
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMassively enhanced ionic transport in irradiated crystalline pyrochlore†
Cortney R.
Kreller
 *a,
James A.
Valdez
b,
Terry G.
Holesinger
c,
Jonathan
Morgan
d,
Yongqiang
Wang
b,
Ming
Tang
b,
Fernando H.
Garzon
e,
Rangachary
Mukundan
a,
Eric L.
Brosha
a and
Blas P.
Uberuaga
b
*a,
James A.
Valdez
b,
Terry G.
Holesinger
c,
Jonathan
Morgan
d,
Yongqiang
Wang
b,
Ming
Tang
b,
Fernando H.
Garzon
e,
Rangachary
Mukundan
a,
Eric L.
Brosha
a and
Blas P.
Uberuaga
b
aMPA-11: Materials Synthesis and Integrated Devices, Los Alamos National Laboratory, Los Alamos, NM 87545, USA. E-mail: ckreller@lanl.gov
bMST-8: Materials Science in Radiation and Dynamics Extremes, Los Alamos National Laboratory, Los Alamos, NM 87545, USA
cMST-16: Nuclear Materials Science, Los Alamos National Laboratory, Los Alamos, NM 87545, USA
dDepartment of Materials Science & Engineering, University of Sheffield, Sheffield S10 2TN, UK
eAdvanced Materials Laboratory, University of New Mexico, Albuquerque, NM 87106, USA
First published on 4th February 2019
Abstract
Crystalline disorder in complex oxides can have a pronounced effect on mass transport, but the role of disorder within the complex landscape of material and microstructural defects is not uniquely understood. In this work, we use radiation damage, not to study the fundamental damage response, but rather as a tool to induce and control the extent of disorder of a single oxide composition in order to explore the relationship between intrinsic disorder and transport. Thin films of the ordered pyrochlore Gd2Ti2O7 (GTO) were irradiated with light ions and transitioned from ordered pyrochlore to disordered crystalline defect fluorite to an amorphous structure, disordering monotonically with increasing fluence, but nonhomogeneously throughout the film. Conductivity increased substantially with increasing disorder, where only slight disordering results in the majority of the change in conductivity. Additionally, while the pristine GTO films exhibited mixed ionic and p-type electronic conductivity, the total conductivity of the material that exhibited maximum disorder prior to the onset of amorphization was ionic in nature.
Introduction
The goal of clean and efficient energy generation and consumption places demanding cost and performance requirements on replacement technologies. While Solid Oxide Fuel Cells (SOFCs) are a low-carbon technology exhibiting fuel flexibility with exceptional efficiency, the high operating temperature of 800–1000 °C presents barriers due to cost and durability. Fast ionic conducting materials are required to reduce the operating temperature to the target 500–700 °C range. Numerous avenues are currently being explored to enhance ionic conductivity in complex oxide materials, including dopants, nano-structuring, and strain engineering.1–3 Their effect on material properties, specifically mobile carrier concentration and mobility, needs to be understood and ultimately exploited to develop advanced functional materials. Crystalline disorder in complex oxides plays an important role in determining mass transport properties, but it is challenging to isolate the effect of order/disorder independent of changes in chemical composition and microstructural defects.Pyrochlores are a model system for studying the role of disorder. Pyrochlore compounds, A2B2O7, are a superstructure of the fluorite phase with ordering on both the cation and anion sublattice. The anion sublattice is characterized by structural vacancies that compensate the reduced valence of the A cation, as compared to a true fluorite. The introduction of anti-site disorder, where the A and B cations switch positions, is accompanied by increasing disorder on the anion sublattice yielding a partially disordered defect-fluorite phase, and complete randomization of the A- and B-site cations yields the fluorite structure. The anion disorder essentially makes the structural vacancies available as carriers for conductivity. The stability of the ordered pyrochlore structure is generally correlated with cation radii ratio. When the cations are of sufficiently different sizes (rA/rB > 1.46) the pyrochlore structure is favored.4 As this ratio decreases and the cations become more similar in size, ordering is less favored and the material adopts a partial to fully disordered fluorite structure depending on composition and processing history. Researchers have previously investigated transport effects in this material system by introducing disorder via doping, temperature, pressure or mechanical milling.5–16 In this work light ion irradiation is used to induce and control the extent of disorder without changing other variables that might impact the transport (such as chemistry).
