peak is decreased, the channel becomes wider and oligomers migrate out of the channel. Combining small-angle and wide-angle XRD analyses, the migration of carbon and nitrogen species during the thermolysis with MCM-41 is illustrated in Fig. S6.† In the manually ground mixture, the contact between melamine and MCM-41 is weak. With the temperature rising up to 350 °C, melamine gradually migrates into MCM-41 and then oligomerizes between 350 °C and 450 °C. When the temperature reaches 450 °C, the channels are maximally filled with carbon and nitrogen species. At higher temperature, carbon and nitrogen species migrate out of the channel, deposit on the opened-up surface and polymerize.
To identify the role of the pore size, MCM-41 with larger channels is used as the template (denoted as MS-x) (x = 1, 2, 3). Nitrogen physical sorption (Fig. S7†) suggests that the three samples have a uniform pore size distribution, ranging from 3.8 to 6.3 nm. With templating by MS-x, the as-prepared g-C3N4 exhibits a three-dimensional morphology (Fig. S8†), demonstrating that melamine migrates into the channel before polymerization. Nevertheless, the larger channel corresponds to the lack of shape selectivity for melamine which leads to the conventional three-dimensional mesoporous structure, rather than a hollow structure. In the case of dense silica with a comparable particle size, the observation of a bulk morphology (Fig. S9†) revealed that g-C3N4 does not polymerize around silica, further suggesting that the channels of MCM-41 serve as the basket to enrich melamine.
The vesicle structure enables the separation of redox active sites on the interior and exterior surfaces, thereby facilitating the charge separation and enhancing the photocatalysis.48 Reducing the wall thickness of the vesicles is beneficial to shorten the migration path of carriers from inside to outside. Therefore, it is beneficial to have a thinner wall. Fig. S10† shows the TEM images of g-C3N4 vesicles with a tunable shell thickness. Through facile tuning of the mass ratio of melamine to MCM-41 between 0.1 and 0.8, the thickness of the vesicle structure can be regulated continuously from 42.1 to 17.5 nm. At the same time, the primary nanoparticles composed of a shell are in close contact with each other, which provides good structural stability even if its thickness is very thin.
Versatile method to tailor the double-shell g-C3N4 and heterojunction
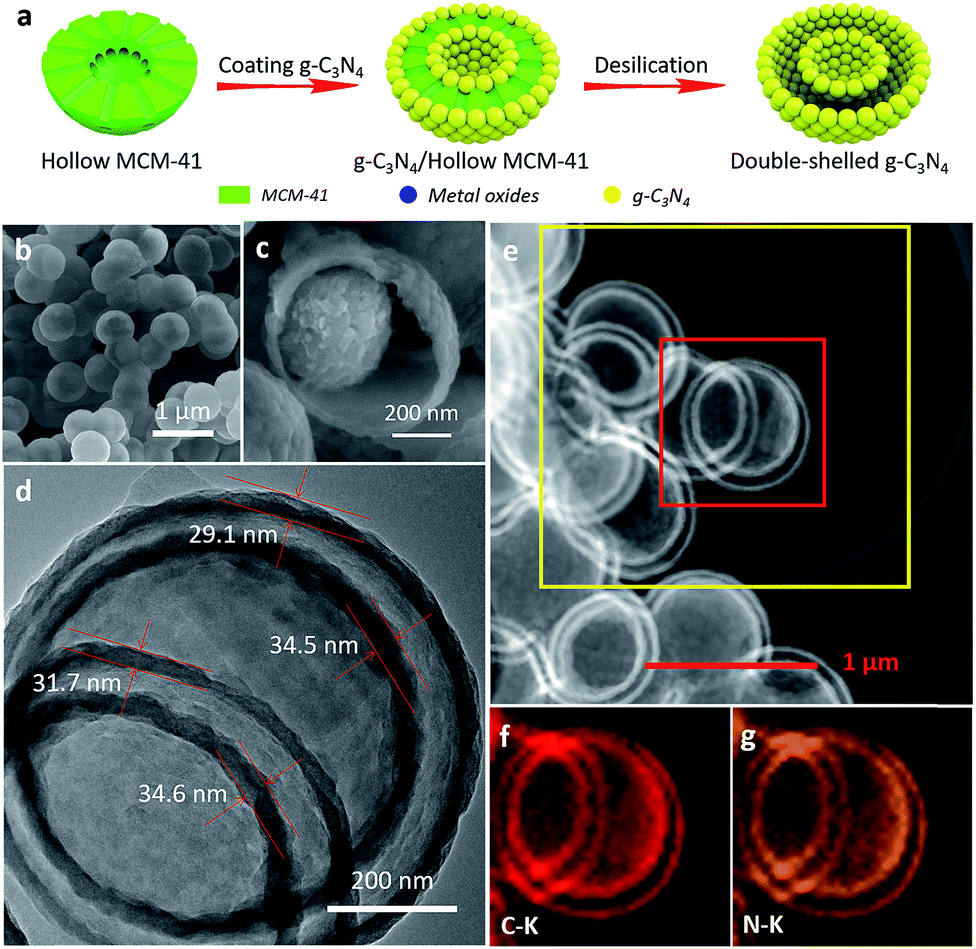
For constructing multi-shell g-C3N4 vesicles, increasing the layers of the opened-up surface of MCM-41 is sufficient when using the solid-state method. For instance, the preparation of double-shell vesicles requires two homocentric opened-up surfaces and hollow MCM-41 meets this. By contrast, the conventional templating method needs a three-layer template that has two voids for nano-casting. However, those three-layer templates are difficult to prepare. Hollow