
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
2D-dual-spacing channel membranes for high performance organic solvent nanofiltration†
Shaofei
Wang
 ,
Dinesh
Mahalingam
,
Burhannudin
Sutisna
and
Suzana P.
Nunes
*
,
Dinesh
Mahalingam
,
Burhannudin
Sutisna
and
Suzana P.
Nunes
*
Biological and Environmental Science and Engineering Division (BESE), Advanced Membranes and Porous Materials Center (AMPM), King Abdullah University of Science and Technology (KAUST), Thuwal, 23955-6900, Saudi Arabia. E-mail: suzana.nunes@kaust.edu.sa
First published on 10th January 2019
Abstract
Two-dimensional (2D) lamellar membranes are highly advantageous in molecular separations. However, the permeance-rejection trade-off is always a major challenge, since the permeant transport mostly occurs in single-spacing channels with undesired microenvironments. Inspired by the structure of aquaporins, we design alternating dual-spacing channel graphene oxide (GO) membranes, with locally tailored chemical microenvironments, that give high permeance, high rejection and high stability in organic solvent nanofiltration. This unique structure is easily constructed by in situ intercalating and cross-linking scattered sub-5 nm silica nanoparticles in the GO interlayers. The hydrophilic nanoparticles locally widen the interlayer channels to enhance the solvent permeance. In the alternating nanoparticle-free areas, the GO layers simultaneously bend and the π–π interactions retain the narrow and hydrophobic channels, promoting high solute rejection. With a 10-fold increase in water permeance and unaffected rejection, the dual-spacing channel membranes exhibit one of the best performances for organic solvent nanofiltration. The methanol permeance reaches 290 L m−2 h−1 bar−1, with more than 90% rejection of dyes larger than 1.5 nm. This new approach of designing hierarchical channels in 2D materials can be used for a wide spectrum of applications.
Introduction
Membranes have a growing impact in water desalination, chemical and pharmaceutical industrial separations.1 A synthetic membrane with nanometer-size channels, characterized by high flux, precise size sieving ability and excellent durability, is the key component in these processes. The separation performance of a membrane is largely dependent on the dimensions, as well as on the physical and chemical microenvironments of the transmembrane channels.2 The development of advanced membranes with engineered nanochannel structures is urgently required to satisfy the industrial demands for filtering large volumes of organic solvents.3,4In the past few decades, the demand for ultra-thin membranes has been fulfilled by the exfoliation of various two-dimensional (2D) materials, including graphene,5 graphene oxide (GO),6–8 MXenes,9,10 BN,11 C3N4,12 WS2,13 2D metal–organic frameworks,14etc. These 2D material membranes, either processed in the form of single porous layers, multilayered stacks, or mixed matrix membranes, feature ultimate separation capabilities.15,16 The rational channel construction is of vital importance in achieving high performances. Ideally, drilling uniform pores in a single layer or vertically aligning of 2D materials could form the mostly desired channel structure.5,17–19 However, these preparation processes are challenging and would be hardly scalable. Multilayered stacked membranes, referred also as lamellar membranes, offer a technically practical pattern for translating 2D materials into membranes. In lamellar membranes, the penetrants flow in a tortuous way, first passing through in-plane gaps, and then through the nanochannels between the adjacent layers. Due to the irregularity of the in-plane gaps, the interlayer nanochannel becomes the path with predominant control of the separation efficiency.20 In this regard, major efforts have been devoted to increase the regularity of these channels by tuning the stacking efficiency,21 adopting rigid nanosheets,22 or crosslinking them with different segments23,24 to obtain single-spacing channels that can execute precise size-sieving. However, considering the long and tortuous in-plane channels, a regular and tight channel size weakens the penetrant permeance. A widely adopted solution to enhance the permeance of the lamellar membranes is to intercalate nano-sized spacers into the interlayers,20 such as copper hydroxide,6 carbon nanotubes,25 nanoparticles,9,26 and organic molecules.27,28 Recently, covalent cross-linking has proved effective in achieving high stability of GO membranes in organic solvents.16,28 An expanded interlayer spacing always leads to a considerably enhanced penetrant permeance. However, in some previous studies, the spacer size, varying from 5 nm to even a hundred nanometers, are always larger than that of the rejected molecules. Also, a high spacer distribution density leads to the entire membrane channel expansion to an undesired size range, weakening their size sieving ability.29 Therefore, a single spacing channel can hardly provide a solution to overcome the trade-off between permeance and rejection.
In nature, biological membranes possess delicate hierarchical channels with a perfectly tuned chemical microenvironment that provides an ultrahigh permselectivity for molecules and ions. Aquaporins are known for promoting a highly efficient water transport in biological membranes. The protein molecules arrange themselves forming an hourglass channel with a large and hydrophilic part to provide the high water permeance, and a narrow hydrophobic site to provide the high ion rejection.30 The discovery of aquaporins has inspired the design of highly efficient liquid separation membranes.31 Learning from the unique structure of aquaporins, we believe that the design of multi-spacing channels with locally tuned microenvironments would be a potential solution to overcome the critical restrictive permeance-rejection trade-off. While there has been considerable work regarding the hierarchical pore construction in polymeric membranes,3,32 analogous reports on laminate 2D material membranes are quite rare. Meanwhile, though there has been quite a lot of work on intercalated 2D membranes, the local channel affinity towards targeted penetrants has attracted less attention than the size control.23,33 The difficulty lies in the lack of effective methods to locally tailor the physical and chemical microenvironments in the specific nanospace. Recently, Abozar et al.34 demonstrated that the interactions between the polar groups along the edges of the GO sheets and solvent play a dominant role in the solvent permeance.
In this study, we propose a novel and simple strategy to fabricate GO membranes with alternating dual-spacing channels, enabled by the in situ generation of silica nanoparticles at specific domains between the GO layers. Different from other post intercalating GO membranes, the in situ method could help the formation of small and more scattered nanoparticles, which would contribute new structures. The expanded interlayer spacing around the hydrophilic nanoparticles contributes to a high permeance. Meanwhile, the combination of sub-5 nm spacers and flexible GO sheets22,35 leads to the local formation of narrow hydrophobic necks, contributing to a high rejection of small molecules (Scheme 1). Compared with unmodified GO membranes, the GO–Si membranes exhibited 10 times higher water permeance with negligible loss of dye rejection. These membranes show excellent performance in organic solvent nanofiltrations, with a high methanol permeance of 290 L m−2 h−1 bar−1, and >90% rejection of dyes larger than 1.5 nm. Furthermore, the spacers also act as cross-linkers and bring excellent stability to the GO membrane in organic solvents.