Because of the potential importance of pyrochlores as nuclear waste forms, the radiation tolerance of a wide range of pyrochlores has been extensively studied. It has been shown that, generally, materials with high anti-site defect formation energy (those that strongly prefer the ordered pyrochlore structure) are readily amorphized under irradiation while materials that can accommodate anti-site disorder (materials that tend to form the defect fluorite phase) exhibit excellent radiation tolerance.17–19 This is due to the fact that disorder is unavoidably introduced via irradiation but the energy penalty associated with that disorder is related to the energy of the disordering defects (the antisites). The Gd2(ZrxTi1−x)2O7 system has been extensively studied from both radiation tolerance and fast ion transport perspectives. The fully ordered Gd2Ti2O7 end member is highly susceptible to irradiation damage.20–22 Substitution of Zr for Ti results in a structure that borders the pyrochlore/defect fluorite phase boundary that is more tolerant to irradiation (rGd/rTi = 1.74, rGd/rZr = 1.46).23 Additionally, as shown in the seminal work by Moon and Tuller, the substitution of Zr on the Ti-site is accompanied by an almost four order of magnitude increase in conductivity at 600 °C.24–27 Other studies have also investigated the role of chemistry-induced disorder with mixed results. For example Yamamura et al.16 examined over 20 compositions of (Ln1−xLn′x)2Zr2O7 (Ln = Gd, Sm, or Nd; Ln′ = Y, Yb, or Gd) and observed a sharp maximum in conductivity at the phase boundary between the pyrochlore and defect fluorite phase (as determined by cation radius ratio). Work by Díaz-Guillén et al.11 examined the Gd2−yLnyZr2O7 (Ln = Er, Y, Dy, Sm, Nd, and La) system and observed a step change in conductivity at the fluorite/pyrochlore phase boundary (also determined by cation radius ratio). Both of these reports find an increase in conductivity with the ordering of the pyrochlore phase, in contrast to the results of Tuller et al. However, as noted, studies such as these convolute the effects of disorder with chemistry and do not unambiguously isolate the role of disorder in enhancing the conductivity. The current work aims to resolve this uncertainty and deconvolute the effect of disorder from that of chemistry to provide a more solid understanding of this relationship.
Results and discussion
In this work, thin films of Gd2Ti2O7 on YSZ were studied as a function of irradiation induced disorder. Details of materials synthesis, irradiation conditions and characterization are given in the ESI.† The change in the diffraction pattern with irradiation in the GTO films is summarized in Fig. 1. The pyrochlore structure rapidly disorders into the defect fluorite with increasing fluence before plateauing at approximately 50% cation disorder. The onset of amorphization occurred between a fluence of 5 × 1016 and 8 × 1016 He cm−2 indicating that GTO can tolerate approximately 50% cation disorder before the high anti-site defect formation energy results in amorphization of the material. In our analysis, we assume that the cation distribution induced by the irradiation is essentially frozen into place at the temperatures of interest while the oxygen defects (beyond oxygen Frenkel pairs, which can be viewed as defects that describe anion disorder), having orders-of-magnitude higher mobility, anneal out, and the oxygen adjusts to that cation structure.46 | ||
| Fig. 1 Normalized XRD peak ratios and percent cation disorder verses fluence. The calculation of percent cation disorder from the XRD peak intensities is described in the ESI.† | ||
It has been reported that local disorder occurs in materials that are nominally of pyrochlore structure,28 that local order can exist in disordered materials (and vice versa),8,29–32 and also that irradiation can induce local disorder within an otherwise ordered structure.33,34 As XRD only provides average structural information, transmission electron microscopy (TEM) was utilized to examine crystallinity as a function of film depth in cross-sections of irradiated films in order to further investigate the nature of the irradiation-induced disorder. A TEM micrograph of a film irradiated at the highest fluence considered in this study, 9 × 1016 He cm−2, is shown in Fig. 2. The insets in Fig. 2 show the selected area electron diffraction patterns (SAED) throughout the film depth between the exposed GTO surface and the GTO/YSZ interface. The material closest to the exposed GTO surface retained the ordered pyrochlore structure. With increasing depth, the superlattice reflections disappear as the material adopts the defect fluorite structure before all of the crystalline reflections disappear as the material becomes amorphous near the GTO/YSZ interface. These are not sharp transitions, but rather a gradient of mixed-phase microdomains. The observed trend of increasing disorder with increasing depth is consistent with the SRIM calculation of DPA as a function of depth (Fig. S1†). The diffraction patterns were obtained from grazing incidence X-ray diffraction at an angle of 0.75°, which corresponds to an averaging of a depth of approximately 135 nm from the surface of the GTO film.
