
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights into a layered hybrid solid electrolyte and its application in long lifespan high-voltage all-solid-state lithium batteries†
Shicheng
Yu‡
 *a,
Sebastian
Schmohl‡
b,
Zigeng
Liu
*ac,
Marija
Hoffmeyer
b,
Nino
Schön
ad,
Florian
Hausen
ad,
Hermann
Tempel
a,
Hans
Kungl
a,
Hans–D.
Wiemhöfer
b and
Rüdiger–A.
Eichel
ad
*a,
Sebastian
Schmohl‡
b,
Zigeng
Liu
*ac,
Marija
Hoffmeyer
b,
Nino
Schön
ad,
Florian
Hausen
ad,
Hermann
Tempel
a,
Hans
Kungl
a,
Hans–D.
Wiemhöfer
b and
Rüdiger–A.
Eichel
ad
aInstitut für Energie- und Klimaforschung (IEK-9: Grundlagen der Elektrochemie), Forschungszentrum Jülich, D-52425 Jülich, Germany. E-mail: s.yu@fz-juelich.de; zigeng.liu@cec.mpg.de
bInstitut für Anorganische und Analytische Chemie, Westfälische Wilhelms-Universität Münster, D-48149 Münster, Germany
cMax-Planck-Institut für Chemische Energiekonversion, D-45470 Mülheim an der Ruhr, Germany
dInstitut für Physikalische Chemie, RWTH Aachen Universität, D-52074 Aachen, Germany
First published on 16th January 2019
Abstract
Direct integration of a metallic lithium anode with the ceramic Li1.3Al0.3Ti1.7(PO4)3 (LATP) electrolyte into an all-solid-state battery is highly challenging due to their chemical and electrochemical incompatibility. Herein, a layered hybrid solid electrolyte is designed by coating the ceramic LATP electrolyte with a protective polymer electrolyte, polyphosphazene/PVDF-HFP/LiBOB. This polymer electrolyte comprises highly Li+ conductive polyphosphazene and mechanically stable PVDF-HFP as the polymer matrix, and the mobile lithium ions in the polymer layer are supplied by LiBOB. Equipped with both polymer and ceramic components, the hybrid electrolyte possesses favorable features, such as a flexible surface, high ionic conductivity, high chemical stability against lithium and wide electrochemical stability window (4.7 V), which all to help realize its application in all-solid-state lithium batteries. The prepared all-solid-state battery with a metallic lithium anode and high-voltage Li3V2(PO4)3/CNT cathode shows high capacity and excellent cycling performance with negligible capacity loss over 500 cycles at 50 °C. Furthermore, the analysis of the hybrid solid electrolyte after long-term cycling demonstrates outstanding electrode/electrolyte interfacial stability. This study suggests that use of solid organic–inorganic hybrid electrolyte is a promising approach to circumvent the mechanical, chemical and electrochemical limitations at the interface of electrodes and ceramic electrolyte for all-solid-state batteries.
Introduction
All-solid-state batteries (ASSBs) are of great interest because of their inherent safety and wide operable temperature range due to the lack of flammable liquid components, as well as potential benefits in terms of gravimetric and volumetric energy density.1,2 Instead of a porous separator soaked with a liquid electrolyte, ASSBs use a solid electrolyte, which acts as an electrical insulator and ionic conductor at once. From the viewpoint of mechanical compatibility, a compact solid electrolyte permits the use of a metallic lithium anode in ASSBs because the solid nature of the electrolyte can effectively block lithium dendrites.3–5 Therefore, an increase in energy density can be achieved compared to lithium-ion batteries with conventional anode materials (e.g., graphite).1 Additionally, the high electrochemical stability of some solid electrolytes facilitates the use of high-capacity (e.g., LiCoO2) or high-voltage (e.g., Li3V2(PO4)3) cathodes,5,6 which also leads to increased energy densities at the cell level. However, unresolved fundamental issues remain in the development of ASSBs with metallic lithium anodes, mainly with respect to electrochemical compatibility at the interfaces.7 Moreover, there are also significant engineering challenges that require methodical effort to enable obtaining a tangible product. Therefore, the scale-up process from academic laboratories to industry to bring ASSBs to the booming sustainable energy market remains unsuccessful so far, owing to the required high cost and complicated processing efforts.8The origin of electrochemical interfacial issues, leading to a variation in composition over the interface and an increment of the interfacial resistance, can be narrowed down to atomic-scale reactions, such as element inter-diffusion and segregation during battery operation,9,10 and the formation of space charge layers and unwanted redox reactions due to electrochemical instabilities.11 Quite a few successful approaches have been reported to mitigate the interfacial issues between solid electrolytes and cathodes, including coating a solid electrolyte on the surface of cathodes, interface softening and adding a solid electrolyte to the cathode composite.6,12–14 However, the interface at the anode side has rarely been explored, especially when metallic lithium is used, due to the fact that many solid electrolytes are incompatible with metallic Li.
Li1.3Al0.3Ti1.7(PO4)3 (denoted as LATP) is an attractive solid electrolyte with a scalable production procedure for the possible commercialization of ASSBs, owing to its competitive ionic conductivity (close to 1 mS cm−1 at room temperature) and its low sensitivity against an oxygen/moisture atmosphere compared to other ceramic ion conductors like sulfide-based glass electrolytes or garnet electrolytes.15–17 Unfortunately, LATP degrades when in contact with the lithium metal since Ti4+ will be reduced to Ti3+, leading to a decomposed electrolyte surface with high resistance for Li+ migration.15 Moreover, like most ceramic electrolytes, poor mechanical contacts and/or micro-cracks may occur between the LATP electrolyte and electrodes during battery preparation and operation, thus blocking percolation pathways for lithium ions. Normally, most mechanical interfacial issues can be circumvented by specialized fabrication methods like spark plasma sintering (for “bulk-type” ASSBs)18 and deposition methods (for “thin-film” ASSBs),19,20 or by applying external pressure on cells.6 But, all these methods usually result in either low cell energy or low energy density at the system level and the required massive fabrication efforts are hardly realized in large-scale production.
Since polymer electrolytes have the features of high mechanical flexibility, large intrinsic electrochemical stability windows and great chemical stability against metallic lithium,21–24 an alternative way to overcome the interfacial and technical issues, and to enable the application of metallic lithium in ASSBs is to sandwich a thin layer of polymer between the electrode and ceramic electrolyte. It is possible to solve the above-mentioned issues with this complementary polymer/ceramic/polymer electrolyte. First, the magnitude of the electric field and trapped positive charge at the Li/polymer interface is reduced due to the blocking of anion transport by the ceramic layer, which facilitates protection of the polymer electrolyte from chemical/electrochemical decomposition and improves the coulombic efficiency of a battery.25 Second, since the polymer is an electronic insulator, direct electron transfer for an interfacial chemical reaction at the ceramic surface is avoided which could significantly enlarge the electrochemical stability window of ceramic electrolytes.26 Third, the interface fabrication process is simplified as compared with that for all-ceramic ASSBs. Finally, introducing the polymer electrolyte as a thin layer between ceramic and electrodes not only prevents the direct contact of metallic lithium with the ceramic electrolyte, which eliminates the possibilities of side reactions, but also forms a favorable interface between rigid ceramic components, which results in a better connection in the solid-state battery. Although the layered hybrid solid electrolyte architecture increases the total resistance of the electrolyte as compared to a single inorganic electrolyte, such a structure is expected to be more suitable for an all-solid-state lithium battery since wetting of the lithium surface by the polymer provides a uniform Li+ flux across the interface and thus suppresses the formation of lithium dendrites.
Herein, we present an all-solid-state test cell with a stable metallic lithium anode which makes full use of the promising manufacturability and high ionic conductivity of LATP. By combining the ceramic LATP solid electrolyte with a solid polymer electrolyte (denoted as SPE), a layered hybrid solid electrolyte (SPE/LATP/SPE, denoted as LHSE) was constructed and it exhibited great advances regarding chemical and electrochemical stability against metallic lithium anodes. Specifically, highly ionic conductive oligoethylene oxide-functionalized polyphosphazene, poly[bis(2-(2-methoxyethoxy)ethoxy)phosphazene] (MEEP) and highly mechanically stable poly(vinylidene fluoride-co-hexafluoropropylene) (PVDF-HFP) were rationally chosen for the fabrication of the SPE to ensure eligible electrochemical properties and interfacial mechanical strength. The prepared LHSEs were (electro-)chemically stable against metallic lithium and were applied in test cells of the type Li|LHSE|Li3V2(PO4)3/CNT.
Experimental section
Preparation of the layered hybrid solid electrolyte
All chemicals for the sample syntheses were purchased from Sigma-Aldrich and were used as received.Poly[bis(2-(2-methoxyethoxy)ethoxy)phosphazene] (MEEP) was synthesized according to the four step synthesis route reported by Wang et al.27,28 and a living cationic polymerization reaction that was first published by Allcock et al.29 with minor modifications.23 After synthesis of the precursor polymer [NPCl2]n, MEEP was obtained by substitution of chlorine atoms with corresponding sodium alcoholates. The details of the synthesis, as well as the electrochemical and thermal properties of MEEP, have already been published before.30
The synthesis of the Li1.3Al0.3Ti1.7(PO4)3 powder and the preparation of ceramic electrolyte pellets were performed as reported in our previous studies.6,16 The ceramic pellets used as substrates for the polymer electrolyte coating were prepared from the LATP powder pre-calcined at 800 °C via a pressing–sintering process, which including uniaxial die pressing, densification by cold isostatic pressing at 504 MPa for 30 seconds, and subsequent sintering at 1100 °C for 8 hours. The thickness and surface roughness of the LATP pellets were controlled by polishing with sandpapers (P800 and P1200). The relative density of the prepared ceramic substrate is higher than 90% of its theoretical density (2.93 g cm−3).