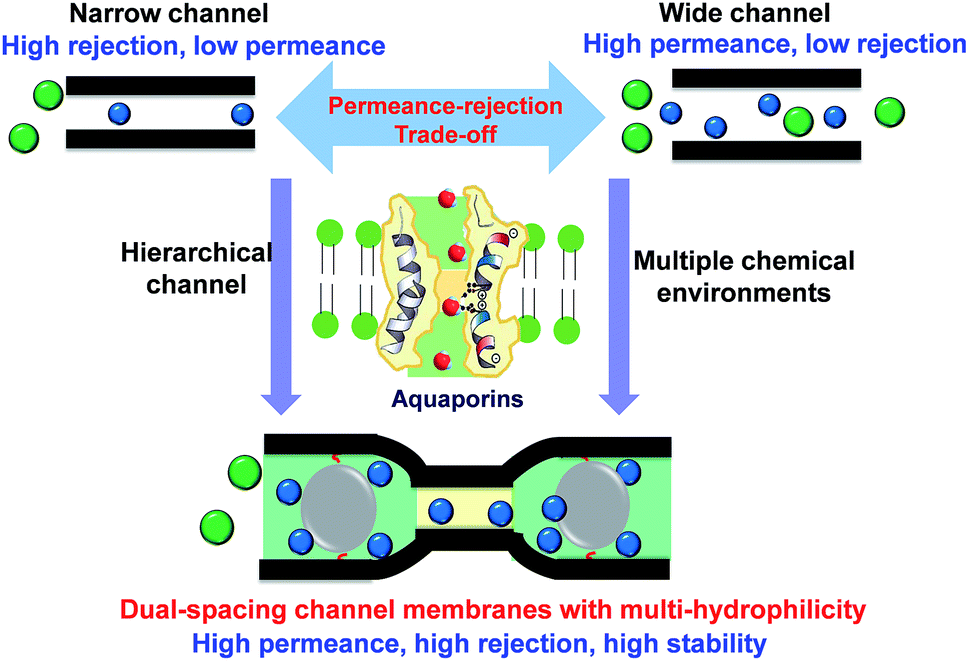 | ||
| Scheme 1 The concept of designing alternating dual-spacing channels with tailored chemical microenvironment in 2D material membranes to overcome the permeance-rejection trade-off. On the bottom: the green colour indicates a hydrophilic domain; the yellow colour indicates the hydrophobic sectors. | ||
Experimental section
Synthesis of GO sheets
All the chemicals were used as received from Sigma-Aldrich without further purification, unless otherwise specified. The GO sheets were synthesized according to an improved synthesis method reported by Tour et al.36 with minor modifications. Typically, 3.0 g of graphite flakes (>100 mesh) and KMnO4 (18.0 g, Fisher Scientific) were added to a three-necked flask in an ice bath. Subsequently, a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of concentrated H2SO4/H3PO4 (360:40 mL) was added to the mixture. The reaction medium was then heated to 50 °C and stirred at 300 rpm for 24 h. The color changed from black to brown. After being cooled to room temperature, the mixture was poured into a beaker containing 400 mL ice with 30% H2O2 (3 mL, VWR Chemicals). The mixture was kept undisturbed to help the deposition of graphite oxide. After decanting, the bottom deposits were centrifuged (10000 rpm for 1 h) and washed in succession with HCl solution (9:1 water:HCl by volume), water and ethanol till the pH reached 7. Then the obtained solid was vacuum-dried at room temperature for 48 h. The GO sheets were exfoliated in ethanol by sonication in a water bath for 1 h. To obtain uniform-sized single layers of GO, the dispersions were further centrifuged at 3000 rpm for 10 min, removing the unexfoliated and excessively large GO sheets. The concentration of the GO dispersion in ethanol was 0.1 mg mL−1.
:1 mixture of concentrated H2SO4/H3PO4 (360:40 mL) was added to the mixture. The reaction medium was then heated to 50 °C and stirred at 300 rpm for 24 h. The color changed from black to brown. After being cooled to room temperature, the mixture was poured into a beaker containing 400 mL ice with 30% H2O2 (3 mL, VWR Chemicals). The mixture was kept undisturbed to help the deposition of graphite oxide. After decanting, the bottom deposits were centrifuged (10000 rpm for 1 h) and washed in succession with HCl solution (9:1 water:HCl by volume), water and ethanol till the pH reached 7. Then the obtained solid was vacuum-dried at room temperature for 48 h. The GO sheets were exfoliated in ethanol by sonication in a water bath for 1 h. To obtain uniform-sized single layers of GO, the dispersions were further centrifuged at 3000 rpm for 10 min, removing the unexfoliated and excessively large GO sheets. The concentration of the GO dispersion in ethanol was 0.1 mg mL−1.
Synthesis of GO–Si membranes
The GO–Si membranes were fabricated by a vacuum filtration method. 5 mL of GO dispersion in stock was diluted to 0.01 mg mL−1, by adding ethanol. After adding 20 μL (3-aminopropyl)triethoxysilane (APTES), the mixture was stirred for 20 min at room temperature to help the attachment of APTES on GO. Then the mixture was filtered through a nylon microfiltration membrane (0.22 μm pore size, 47 mm diameter, supplied by GVS Filtration Inc) or an anodic aluminum oxide (AAO) membrane (0.2 μm pore size, 47 mm diameter, supplied by Whatman). After the filtration, 200 mL NaOH solution (pH = 11) was added. The filtration was terminated when about 10 mL solution had passed through. In this case, the membrane would be soaked in NaOH solution. The system was maintained in this way for 8 h, and then the filtration continued to eliminate the NaOH. The membrane was then filtered with 100 mL ethanol to wash the unreacted APTES. These membranes are referred to as the GO–Si2 membrane. GO–Si2 membranes with different thicknesses were also prepared, by proportionally increasing the GO and APTES amount. Other membranes were also prepared by following the same procedure without further filtration of NaOH, the membranes were denoted as the GO–Si1 membrane.Characterization
The size of the GO sheets and the cross-sectional morphology of the composite membranes were analyzed by scanning electron microscopy, SEM (FEI Magellan™ XHR SEM coupled with energy-dispersive X-ray spectroscopy (EDS)). Samples for cross-sectional imaging were prepared by freeze fracturing the membranes in liquid nitrogen, and then coating with a 3 nm thin film of Ir. Transmission electron microscopy (TEM) images of the GO layer were obtained on an FEI Titan G2 80–300 ST, operated at 300 kV. For that, a thin layer of GO–Si (∼60 nm) was fabricated on an anodic alumina (AAO) porous support. After dissolving the support with HCl, the GO layer was gently transferred to a TEM carbon grid. The thickness of the GO sheets and membranes was measured by AFM (Bruker Dimension Icon SPM). For that, the samples were filtered onto an AAO support and transferred to a clean silicon wafer, after dissolving the support with HCl. The X-ray Diffraction (XRD) investigation was carried out using a powder XRD system (Bruker D8 advance) with CuKα (λ = 0.154 nm), operating at 40 keV with a current of 20 mA. The chemical analysis was performed using Attenuated Total Reflectance (ATR) coupled with Fourier Transform Infra-Red Spectroscopy (FTIR) (FTIR-iS10) in the wavenumber range of 500–4000 cm−1 for an average of 32 scans with 4 cm−1 resolution. X-ray Photoelectron Spectroscopy (XPS) was performed on Amicus equipment to measure the elemental composition of the surface of the synthesized GO sheets and membranes.Membrane nanofiltration
The membrane nanofiltration performances were tested in dead-end stainless-steel cells with a driving force of 1 bar at room temperature. All the dyes used in this study were supplied by Sigma-Aldrich, except for brilliant blue R250, supplied by Fisher Scientific. The membranes were pre-soaked in the testing solvents for 1 h before testing. The analysis of the dye concentration was conducted with a NanoDrop UV-vis spectrophotometer. All the collected data are averages of three parallel tests. The pure solvent permeance (F) was calculated using eqn (1):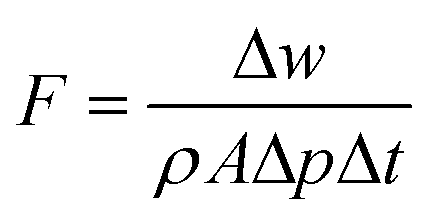 | (1) |
The dye rejection was determined by eqn (2), where Cp and Cf represent the concentration of the permeate and feed solutions, respectively.
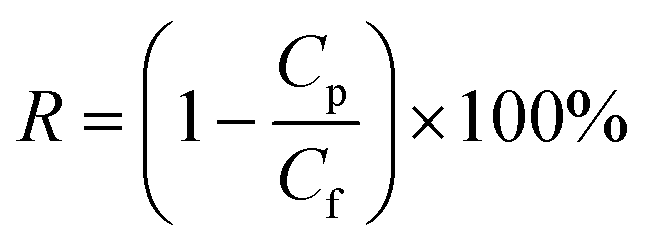 | (2) |
Results and discussion
GO sheets, the building blocks, were synthesized by the exfoliation of graphite oxide prepared by a mild improved GO synthesis method. By successive centrifugation steps, we removed the large or non-exfoliated GO units and obtained GO nanosheets with a thickness of ∼1 nm (Fig. 1a) and relatively uniform lateral size, around 500–1000 nm (atomic force microscopy (AFM) image in Fig. 1b; scanning electron microscope (SEM) image in Fig. S1, ESI†). The nano- rather than micrometer-sized GO sheets are expected to lead to membranes with more in-plane pores, which favor a faster solvent transport, as reported by Zhu et al.,37 and Wang et al.9 The Fourier-transform infrared spectroscopy (FTIR) spectra of GO (Fig. S2, ESI†) show the characteristic peaks of C–O, C–OH, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibrations, confirming the successful oxidation of GO.36 The C/O ratio calculated from X-ray photoelectron spectroscopy (XPS) (Fig. S3, ESI†) is about 7/3, which indicates a medium degree of oxidation. The four deconvoluted peaks in Fig. 1c show the four kinds of carbon bonds and their percentage, which is a good indication of epoxy, carboxylic and hydroxyl oxygen on the GO. In addition, the X-ray diffraction (XRD) curves in Fig. 1d demonstrate the interlayer spacing were expanded from graphite 0.34 nm to graphene oxide 0.86 nm. All the pieces of evidence confirm the successful exfoliation and oxidation of GO.