 | ||
| Fig. 2 Cross-sectional TEM image of film exposed to 9 × 1016 He cm−2. SAED insets show non-uniform damage throughout film thickness. | ||
Referring to Fig. S1,† this near surface region is expected to have the lowest, and relatively constant, DPA for a given fluence. The extent of disorder calculated from the peak ratios of this region of the film reported in Fig. 1 therefore represents a lower limit of the extent of irradiation-induced disorder of the film in its entirety. The extreme sensitivity of the GTO material to DPA is not entirely surprising as this material is well within the pyrochlore stability field with rA/rB = 1.74 and possesses a relatively large anti-site defect formation energy.17,35–37 The details of disordering at low irradiation doses are generally of little interest from a radiation-tolerant materials perspective, so it has not been studied in detail. However, reports on swift heavy ion irradiation have found a disordered shell around an amorphous core in these types of materials. The transition is too sharp to say that there is a gradient in disorder, but it does show that GTO can be disordered, at least in part, in some cases.38–40
The conductivity of films exposed to varying He fluence is plotted in Arrhenius form in Fig. 3 and is observed to increase with increasing fluence, i.e. disorder. This is more clearly evident in Fig. 4 where the isothermal conductivity is plotted against the calculated disorder. A marked increase in conductivity is observed immediately upon introducing very small amounts of anti-site disorder into the ordered pyrochlore structure, corresponding to fluences of 1013 to 1014 He cm−2. The conductivity increases by almost 2 orders of magnitude at 500 °C and by almost one order of magnitude at 800 °C at disorder levels <2% before plateauing, then increasing slightly with further disordering up to 20%. The data point shown for ∼25% disorder was obtained at a fluence of 1 × 1016 He cm−2—this was the highest fluence at which conductivity measurements were made on GTO that fully retained a crystalline structure. The next damage condition measured was 8 × 1016 He cm−2, a condition under which the material had begun to amorphize at the depths probed by XRD. A disorder level of ∼50% is assigned to data at this fluence because our XRD analysis suggests that this is the maximum level of crystalline anti-site disorder the material can accommodate. The conductivity measurements for this film are shown as open symbols in Fig. 4 to indicate that in addition to a disordered crystalline structure, it also contains some amorphous material and thus starts to probe a different structural regime. The introduction of the amorphous phase coincided with a further increase in conductivity by roughly another order of magnitude. At a fluence of 9 × 1016 He cm−2, the intermediate frequency impedance feature attributed to the GTO film disappeared and only a high frequency intercept was observed. Thus the conductivity of the GTO film exposed to this fluence could not be deconvoluted from that of the YSZ substrate.
 | ||
| Fig. 3 Arrhenius plot showing temperature dependence of conductivity for thin films after varying irradiation exposure (fluence in He cm−2). Data represents average of multiple electrodes and at least two temperature cycles. | ||
 | ||
| Fig. 4 Log conductivity verses percent disorder for selected temperatures. | ||
The conductivity of a pristine film and a film irradiated at 1016 He cm−2 were also measured as a function of pO2 (1–10−6) in order to evaluate ionic verses electronic contributions to the total impedance, as described in the ESI. The conductivity of the pristine film exhibited a slight pO2 dependence, increasing with increasing pO2. The pO2 dependence decreased with increasing temperature. The slight positive slope observed is consistent with mixed ionic and p-type electronic conductivity. The defect chemical model derived by Moon predicts a positive ¼ or 1/6 slope to corresponding to p-type conductivity at high pO2 for the pristine GTO material. The highest slope that we observed at 600 °C was only approximately 1/10, indicating that there is also a contribution of a pO2 independent ionic conductivity in this regime. On the other hand, the irradiated film exhibited no dependence on pO2 even at 600 °C, indicating that the total conductivity is dominated by ionic contributions in this film. In the pristine film, ionic conductivity is low and therefore the total conductivity is dominated by extrinsic defects resulting from background impurities.41 As disorder is introduced in the material, ionic conductivity increases and overwhelms the small electronic contribution arising from extrinsic defects. A future publication will report on the disorder dependent transition from electronic to ionic dominated conductivity in detail.