Mechanically stable polymer electrolytes were prepared in an argon filled glove box. A mixture of 65.3 wt% MEEP polymer, 21.7 wt% poly(vinylidene fluoride-co-hexafluoropropylene) (PVDF-HFP, Mw = ∼400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) and 13.0 wt% lithium bis(oxalato)borate (LiBOB) were prepared and stirred in acetone (≥99.9%) overnight to form a homogenous solution. Then the solution was spread onto a glass plate, stabilized at room temperature overnight, dried at 50 °C for 24 hours, and further dried under vacuum at 100 °C for one day to form the polymer electrolyte. Furthermore, the thickness of the solid polymer electrolyte was varied by using different volumes of the coating solution. For example, ∼70 μL slurry eventually resulted in a ∼120 μm dry polymer layer.
000) and 13.0 wt% lithium bis(oxalato)borate (LiBOB) were prepared and stirred in acetone (≥99.9%) overnight to form a homogenous solution. Then the solution was spread onto a glass plate, stabilized at room temperature overnight, dried at 50 °C for 24 hours, and further dried under vacuum at 100 °C for one day to form the polymer electrolyte. Furthermore, the thickness of the solid polymer electrolyte was varied by using different volumes of the coating solution. For example, ∼70 μL slurry eventually resulted in a ∼120 μm dry polymer layer.
The layered hybrid solid electrolyte was prepared by coating the polymer layers on LATP pellets in an argon filled glovebox. As schematically shown in route (a) in Fig. 1, the polymer electrolyte was coated on both sides of the LATP pellets by repeating the polymer layer formation process described above.
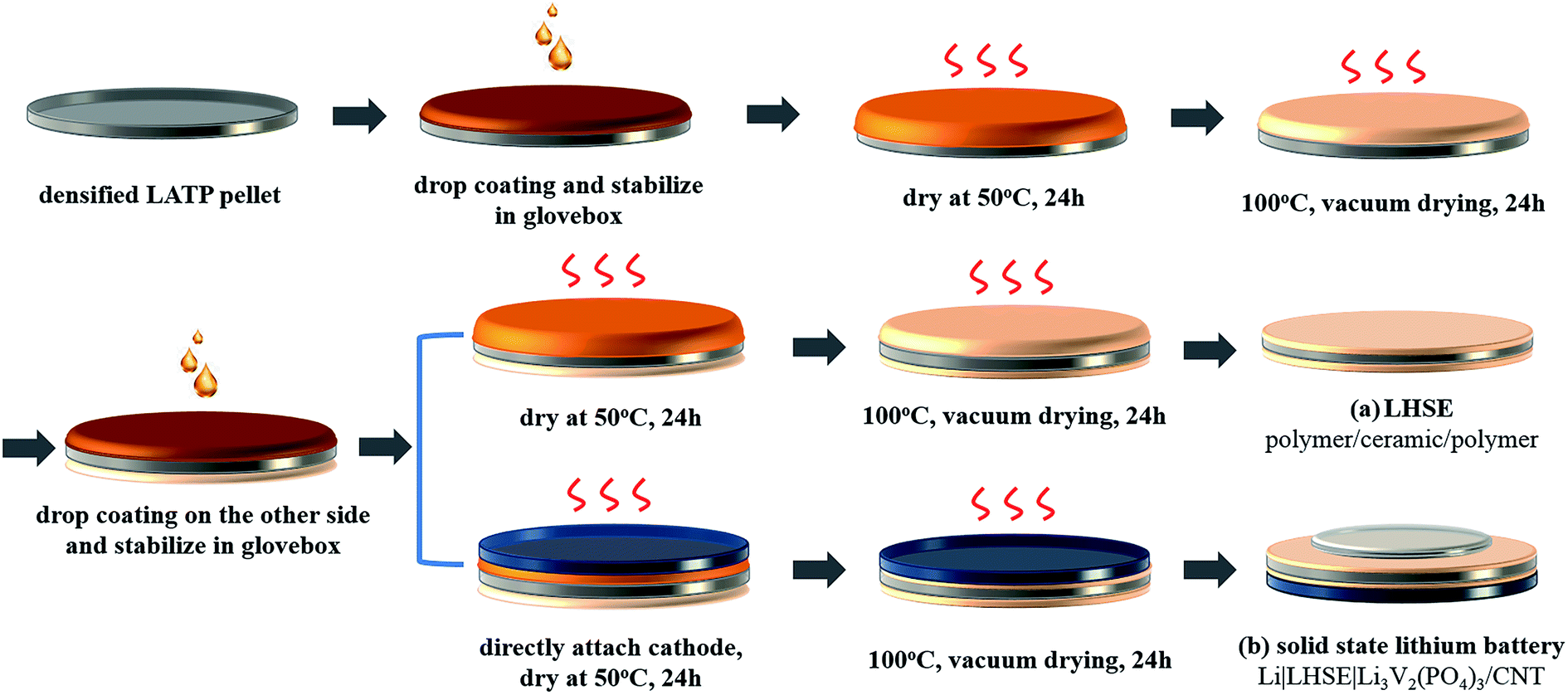 | ||
| Fig. 1 Illustration of the preparation process for the polymer/ceramic/polymer layered hybrid solid electrolyte (LHSE) (a) and the solid-state Li|LHSE|Li3V2(PO4)3/CNT battery (b). | ||
Characterization of the layered hybrid solid electrolyte
The crystal structure and phase analysis was carried out using powder X-ray diffraction (XRD) measurements using an EMPYREAN (Panalytical, Netherlands) X-ray diffractometer with Cu-Kα radiation, operated at 40 kV, 40 mA and a scan rate of 1° min−1. The FTIR spectra of the membranes were obtained using the attenuated total reflection (ATR) attachment on a Frontier FTIR spectrometer (Alpha, Bruker) from 500 to 2000 cm−1, with 32 scans at a resolution of 4 cm−1. Scanning electron microscopy (SEM) and EDX mapping images were taken using a Quanta FEG 650 (FEI, USA) environmental scanning electron microscope equipped with an EDAX-TSL detector. Transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HR-TEM) measurements were conducted on a Tecnai F20 (FEI, USA) transmission electron microscope using an accelerating voltage of 200 kV. Thermogravimetric analysis (TGA) was performed on a NETZSCH TGA/STA-QMS 403D thermoanalyzer (Germany) between 40 and 600 °C with a heating rate of 3 °C min−1 under constant O2 flow. Atomic force microscope (AFM) images of LATP pellets were acquired on an AFM microscope (Bruker, Santa Barbara, USA, Dimension Icon Microscope) operating inside a glovebox with electrochemical strain microscopy (ESM) mode, which is suitable to qualitatively detect local variations in ionic mobility. The contact resonance frequency and the amplitude were tracked with a phase-locked loop (HF2LI, Zurich Instruments, Switzerland). Further information on the sample polishing and instrument setup is described elsewhere.31 Topographical images as well as the change in the amplitude signal were recorded simultaneously.The cyclic voltammetry (CV) curves were recorded with a computer-controlled potentiostat (Autolab PGStat302N, Metrohm). The anodic oxidation limit was measured with a scan rate of 1 mV s−1 in the range from 2.6 V to 7 V using an inert polarizable platinum working electrode. The cathodic reduction limit was determined analogously between 2.8 V and −0.4 V by choosing an inert copper electrode as the polarizable working electrode. Both working electrodes had a diameter of 1 mm and were polished with abrasive paper down to grain P4000 before use.
The AC impedance measurements were performed in symmetric cells with either two Pt or two lithium electrodes using a potentiostat with a built-in frequency response analyzer (VSP-300, Bio-Logic). Measurements were conducted from 7 MHz to 1 Hz with a peak-to-peak voltage amplitude of 10 mV. The temperature was varied from 10 to 50 °C in a climate chamber (Binder KB 115). The equation for calculating ionic conductivity is
| σ = L/RbS | (1) |
Preparation of the free-standing Li3V2(PO4)3/CNT electrode
Binder-free, current collector-free, carbon nanotube (CNT)-enhanced Li3V2(PO4)3 electrodes were prepared by a conventional sol–gel route on CNT fabrics. First, ammonium metavanadate (NH4VO3, ≥99.0%) and oxalic acid (H2C2O4, ≥99.0%) in a weight ratio of 1:3 were first dissolved in deionized water and magnetically stirred at 50 °C for 12 hours until a dark blue solution was formed. Second, stoichiometric amounts of lithium acetate dihydrate (LiCOOCH3·2H2O, ≥99.0%) and ammonium dihydrogenphosphate (NH4H2PO4, ≥99.999%) were dissolved in deionized water, separately, and then the dark blue solution was slowly added under continuous stirring for 12 hours to form the precursor solution. A4-size CNT fabrics (thickness = 115 μm, Tortech Nano-Fibers Ltd, Israel) were cut into desired sizes, rinsed with concentrated hydrochloric acid and an 1:1 (vol./vol.) ethanol/water mixture several times before use to remove the impurities as well as to improve their hydrophilicity, and immersed in the prepared precursor solution. After refluxing at 120 °C overnight, the CNT fabrics were transferred into a vacuum oven and dried at 50 °C overnight to form Li3V2(PO4)3/CNT solid substrates. With the purpose of increasing the mass loading, the precursor solution was uniformly dropped onto the solid substrates with appropriate amounts (∼0.5 mL cm−2). Once the solvent was slowly evaporated, the solid substrates were further dried at 100 °C under vacuum. The CNT enhanced Li3V2(PO4)3 electrodes were obtained by annealing the solid substrates at 850 °C under N2 flow in a tube furnace for 8 hours, removing the loosely bound powder on their surface, and pressing uniaxially at 20 MPa. Then the Li3V2(PO4)3/CNT electrode was directly transferred into a glovebox for the battery assembly. The mass loading obtained from TGA measurements of the prepared free-standing Li3V2(PO4)3/CNT electrode is about 10 mg cm−2.