MCM-41 with a pore size of 2.2 nm (Fig. S11†) is facilely prepared by the surface-protected etching method,53 which retains the shape selectivity for melamine.Double-shell g-C3N4 vesicles (DSCN) exhibit a uniform spherical structure without a bulk morphology (Fig. 2b), which further confirms that the solid-state templating method is effective to tailor the morphology. The SEM image of the broken DSCN in Fig. 2c clearly shows a double-shell structure, which is composed of primary fragmental nanoparticles inconsistent with SSCN. The TEM image in Fig. 2d further evidences the double-shell vesicle structure, with an interior and exterior wall thickness of ca. 30 nm. Fig. 2e–g present the TEM-mapping images of DSCN. They clearly show the uniform elemental distribution of carbon and nitride, corresponding to the heptazine heterocycle ring (C6N7) units of g-C3N4.
 | ||
| Fig. 2 (a) Schematic illustration of the preparation and (b, c) SEM, (d) TEM and (e–g) TEM-mapping images of DSCN. | ||
Apart from morphology control, the method can also realize the encapsulation of the secondary component for heterojunction construction. The channel of MCM-41 is not occupied in the g-C3N4/MCM-41 composites; if a certain amount of the secondary component is placed in the channel of MCM-41 in advance (Fig. 3a), metal and metal oxide encapsulated g-C3N4 vesicles could be prepared. Metals or metal oxides were introduced into MCM-41 through a highly reproducible impregnation method, followed by thermal calcination to convert the metal ions into metals or metal oxides. MCM-41 can act not only as molecular sieves to produce vesicles but also as a basket to contain the secondary component to realize encapsulation.
 | ||
| Fig. 3 (a) Schematic illustration of the encapsulation of the secondary component inside SSCN. (b) SEM, (c) particle size distribution, (d, e) TEM and (f) TEM-mapping images of Ag@SSCN capsules. Red, yellow and blue represent carbon, nitride and silver, respectively. | ||
The silver encapsulated g-C3N4 vesicle composites (denoted as Ag@SSCN) were prepared through the same method except for the use of Ag/MCM-41 templates. The as-prepared Ag@SSCN has a uniform 637 nm spherical structure without a bulk morphology (Fig. 3b and c), which further confirms that the new method is universal for facile morphology control. The TEM images (Fig. 3d and e) reveal Ag@SSCN having a similar vesicle structure to SSCN, suggesting that silver nanoparticles in the channel of MCM-41 do not affect the selective deposition of g-C3N4. No apparent silver nanoparticles were observed, suggesting its high dispersion, which is attributed to the anchoring of silver by nitrogen atoms. Meanwhile, the co-existence of carbon, nitride and silver was confirmed by the TEM-mapping images (Fig. 3f). It is clear that carbon and nitrogen constituted the shell and silver dispersed evenly inside the shell as an encapsulation structure.