Early work by Tuller et al. is frequently cited in the discussion of disorder vs. transport, so it is instructive to compare the results of disorder introduced via anti-site defects in a constant composition system reported herein with that of disorder introduced via isovalent substitution.24–27 The data in Fig. 4 compellingly indicates that crystalline disorder alone significantly impacts the transport properties of the GTO material. The orders of magnitude increase observed upon introducing disorder with irradiation is qualitatively similar to what has been reported in the literature for disorder introduced via Zr substitution on the B site in Gd2(ZrxTi1−x)2O7. Moon and Tuller report an approximate 3.5 order of magnitude increase in conductivity at 600 °C when x is increased from 0 to 0.4.25 This coincides with a reduction to 0.94 in their reported cation order parameter.24 A decrease in order parameter of 0.06 corresponds to approximately 3.4% disorder in the GTO films discussed herein. We find approximately 2% disorder in the film irradiated at a fluence of 1 × 1015 He cm−2, at which point we observe a 1.2 order of magnitude increase in the conductivity of the film relative to pristine at 600 °C.
The observation that introducing disorder via Zr substitution results in a larger increase in conductivity than introducing disorder via anti-site defect formation is also telling, but not surprising. Substitution with Zr in Gd2(ZrxTi1−x)2O7 reduces the cation radius ratio from 1.74 to 1.47 with increasing x from 0 to 1. As with conducting oxides in general, mismatch in ionic radii on a given lattice site typically imparts lattice strain that hinders ionic conductivity.42–44 This was specifically shown to be true in the GTO system where introduction of Ca2+ (rCa/rGd = 1.06) as an aliovalent dopant on the Gd3+ A-site yielded the largest increase in conductivity.45 On the other hand, when anti-site defects are introduced by swapping the Gd A-site and the Ti B-site cations, the relative radii ratio defining the structure may change with the change in cation coordination number that accompanies anti-site formation, but not to as large a degree as substituting with a larger cation such as Ca. Additionally, theoretical studies have found that for all levels of disorder, Gd2Zr2O7 (GZO) exhibits higher conductivity than GTO,46 underscoring the importance of studying disorder independent of chemistry effects.
Conductivity is typically described by a combination of the activation energy EA for diffusion and a pre-exponential factor σo. The values of these parameters are extracted from the Arrhenius plots shown in Fig. 4 and plotted against percent cation disorder in Fig. 5. Error bars are propagated from the raw data set. The pre-exponential constant σo is proportional to both carrier concentration and the pre-exponential mobility factor, while EA is the activation energy of the bulk conduction process. Because the conductivity measurements as a function of pO2 show that, at least in the pristine film, the material exhibits mixed-ionic electronic conductivity, we do not define the nature of the charge carrier in the following discussion. A defect-chemical model is being developed based on pO2 dependent measurements on films exposed to all irradiation conditions that will shed more light on the precise nature of the charge carrier. As shown in Fig. 5a, at low levels of disorder σo is approximately constant within the large associated error, changing at most by 25%. At this same level of disorder, EA drops precipitously, as shown in Fig. 5b. Both σo and EA increase only slightly with increasing disorder up to ∼25% disorder (1 × 1016 He cm−2), before both increase markedly with the onset of amorphization (8 × 1016 He cm−2). At low levels of disorder, the conductivity increase is thus attributable to a disorder-dependent decrease in activation energy, a validation of recent MD simulations.46 The plateau in conductivity up to 25% disorder is accompanied by a corresponding plateau in both σo and EA. At high levels of disorder after the onset of amorphization, the increase in conductivity is attributable to a large enhancement in carrier concentration that offsets the accompanying increase in EA.