Battery assembly and electrochemical characterization
The solid-state lithium ion batteries were fabricated as schematically shown in route (b) in Fig. 1. Once one side of the LATP pellet was coated with the dry polymer electrolyte, drop coating was performed on the other side of the pellet following attaching of a free-standing Li3V2(PO4)3/CNT cathode (Ø = 12 mm) directly on this side when the fresh coated polymer layer is still wet. Then the hybrid electrolyte, attached with the Li3V2(PO4)3/CNT cathode, was stabilized for 12 hours in a glovebox, further dried in a vacuum oven and transferred back to the glovebox to attach a 10 mm metallic lithium disk as the anode in a Swagelok®-type cell. It is worth mentioning that, in order to improve the rate performance, the thickness of the LATP pellet used in the full cell was polished down to 250 μm and the polymer layer between the Li3V2(PO4)3/CNT cathode and LATP is around 50 μm. The thickness of the polymer layer between the lithium anode and LATP is around 120 μm.In order to compare with the electrochemical performance of the as-prepared solid-state lithium batteries, Swagelok cells with liquid electrolyte were also assembled using the Li3V2(PO4)3/CNT electrode, LP30 electrolyte (1 M LiPF6 in EC: DMC (1:1 vol./vol.)) soaked in glass fiber and metallic lithium anode.
CV and galvanostatic charge/discharge tests were carried out using a Bio-Logic potentiostat (VMP3). CV tests were performed in the potential windows of 3.0–4.3 V and 3.0–5.0 V vs. Li/Li+, respectively, at a scan rate of 0.05 mV s−1 at 50 °C in a climate chamber. The galvanostatic tests were performed at different current rates between 3.0 and 4.3 V vs. Li/Li+.
Results and discussion
Structural and morphological analysis of the layered hybrid solid electrolyte
Structural studies of the as-prepared SPE, LATP and LHSE were performed by ATR-IR and XRD. And the results showed that the specimens are obtained with satisfactory phase purity and the components in LHSE are chemically stable with each other. More details regarding the results and additional discussions are presented in the ESI† of this article.The quality of adhesion between the polymer and the ceramic pellets was evaluated using cross-sectional SEM and EDX, and the corresponding interfacial microstructure images and element maps are shown in Fig. 2. A clear layer separation can be identified between the smooth and dense SPE and LATP (cf.Fig. 2(a) and (b)), where the LATP ceramic consists of particles with a length around 3 to 5 μm (cf.Fig. 2(a) and (c)). At the interface between the polymer layer and LATP ceramic (cf.Fig. 2(d)), the SPE has perfectly coated the surface of the LATP particles, benefitting from its low viscosity and sufficient malleability, respectively. As evidenced by the SEM images, very few pores can be observed in the bulk and at the interface of LHSE, which guarantees continuous and efficient Li+ pathways in the LHSE. The good contact at the interface between LATP ceramic and SPE is further substantiated by data regarding the elemental mapping of Ti, O, Al, C, P and N as displayed in Fig. 2(e)–(j). Both LATP and SPE contain O and P but in different concentrations. Ti and Al, the characteristic elements of LATP, are well separated from the SPE, where C and N are characteristic elements, indicating the high stability of the polymer/ceramic interface. Furthermore, it can be concluded that the SPE is able to diffuse into the open pores of the ceramic phase and create intimate contacts with LATP. The heterogeneous distribution of Al in the LATP region owing to the formation of AlPO4 at grain boundaries of LATP during the sintering process is of note, which also confirms the results from XRD and agrees with previous reports.6,16,31 Such a secondary phase could positively affect the ionic conductivity regarding the improvement of relative density of the LATP layer.16
 | ||
| Fig. 2 Cross-sectional SEM images of the layered hybrid solid electrolyte: (a) field of view image of the polymer–ceramic interface. High magnification images of (b) the solid polymer layer, (c) Li1.3Al0.3Ti1.7(PO4)3 (LATP) and (d) the interface region between the LATP and polymer layer. (e)–(j) EDX maps (Ti, O, Al, P, C and N) at the polymer–ceramic interface of the prepared layered hybrid solid electrolyte. The field of view image is shown in (a). | ||
Thermal, chemical and electrochemical stabilities of the layered hybrid solid electrolyte
Thermal, chemical and electrochemical stabilities of the LHSE are important for the feasibility of practical application.The thermal stability of the LATP ceramic electrolyte and SPE was investigated by TGA, as shown in Fig. 3(a). No mass change was observed for the LATP from 40 °C up to 600 °C whereas for the SPE a large mass loss corresponding to the decomposition, carbonization and evaporation of polymer components started at 178 °C. The second mass loss step began at 330 °C for the SPE which is considered to be the residues burning into COx and NOx. Therefore, the LHSE is supposed to have an intrinsic thermal stability up to at least 170 °C, which is much lower than that of the pure ceramic electrolyte but higher than that of most liquid electrolytes. Therefore, this hybrid solid electrolyte allows higher operating temperature of lithium-ion batteries compared to standard cells with liquid electrolytes which start to degrade at temperatures above 65 °C.1
 | ||
| Fig. 3 (a) TGA curves of Li1.3Al0.3Ti1.7(PO4)3 (LATP) and the solid polymer electrolyte (SPE). (b) Impedance spectra of LATP and the layered hybrid solid electrolyte (LHSE) measured in a lithium symmetric cell with a time period of 2 to 60 h at 25 °C. (c) Constant current polarization curve of LHSE measured in a lithium symmetric cell with a current density of 0.13 mA cm−2 at 50 °C. (d) Cyclic voltammetry curves of LHSE with a scan rate of 1 mV s−1. Working electrodes are Pt and Cu. | ||
To investigate the chemical stability of the as-prepared electrolytes against metallic lithium, LATP and LHSE disks, respectively, sandwiched between the lithium metal, were analyzed using electrochemical impedance spectroscopy (EIS) at 25 °C over 60 hours. As shown in Fig. 3b, the total resistance of the Li|LATP|Li cell constantly increased with time and dark grey side products were found on the LATP pellet surface as shown in the inserted photo in Fig. 3b, indicating that continuous side reactions took place at the interface between LATP and Li. In sharp contrast, there was no difference between the initial and final EIS spectra after 60 hours for the Li|LHSE|Li symmetric cell, proving the chemical stability of the LHSE against the lithium metal. The compatibility of lithium and LHSE is further examined by constant current polarization of the Li|LHSE|Li symmetric cell at 50 °C for a period of 1500 hours with 0.13 mA cm−2 current density. No significant change in cell voltage was observed, as shown in Fig. 3c, indicating the high electrochemical stability of the prepared LHSE against the lithium metal. On the basis of these results, the polymer layer can prevent chemical and electrochemical decompositions of LATP when using with the lithium anode by avoiding their direct contacts. Thus, it is possible to fabricate an operable all-solid-state lithium battery with the LHSE regarding the stability of the anode-electrolyte interface.
We also studied the effect of the SPE protection layers on the electrochemical stability window of the LATP ceramic electrolyte by CV experiments with a scan rate of 1 mV s−1 at 60 °C. The results are shown in Fig. 3d. At higher potentials (above 4.7 V), the oxidative degradation of oligoether side chains in the MEEP polymer begins. In the low potential side at around 0 V, reversible lithium deposition and dissolution were observed with the inert copper working electrode. The plating/stripping process is not completely reversible (∼53%) which is attributed to SEI formation. Hence, based on the large electrochemical stability window of LHSE (0–4.7 V), we expect that our hybrid solid electrolyte will also be suitable for a lithium battery with the high-potential cathode material Li3V2(PO4)3 as the positive electrode.