O vibrations, confirming the successful oxidation of GO.36 The C/O ratio calculated from X-ray photoelectron spectroscopy (XPS) (Fig. S3, ESI†) is about 7/3, which indicates a medium degree of oxidation. The four deconvoluted peaks in Fig. 1c show the four kinds of carbon bonds and their percentage, which is a good indication of epoxy, carboxylic and hydroxyl oxygen on the GO. In addition, the X-ray diffraction (XRD) curves in Fig. 1d demonstrate the interlayer spacing were expanded from graphite 0.34 nm to graphene oxide 0.86 nm. All the pieces of evidence confirm the successful exfoliation and oxidation of GO.
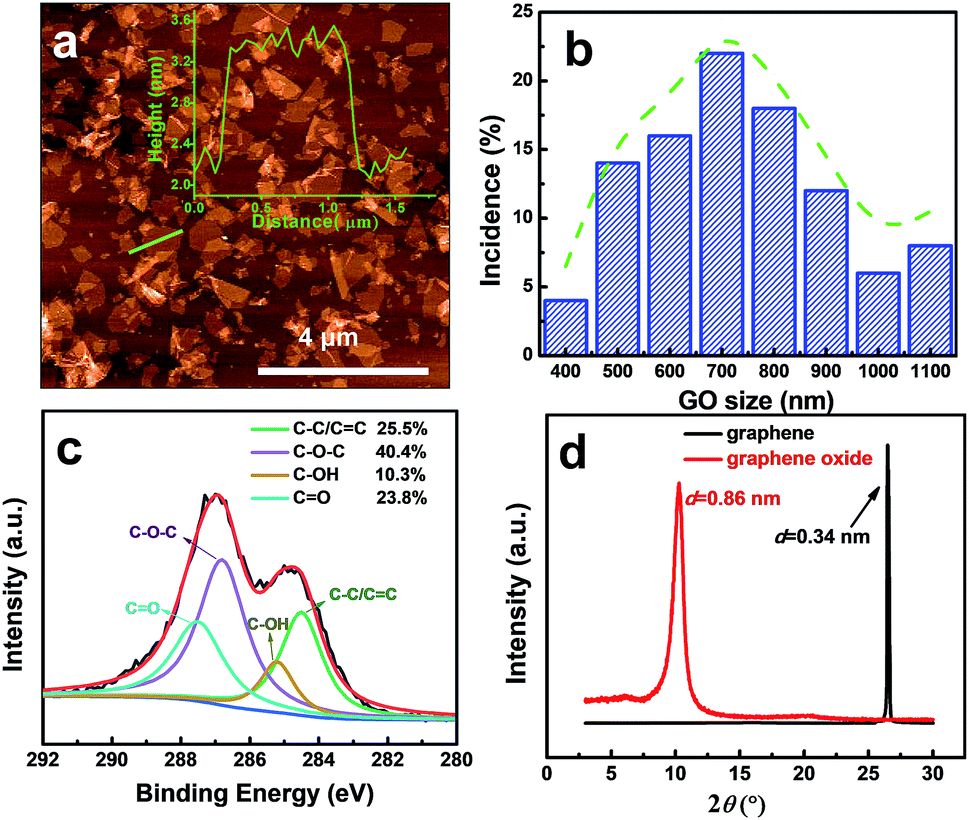 | ||
| Fig. 1 Characterization of graphene oxide. (a) AFM image of the synthesized GO sheets. The inset shows the corresponding height profile of the green line traced on the AFM image. (b) Size distribution of the GO sheets. (c) XPS (C1s) of graphene oxide. (d) XRD of graphene and graphene oxide powder. | ||
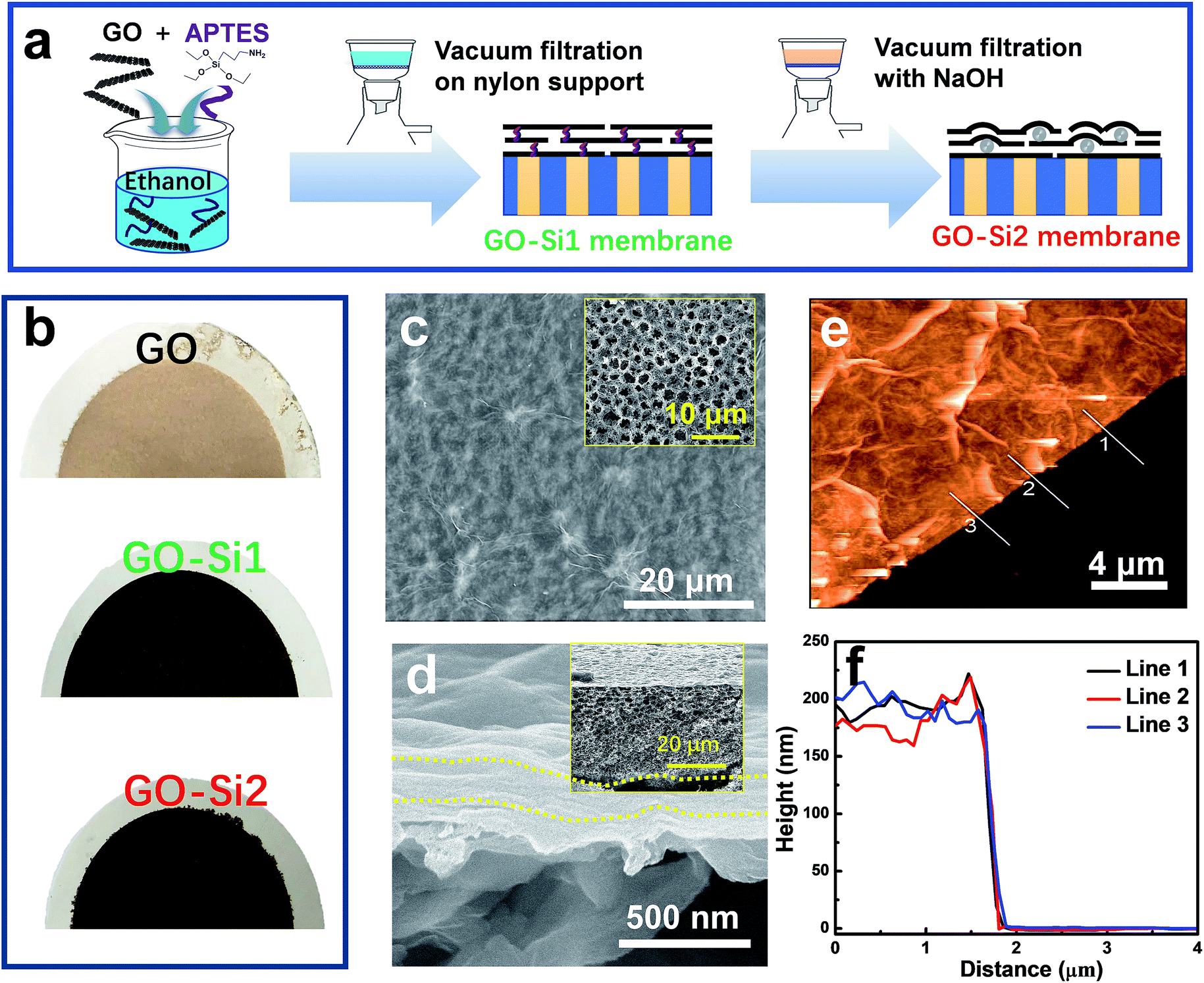 | ||
| Fig. 2 Dual-spacing channel GO membranes intercalated with silica nanoparticles. (a) Preparation procedure. (b) Photograph of different membranes. (c) SEM image of the GO–Si2 membrane surface. The inset shows the surface of the nylon support. (d) Cross-sectional SEM image of the GO–Si2 membrane, prepared on the nylon support. The inset shows a lower magnification of the whole membrane cross-section. (e) AFM image of the GO–Si2 membrane. (f) The corresponding height profile of (e). | ||
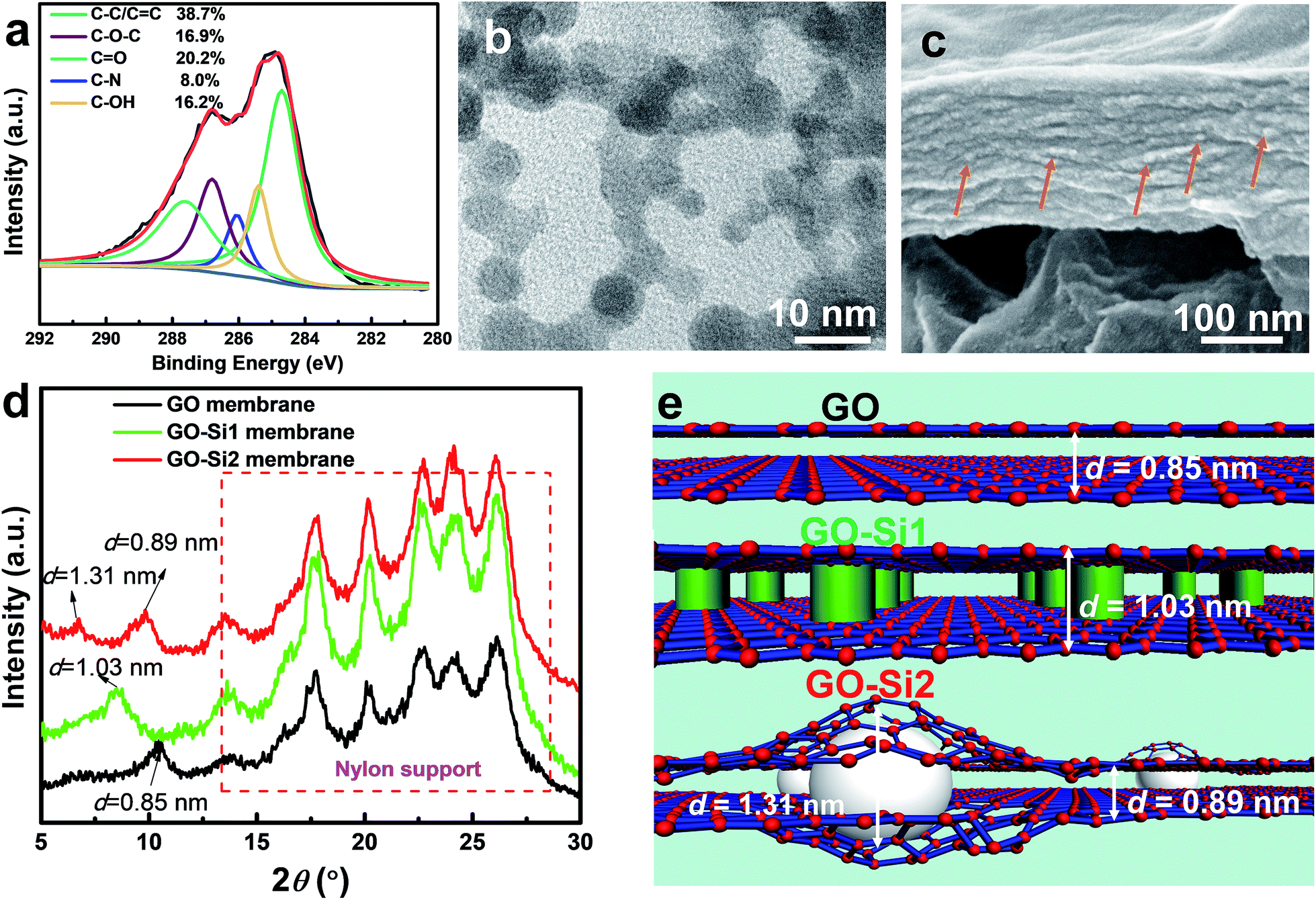 | ||
| Fig. 3 Chemical and physical structures of the membranes. (a) XPS (C 1s), (b) TEM image from the top surface, (c) cross sectional SEM image of GO–Si2 membranes. The orange arrows denote the SiO2 nanoparticles. (d) XRD diffractogram of the membranes, showing the different interlayer d-spacings. Peaks from 13 to 26° are from the nylon support membrane. (e) Schematic diagram illustrating the different GO membrane structures. | ||
The membranes were prepared by the simple and commonly used vacuum filtration method (Fig. 2a). In an optimized procedure, we mixed a dilute concentration of GO (0.01 mg mL−1) with (3-aminopropyl)triethoxysilane (APTES). In the mixtures, APTES may attach on the GO through hydrogen bonding. Ethanol, rather than water, was applied as the dispersing solvent to avoid the APTES hydrolysis. The mixture was filtered through a highly porous nylon support with an average pore size of 0.22 μm. The membranes, referred to here as GO–Si1, have the APTES uniformly distributed through all GO interlayer nanochannels. By further treatment with NaOH aqueous solution, the GO–Si2 membranes were obtained. In this step, the SiO2 nanoparticles should be produced between the GO layers, by hydrolysis and condensation reactions. APTES modified GO has been reported in applications, such as thermally conductive materials,38 reinforcing components,39 and gas separation membranes.40 In our case, APTES modification could greatly improve the mechanical stability of the membranes. Moreover, APTES works as a silica precursor that induces the in situ formation of nanoparticles. The photographs of different membranes are shown in Fig. 2b, in which pristine GO membranes show a light brown color, the GO–Si1 and GO–Si2 membranes exhibit a darker color. The pristine nylon support has a highly porous structure (Fig. 2c inset and Fig. S4 in the ESI†). With a GO loading of 440 mg m−2, the nylon support is uniformly covered without defects (GO–Si2 in Fig. 2c). The intercalation of APTES and the further treatment do not affect the integrity of the GO membranes (Fig. S5a–h, ESI†). For some characterization, we also prepared membranes by using anodic aluminum oxide (AAO) membranes as the support. As seen in the photograph in Fig. S6 in the ESI,† due to the low thickness of the GO layer, the membrane is transparent. The SEM cross-sectional image of the composite membranes display a laminar structure with a thickness of 180 nm (Fig. 1d), which can be confirmed by the AFM height profile in Fig. 1e and f. The thickness could be feasibly tuned from less than 100 nm to larger than 1 μm, by increasing the GO loading amount on the support (Fig. S7a–e, ESI†).