Fig. S12† presents the XPS spectra of Ag@SSCN. It is revealed that the C 1s and N 1s XPS spectra are corresponding to g-C3N4 as analysed previously. The O 1s peak (Fig. S12b†) can be divided into two peaks at 532.6 and 531.6 eV, which belong to lattice oxygen and adsorbed oxygen species, respectively. The Ag 3d5/2 XPS spectra can be divided into two peaks at 368.3 and 367.7 eV, which are assigned to Ag0 and Ag+, respectively. The fitting results showed that the contents of Ag0 and Ag+ were 59.2% and 40.8%, respectively. The results show that the structure of Ag@SSCN retains the structure of g-C3N4 and the encapsulation of Ag is realized successfully.
The content of the second metal component in the encapsulated vesicles can be controlled through changing the impregnation amount of silver in the template. Fig. S13† shows the TEM images of the encapsulated vesicles with different amounts of silver. It suggests that when the content of silver in the template is lower than 6.0 wt%, the morphology of Ag@SSCN does not change significantly, and the single-thin-shell vesicle structure remains. At the same time, the fact that Ag nanoparticles were not visible in TEM suggests that Ag is highly dispersed. When the content of Ag in the template is higher than 10.0 wt%, the morphology of Ag@SSCN dramatically changes, from the regular vesicle structure to a wrinkled vesicle structure, which can provide more active sites for the reaction.
In addition to encapsulating noble metals, base metal oxides can also be encapsulated through the same strategy. The XPS spectra (Fig. S14†) evidence the existence of the metal oxides (Fe2O3, Co2O3, NiO and CuO). Meanwhile the intact vesicle structure (Fig. S15†) evidences that the introduction of metal oxides does not significantly affect the melamine polymerizing into g-C3N4.
Photocatalytic performances
The photocatalytic performance was evaluated through hydrogen evolution from water under visible light (λ > 420 nm) with 10.0 vol% triethanolamine (TEOA) as the electron donor and 3.0 wt% Pt as the co-catalyst. As shown in Fig. 4a, no hydrogen was detected in the absence of photocatalysts. BCN shows a very low photocatalytic H2 evolution rate (HER) of 4.1 μmol h−1, which can be attributed to the low SBET, poor visible light absorption and fast carrier recombination rate. The apparent quantum yield (AQY) of BCN shows only 0.17% at 420 nm, which is very low. The tailored SSCN exhibits significantly enhanced photocatalytic activity with the HER reaching 14.3 μmol h−1, almost 3.5-fold of that with BCN. Such enhanced performance of SSCN is believed to be related to the suppressed carrier recombination (evidenced by the PL spectra in Fig. S16†). In comparison, Ag@SSCN exhibits a further increase of the HER to 18.6 μmol h−1. On the other hand, DSCN exhibits further dramatically enhanced performance and the HER gains a 6.7-fold increase and reaches 27.5 μmol h−1, with the AQY at 420 nm improved to 1.20% (Fig. S17†). The dramatically enhanced photocatalytic activity can be mainly ascribed to the further increased photoabsorption due to the multi-scattering effect.45,54,55 DSCN also maintains a highly reproducible photoactivity with no apparent deterioration during a 20 h cycle test (Fig. 4a), suggesting the great stability of the thin-shell vesicle structure. | ||
| Fig. 4 (a) Time course of hydrogen evolution (i) without photocatalysts and in the presence of (ii) BCN, (iii) SSCN, (iv) Ag@SSCN, and (v) DSCN under visible light (λ > 420 nm). (b) Transient photocurrent response and (c) EIS Nyquist plots of BCN, SSCN, Ag@SSCN and DSCN loaded on the FTO glass. (d) Photocatalytic decoloration of RhB under visible light (λ > 420 nm). | ||
Fig. 4b shows the transient photocurrent response of the catalysts loaded on the FTO glass measured under visible light irradiation. Generally, photocurrent forms mainly through separation of photogenerated electron–hole pairs and diffusion from the interior to the free charge acceptors on its surface or in the electrolyte.56 Among the tailored catalysts, DSCN displays the largest photocurrent response, suggesting less charge recombination and more efficient transition of charge carriers than those of others, in agreement with the change of the photocatalytic activity. Fig. 4c displays the electrochemical impedance spectroscopy (EIS) results, which can reflect the rate of the reaction occurring on the surface of the working electrode by the arc radius. The smaller arc radius than BCN for SSCN suggests a more effective separation efficiency of photogenerated electron–hole pairs and a faster interfacial charge transfer, implying that morphological control could obviously favour the separation and transfer of photogenerated carriers in g-C3N4 and then enhance the photocatalytic activity. Furthermore, DSCN displays the smallest arc radius, suggesting that the double shell structure could change the charge distribution of g-C3N4 and favour charge transfer.