Ion transport in the layered hybrid solid electrolyte
Complex Li+ transport pathways are expected in the LHSE since it enables two ion-transference mechanisms of ion hopping in the LATP crystal structure and Li+ transport related to the segmental motion of polymer chains in SPE.32 As presented in the SEM image and the schematic diagram of the interface between SPE and LATP in Fig. 4a, distinguishable regions for the Li+ migration can be defined as (i) inside the LATP grain, (ii) along/cross the LATP grain boundary, (iii) inside the bulk polymer and (iv) along/cross the boundary of LATP and SPE.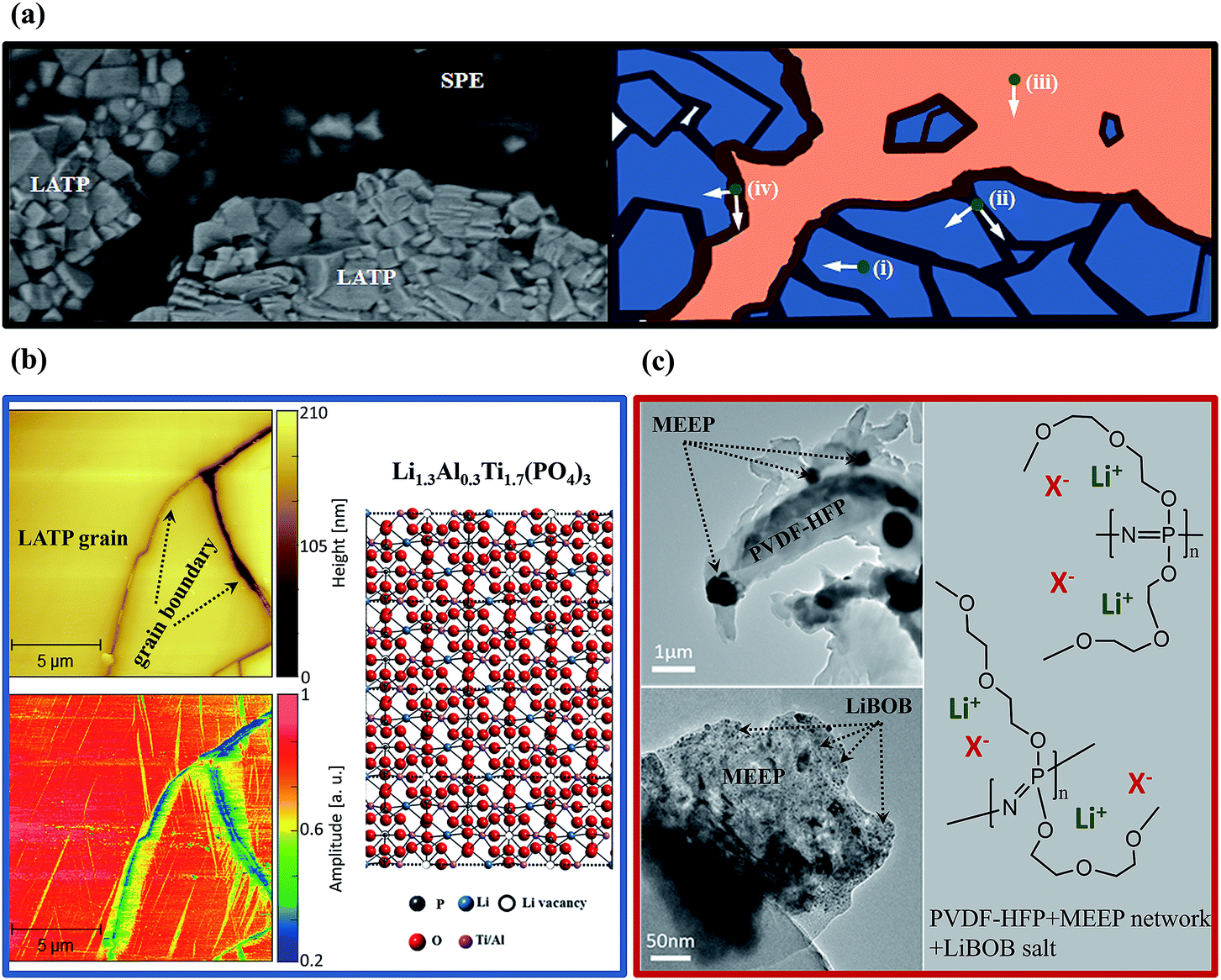 | ||
| Fig. 4 (a) SEM image and schematic drawing of the Li+ transport pathways in the prepared layered hybrid solid electrolyte (LHSE). (b) AFM topography and electrochemical strain microscopy amplitude signal of Li1.3Al0.3Ti1.7(PO4)3 (LATP). The benchball crystal structure image of LATP showing the crystal sites of lithium atoms in the phosphate framework. (c) TEM images of the solid polymer electrolyte (SPE, MEEP/PVDF-HFP/LiBOB) and correlative illustration of the lithium transport in the electrolyte network. | ||
When considering the ion transport in the LATP region of LHSE, the Li+ mobility in the LATP grain and grain boundary is significantly different due to the different energy barriers in the ordered crystal structure and at boundaries with mismatched conductive channels from different grains. The different Li+ mobilities inside the LATP grain and at the grain boundary can be clearly seen in Fig. 4b from which the correlative images of AFM topography and electrochemical strain microscopy (ESM) of identical regions are illustrated. The AFM images reflect the same LATP surface features as observed by SEM, while only a few grains and their relative grain boundary regions are focused. Ion mobility in the indicated regions is shown in the ESM image as the changes in the amplitude can be correlated to local interactions between the applied electric field and the LATP material in the respective areas.31 A strong amplitude signal was found for LATP grains, whereas the grain boundaries exhibit considerably smaller amplitudes. The quantification for the ion transport in the LATP material was resolved in our previous work by analyzing the impedance data of LATP in defined frequency ranges with the method of distribution of relaxation time analysis in two dimensions.17 Specifically, the conductivities of grains ((i) in Fig. 4a) and grain boundaries ((ii) in Fig. 4a) of LATP at 50 °C reach 3.5 mS cm−1 and 4.0 μS cm−1, respectively.17 A total conductivity of 0.45 mS cm−1 at 50 °C for the LATP pellet is achieved, as shown in Fig. S3†.
When considering the ion transport in the SPE region of LHSE, comprehensive observation of the microstructure of SPE could help elucidate the Li+ transportation mechanism of the SPE. In the coated polymer layer, ideally, PVDF-HFP serves as the matrix to maintain the solid structure and offers relatively sluggish Li+ transportation pathways (σLi+ = ∼10−5 S cm−1, at 50 °C).33,34 Faster Li+ migration take places in the MEEP polymer owing to its low crystallinity and low dissociation energy with Li+.24,30 Therefore, a well-mixed MEEP, PVDF-HFP and LiBOB, as can be seen in the TEM images in Fig. 4c, are preferable for obtaining high ionic conductivity. As demonstrated in the low magnification TEM image, the black dots are considered to be the folded MEEP polymer since P and N have a higher atomic mass than C. Such a distribution of the MEEP polymer on the PVDF-HFP matrix could generate the local disordered structure that possibly decreases the crystallinity of PVDF-HFP, resulting in a faster segmental motion of polymer chains for the transportation of Li+ in PVDF-HFP. Partially dissociated lithium ions and anions are transported by the chain movement in PVDF-HFP and MEEP polymers, which is the conventional mechanism of polymer electrolytes, resulting in a significantly low lithium ion transference number of 0.06 for SPE (cf. Fig. S4†) compared with ceramic electrolytes like LATP (t+ ≈ 1). At 50 °C, the ionic conductivity inside the bulk polymer ((iii) in Fig. 4a) can be considered as the SPE conductivity of 0.26 mS cm−1 (cf. Fig. S3†) which is highly consistent with the cross-linked MEEP solid polymer electrolyte.24
When considering the ion transport along or across the interface of LATP and SPE ((iv) in Fig. 4a), lithium ion migration cross the interface of LATP and SPE is still limited by the LATP grain boundary which has significantly lower ionic conductivity. Thus, at 50 °C, the ionic conductivities along and across the ceramic/polymer interface are assessed to be similar to the polymer conductivity (0.26 mS cm−1) and roughly equal to the LATP grain boundary conductivity (≈4.0 μS cm−1), respectively. However, both assumptions need further experimental evidence and cannot be answered based on the present data.
Due to the fact that the concentration gradient during ion transport in polymer layers of LHSE are influenced by the layer thickness and the contact area between the LATP and polymer, it is more precise to discuss the overall ionic conductivity of LHSE based on a specific geometry. The thickness of LATP in LHSE for battery assembly is 250 μm in order to maintain sufficient mechanical support and to minimize the total resistance. The resistance for such a LATP pellet is 64 Ω at 50 °C, obtained from fitting the impedance result with the equivalent circuit shown in Fig. 5a. The thickness of polymer layers in LHSE, on the other hand, can be varied. As shown in Fig. 5b, resistance increases along with the thickness, whereas a thin polymer layer may be penetrated by lithium dendrites during long term cycling. Hence, the thicknesses of the polymer layer between the cathode and LATP and between the lithium anode and LATP are 50 μm and 120 μm, respectively, in order to minimize the inner resistance and prevent dendrite penetration. The intrinsic resistance of these two polymer layers is measured to be 25 Ω and 56 Ω at 50 °C, respectively.