As for the chemical structures, based on the FTIR spectrum in Fig. S8 in the ESI,† the newly formed C–Si (1354 cm−1) and Si–O–Si (1040 cm−1 and 860 cm−1) bonds reveal the chemical functionalization of APTES on GO. The XPS analysis was conducted to confirm the completion of the reactions to form the GO–Si2 membrane (Fig. 3a; S9, ESI†). The curve of the GO–Si2 membrane has N and Si peaks at 401.3 eV and 102.8 eV, respectively. From the deconvoluted C 1s peaks (Fig. 3c), the epoxy C–O ratio is considerably reduced compared with that of pure GO. Therefore, under alkaline conditions, these silane moieties are grafted onto the GO sheets via the SN2 nucleophilic displacement reaction between epoxy groups of GO and amino moieties of APTES.39,41
For the physical structures, the AFM membrane images in Fig. S10† display a wrinkled surface, which can be attributed to the in-plane interactions of GO nanosheets during stacking.42,43 By magnifying the flat region of the membranes, it can be observed that the GO and GO–Si1 membranes have a smooth morphology. In contrast, the surface of the GO–Si2 membrane is decorated with ultra-small nanoparticles. The size of the nanoparticles was further estimated by transmission electron microscopy (TEM), for which the samples were prepared by peeling off the sample from an AAO support and transferring a thin (∼60 nm) GO–Si2 film onto a copper grid. As seen from Fig. 3b, from the top view of the membrane, the particle size is about 3–5 nm. The difference in the nanoparticle darkness is attributed to the different layer location in GO multilayers. The overlap of nanoparticles in different layers may indicate an apparent aggregation. TEM and SEM images in Fig. S11 in the ESI† further confirm this. The high-resolution cross-sectional SEM image of the GO–Si2 membrane displays scattered white dots, which are attributed to the intercalated SiO2 nanoparticles. These pieces of evidence prove the successful formation of sub-5 nm SiO2 nanoparticles between the interlayers. The Si mapping performed by EDS on the membrane surface indicates that there is no aggregation of SiO2 particles (Fig. S12, ESI†). Compared with the pristine brown-colored GO membrane in Fig. 2b, the GO–Si1 and GO–Si2 membranes are clearly darker. This is an indication of the progress of GO reduction.44 The partial reduction of GO can be confirmed by analyzing the deconvoluted part of the C1s peak in the XPS spectra that corresponds to C–O–C. The C–O–C ratio decreases from 40.4% in the pristine GO to 16.9% in the GO–Si2 membranes. During the membrane preparation, the APTES molecules are first uniformly distributed, presumably occupying the GO interlayer spaces. With the NaOH solution treatment, two kinds of reaction take place: the APTES grafting on the original GO epoxy sites and the sol–gel reaction around the grafted sites to form the SiO2 nanoparticles. Since the interlayer space is limited to only 1–2 nm, the formed SiO2 particles are smaller than other intercalated membranes. In addition to their role in expanding the layers, the intercalated SiO2 nanoparticles are covalently bonded to the epoxy groups in different GO layers and act as crosslinkers, bringing stability to the membranes.