The photocatalytic properties were also evaluated through the decoloration of RhB under visible-light irradiation (λ > 420 nm), which presents similar trends to the hydrogen production reaction. Before visible light irradiation, the suspension was stirred in the dark for 30 min to reach the adsorption–desorption equilibrium. As shown in Fig. 4d, SSCN shows a much higher adsorption capability for RhB, mainly attributed to the much larger SBET of 42.4 m2 g−1. By contrast, DSCN with a similar SBET to SSCN displays a higher RhB adsorption ability, suggesting that the double shell structure could enrich the reactant. Notably, photocatalytic decoloration of RhB is actually a deamino process but not mineralization in most cases.57 With visible light irradiation, in the presence of the bulk g-C3N4 (BCN), about 40% of the initial RhB was decolored within 120 min, corresponding to a low first-order kinetic constant of 0.005 min−1 (Fig. S20†). Compared with BCN, SSCN presents a photoactivity enhancement and can totally decolor the initial RhB within the same reaction time. The enhanced photoactivity is attributed not only to the suppressed charge recombination efficiency as evidenced by the steady-state PL spectra but also to the enhanced photoabsorption derived from the multi scatting effect inside the shell. The suppressed charge recombination efficiency originates from the shortened charge carrier migration length, which makes the photogenerated charge carriers more likely to transfer to the surface and be captured by the RhB substrates. Furthermore, DSCN can totally decolor the initial RhB within 40 min and presents the most superior photoactivity. Compared with BCN and SSCN, the first-order kinetic constant improved almost 15.6 and 6.0 fold, respectively. As the SBET values of SSCN and DSCN are similar to each other, the differences of photocatalysis for DSCN are predominantly attributed to the photo scattering effect inside the multiple shells. Notably, as the pore size distribution of the shell is larger than the RhB molecule, the surface including the internal shell is accessible and can be fully utilized for photocatalytic reaction.
Conclusions
In summary, a versatile shape-selective solid-state templating method using MCM-41 sieves has been successfully developed to fabricate stable thin-shell g-C3N4 vesicles and achieve their encapsulation. The proper pore channel of MCM-41 makes it possible for melamine to selectively deposit and transform on the open-up surface but not inside the channel and form a stable and tunable thin-shell morphology. The tailored g-C3N4 vesicles can suppress the recombination of photo-induced electrons and positive holes and enhance photoabsorption, contributing to the enhanced hydrogen evolution rate in water splitting over double-shell g-C3N4 vesicles by 6.7-fold, higher than that with bulk g-C3N4. This study provides a versatile and green method for morphological engineering and secondary component encapsulation. We believe that this facile and versatile method can be used to optimize the electronic structure of g-C3N4 and assemble carbon-based morphology-tailored materials through the proper selection of the template and precursor to induce shape selectivity.Methods
Reagents
Melamine, cetyltrimethyl ammonium bromide (CTAB), tetraethyl orthosilicate (TEOS) and NH3·H2O (27.0 wt%) are of analytical grade and used as received. Distilled water is used at all times.Preparation of MCM-41 and hollow MCM-41
The spherical and hollow MCM-41 sieves are prepared according to the literature.53 The Stöber method is used to prepare spherical MCM-41. The surface-protected etching method is used to prepare hollow MCM-41. In a typical experiment, 1.0 g spherical MCM-41 without calcination was suspended in 160 mL distilled water at 70 °C. The suspension was stirred gently for 12 h to generate a hollow structure. The mechanism of the surface-protected etching method can be found in the literature.53 Spherical and hollow MCM-41 sieves then undergo calcination at 550 °C for 6 h to eliminate CTAB.Preparation of single and double-shell g-C3N4