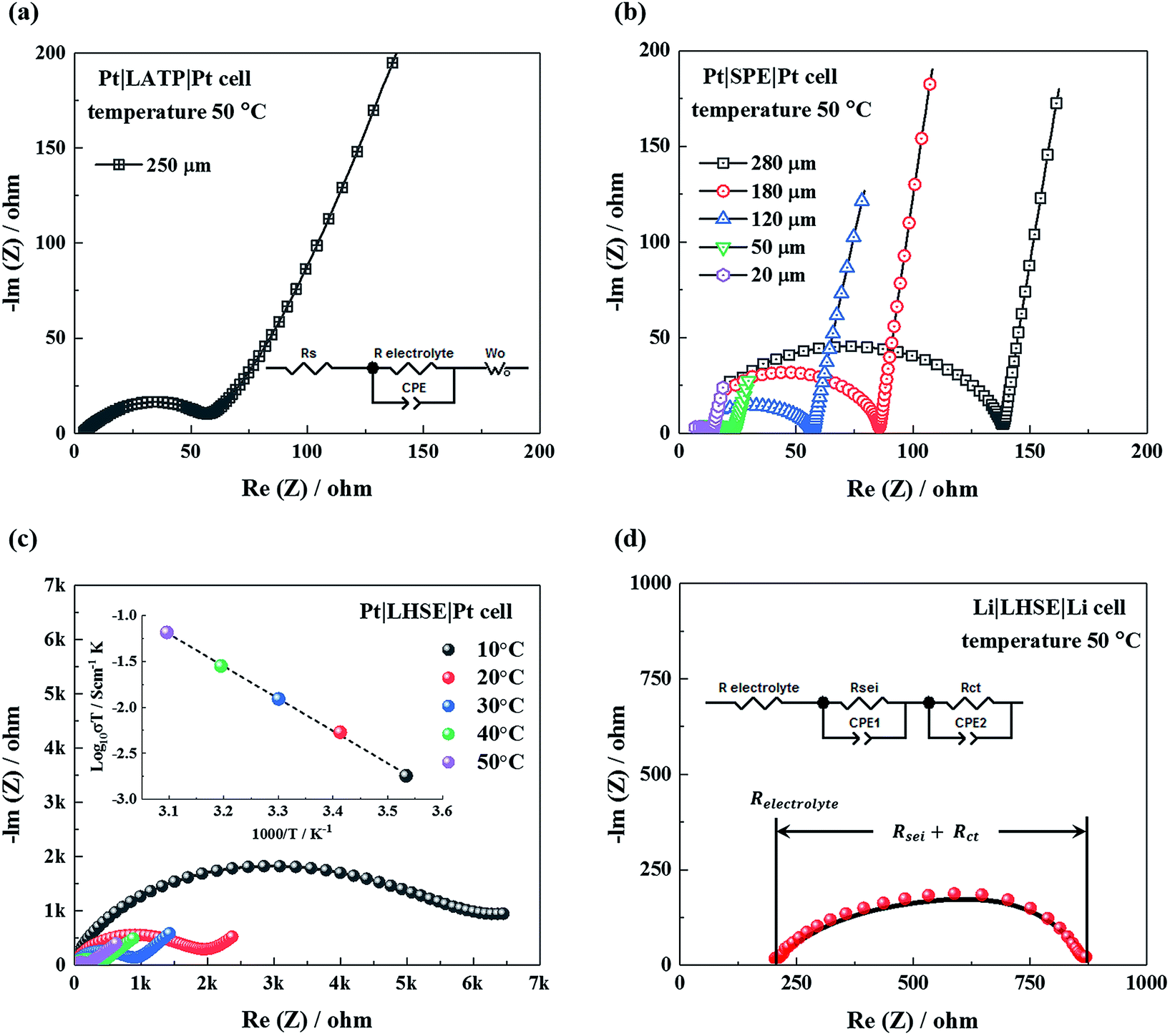 | ||
| Fig. 5 Nyquist plots of the (a) Li1.3Al0.3Ti1.7(PO4)3 (LATP) electrolyte with a thickness of 250 μm and (b) solid polymer electrolyte (SPE), with different thicknesses of 20, 50, 120, 180 and 280 μm, measured in symmetric cells with Pt electrodes at 50 °C. (c) Nyquist and Arrhenius plots of the layered hybrid solid electrolyte (LHSE) measured in a symmetric cell with Pt electrodes. And (d) Nyquist plot of LHSE measured in a symmetric cell with lithium electrodes. The LHSE is prepared by coating a 120 μm polymer layer on one side and a 50 μm polymer layer on the other side of a 250 μm thick LATP pellet. The impedance data obtained from Pt symmetric cells are fitted with the equivalent circuit in Fig. 5a; the equivalent circuit in Fig. 5d is used for fitting the impedance data obtained from the lithium symmetric cell. | ||
As shown in Fig. 5c, the total resistance of the LHSE at 50 °C is 192 Ω, about 50 Ω larger than the accumulated resistance of all three individual layers, indicating the presence of interfacial resistance between the LATP and polymer in LHSE. For the LHSE with the defined thicknesses of each layer, an overall ionic conductivity of 0.2 S cm−1 at 50 °C is achieved, which is sufficient for a solid electrolyte in ASSBs.1 At lower temperatures, the total ionic conductivity of LHSE decreases by orders of magnitude, which will significantly impede the rate performance of an all-solid-state battery. Although the total ionic conductivity of LHSE is limited due to the higher resistivity of SPE layers, the viscoelastic SPE material can, not only prevent a direct contact between the LATP and lithium anode, but also expand the net contact area and improve the adhesion between the electrolyte and electrodes by infiltrating into the body of LATP through the cracks or pores between LATP grains.
The lithium-LHSE interface charge-transfer kinetics are studied by analyzing the impedance data measured in the Li|LHSE|Li symmetric cell at 50 °C. As shown in the Nyquist plot in Fig. 5d, the LHSE resistance is given by the high frequency intersection that the value is consistent with the result obtained from the Pt symmetric cell (cf.Fig. 5c). The diameter of the semicircle has two contributions of (a) the resistance of the SEI formed on the lithium electrode by its reaction with the electrolyte (Rsei) and (b) the charge-transfer resistance (Rct).35,36 The additional presence of SEI will superimpose an additional contribution, which cannot be separated from the charge transfer. Even so, the overall resistivity of the lithium-LHSE interface can be estimated from the result and is believed not to be the limitation for fabricating an operational all-solid-state battery.
Electrochemical performance of the all-solid-state lithium battery
Electrochemical properties are evaluated for the all-solid-state Li|LHSE|Li3V2(PO4)3/CNT batteries with following specifications of the electrodes: metallic lithium is used as the anode to maximize the energy density of the battery. A 95 μm thick free-standing Li3V2(PO4)3/CNT, as shown in the SEM photographs in Fig. S5,† is cut into disks as a high-voltage cathode. The CNT fabrics serve as the 3D electronic conducting network as well as the flexible frame for Li3V2(PO4)3. The Li3V2(PO4)3 particles, with a size below 500 nm, are located on the surface of CNT fabrics and also embedded in the bulk of the CNT matrix, thus resulting in a high mass loading of 10 mg cm−2. The preparation and structural features of the Li3V2(PO4)3/CNT are similar to those of previously reported CNT enhanced NaTi2(PO4)3 and Na3V2(PO4)3 electrodes for sodium-ion batteries.37The schematic of the electrochemical compatibility of the battery components regarding voltage and chemical potential is presented in Fig. 6a. The LATP ceramic electrolyte has an intrinsic electrochemical stability window referring to the lowest reduction potential of 2.17 V vs. Li/Li+ and the highest oxidation potential of 4.21 V vs. Li/Li+,38,39 which is relatively narrow compared with the operating voltage range of the electrode pair of the lithium anode and Li3V2(PO4)3 cathode. According to the CV result of LHSE shown in Fig. 3d, the SPE coating significantly improved the electrochemical stability of LATP and thus extended the voltage window of LHSE to a range of 0–4.7 V. Therefore, the electrochemical stability window of LHSE covers the phase transition potential of Li3V2(PO4)3 → LiV2(PO4)3 (4.11 V vs. Li/Li+) and even the complete phase transition of Li3V2(PO4)3 → V2(PO4)3 (4.58 V vs. Li/Li+).40
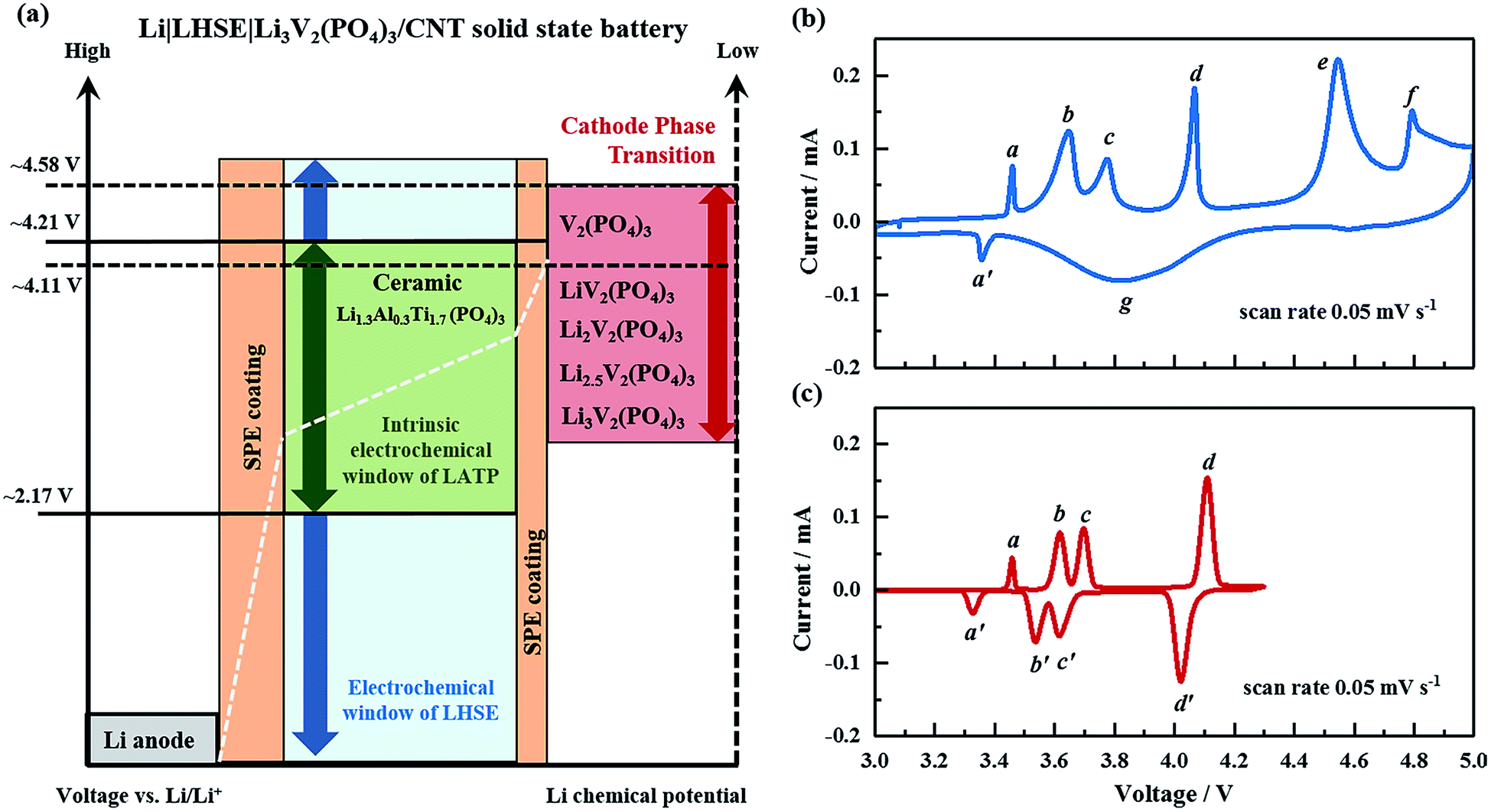 | ||
| Fig. 6 (a) Schematic of the Li|layered hybrid solid electrolyte (LHSE)|Li3V2(PO4)3/CNT all-solid-state lithium battery. The narrow intrinsic electrochemical stability window of Li1.3Al0.3Ti1.7(PO4)3 is extended in the LHSE. Cyclic voltammetry profiles of the all-solid-state lithium battery measured in the voltage ranges of 3.0–5.0 V and 3.0–4.3 V vs. Li/Li+, respectively, with a scan rate of 0.05 mV s−1 at 50 °C ((b) and (c)). | ||