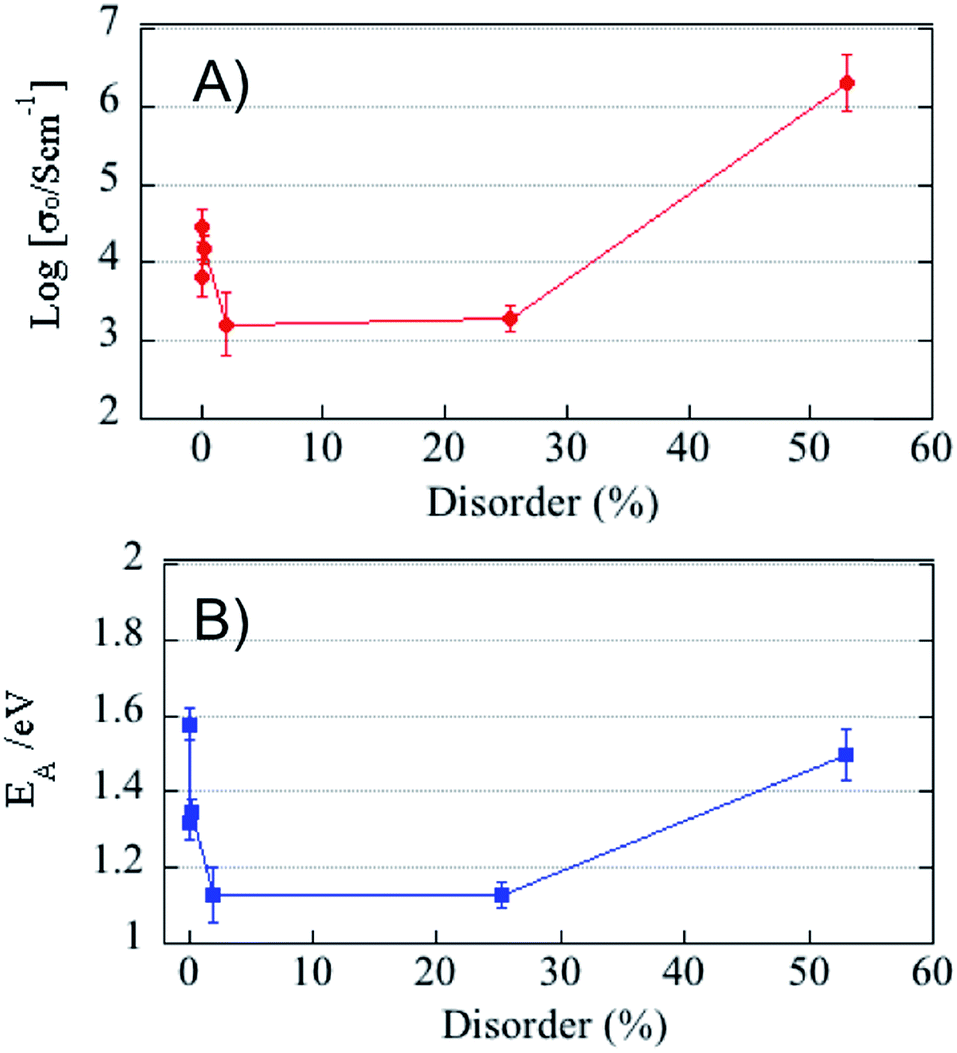 | ||
| Fig. 5 Log of pre-exponential factor, σo, (A) and activation energy, EA, (B) verses percent cation disorder. | ||
As disorder is introduced on the cation sublattice, so is it induced on the anion sublattice, thus freeing oxygen vacancies from their fully ordered lattice positions and introducing increased charge carriers in the form of 48f–8b Frenkel pairs. While we expect the carrier concentration to increase with increasing disorder, the non-uniform nature of the irradiation induced anti-site defects, as well as the pyrochlore structure's inherent ability to accommodate local disorder within a long range ordered structure, may be leaving Frenkel pairs stranded in local and isolated islands of disorder unable to contribute to long-range anion transport in the limit of low disorder <2%. Indeed, such potential percolation effects have been observed in simulations of cation transport in disordered pyrochlores,47 indicating that one would require a connected network of disordered domains to achieve high conductivity in these materials.
Within this region of low disorder, it is the reduction in activation energy that is driving the conductivity enhancement. This observation is in contrast to some reports on order-to-disorder transitions modulated by chemistry,9,16 as well as thermal processing,8,48 where the ordered structure is reported to yield the lowest activation energy. However, other studies do show a decrease in activation energy with increasing disorder. Díaz-Guillén observed a monotonic decrease in activation energy in Gd2−yLnyZr2O7 with increasing RLn (i.e. increasing disorder), though an almost step change increase in conductivity was observed at the disordered fluorite/pyrochlore phase boundary.11 Tuller reported a decrease in activation energy in the Y2(ZrxTi1−x)2O7 system at x < 0.2, though the conductivity is not reported in this region. On the other hand, previous density functional theory (DFT) calculations have examined the lattice expansion introduced by pairs of anti-sites in a related compound, Lu2Ti2O7.49 Regardless of the configuration, those calculations reveal that anti-site pairs induce an increase in the volume of the material 17–20 Å3 per anti-site pair. Similarly, DFT calculations of fully disordered pyrochlores show consistent increases in volume in the disordered state.19 Thus, it is expected that, as damage is introduced into the material in the form of anti-site disorder, the material will expand, potentially leading to faster pathways as more open space is created. At very low levels of disorder these local, non-percolated anti-sites do not lead to an overall increase in lattice parameter as measured by GIXRD, though it is possible that our measurements are not capturing the lattice expansion due to the non-uniform nature of the damage profile in the thin films. The difference that we observe in the GTO system studied herein verses some prior work is likely attributable to differences in composition. Both geometric effects (lattice expansion) as well as the nature of the metal–oxygen bond influence transport. Shlyakhtina et al. note that the geometric factor tends to dominate in titanate pyrochlores while in the zirconates, the Zr–O bond energy has greater influence.50 It is also worth noting that the way in which disorder is introduced by substitution is fundamentally different, in that at low substitution levels the cation will occupy the A/B site with the closest match in ionic radii and anti-site formation, with A/B site cation mixing, will only occur at high substitution levels (if at all). Additionally, the transition from mixed ionic–electronic to ionic conductivity also must be considered, as the charge carriers contributing to the total measured conductivity are different between the pristine and irradiated films. Finally, other complicating effects might occur, including the presence of short-range order51 and, in chemically complex systems, the formation of multiple structural domains29,52 that, depending on composition, can lead to more complex structures that are not easy to interpret.