The interlayer d-spacings govern the separation performances of the membranes. The diffraction peaks were obtained by XRD and the d-spacing further calculated using the Bragg's law. As shown in Fig. 3d, the peaks ranging from 13 to 26° are relative to those of nylon support membranes.45 The XRD pattern of the unmodified GO membrane indicates a single d-spacing of 0.85 nm. After treating with APTES (GO–Si1 membrane), the high content of APTES helped to expand the single interlayer d-spacing to 1.03 nm, while the XRD pattern of the GO–Si2 membrane indicates a dual d-spacing, with peaks corresponding to d values of 1.31 and 0.89 nm. Although there have been dual d-spacing reports for GO membranes before,27,46 they refer to regions with a distinct degree of partial reduction. In most of the cases, single d-spacings are observed in the nanoparticle intercalated GO membranes6,9,26 relative to narrow original GO interlayers or enlarged modified ones. The dual peaks in our study should be assigned to the different flake regions with and without nanoparticles. By considering the low atomic ratio of Si (∼4%), the number of nanoparticles in the membrane is quite small. This explains the low peak intensity at d = 1.31 nm. Since the uncrosslinked APTES is washed away (confirmed by the lower Si content in GO–Si2 compared with that of the GO–Si1 membrane), the silica is formed only around scattered GO epoxy sites. Due to the flexible nature of GO nanosheets,35,47 sections of the pristine GO interlayer are preserved with the d-spacing increasing only slightly to 0.89 nm (Fig. 3e). Therefore, the result is a hierarchical structure containing 2D channels with dual interlayer spacings. The change in d-spacing also contributes to different total membrane thicknesses with the same GO loading (Fig. S13, ESI†). The pristine GO membrane exhibits a thickness of 170 nm. With localized spacers, the thickness of the GO–Si2 membrane slightly increased to 180 nm, while the GO–Si1 membrane with uniformly distributed molecular spacers has a much thicker GO layer of 216 nm. The requirements to achieve such a dual-channel structure are schematically illustrated in Fig. S14.† Besides these factors, the in situ method helps to preserve the membrane structure, which leads to the different structure from other particle intercalated membranes.
Having the intercalated nanoparticles in the interlayer channels could potentially block molecular pathways through the channels. To check whether the intercalated nanoparticles could hinder any molecular transport, the permeances of the single gases, N2, CH4 and CO2, through the membranes were evaluated (Table S1, ESI†). Due to the expanded channel sizes, the GO–Si2 membrane has almost two-fold higher gas permeance than the pristine GO membrane. This indicates that the presence of SiO2 nanoparticles creates more free space around the nanoparticles rather than blocking some pathways for molecular transport. The membrane performance for liquid separation was then investigated. The water permeances and rose bengal rejections (Fig. 4a) were tested. Due to the uniform stacking and small interlayer spacing, the pristine GO membrane exhibits a water permeance of 18.7 L m−2 h−1 bar−1 and a rose bengal rejection of 99.3%. After intercalating APTES, the channel expands and the water permeance increases to 225 L m−2 h−1 bar−1, whereas the rose Bengal rejection decreases to 48.6%. Following Geim and co-workers,48 we used the Hagen–Poiseuille equation (eqn (3)) for 2D laminar membranes to explain the permeation increase:
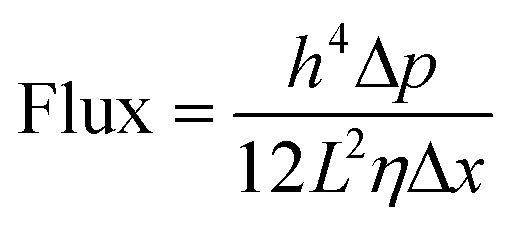 | (3) |
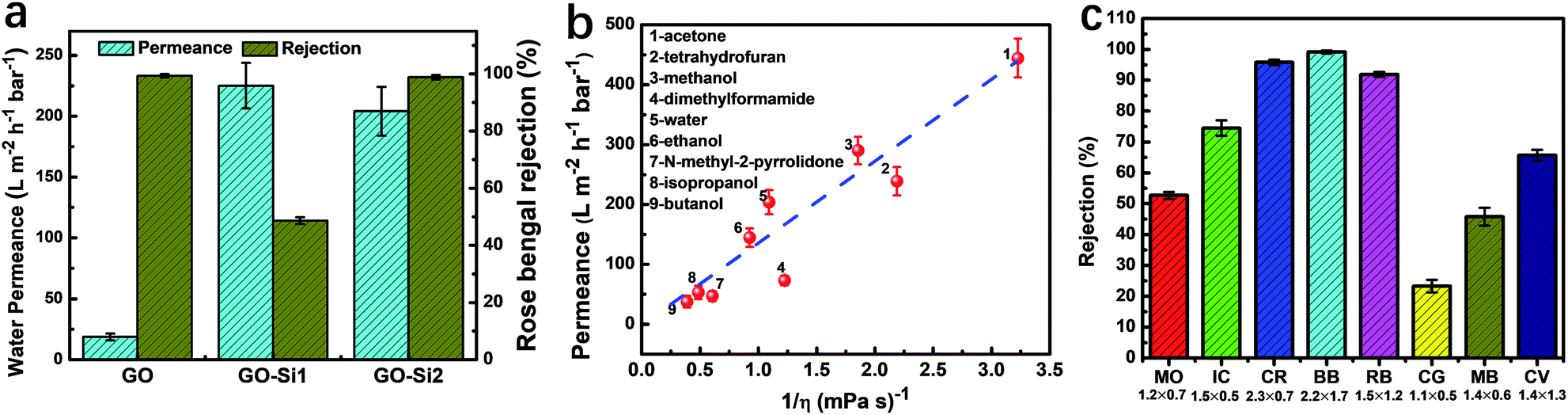 | ||
| Fig. 4 Membrane nanofiltration performance. (a) Water permeance and rose bengal rejection of GO, GO–Si1 and GO–Si2 membranes. (b) Pure solvent permeance as a function of their inverse viscosity for GO–Si2 membranes. The used solvents are numbered and named on the left. The blue dashed line indicates the hypothetical linear relationship. (c) Dye rejection of the GO–Si2 membranes. The dyes were dissolved in methanol. The dyes used were methyl orange (MO), indigo carmine (IC), Congo red (CR), brilliant blue R250 (BB), rose bengal (RB), chrysoidine G (CG), methylene blue (MB), and crystal violet (CV). The corresponding sizes (in nm) are mentioned under their abbreviations. | ||
By taking advantage of the high stability of GO, our goal is to use the dual-spacing channel GO–Si2 membranes for separations in organic solvents. The permeances of various solvents were evaluated by using a dead-end filtration setup under a pressure of 1 bar. In Fig. 4b, we plot the permeance of different solvents as a function of the reciprocal of the viscosity (η). For most solvents, as the viscosity decreases, the GO–Si2 membrane exhibits an increase of permeance. Notably, the membrane has an acetone permeance of 444 L m−2 h−1 bar−1 and a methanol permeance of 290 L m−2 h−1 bar−1, which are quite high values compared with those of the pristine GO and other types of membranes.21,45,51 There is a linear correlation between the methanol permeance and 1/thickness of the GO layer in the GO–Si2 membrane (Fig. S15, ESI†), indicating the uniform structure along the thickness axis. Unlike the GO membrane reported by Nair and co-workers,21 for which the solvent permeances are linearly dependent on 1/η, our GO–Si2 membrane shows some deviation of the linear correlations (the dashed line in Fig. 4b). The solvent properties including the viscosity, relative polarity, kinetic diameter, and total Hansen solubility parameter are listed in Table S2 in the ESI.† In the GO–Si2 membrane, the SiO2 nanoparticles are distributed in the membrane channels with abundant hydrophilic –OH groups. The interaction with hydrophilic or polar solvents is therefore favored.52 Therefore, we assume that while the viscosity plays the predominant role, the relative polarity also contributes to the solvent permeance. This assumption can be proved by the lower permeance of low polarity solvents, such as tetrahydrofuran, dimethylformamide and N-methyl-2-pyrrolidone, being located under the dashed line, while highly polar solvents, such as methanol, water and ethanol, have permeance values well above the dashed line. In this regard, the hydrophilic nanoparticle domains act like a pump that efficiently gathers the polar molecules and promotes the fast transport through the hydrophobic graphite channel sectors. For a channel with uniform chemical potential, the single spacing channel could achieve high permeance and rejection for molecular separations.22 But for 2D materials with non-uniform chemical potential, such as GO, the synergy of solution and diffusion is more effective in achieving high molecular transport.