To gain insight into the electrochemical behavior of the Li|LHSE|Li3V2(PO4)3/CNT all-solid-state battery, CV was performed first in a broad voltage range of 3.0 to 5.0 V at a scan rate of 0.05 mV s−1 at 50 °C (cf.Fig. 6b). Four oxidation peaks corresponding to a sequence of phase transitions of LixV2(PO4)3 where x = 3.0, 2.5, 2.0, 1.0 and 0 are observed at about 3.65 V (b), 3.77 V (c), 4.11 V (d), and 4.58 V (e) vs. Li/Li+. At the potential range above 4.7 V, the oxidation peak at 4.79 V (f) is assigned to the decomposition of LHSE, which is also accompanied by a strong polarization effect at higher potential. In lithium free vanadium phosphate, the V sites exhibit a fairly close average bond distance and an average vanadium valence of +4.5,40 indicating that the mixed V4+ and V5+ states do not display charge ordering in this phase. Such phenomena will result in a disordered lithium reinsertion, as is evidenced by the solid solution behavior during the reduction process, which has also merged with the reduction peaks of Li2V2(PO4)3 → Li2.5V2(PO4)3 and Li2.5V2(PO4)3 → Li3V2(PO4)3 owing to the large polarization caused by the electrolyte decomposition (together denoted as region (g)). Additionally, a pair of small redox peaks, (a) and (a′), is presented at a potential below 3.5 V, which is characterized as the LiFePO4 impurity in the cathode that formed due to the presence of Fe particles in the carbon nanotubes.41 A tiny amount of impurity (below 3.5 wt%) will influence the battery performance not strongly, as is evidenced by the discharge plateau at the corresponding voltage in the charge–discharge profiles in the following part.
When performing a CV test for a battery in the voltage range of 3.0–4.3 V vs. Li/Li+ (cf.Fig. 6c), the cell exhibits four distinguishable pairs of redox peaks, being associated with the reversible phase transitions of Li3V2(PO4)3 ↔ Li2.5V2(PO4)3 ((b) and (b′)), Li2.5V2(PO4)3 ↔ Li2V2(PO4)3 ((c) and (c′)) and Li2V2(PO4)3 ↔ LiV2(PO4)3 ((d) and (d′)), as well as the redox peaks of (a) and (a′) for LiFePO4. Importantly, no solid solution behavior, electrolyte decomposition or strong polarization can be observed from the CV curve. Comparing the CV curves in the two relevant voltage ranges, it's obvious that the Li|LHSE|Li3V2(PO4)3/CNT solid-state cells are preferable to be operated in the voltage range of 3.0–4.3 V to avoid the formation of kinetically sluggish V2(PO4)3 phase.42,43
The electrochemical characteristics of the high-voltage Li3V2(PO4)3/CNT cathode in the voltage range of 3.0–4.3 V were initially assessed in liquid electrolyte batteries by using the conventional LP30 electrolyte and metallic lithium anode, and the results are shown in Fig. S6.† The charge–discharge profiles of the second to fifth cycles at 0.05C demonstrated well defined voltage plateaus (cf. Fig. S6a†) consistent with previous reports.40,44,45 The tiny reversible plateau at about 3.3 V is the LiFePO4 impurity, whose capacity is less than 5% of the total discharge capacity. The specific discharge capacity is around 130 mA h g−1, corresponding to 98% of its theoretical capacity (131.5 mA h g−1) based on two lithium extractions. The electrochemical behavior before and after formation cycles was investigated by EIS in the frequency range from 100 kHz to 10 mHz. As shown in Fig. S6b,† all Nyquist plots of the cells exhibited a semicircle in the high frequency region which is ascribed to the charge-transfer process of lithium ions from the surface layer of the electrode through the interface into the electrolyte. In addition, a straight line appeared in the low-frequency region which is attributed to the diffusion of lithium ions in the Li3V2(PO4)3 cathode.46 The impedance plots were fitted by using the equivalent circuit shown in Fig. S6b.† Their analyses showed that after formation cycles, the charge-transfer resistances (Rct) of the cathode decreased from 114 Ω to 79 Ω, hinting at the structural and electrical integrity of the electrode and its interface with the liquid electrolyte after battery formation. The cycling performance and rate capability of the Li3V2(PO4)3/CNT electrode in liquid electrolyte cells at current rates of 0.5, 1 and 10C are shown in Fig. S6c.† The electrode could be stably cycled for up to 1000 times at each current rate with the coulombic efficiency close to 100% for every cycle, indicating the good reversibility of lithiation/delithiation in the Li3V2(PO4)3/CNT electrode. The capacity retentions are 94, 96 and 88% after long-time cycling at 0.5, 1 and 10C current rates, respectively. The discharge capacities after 1000 loops at 0.5 and 1C are still higher than 120 mA h g−1, corresponding to more than 90% of the theoretical capacity. Even at a high current rate of 10C, the battery still delivered 72% of its theoretical capacity after 1000 cycles. Therefore, the free-standing electrode, with its novel morphology,37 exhibits outstanding electrochemical performance in liquid electrolyte cells in the relevant voltage range. Based on the above results, the electrochemical properties of the Li3V2(PO4)3/CNT cathode are expected not to hinder the overall performance of the solid-state lithium battery with the LHSE electrolyte in the relevant voltage range.
In contrast to the liquid electrolyte cells which were cycled at room temperature, the temperature was set at 50 °C for all the electrochemical measurements of the prepared Li|LHSE|Li3V2(PO4)3/CNT solid-state lithium batteries to take advantage of the improved ionic conductivity of LHSE at elevated temperature. The formation cycles of the solid-state lithium batteries were performed at a current rate of 0.05C in the voltage range of 3.0–4.3 V for 5 times. The charge–discharge profiles of the formation cycles are presented in Fig. 7a, showing a continuous increasing trend of coulombic efficiency and slight capacity decrease along with cycling numbers. The initial discharge capacity of the battery is 130.5 mA h g−1 and stabilized at about 123 mA h g−1 after the formation cycles. Three main charge–discharge plateaus corresponding to three reversible phase transitions of Li3V2(PO4)3 ↔ LiV2(PO4)3 and an extra tiny plateau for the LiFePO4 impurity are observed. It's worth noting that these plateaus, compared with the plateaus of the battery with the liquid electrolyte,40 are tilted, owing to the more sluggish diffusion kinetics at the interfaces as well as the relatively higher overall electrolyte resistance in the all solid-state battery than that with the liquid electrolyte.