With further increases in crystalline disorder, an increase in σo is reasonably expected as it is likely that regions of anion sublattice disorder would overlap and form continuous conduction pathways at higher disorder levels.34 While not entirely understood, the plateau in conductivity with increasing crystalline disorder is qualitatively similar with that experimentally observed by Tuller in the Zr substitution studies as well as recent modeling results that show a saturation in the dependence of oxygen diffusivity on disorder.46 At high levels of disorder after the onset of amorphization, the increase in conductivity is attributable to a large enhancement in carrier concentration that offsets the accompanying increase in EA.
The trends in conductivity with increasing disorder identified in this study are in agreement with theoretical studies reported in the literature. Catlow and Wilde53 used molecular dynamics simulations to identify the energy of formation of oxygen Frenkel defects as the controlling factor in ionic conductivity in pyrochlore oxides. They found that disorder was necessary in both GTO and GZO materials in order to decrease the anion Frenkel defect formation energies to allow for intrinsic mobile oxygen vacancies. More recently Perriot et al.46 expanded on these results and found that disorder generally enhances ionic conductivity, and this work identified increased carrier concentration as well as an increase in their mobility contributed to the improved diffusivity. This work additionally predicts a plateau in conductivity with increasing disorder, in agreement with what was observed prior to the onset of amorphization in our study. Analysis of the radial distribution function of oxygen as a function of disorder showed that in GTO, the oxygen becomes completely detached from the cation sublattice exhibiting an amorphous-like structure, even though the cation sublattice remains crystalline with a defect-fluorite structure. In the numerical analysis, modest gains were observed in oxygen diffusivity in GTO beyond cation disorder of 0.5 compared to the increase observed at lower levels of disorder.
Conclusions
Our results unequivocally show that disorder alone, independent of chemical doping, has a marked influence on transport in pyrochlore oxides and suggest that the conflicting reports on the role of order and disorder on transport in these materials may be due, in part, to convoluting effects involving changes in chemistry, including, perhaps, a more complex microstructure than simple solid-solution mixing. Our results offer new insight into the relationship between anti-site disorder and conductivity, providing new avenues for tailoring materials with enhanced conductivity for fast ion conducting applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Materials Sciences and Engineering Division grant number LANLE4BU. LANL, an affirmative action/equal opportunity employer, is operated by Los Alamos National Security, LLC, for the National Nuclear Security Administration of the U.S. Department of Energy under contract DE-AC52-06NA25396. The data reported in the paper were presented at the 232nd Meeting of the Electrochemical Society.Notes and references
- H. L. Tuller and S. R. Bishop, Annu. Rev. Mater. Res., 2011, 41, 369–398 CrossRef CAS.
- J. L. M. Rupp, Solid State Ionics, 2012, 207, 1–13 CrossRef CAS.
- A. J. Jacobson, Chem. Mater., 2010, 22, 660–674 CrossRef CAS.
- M. A. Subramanian, G. Aravamundan and G. V. Subba Rao, Prog. Solid State Chem., 1983, 15, 55–143 CrossRef CAS.
- K. J. Moreno, A. F. Fuentes, M. Maczka, J. Hanuza, U. Amador, J. Santamaría and C. León, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 184303 CrossRef.
- A. F. Fuentes, K. Boulallya, M. Maczka, J. Hanuza and U. Amador, Solid State Sci., 2005, 7, 343–353 CrossRef CAS.
- F. X. Zhang, B. Manoun, S. K. Saxena and C. S. Zha, Appl. Phys. Lett., 2005, 86, 3 Search PubMed.
- M. P. van Dijk, K. J. de Vries and A. J. Burggraaf, Solid State Ionics, 1983, 9&10, 913–920 Search PubMed.
- H. Nishino, H. Yamamura, T. Arai, K. Kakinuma and K. Nomura, J. Ceram. Soc. Jpn., 2004, 112, 541–546 CrossRef CAS.
- K. V. G. Kutty, C. K. Mathews, T. N. Rao and U. V. Varadaraju, Solid State Ionics, 1995, 80, 99–110 CrossRef.
- J. A. Díaz-Guillén, A. F. Fuentes, M. R. Díaz-Guillén, J. M. Almanza, J. Santamaría and C. León, J. Power Sources, 2009, 186, 349–352 CrossRef.
- J. García-Barriocanal, K. J. Moreno, G. Mendoza-Suárez, A. F. Fuentes, J. Santamaría and C. León, J. Non-Cryst. Solids, 2005, 351, 2813–2818 CrossRef.
- S. T. Norberg, S. Hull, S. G. Eriksson, I. Ahmed, F. Kinyanjui and J. J. Biendicho, Chem. Mater., 2012, 24, 4294–4300 CrossRef CAS.
- A. N. Radhakrishnan, P. Prabhakar Rao, K. S. Mary Linsa, M. Deepa and P. Koshy, Dalton Trans., 2011, 40, 3839–3848 RSC.
- B. J. Wuensch, K. W. Eberman, C. Heremans, E. M. Ku, P. Onnerud, E. M. E. Yeo, S. M. Haile, J. K. Stalick and J. D. Jorgensen, Solid State Ionics, 2000, 129, 111–133 CrossRef CAS.
- H. Yamamura, Solid State Ionics, 2003, 158, 359–365 CrossRef CAS.
- K. E. Sickafus, Science, 2000, 289, 748–751 CrossRef CAS PubMed.
- K. E. Sickafus, R. W. Grimes, J. A. Valdez, A. Cleave, M. Tang, M. Ishimaru, S. M. Corish, C. R. Stanek and B. P. Uberuaga, Nat. Mater., 2007, 6, 217–223 CrossRef CAS PubMed.
- G. Pilania, K. R. Whittle, C. Jiang, R. W. Grimes, C. R. Stanek, K. E. Sickafus and B. P. Uberuaga, Chem. Mater., 2017, 29, 2574–2583 CrossRef CAS.
- W. J. Weber and N. J. Hess, Nucl. Instrum. Methods Phys. Res., Sect. B, 1993, 80/81, 1245–1248 CrossRef.
- S. X. Wang, B. D. Begg, L. M. Wang, R. C. Ewing, W. J. Weber and K. V. Govidan Kutty, J. Mater. Res., 1999, 14, 4470–4473 CrossRef CAS.
- J. Lian, J. Chen, L. M. Wang, R. C. Ewing, J. M. Farmer, L. A. Boatner and K. B. Helean, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 9 CrossRef.
- R. D. Shannon and C. T. Prewitt, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1969, 25, 925–946 CrossRef CAS.
- P. K. Moon, PhD thesis, Massachusetts Institute of Technology, 1988.
- P. K. Moon and H. L. Tuller, Mater. Res. Soc. Symp. Proc., 1989, 135, 149–163 CrossRef CAS.
- P. K. Moon and H. L. Tuller, Solid State Ionics, 1988, 28–30, 470–474 CrossRef.
- H. L. Tuller and P. K. Moon, Mater. Sci. Eng., B, 1988, 1, 171–191 CrossRef.
- P. E. Blanchard, R. Clements, B. J. Kennedy, C. D. Ling, E. Reynolds, M. Avdeev, A. P. Stampfl, Z. Zhang and L. Y. Jang, Inorg. Chem., 2012, 51, 13237–13244 CrossRef CAS PubMed.
- M. P. van Dijk, F. C. Mijlhoff and A. J. Burggraaf, J. Solid State Chem., 1986, 62, 377–385 CrossRef CAS.
- M. L. Sanjuán, C. Guglieri, S. Díaz-Moreno, G. Aquilanti, A. F. Fuentes, L. Olivi and J. Chaboy, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 104207 CrossRef.
- J. Shamblin, M. Feygenson, J. Neuefeind, C. L. Tracy, F. X. Zhang, S. Finkeldei, D. Bosbach, H. D. Zhou, R. C. Ewing and M. Lang, Nat. Mater., 2016, 15, 507–512 CrossRef CAS PubMed.
- L. Martel, M. Naji, K. Popa, J.-F. Vigier and J. Somers, Sci. Rep., 2017, 7, 12269 CrossRef PubMed.