Besides the permeance of solvents, molecular selectivity is essential for the application in nanofiltration. To evaluate the membrane ability to reject low molecular weight solutes, the rejection of different dyes was evaluated in methanol, a commonly applied organic solvent for pharmaceuticals. Both positively and negatively charged dyes with different molecular sizes were tested. The chemical structure, molecular weight and molecular dimensions of the dyes are displayed in Fig. S16,† as estimated using a Molecular Mechanics 2 method in Chem3D. The values agree with other previous reports11,53,54 and have slight differences with those reported by Shi et al.45 The dye rejections are plotted in Fig. 4c, and the corresponding sizes are mentioned below. Colored feed and permeate samples are shown in Fig. S17 in the ESI† and the corresponding UV-vis spectra are shown in Fig. S18.† Theoretically, three major phenomena could contribute to the dye rejection: absorption on the membrane surface, physical sieving by the 2D nanochannels, and electrostatic interactions. In all rejection tests, we used 80 mL of dye solutions with a concentration of 100 ppm (Cf) as feed. After filtering 40 mL, we measured the concentration of both the permeate and retentate, Cp and Cr, respectively. In Table S3,† we calculated the expected theoretical Cr/Cf values to achieve the mass balance, assuming that the dyes do not absorb on the membranes. For both positively and negatively charged dyes the actual and theoretical values are quite close, indicating low dye absorption. Also, the thin membrane thickness will allow only small amount absorption. The channel size is therefore the predominant parameter to achieve the dye rejection.
The GO–Si2 membrane rejects more than 90% of large molecules, such as brilliant blue R250 (BB, 2.2 nm × 1.7 nm, 99.2% rejection), Congo red (CR, 2.3 nm × 0.7 nm, 95.8% rejection) and rose bengal (RB, 1.5 nm × 1.2 nm, 91.9% rejection). In contrast, the rejection of dyes smaller than 1.5 nm is quite low. For instance, the rejection of chrysodine G (CR) with a size of 1.1 nm × 0.5 nm is only 23.3%. For methylene blue (MB) with a size of 1.4 nm × 0.6 nm, the rejection is 45.8%, and for crystal violet (CV, 1.4 nm × 1.3 nm) it is just below 70%. Therefore, the channels in the GO–Si2 membrane are highly size-selective, with a sharp limit for permeation of dyes only smaller than 1.5 nm. Nevertheless, charge is also an important factor in this size range. For the much higher rejection of rose bengal, compared to crystal violet of similar size, the negatively charged carboxylic groups46 might play an auxiliary role, leading to the dye repulsion by the negatively charged GO–Si2 membrane.
By considering the permeance and rejection properties, the alternating dual-spacing channel structure of the GO–Si2 membranes, with small-sized GO building blocks, enables an excellent performance for nanofiltration in organic solvents. As illustrated in Fig. 5 and detailed in Table S4 in the ESI,† the performance of our membrane is highly competitive with most GO membranes in the literature, being among the best reported data for organic solvent resistant ones.
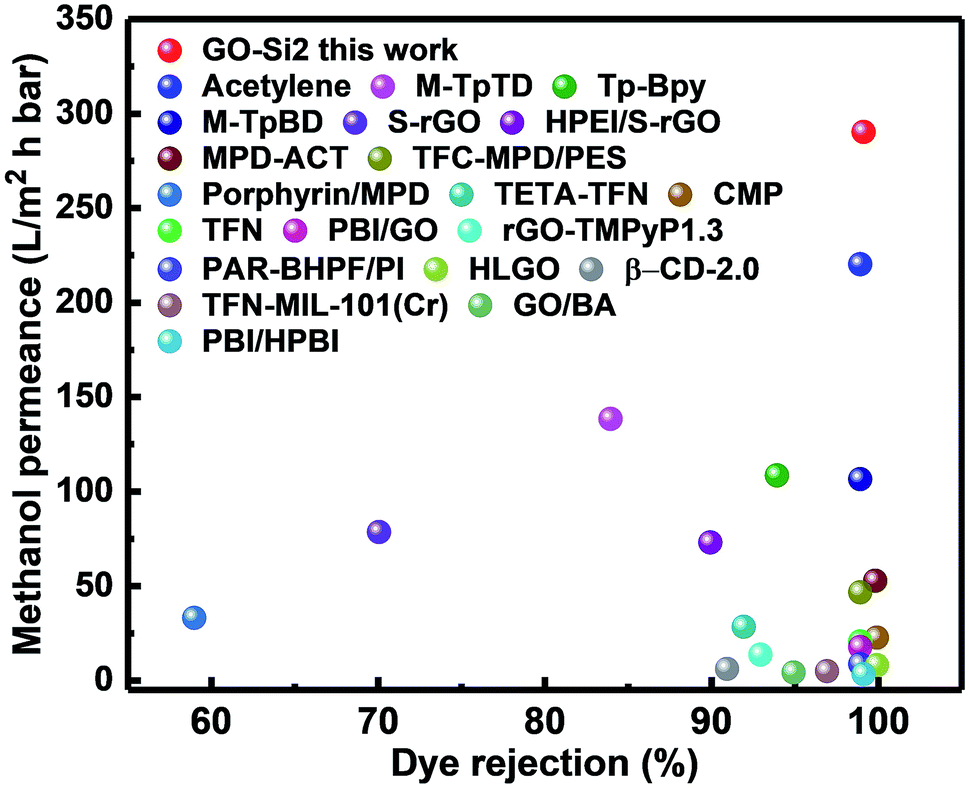 | ||
| Fig. 5 Comparison of the separation performance of the GO–Si2 membrane versus other reported membranes in methanol media. Detailed information can be found in Table S4.† | ||
The stability of membranes in organic solvents is another major issue for OSN membranes. The flux, exemplified for methanol in Fig. 6a, linearly increases with the applied pressure. The permeance, defined by flux/pressure, showed only a slight decrease. This indicates that the membrane is mechanically stable and does not suffer any compaction and the fluidic channels in the GO layer are less affected by pressure, at least up to the tested value of 10 bar. This is quite different from previously reported GO membranes, which frequently suffer an elastic deformation and consequent permeance loss under higher pressure.6,55 The stability is given by the intercalated and cross-linked SiO2 nanoparticles, which help to maintain a stable channel size and makes the GO membrane quite robust. In a long-term test the GO–Si2 membrane demonstrated excellent stability with the different solvent permeances being constant after 72 h operation (Fig. 6b). For the unmodified GO membrane, after 72 h testing, the integrity was partially destroyed. This may be because GO membranes with highly charged surfaces can be easily re-exfoliated in polar solvents,28 while GO–Si2 membranes kept the uniform structure (Fig. S19, ESI†). The stability arises from the cross-linking and partial reduction in the alkaline treatment process, which enhance the covalent force and π–π stacking between adjacent layers. Due to the presence of SiO2 nanoparticles, the membranes may not be stable in high pH solvents, which needs to be addressed in future work. However, bearing the advantages of high permeance, high rejection, and excellent stability, our GO–Si2 membrane shows great potential in OSN.
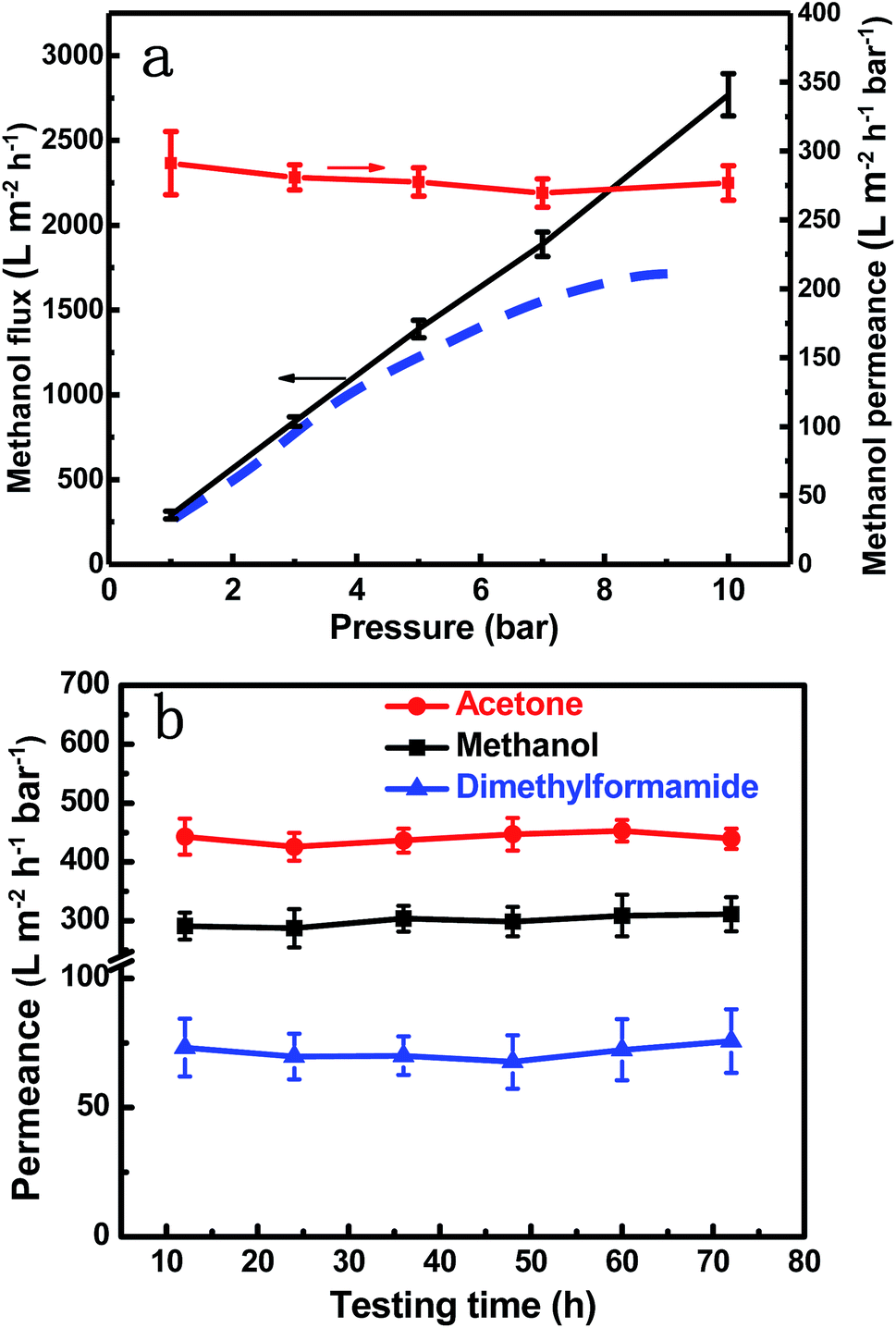 | ||
| Fig. 6 Membrane stability. (a) Methanol flux and permeance of the GO–Si2 membrane at different pressures. The blue dashed line shows the trend for some other GO membranes, in which the flux slowly increases as the pressure increases. (b) Long-term solvent permeance of GO–Si2 membranes. | ||
In our system, we used a vacuum filtration method to obtain flat sheet GO composite membranes. Vacuum filtration is the most common and straightforward route for the large-scale fabrication of GO membranes.56 Besides flat sheet membranes, a more potential scalable configuration may be hollow fiber composite membranes prepared by the vacuum filtration method. The challenges may lie in achieving suitable support surfaces with optimized surface roughness and wettability to obtain a homogeneous and highly ordered laminate GO membrane. And these issues can be resolved by pre-modification on the support membrane surface22 or a more delicate filtration method.51
Conclusions
In summary, we developed a 2D lamellar membrane with alternating dual-spacing nanochannels by intercalating in situ formed SiO2 nanoparticles, which can overcome the well-known permeance-rejection trade-off for nano- and ultrafiltration. The small nanoparticles work as spacers that tune both the physical microenvironment, by expanding the channel spacing, and the chemical microenvironment, by locally providing rich hydrophilic groups, enabling a fast solvent transport. Simultaneously, the channel sectors free of nanoparticles maintain a narrow-sized and hydrophobic path, enabling a high rejection of small molecules. Notably, a high methanol permeance of 290 L m−2 h−1 bar−1, and a higher than 90% rejection of dyes with sizes larger than 1.5 nm was reported for the membrane, making it highly attractive for nanofiltration in pharmaceutical processes. This approach of creating hierarchical channels with tailored physical and chemical microenvironments in 2D lamellar membranes opens a door for the development of highly efficient membranes and other applications, such as adsorption, drug delivery and catalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank King Abdullah University of Science and Technology for the financial support, in particular the Water Desalination and Reuse Center for the grants URF/1/1971-32-599 01 and URF/1/1971-33-01 and OSR for CRG6 grant URF/1/3441-01-01.Notes and references
- P. Marchetti, M. F. Jimenez Solomon, G. Szekely and A. G. Livingston, Chem. Rev., 2014, 114, 10735–10806 CrossRef CAS PubMed.
- L. Cao, X. He, Z. Jiang, X. Li, Y. Li, Y. Ren, L. Yang and H. Wu, Chem. Soc. Rev., 2017, 46, 6725–6745 RSC.
- W. J. Koros and C. Zhang, Nat. Mater., 2017, 16, 289–297 CrossRef CAS PubMed.
- S. Karan, Z. Jiang and A. G. Livingston, Science, 2015, 348, 1347–1351 CrossRef CAS PubMed.
- K. Celebi, J. Buchheim, R. M. Wyss, A. Droudian, P. Gasser, I. Shorubalko, J.-I. Kye, C. Lee and H. G. Park, Science, 2014, 344, 289–292 CrossRef CAS PubMed.
- H. Huang, Z. Song, N. Wei, L. Shi, Y. Mao, Y. Ying, L. Sun, Z. Xu and X. Peng, Nat. Commun., 2013, 4, 2979 CrossRef PubMed.
- R. K. Joshi, P. Carbone, F. C. Wang, V. G. Kravets, Y. Su, I. V. Grigorieva, H. A. Wu, A. K. Geim and R. R. Nair, Science, 2014, 343, 752–754 CrossRef CAS PubMed.
- H. Li, Z. Song, X. Zhang, Y. Huang, S. Li, Y. Mao, H. J. Ploehn, Y. Bao and M. Yu, Science, 2013, 342, 95–98 CrossRef CAS PubMed.
- L. Ding, Y. Wei, Y. Wang, H. Chen, J. Caro and H. Wang, Angew. Chem., Int. Ed., 2017, 56, 1825–1829 CrossRef CAS PubMed.
- L. Ding, Y. Wei, L. Li, T. Zhang, H. Wang, J. Xue, L.-X. Ding, S. Wang, J. Caro and Y. Gogotsi, Nat. Commun., 2018, 9, 155 CrossRef PubMed.
- C. Chen, J. Wang, D. Liu, C. Yang, Y. Liu, R. S. Ruoff and W. Lei, Nat. Commun., 2018, 9, 1902 CrossRef PubMed.
- Y. Wang, L. Li, Y. Wei, J. Xue, H. Chen, L. Ding, J. Caro and H. Wang, Angew. Chem., Int. Ed., 2017, 56, 8974–8980 CrossRef CAS PubMed.
- L. Sun, Y. Ying, H. Huang, Z. Song, Y. Mao, Z. Xu and X. Peng, ACS Nano, 2014, 8, 6304–6311 CrossRef CAS PubMed.
- Y. Peng, Y. Li, Y. Ban, H. Jin, W. Jiao, X. Liu and W. Yang, Science, 2014, 346, 1356–1359 CrossRef CAS PubMed.
- J. Zhu, J. Hou, A. Uliana, Y. Zhang, M. Tian and B. Van der Bruggen, J. Mater. Chem. A, 2018, 6, 3773–3792 RSC.
- F. Fei, L. Cseri, G. Szekely and C. F. Blanford, ACS Appl. Mater. Interfaces, 2018, 10, 16140–16147 CrossRef CAS PubMed.
- S. P. Surwade, S. N. Smirnov, I. V. Vlassiouk, R. R. Unocic, G. M. Veith, S. Dai and S. M. Mahurin, Nat. Nanotechnol., 2015, 10, 459–464 CrossRef CAS PubMed.
- S. P. Koenig, L. Wang, J. Pellegrino and J. S. Bunch, Nat. Nanotechnol., 2012, 7, 728 CrossRef CAS PubMed.
- Y. Xia, T. S. Mathis, M. Q. Zhao, B. Anasori, A. Dang, Z. Zhou, H. Cho, Y. Gogotsi and S. Yang, Nature, 2018, 557, 409–412 CrossRef CAS PubMed.
- B. Mi, Science, 2014, 343, 740–742 CrossRef CAS PubMed.
- Q. Yang, Y. Su, C. Chi, C. Cherian, K. Huang, V. Kravets, F. Wang, J. Zhang, A. Pratt and A. Grigorenko, Nat. Mater., 2017, 16, 1198 CrossRef CAS PubMed.
- J. Wang, P. Chen, B. Shi, W. Guo, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2018, 57, 6814–6818 CrossRef CAS PubMed.
- S. Wang, Y. Xie, G. He, Q. Xin, J. Zhang, L. Yang, Y. Li, H. Wu, Y. Zhang, M. D. Guiver and Z. Jiang, Angew. Chem., Int. Ed., 2017, 56, 14246–14251 CrossRef CAS PubMed.
- Z. Zhang, N. Li, Y. Sun, H. Yang, X. Zhang, Y. Li, G. Wang, J. Zhou, L. Zou and Z. Hao, ACS Appl. Mater. Interfaces, 2018, 10, 27205–27214 CrossRef CAS PubMed.
- Y. Han, Y. Jiang and C. Gao, ACS Appl. Mater. Interfaces, 2015, 7, 8147–8155 CrossRef CAS PubMed.
- M. Zhang, K. Guan, J. Shen, G. Liu, Y. Fan and W. Jin, AIChE J., 2017, 63, 5054–5063 CrossRef CAS.
- K. H. Thebo, X. Qian, Q. Zhang, L. Chen, H.-M. Cheng and W. Ren, Nat. Commun., 2018, 9, 1486 CrossRef PubMed.
- T. Gao, H. Wu, L. Tao, L. Qu and C. Li, J. Mater. Chem. A, 2018, 6, 19563–19569 RSC.
- X. Tang, Y. Qu, S.-L. Deng, Y.-Z. Tan, Q. Zhang and Q. L. Liu, J. Mater. Chem. A, 2018, 6, 22590–22598 RSC.
- K. Murata, K. Mitsuoka, T. Hirai, T. Walz, P. Agre, J. B. Heymann, A. Engel and Y. Fujiyoshi, Nature, 2000, 407, 599–605 CrossRef CAS PubMed.
- R. Taheri, A. Razmjou, G. Szekely, J. Hou and G. R. Ghezelbash, Bioinspiration Biomimetics, 2016, 11, 041001 CrossRef PubMed.
- I. Rose, C. G. Bezzu, M. Carta, B. Comesaña-Gándara, E. Lasseuguette, M. C. Ferrari, P. Bernardo, G. Clarizia, A. Fuoco and J. C. Jansen, Nat. Mater., 2017, 16, 932 CrossRef CAS PubMed.
- J. Shen, G. Liu, Y. Ji, Q. Liu, L. Cheng, K. Guan, M. Zhang, G. Liu, J. Xiong and J. Yang, Adv. Funct. Mater., 2018, 28, 1801511 CrossRef.
- A. Akbari, S. E. Meragawi, S. T. Martin, B. Corry, E. Shamsaei, C. D. Easton, D. Bhattacharyya and M. Majumder, ACS Appl. Mater. Interfaces, 2018, 10, 2067–2074 CrossRef CAS PubMed.
- J. Kim, L. J. Cote and J. Huang, Acc. Chem. Res., 2012, 45, 1356–1364 CrossRef CAS PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- P. Sun, F. Zheng, M. Zhu, Z. Song, K. Wang, M. Zhong, D. Wu, R. B. Little, Z. Xu and H. Zhu, ACS Nano, 2014, 8, 850–859 CrossRef CAS PubMed.
- X. Pu, H.-B. Zhang, X. Li, C. Gui and Z.-Z. Yu, RSC Adv., 2014, 4, 15297–15303 RSC.
- H. Yang, F. Li, C. Shan, D. Han, Q. Zhang, L. Niu and A. Ivaska, J. Mater. Chem., 2009, 19, 4632–4638 RSC.
- J. Zhang, Q. Xin, X. Li, M. Yun, R. Xu, S. Wang, Y. Li, L. Lin, X. Ding and H. Ye, J. Membr. Sci., 2019, 570, 343–354 CrossRef.
- S. Wang, P. J. Chia, L. L. Chua, L. H. Zhao, R. Q. Png, S. Sivaramakrishnan, M. Zhou, R. G. S. Goh, R. H. Friend and A. T. S. Wee, Adv. Mater., 2008, 20, 3440–3446 CrossRef CAS.
- L. J. Cote, J. Kim, Z. Zhang, C. Sun and J. Huang, Soft Matter, 2010, 6, 6096–6101 RSC.
- X. Shen, X. Lin, N. Yousefi, J. Jia and J.-K. Kim, Carbon, 2014, 66, 84–92 CrossRef CAS.
- H. Liu, H. Wang and X. Zhang, Adv. Mater., 2015, 27, 249–254 CrossRef CAS PubMed.
- L. Huang, J. Chen, T. Gao, M. Zhang, Y. Li, L. Dai, L. Qu and G. Shi, Adv. Mater., 2016, 28, 8669–8674 CrossRef CAS PubMed.
- Y. Han, Z. Xu and C. Gao, Adv. Funct. Mater., 2013, 23, 3693–3700 CrossRef CAS.
- Y. Liu, Y. Su, J. Guan, J. Cao, R. Zhang, M. He, K. Gao, L. Zhou and Z. Jiang, Adv. Funct. Mater., 2018, 28, 1706545 CrossRef.
- R. Nair, H. Wu, P. Jayaram, I. Grigorieva and A. Geim, Science, 2012, 335, 442–444 CrossRef CAS PubMed.
- G. Liu, W. Jin and N. Xu, Chem. Soc. Rev., 2015, 44, 5016–5030 RSC.
- W. Wang, E. Eftekhari, G. Zhu, X. Zhang, Z. Yan and Q. Li, Chem. Commun., 2014, 50, 13089–13092 RSC.
- F. Zhou, H. N. Tien, W. L. Xu, J. T. Chen, Q. Liu, E. Hicks, M. Fathizadeh, S. Li and M. Yu, Nat. Commun., 2017, 8, 2107 CrossRef PubMed.
- Z. Yuan, X. Wu, Y. Jiang, Y. Li, J. Huang, L. Hao, J. Zhang and J. Wang, J. Membr. Sci., 2018, 549, 1–11 CrossRef CAS.
- S. Kandambeth, B. P. Biswal, H. D. Chaudhari, K. C. Rout, S. Kunjattu H, S. Mitra, S. Karak, A. Das, R. Mukherjee and U. K. Kharul, Adv. Mater., 2017, 29, 1603945 CrossRef PubMed.
- P.-Y. Du, H. Li, X. Fu, W. Gu and X. Liu, Dalton Trans., 2015, 44, 13752–13759 RSC.
- L. Sun, H. Huang and X. Peng, Chem. Commun., 2013, 49, 10718–10720 RSC.
- G. Liu, W. Jin and N. Xu, Angew. Chem., Int. Ed., 2016, 55, 13384–13397 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ta10872b |
| This journal is © The Royal Society of Chemistry 2019 |