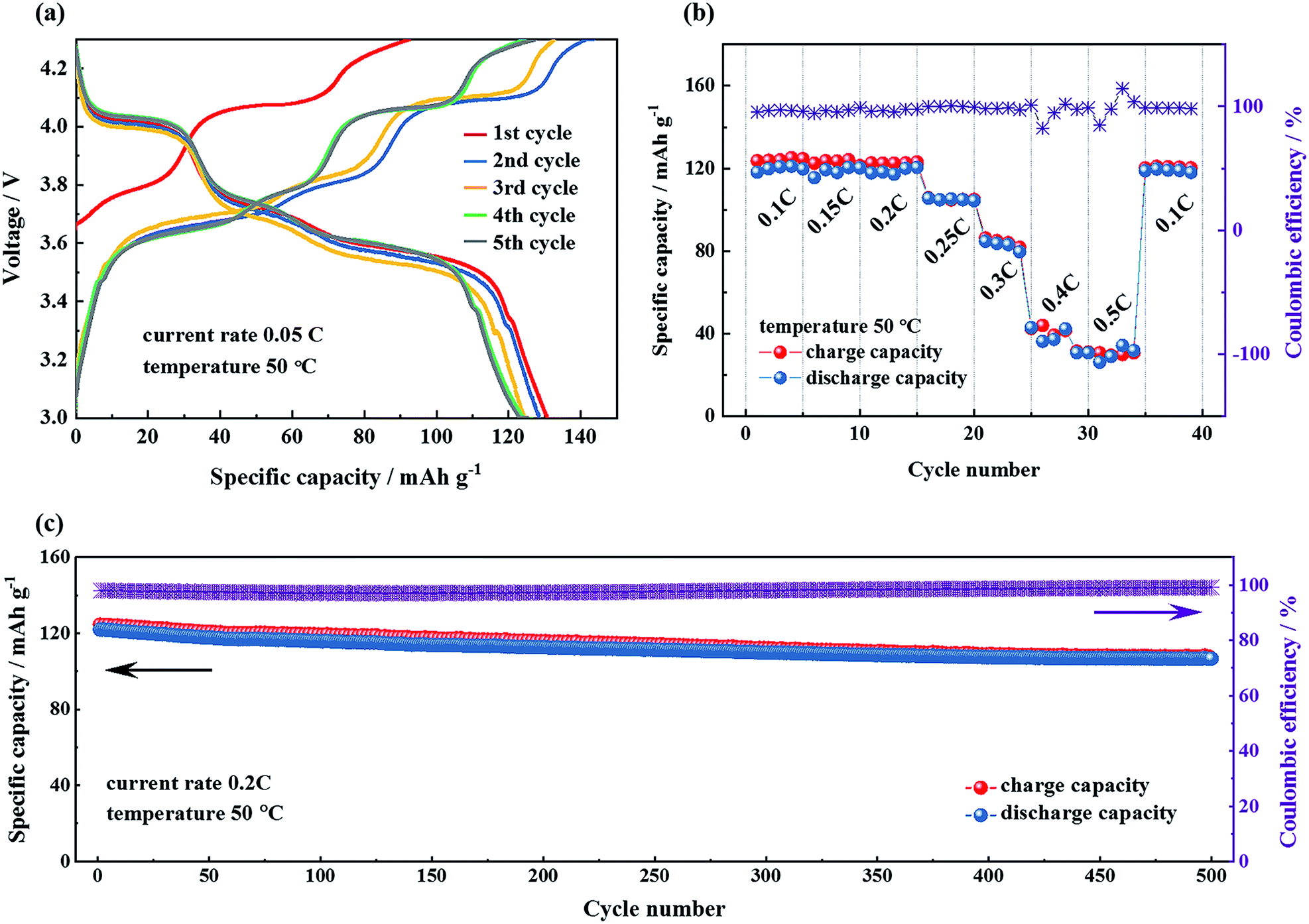 | ||
| Fig. 7 Electrochemical performance of the Li|layered hybrid solid electrolyte|Li3V2(PO4)3/CNT all-solid-state lithium battery at 50 °C in the voltage range of 3.0–4.3 V vs. Li/Li+: (a) charge–discharge curve of the initial five cycles at a current rate of 0.05C, (b) rate capability at current rates from 0.1C to 0.5C, respectively, and (c) long-term cycling measurement at a current rate of 0.2C. | ||
The rate capability and the corresponding charge–discharge profiles of the Li|LHSE|Li3V2(PO4)3/CNT solid-state lithium battery, cycled between 3.0 and 4.3 V with current rates from 0.1C up to 0.5C at 50 °C, are shown in Fig. 7b and S7,† respectively. The Li|LHSE|Li3V2(PO4)3/CNT battery demonstrates low polarization, a high coulombic efficiency and reasonable charge/discharge capacity when the current rate is below 0.2C, which is attributed to the sufficient ionic conductivity of LHSE and the favorable interfacial properties between the electrodes and the electrolyte. When the current rate is above 0.2C, the obtained discharge capacities drop very quickly, which indicates the kinetic limitation of Li+ in LHSE impeding the cycling capability of the battery. Moreover, as shown in Fig. S7,† overpotential increases dramatically and capacity decreases sharply with the current rate increasing, meaning the polarization significantly influences the electrochemical behavior of the battery. However, the electrochemical performance can be recovered to the initial values when the current rate was returned back to 0.1C after cycling at higher rates, which suggests the good durability of the LHSE at high overpotential.
To explore the cycling stability of the Li|LHSE|Li3V2(PO4)3/CNT battery, a charge–discharge test was performed at 0.2C and the results are shown in Fig. 7c. The discharge capacity of the battery after 500 cycles is 107.6 mA h g−1, corresponding to 88.2% of its initial discharge capacity and 81.8% of the theoretical capacity of the Li3V2(PO4)3 cathode. Thus, the battery is at least able to be cycled for up to 500 times with minor capacity fading. The impedance measurement was performed on the battery in the pristine state, after 100 cycles and after 500 cycles, respectively, and the obtained Nyquist plots are shown in Fig. S8.† The overall battery resistance slightly decreased after the first hundred cycles and kept basically constant until the end of long-term cycling, indicating the high electrochemical stability of the prepared battery. Moreover, post-test characterization was carried out on the lithium anode, the SPE layer and LATP in LHSE after disassembling this long–term cycling battery. The results and detailed discussion are shown in the ESI (cf. Fig. S9†), representing the success in achieving the high electrochemical stability and compatibility of the prepared LHSE electrolyte with the metallic lithium anode in the relevant potential range.
The prepared Li|LHSE|Li3V2(PO4)3/CNT cell exhibits a gravimetric energy density of ∼460 W h kg−1 based on the weight of two electrodes, which is highly comparable with that of the conventional lithium-ion batteries with the liquid electrolyte. In addition to the advantages of using lithium anode and high-voltage Li3V2(PO4)3 cathode, the structural features of the free-standing Li3V2(PO4)3/CNT electrode also play an important role in the improvement of the gravimetric energy density. Unlike the recently used composite electrode for bulk-type solid-state batteries,6 this free-standing Li3V2(PO4)3/CNT electrode requires no binder, additive carbon, pre-added electrolyte or metal current collector. The Li3V2(PO4)3 mass loading reaches 71 wt% of the complete cathode. Finally, for the free-standing cathode, high electronic conductivity and sufficient ionic conductivity are ensured respectively by the CNT framework37 and the penetration of the polymer electrolyte into the cathode pores during battery assembly. The rate capability and cycling performance of the Li|LHSE|Li3V2(PO4)3/CNT cells are worse than those with the liquid electrolyte, owing to the intrinsic limitation of ionic conductivity in LHSE and the small contact areas between the electrolyte and electrode in the solid-state battery. When comparing the Li|LHSE|Li3V2(PO4)3/CNT battery with a previously reported solid-state battery using LATP electrolyte with LiTi2(PO4)3 anode and Li3V2(PO4)3 cathode,6 the Li|LHSE|Li3V2(PO4)3/CNT battery (i) can be operated without high external pressure, which was used to maintain the mechanical contacts between ceramic electrodes and the ceramic electrolyte, (ii) is no longer limited in a narrow voltage window, and (iii) exhibited higher energy density using the metallic lithium anode while the LATP is protected by the polymer layer from chemical and electrochemical decomposition with the lithium metal. In addition, the polymer protection layer for solid-state batteries, as well as the high mass loading of the Li3V2(PO4)3/CNT electrode, which can be directly formed with the crystallization of active materials, are easily scalable and relatively cheap in terms of energy per gram.
Conclusions
In this work, for lithium batteries, a layered hybrid solid electrolyte (LHSE) with LATP ceramic and polymer coating layers was prepared and its structural, thermal, chemical and electrochemical properties were systematically investigated. The advantages of coating the polymer electrolyte on the LATP pellet are (i) the increased contact area between the electrode and electrolyte, (ii) protection of LATP ceramic, resulting in a wide electrochemical stability window, (iii) the improved (electro-)chemical compatibility of LATP with metallic lithium and (iv) the simplified interface fabrication process for all-solid-state batteries. We demonstrated an all-solid-state lithium battery, Li|LHSE|Li3V2(PO4)3/CNT, which can be cycled for at least 500 times at 50 °C with slight capacity fading. Post-test characterization of the lithium surface, polymer coating layer and LATP pellet after long term cycling indicated the high mechanical, chemical and electrochemical stability of the Li|LHSE|Li3V2(PO4)3/CNT battery. This design opened the possibility for manufacturing all-solid-state lithium batteries with a high-voltage cathode and metallic lithium anode to enhance their energy density. Further tuning the LHSE to improve the rate capability of all-solid-state batteries is still under investigation to pave the way for its large-scale practical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the projects of “Materials and Components to Meet High Energy Density Batteries II” of the funding program “Excellent battery” from the Bundesministerium für Bildung und Forschung (BMBF) within the framework of the program “Materials Innovations for Industry and Society” (Project No.: 03XP0084C). The carbon nanotube fabrics were supplied by Tortech Nano-Fibers Ltd. (Israel) free of charge, for which the authors are grateful. S. Yu is grateful to Dr Roland Schierholz and Daniel Kröger who helped with SEM and TEM experiments. Open Access funding provided by the Max Planck Society.Notes and references
- J. Janek and W. G. Zeier, Nat. Energy, 2016, 1, 16141 CrossRef.