- J. Lian, L. M. Wang, S. X. Wang, J. Chen, L. A. Boatner and R. C. Ewing, Phys. Rev. Lett., 2001, 87, 145901 CrossRef CAS PubMed.
- D. Simeone, G. J. Thorogood, D. Huo, L. Luneville, G. Baldinozzi, V. Petricek, F. Porcher, J. Ribis, L. Mazerolles, L. Largeau, J. F. Berar and S. Surble, Sci. Rep., 2017, 7, 7 CrossRef PubMed.
- G. R. Lumpkin, M. Pruneda, S. Rios, K. L. Smith, K. Trachenko, K. R. Whittle and N. J. Zaluzec, J. Solid State Chem., 2007, 180, 1512–1518 CrossRef CAS.
- X. J. Wang, H. Y. Xiao, X. T. Zu, Y. Zhang and W. J. Weber, J. Mater. Chem. C, 2013, 1, 1665 RSC.
- Y. Li, P. M. Kowalski, G. Beridze, A. R. Birnie, S. Finkeldei and D. Bosbach, Scr. Mater., 2015, 107, 18–21 CrossRef CAS.
- J. Zhang, M. Lang, R. C. Ewing, R. Devanathan, W. J. Weber and M. Toulemonde, J. Mater. Res., 2010, 25, 1344–1351 CrossRef CAS.
- R. Sachan, V. R. Cooper, B. Liu, D. S. Aidhy, B. K. Voas, M. Lang, X. Ou, C. Trautmann, Y. W. Zhang, M. F. Chisholm and W. J. Weber, J. Phys. Chem. C, 2017, 121, 975–981 CrossRef CAS.
- D. S. Aidhy, R. Sachan, E. Zarkadoula, O. Pakarinen, M. F. Chisholm, Y. Zhang and W. J. Weber, Sci. Rep., 2015, 5, 16297 CrossRef CAS PubMed.
- H. L. Tuller, J. Phys. Chem. Solids, 1994, 55, 1393–1404 CrossRef CAS.
- J. A. Kilner, Solid State Ionics, 2000, 129, 13–23 CrossRef CAS.
- D. J. Kim, J. Am. Ceram. Soc., 1989, 72, 1415–1421 CrossRef CAS.
- M. Mogensen, D. Lybye, N. Bonanos, P. V. Hendriksen and F. W. Poulsen, Solid State Ionics, 2004, 174, 279–286 CrossRef CAS.
- S. Kramer, M. Spears and H. L. Tuller, Solid State Ionics, 1994, 72, 59–66 CrossRef CAS.
- R. Perriot and B. P. Uberuaga, J. Mater. Chem. A, 2015, 3, 11554–11565 RSC.
- R. Perriot, B. P. Uberuaga, R. J. Zamora, D. Perez and A. F. Voter, Evidence for Percolation Diffusion of Cations and Material Recovery in Disordered Pyrochlore from Accelerated Molecular Dynamics Simulations, 2016, arXiv:1607.07101 Search PubMed.
- A. V. Shlyakhtina, A. V. Knotko, M. V. Boguslavskii, S. Y. Stefanovich, D. V. Peryshkov, I. V. Kolbanev and L. G. Shcherbakova, Solid State Ionics, 2005, 176, 2297–2304 CrossRef CAS.
- Y. H. Li, B. P. Uberuaga, C. Jiang, S. Choudhury, J. A. Valdez, M. K. Patel, J. Won, Y. Q. Wang, M. Tang, D. J. Safarik, D. D. Byler, K. J. McClellan, I. O. Usov, T. Hartmann, G. Baldinozzi and K. E. Sickafus, Phys. Rev. Lett., 2012, 108, 195504 CrossRef CAS PubMed.
- A. V. Shlyakhtina and L. G. Shcherbakova, Solid State Ionics, 2011, 192, 200–204 CrossRef CAS.
- M. de los Reyes, K. R. Whittle, Z. Zhang, S. E. Ashbrook, M. R. Mitchell, L.-Y. Jang and G. R. Lumpkin, RSC Adv., 2013, 3, 5090–5099 RSC.
- K. R. Whittle, L. M. D. Cranswick, S. A. T. Redfern, I. P. Swainson and G. R. Lumpkin, J. Solid State Chem., 2009, 182, 442–450 CrossRef CAS.
- P. J. Wilde and C. R. A. Catlow, Solid State Ionics, 1998, 112, 173–183 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ta10967b |
| This journal is © The Royal Society of Chemistry 2019 |