In a typical experiment for the preparation of the single-shell g-C3N4, 1.0 g melamine and a certain amount of MCM-41 were ground to uniform phase, followed by thermal polymerization at 550 °C for 3 h at a heating rate of 10 °C min−1. After cooling down, the as-obtained yellow composites were treated with 4 M NH4HF2 aqueous solution to eliminate the silica templates. The solid was washed with water several times and dried at 100 °C overnight. In the case of the double-shell g-C3N4 preparation, an identical process was conducted but with the hollow MCM-41 as the sacrificial template. The as-obtained single- and double-shell g-C3N4 were labelled SSCN and DSCN.Photocatalytic hydrogen evolution
Visible light photocatalytic water splitting for hydrogen production was conducted in a Pyrex top-irradiation reaction vessel connected to a glass closed gas system to evaluate the photocatalytic performances of the tailored g-C3N4. Visible light comes from a 300 W Xe lamp with an optical cut-off filter (λ > 420 nm). In a typical experiment, 50 mg of the tailored g-C3N4 was mixed with 10 mL TEOA and 90 mL H2O and ultrasonicated for 5 min. Then, meanwhile, the system was evacuated several times to completely remove the air, and aqueous H2PtCl6 with 3.0 wt% Pt was loaded and photoreduced for 30 min. After stopping the vacuum operation, the evolved gases were in situ analysed every 60 min using a gas chromatograph (GC-14) equipped with a TCD and a 5 Å molecular sieve column. Moreover, the apparent quantum yield (AQY) under monochromatic light was calculated by a similar procedure from the following equation:
Photocatalytic decoloration of RhB
The visible light photocatalytic activity of the catalysts was evaluated with photodecoloration of RhB. Visible light comes from a 300 W Xe lamp with an optical cut-off filter (λ > 420 nm). In a typical experiment, 50 mg of the tailored g-C3N4 were mixed with 50 mL RhB solution (50 mg L−1) and ultrasonicated for 1 min. Before visible light irradiation, the suspension was stirred at 35 °C in the dark for 30 minutes to reach the adsorption–desorption equilibrium. After turning on the visible light, the suspension was sampled with 5 minute intervals and centrifuged to measure the concentration of RhB with an ultraviolet spectrophotometer.Characterization
TEM images were taken on a Tecnai G2 20 S-twin instrument (FEI Company). The nitrogen sorption isotherms were measured with a Quantachrome autosorb-iQ2 gas adsorption analyzer at 77 K. XRD patterns were recorded on a Smart Lab 9 X-ray diffractometer (Rigaku Corporation). FT-IR spectra were measured on an EQUINOX55 FT-IR spectrometer. XPS spectra were recorded on a Thermo VG ESCALAB250 instrument. Steady-state PL spectra were acquired on a JASCO FP-6200 spectrofluorometer with an excitation wavelength of 310 nm. UV-DSR spectra were recorded on a UV-3600Plus spectrophotometer with BaSO4 as the reference sample. The photoelectrochemical experiments were performed in a standard three-electrode cell on a CHI660E work station with the synthesized samples loaded on FTO glass as the working electrode. Ag/AgCl was the reference electrode and a platinum plate was the auxiliary electrode. The working electrode was placed in the middle of 0.1 M Na2SO4 aqueous solution. A 300 W Xe lamp with a 420 nm cut-off filter provided visible-light irradiation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the State Key Program of National Natural Science Foundation of China (Grant no. 21306018), the QianRen program, and the Fundamental Research Funds for the Central Universities (Grant No. DUT16LK12).References
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed.
- W. J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159 CrossRef CAS PubMed.
- Y. Zheng, L. Lin, B. Wang and X. Wang, Angew. Chem., Int. Ed., 2015, 54, 12868 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, M. Jaroniec, Y. Jin and S. Z. Qiao, Small, 2012, 8, 3550 CrossRef CAS PubMed.
- D. T. Martin, K. Qiu, S. A. Shevlin, A. D. Handoko, X. Chen, Z. Guo and J. Tang, Angew. Chem., Int. Ed., 2014, 126, 9394 CrossRef.
- D. J. Martin, P. J. Reardon, S. J. Moniz and J. Tang, J. Am. Chem. Soc., 2014, 136, 12568 CrossRef CAS PubMed.
- G. Zhang, Z. A. Lan and X. Wang, Chem. Sci., 2017, 8, 5261 RSC.
- G. Zhang, Z.-a. Lan and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 15712 CrossRef PubMed.
- S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Z.-X. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731 RSC.
- J. Fang, H. Fan, M. Li and C. Long, J. Mater. Chem. A, 2015, 3, 13819 RSC.
- A. Naseri, M. Samadi, A. Pourjavadi, A. Z. Moshfegh and S. Ramakrishna, J. Mater. Chem. A, 2017, 5, 23406 RSC.
- J. Ran, T. Y. Ma, G. Gao, X.-W. Du and S. Z. Qiao, Energy Environ. Sci., 2015, 8, 3708 RSC.
- Y. Fang, X. Li and X. Wang, ACS Catal., 2018, 8, 8774 CrossRef CAS.
- Y. Fang, Y. Xu, X. Li, Y. Ma and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 9749 CrossRef CAS PubMed.
- K. Wang, Q. Li, B. Liu, B. Cheng, W. Ho and J. Yu, Appl. Catal., B, 2015, 176–177, 44 CAS.
- P. Niu, Y. Yang, J. C. Yu, G. Liu and H. M. Cheng, Chem. Commun., 2014, 50, 10837 RSC.
- W. Yu, D. Xu and T. Peng, J. Mater. Chem. A, 2015, 3, 19936 RSC.
- Y.-L. Wang, Y. Tian, Z.-L. Lang, W. Guan and L.-K. Yan, J. Mater. Chem. A, 2018, 6, 21056 RSC.
- J. Zhou, H. Wu, C.-Y. Sun, C.-Y. Hu, X.-L. Wang, Z.-H. Kang and Z.-M. Su, J. Mater. Chem. A, 2018, 6, 21596 RSC.
- P. Yang, H. Zhuzhang, R. Wang, W. Lin and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 1134 CrossRef CAS PubMed.
- C. Ling, X. Niu, Q. Li, A. Du and J. Wang, J. Am. Chem. Soc., 2018, 140, 14161 CrossRef CAS PubMed.
- M. Zheng, J. Shi, T. Yuan and X. Wang, Angew. Chem., Int. Ed., 2018, 57, 5487 CrossRef CAS PubMed.
- Y. Zhang, Z. Zhou, Y. Shen, Q. Zhou, J. Wang, A. Liu, S. Liu and Y. Zhang, ACS Nano, 2016, 10, 9036 CrossRef CAS PubMed.
- X. Wang, X. Chen, A. Thomas, X. Fu and M. Antonietti, Adv. Mater., 2009, 21, 1609 CrossRef CAS.
- M. Zhang, W. Yao, Y. Lv, X. Bai, Y. Liu, W. Jiang and Y. Zhu, J. Mater. Chem. A, 2014, 2, 11432 RSC.
- X. Bai, C. Sun, S. Wu and Y. Zhu, J. Mater. Chem. A, 2015, 3, 2741 RSC.
- Q. Han, B. Wang, J. Gao, Z. Cheng, Y. Zhao, Z. Zhang and L. Qu, ACS Nano, 2016, 10, 2745 CrossRef CAS PubMed.
- Z. Zhou, Y. Shen, Y. Li, A. Liu, S. Liu and Y. Zhang, ACS Nano, 2015, 9, 12480 CrossRef CAS PubMed.
- B. V. Lotsch and W. Schnick, Chemistry, 2007, 13, 4956 CrossRef CAS PubMed.
- M. B. Ansari, B.-H. Min, Y.-H. Mo and S.-E. Park, Green Chem., 2011, 13, 1416 RSC.
- J. Chen, Z. Mao, L. Zhang, D. Wang, R. Xu, L. Bie and B. D. Fahlman, ACS Nano, 2017, 11, 12650 CrossRef CAS PubMed.
- L. Shi, L. Liang, F. Wang, M. Liu, K. Chen, K. Sun, N. Zhang and J. Sun, ACS Sustainable Chem. Eng., 2015, 3, 3412 CrossRef CAS.
- J. Xu, Y. Wang and Y. Zhu, Langmuir, 2013, 29, 10566 CrossRef CAS PubMed.
- D. J. Martin, K. Qiu, S. A. Shevlin, A. D. Handoko, X. Chen, Z. Guo and J. Tang, Angew. Chem., Int. Ed., 2014, 53, 9240 CrossRef CAS PubMed.
- J.-H. Zhang, Y.-J. Hou, S.-J. Wang, X. Zhu, C.-Y. Zhu, Z. Wang, C.-J. Li, J.-J. Jiang, H.-P. Wang, M. Pan and C.-Y. Su, J. Mater. Chem. A, 2018, 6, 18252 RSC.
- X. Wang, Y. Zhang, W. Luo, A. A. Elzatahry, X. Cheng, A. Alghamdi, A. M. Abdullah, Y. Deng and D. Zhao, Chem. Mater., 2016, 28, 2356 CrossRef CAS.
- X. Du and S. Z. Qiao, Small, 2015, 11, 392 CrossRef CAS PubMed.
- J. Zhang, M. Zhang, C. Yang and X. Wang, Adv. Mater., 2014, 26, 4121 CrossRef CAS PubMed.
- C. Dai, A. Zhang, M. Liu, L. Gu, X. Guo and C. Song, ACS Nano, 2016, 10, 7401 CrossRef CAS PubMed.
- C. Dai, A. Zhang, M. Liu, X. Guo and C. Song, Adv. Funct. Mater., 2015, 25, 7479 CrossRef.
- D. Zheng, G. zhang and X. Wang, Appl. Catal., B, 2015, 179, 479 CrossRef CAS.
- Z. Tong, D. Yang, Z. Li, Y. Nan, F. Ding, Y. Shen and Z. Jiang, ACS Nano, 2017, 11, 1103 CrossRef CAS PubMed.
- J. Sun, J. Zhang, M. Zhang, M. Antonietti, X. Fu and X. Wang, Nat. Commun., 2012, 3, 1139 CrossRef.
- L. Zhou, Z. Zhuang, H. Zhao, M. Lin, D. Zhao and L. Mai, Adv. Mater., 2017, 29, 1602914 CrossRef PubMed.
- B. Qiu, Q. Zhu, M. Du, L. Fan, M. Xing and J. Zhang, Angew. Chem., Int. Ed., 2017, 56, 2684 CrossRef CAS PubMed.
- Y. Zhao and L. Jiang, Adv. Mater., 2009, 21, 3621 CrossRef CAS.
- Y. Yang, X. Liu, X. Li, J. Zhao, S. Bai, J. Liu and Q. Yang, Angew. Chem., 2012, 51, 9164 CrossRef CAS PubMed.
- D. Zheng, X. N. Cao and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 11512 CrossRef CAS PubMed.
- Q. Zhang, D. Q. Lima, I. Lee, F. Zaera, M. Chi and Y. Yin, Angew. Chem., 2011, 50, 7088 CrossRef CAS PubMed.
- T. Ikuno, J. Zheng, A. Vjunov, M. Sanchez-Sanchez, M. A. Ortuño, D. R. Pahls, J. L. Fulton, D. M. Camaioni, Z. Li, D. Ray, B. L. Mehdi, N. D. Browning, O. K. Farha, J. T. Hupp, C. J. Cramer, L. Gagliardi and J. A. Lercher, J. Am. Chem. Soc., 2017, 139, 10294–10301 CrossRef CAS PubMed.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710 CrossRef CAS.
- B. Smit, Chem. Rev., 2008, 108, 4125 CrossRef CAS PubMed.
- Z. Teng, X. Su, Y. Zheng, J. Sun, G. Chen, C. Tian, J. Wang, H. Li, Y. Zhao and G. Lu, Chem. Mater., 2013, 25, 98 CrossRef CAS.
- B. Jin, E. Jung, M. Ma, S. Kim, K. Zhang, J. I. Kim, Y. Son and J. H. Park, J. Mater. Chem. A, 2018, 6, 2585 RSC.
- H. Li, Z. Bian, J. Zhu, D. Zhang, G. Li, Y. Huo, H. Li and Y. Lu, J. Am. Chem. Soc., 2007, 129, 8406 CrossRef CAS PubMed.
- D. Zhang, Y. Guo and Z. Zhao, Appl. Catal., B, 2018, 226, 1 CrossRef CAS.
- S. Bae, S. Kim, S. Lee and W. Choi, Catal. Today, 2014, 224, 21 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ta01521c |
| This journal is © The Royal Society of Chemistry 2019 |