- C. Cao, Z. –B. Li, X. –L. Wang, X. –B. Zhao and W. –Q. Han, Frontiers in Energy Research, 2014, 2, 1–10 CrossRef.
- A. Varzi, R. Raccichini, S. Passerini and B. Scrosati, J. Mater. Chem. A, 2016, 4, 17251–17259 RSC.
- M. Kotobuki, K. Kanamura, Y. Sato and T. Yoshida, J. Power Sources, 2011, 196, 7750–7754 CrossRef CAS.
- Y. Kato, S. Hori, T. Saito, K. Suzuki, M. Hirayama, A. Mitsui, M. Yonemura, H. Iba and R. Kanno, Nat. Energy, 2016, 1, 16030 CrossRef CAS.
- S. Yu, A. Mertens, H. Tempel, R. Schierholz, H. Kungl and R. A. Eichel, ACS Appl. Mater. Interfaces, 2018, 10, 22264–22277 CrossRef CAS PubMed.
- X. Han, Y. Gong, K. Fu, X. He, G. T. Hitz, J. Dai, A. Pearse, B. Liu, H. Wang, G. Rubloff, Y. Mo, V. Thangadurai, E. D. Wachsman and L. Hu, Nat. Mater., 2017, 16, 572–579 CrossRef CAS PubMed.
- K. Kerman, A. Luntz, V. Viswanathan, Y. –M. Chiang and Z. Chen, J. Electrochem. Soc., 2017, 164, A1731–A1744 CrossRef CAS.
- D. Santhanagopalan, D. Qian, T. McGilvray, Z. Wang, F. Wang, F. Camino, J. Graetz, N. Dudney and Y. S. Meng, J. Phys. Chem. Lett., 2014, 5, 298–303 CrossRef CAS.
- Z. Wang, D. Santhanagopalan, W. Zhang, F. Wang, H. L. Xin, K. He, J. Li, N. Dudney and Y. S. Meng, Nano Lett., 2016, 16, 3760–3767 CrossRef CAS PubMed.
- K. Takada, Langmuir, 2013, 29, 7538–7541 CrossRef CAS PubMed.
- K. Takada, N. Ohta, L. Zhang, X. Xu, B. T. Hang, T. Ohnishi, M. Osada and T. Sasaki, Solid State Ionics, 2012, 225, 594–597 CrossRef CAS.
- A. Sakuda, H. Kitaura, A. Hayashi, M. Tatsumisago, Y. Hosoda, T. Nagakane and A. Sakamoto, Chem. Lett., 2012, 41, 260–261 CrossRef CAS.
- Y. Seino, T. Ota and K. Takada, J. Power Sources, 2011, 196, 6488–6492 CrossRef CAS.
- S. Zekoll, C. Marriner-Edwards, A. K. O. Hekselman, J. Kasemchainan, C. Kuss, D. Armstrong, D. Cai, R. Wallace, F. H. Richter, J. Thijssen and P. G. Bruce, Energy Environ. Sci., 2017, 11, 185–201 RSC.
- S. Yu, A. Mertens, X. Gao, D. C. Gunduz, R. Schierholz, S. Benning, F. Hausen, J. Mertens, H. Kungl, H. Tempel and R. -A. Eichel, Funct. Mater. Lett., 2016, 09, 1650066 CrossRef CAS.
- A. Mertens, S. Yu, N. Schön, D. C. Gunduz, H. Tempel, R. Schierholz, F. Hausen, H. Kungl, J. Granwehr and R. –A. Eichel, Solid State Ionics, 2017, 309, 180–186 CrossRef CAS.
- F. Lalère, J. B. Leriche, M. Courty, S. Boulineau, V. Viallet, C. Masquelier and V. Seznec, J. Power Sources, 2014, 247, 975–980 CrossRef.
- J. Xie, J. F. M. Oudenhoven, P. –P. R. M. L. Harks, D. Li and P. H. L. Notten, J. Electrochem. Soc., 2014, 162, A249–A254 CrossRef.
- A. A. Talin, D. Ruzmetov, A. Kolmakov, K. McKelvey, N. Ware, F. El Gabaly, B. Dunn and H. S. White, ACS Appl. Mater. Interfaces, 2016, 8, 32385–32391 CrossRef CAS PubMed.
- F. Croce, G. B. Appetecchi, L. Persi and B. Scrosati, Nature, 1998, 394, 456–458 CrossRef CAS.
- T. Zhang, N. Imanishi, Y. Shimonishi, A. Hirano, Y. Takeda, O. Yamamoto and N. Sammes, Chem. Commun., 2010, 46, 1661–1663 RSC.
- J. Paulsdorf, N. Kaskhedikar, M. Burjanadze, S. Obeidi, N. A. Stolwijk, D. Wilmer and H. D. Wiemhöfer, Chem. Mater., 2006, 18, 1281–1288 CrossRef CAS.
- S. Jankowsky, M. M. Hiller, O. Fromm, M. Winter and H. D. Wiemhöfer, Electrochim. Acta, 2015, 155, 364–371 CrossRef CAS.
- W. Zhou, S. Wang, Y. Li, S. Xin, A. Manthiram and J. B. Goodenough, J. Am. Chem. Soc., 2016, 138, 9385–9388 CrossRef CAS PubMed.
- J. Zhang, X. Zang, H. Wen, T. Dong, J. Chai, Y. Li, B. Chen, J. Zhao, S. Dong, J. Ma, L. Yue, Z. Liu, X. Guo, G. Cui and L. Chen, J. Mater. Chem. A, 2017, 5, 4940–4948 RSC.
- B. Wang, E. Rivard and I. Manners, Inorg. Chem., 2002, 41, 1690–1691 CrossRef CAS.
- Z. Wang and I. Manners, Macromolecules, 2005, 38, 5047–5054 CrossRef CAS.
- H. R. Allcock, S. D. Reeves, C. R. De Denus and C. A. Crane, Macromolecules, 2001, 34, 748–754 CrossRef CAS.
- S. Jankowsky, M. M. Hiller and H. D. Wiemhöfer, J. Power Sources, 2014, 253, 256–262 CrossRef CAS.
- N. Schön, D. C. Gunduz, S. Yu, H. Tempel, R. Schierholz and F. Hausen, Beilstein J. Nanotechnol., 2018, 9, 1564–1572 CrossRef.
- A. Manthiram, X. Yu and S. Wang, Nat. Rev. Mater., 2017, 2, 16103 CrossRef CAS.
- A. Manuel Stephan and K. S. Nahm, Polymer, 2006, 47, 5952–5964 CrossRef.
- S. Yu, L. Chen, Y. Chen and Y. Tong, Appl. Surf. Sci., 2012, 258, 4983–4989 CrossRef CAS.
- J. Evans, C. A. Vincent and P. G. Bruce, Polymer, 1987, 28, 2324–2328 CrossRef CAS.
- M. M. Hiller, M. Joost, H. J. Gores, S. Passerini and H. D. Wiemhöfer, Electrochim. Acta, 2013, 114, 21–29 CrossRef CAS.
- S. Yu, Z. Liu, H. Tempel, H. Kungl and R. Eichel, J. Mater. Chem. A, 2018, 18304–18317 RSC.
- X. Xu, Z. Wen, X. Yang, J. Zhang and Z. Gu, Solid State Ionics, 2006, 177, 2611–2615 CrossRef CAS.
- Y. Zhu, X. He and Y. Mo, ACS Appl. Mater. Interfaces, 2015, 7, 23685–23693 CrossRef CAS PubMed.
- S. Yu, A. Mertens, H. Kungl, R. Schierholz, H. Tempel and R. –A. Eichel, Electrochim. Acta, 2017, 232, 310–322 CrossRef CAS.
- Y. Huang and J. B. Goodenough, Chem. Mater., 2008, 20, 7237–7241 CrossRef CAS.
- S. C. Yin, H. Grondey, P. Strobel, H. Huang and L. F. Nazar, J. Am. Chem. Soc., 2003, 125, 326–327 CrossRef CAS PubMed.
- S. C. Yin, P. S. Strobel, H. Grondey and L. F. Nazar, Chem. Mater., 2004, 16, 1456–1465 CrossRef CAS.
- C. Liu, R. Massé, X. Nan and G. Cao, Energy Storage Materials, 2016, 4, 15–58 CrossRef.
- S. Yu, A. Mertens, R. Schierholz, X. Gao, Ö. Aslanbas, J. Mertens, H. Kungl, H. Tempel and R. –A. Eichel, J. Electrochem. Soc., 2017, 164, A370–A379 CrossRef CAS.
- S. Yu, H. Tempel, R. Schierholz, Ö. Aslanbas, X. Gao, J. Mertens, L. G. J. de Haart, H. Kungl and R. –A. Eichel, ChemElectroChem, 2016, 3, 1157–1169 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ta11259b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |