
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOperando EXAFS study reveals presence of oxygen in oxide-derived silver catalysts for electrochemical CO2 reduction†
Nienke J.
Firet
 a,
Marijn A.
Blommaert
a,
Thomas
Burdyny
a,
Anirudh
Venugopal
a,
Divya
Bohra
a,
Alessandro
Longo
*bc and
Wilson A.
Smith
*a
a,
Marijn A.
Blommaert
a,
Thomas
Burdyny
a,
Anirudh
Venugopal
a,
Divya
Bohra
a,
Alessandro
Longo
*bc and
Wilson A.
Smith
*a
aMaterials for Energy Conversion and Storage (MECS), Department of Chemical Engineering, Faculty of Applied Sciences, Delft University of Technology, van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: w.smith@tudelft.nl
bNetherlands Organization for Scientific Research (NWO), The European Synchrotron Radiation Facility (ESRF), CS40220, 38043 Grenoble Cedex 9, France. E-mail: alessandro.longo@esrf.fr
cISMN-CNR, UOS Palermo, Via Ugo La Malfa, 153, 90146 Palermo, Italy
First published on 10th January 2019
Abstract
Electrocatalysis of carbon dioxide can provide a valuable pathway towards the sustainable production of chemicals and fuels from renewable electricity sources. One of the main challenges to enable this technology is to find suitable electrodes that can act as efficient, stable and selective CO2 reduction catalysts. Modified silver catalysts and in particular, catalysts electrochemically derived from silver-oxides, have shown great promise in this regard. Here, we use operando EXAFS analysis to study the differences in surface composition between a pure silver film and oxide-derived silver catalysts – a nanostructured catalyst with improved CO2 reduction performance. The EXAFS analysis reveals the presence of trace amounts of oxygen in the oxide-derived silver samples, with the measured oxygen content correlating well with experimental studies showing an increase in CO2 reduction reactivity towards carbon monoxide. The selectivity towards CO production also partially scales with the increased surface area, showing that the morphology, local composition and electronic structure all play important roles in the improved activity and selectivity of oxide-derived silver electrocatalysts. Earlier studies based on X-ray photoelectron spectroscopy (XPS) were not able to identify this oxygen, most likely because in ultra-high vacuum conditions, silver can self-reduce to Ag0, removing existing oxygen species. This operando EXAFS study shows the potential for in situ and operando techniques to probe catalyst surfaces during electrolysis and aid in the overall understanding of electrochemical systems.
Introduction
In recent years, several research efforts have been made to expedite the implementation of renewable energy into society. While renewable electricity can be generated at large scales through solar and wind energy with continually decreasing costs, challenges remain in effectively utilizing the intermittent electricity that is generated. Storing energy in the form of chemicals is one means of using this electricity that may both increase its value and provide an energy-dense transportable product. The conversion of renewable electricity into carbon-based chemicals and fuels via electrochemical CO2 reduction can directly help both these efforts.Depending on the composition and structure of the catalyst used in the electrochemical CO2 reduction reaction, the product formed from CO2 can be tuned towards different carbon-based compounds. In particular, d-band metals such as gold, silver and zinc are suitable for the selective production of CO, whereas copper binds the CO reaction intermediate more strongly, enabling further proton/electron transfers that drive the formation of higher order products.1 Among these metals, silver is a promising candidate for the industrial synthesis of CO since it has a good balance between its relatively low cost and reasonable performance and stability.2 To improve the performance of silver, a number of modified silver catalysts have been investigated which show significantly improved selectivity, activity, onset potential and stability versus their pure metallic counterpart. Strategies to improve the catalytic performance of silver have included doping with p-block elements and various nanostructuring approaches, among others.3–5 Among these promising candidates are oxide-derived silver (ODAg) catalysts.6 During the fabrication of oxide-derived silver, an anodic pulse is applied to the silver electrode, creating an oxide layer on top of the bulk metal. During CO2 reduction, the silver oxide layer at the surface rapidly reduces back to metallic silver. For other metals, such as gold, copper and lead, oxide-derived techniques have also demonstrated improved performance metrics including increased CO2 reduction selectivity and reduced overpotentials required to reach >1 mA cm−2.7–9 These treatments result in highly-porous electrode surfaces which impact the mass transport of reactants and products while altering the local pH and buffering abilities of the electrolyte near the catalyst's surface. Other research has suggested that the intrinsic activity of the oxide-derived metals are affected by sub-surface oxides10,11 or new coordination sites12 due to the formation of preferably-oriented surfaces and defect sites that form as a result of the OD treatment.
In the case of silver, a higher local pH near the electrode surface is further thought to influence the selectivity for CO formation over that of hydrogen.6,13 When mass transport is limited by the porous nature of the electrode, hydroxide ions (OH−) that are formed as a result of CO2 and H2O reduction accumulate near the surface resulting in a higher local pH than that of a flat surface. While a high local pH favours the CO2 reduction reaction, it simultaneously suppresses hydrogen evolution, providing an avenue to improve product selectivity on roughened nanoporous silver electrodes.
Apart from the changed electrolyte transport dynamics at the catalysts' surface, an alteration in the material properties as a result of the oxide-derived treatment, such as electronic, structural or compositional changes, can also give rise to the improved catalytic properties. Since oxide-derived silver catalysts are formed from a silver oxide, the potential presence of traces of oxygen and its role in improving the CO2 reduction reaction were proposed in several papers.10,11 If present, oxygen could help stabilize the CO2 molecule when it arrives at the electrode surface, and thus may contribute to the enhanced selectivity towards CO2 reduction. Since the rate-determining step for CO2 reduction to CO on silver is the adsorption of CO2, reducing the overpotential of this reaction step is then very important for increasing the overall efficiency and activity of the reaction.
Controversy exists regarding this hypothesis, however, as other studies using surface-characterization techniques found no traces of oxygen in oxide-derived silver catalysts.12 It is important to emphasize that many techniques capable of measuring trace amounts of elements, such as XPS, do so under high vacuums, which can remove oxygen from a silver oxide species. Other techniques such as Kelvin probe force microscopy (KPFM) that measure the work function can only give an indication as to whether trace amounts of oxygen might exist, but do not provide definitive proof or indications of how oxygen may be incorporated into the Ag catalyst. In situ and operando techniques are therefore needed to probe the catalyst's electronic, compositional, and atomic structure during electrolysis to distinguish any surface related affects that may lead to improved performance, from the beneficial effects caused by the nanostructured morphology and high local pH.
In this work, we use a combination of operando extended X-ray absorption fine structure (EXAFS), electrochemical methods, DFT calculations and ex situ characterization such as grazing incidence X-ray diffraction (GIXRD) to examine the surface properties and catalytic activity of both silver and oxide-derived silver catalysts for electrochemical CO2 reduction. We find a correlation between the faradaic efficiency for CO formation with the measured concentration of oxygen in the silver lattice. These results suggest that the improved performance for oxide-derived silver catalysts may arise from a combination of increased activity of Ag induced by the presence of surface oxygen, and from a high local pH created through the nanostructured morphology. From these results, we propose several mechanisms via which the inclusion of atomic oxygen near the surface can aid the CO2 reduction reaction over silver electrodes. In operando EXAFS data shows how vital in situ and operando techniques are to properly understand electrochemical systems.
Results and discussion
Catalyst characterization
Oxide-derived silver catalysts were fabricated according to an earlier published method (Fig. 1).6 All experimental details are presented in the ESI.† From electrochemical measurements, the ODAg catalyst has a lower onset potential, higher CO selectivity, higher activity (current density at a given potential) and improved stability compared to silver foil catalysts (Fig. S2a and b†). To make ODAg, a Ag foil is strongly oxidised by an oxidation/reduction-cycling treatment in an alkaline electrolyte. This treatment produces a silver oxide that quickly reduces back to metallic silver once a reducing potential is applied to perform CO2 reduction. The initial few seconds of the CO2 reduction experiment thus produces the final oxide-derived silver catalyst. In Fig. 1a, the progression of the oxide-derived treatment is shown schematically (Ag foil, silver oxide and ODAg). We will denote the oxide precursor as AgOx, since this oxide consists of a mixture of Ag2O, AgO and Ag as appears from the XRD pattern (Fig. S3†). | ||
| Fig. 1 (a) Fabrication scheme of oxide-derived silver (ODAg), silver is denoted by green, silver oxide by dark orange. SEM images show how the surface morphology changes drastically when (b) silver is (c) oxidised to AgOx and (d) reduced to form ODAg. | ||
To understand the underlying reasons for the difference in CO2 reduction performance between Ag and ODAg, a number of materials characterization techniques were performed. While the morphology of ODAg (Fig. 1b and d) is vastly different than that of Ag foil, the surface composition as measured by XPS is similar for the two catalysts (Fig. 2a–d). XPS spectra were recorded ex situ, so contamination from air could not be avoided due to the transport from the electrochemical cell to the XPS instrument. The silver foil and ODAg have very similar Ag 3d spectra with a peak at 368.2 eV and a satellite peak at 371.9 eV (Fig. 2a). The combination of these peaks is ascribed to metallic silver.14 The AgOx precursor was also examined by XPS, and in this sample the silver binding energy shifted to 367.8 eV, along with a disappearance of the satellite peak. This agrees well with literature values for Ag2O.14 Since AgO is notoriously unstable in ultra-high vacuum, its presence cannot be confirmed nor rejected.14,15 Detailed scans of the O 1s region were also recorded but not very insightful (Fig. 2b–d) because Ag–O contributions can overlap with carbonate groups from air contamination in the O 1s region.16,17 The only definitive assignment that can be made in the O 1s spectrum in Fig. 2c is the small peak at 529.3 eV, ascribed to Ag2O.18 It is surprising that this peak is so small since the shift in the corresponding Ag 3d spectrum seems to indicate pure silver(I) oxide. The larger peak at 530.8 eV of the O 1s spectrum could be ascribed to oxygen chemisorbed on a silver surface17 or Ag–O–Ag bonds19 but not to pure Ag2O. The higher binding energies of the O 1s in the Ag and ODAg sample can mainly be ascribed to contamination from air (note that these peaks are 3 and 10 times smaller than the AgOx sample, respectively), although their binding energy is also very close to a species described as subsurface O in silver at 531.2 eV.16 Since the binding energies of contamination and silver–oxygen bonds overlap, it is difficult to draw significant conclusions from the O 1s spectra. It is therefore necessary to use other characterization techniques to properly investigate the electronic and compositional differences between Ag and ODAg.
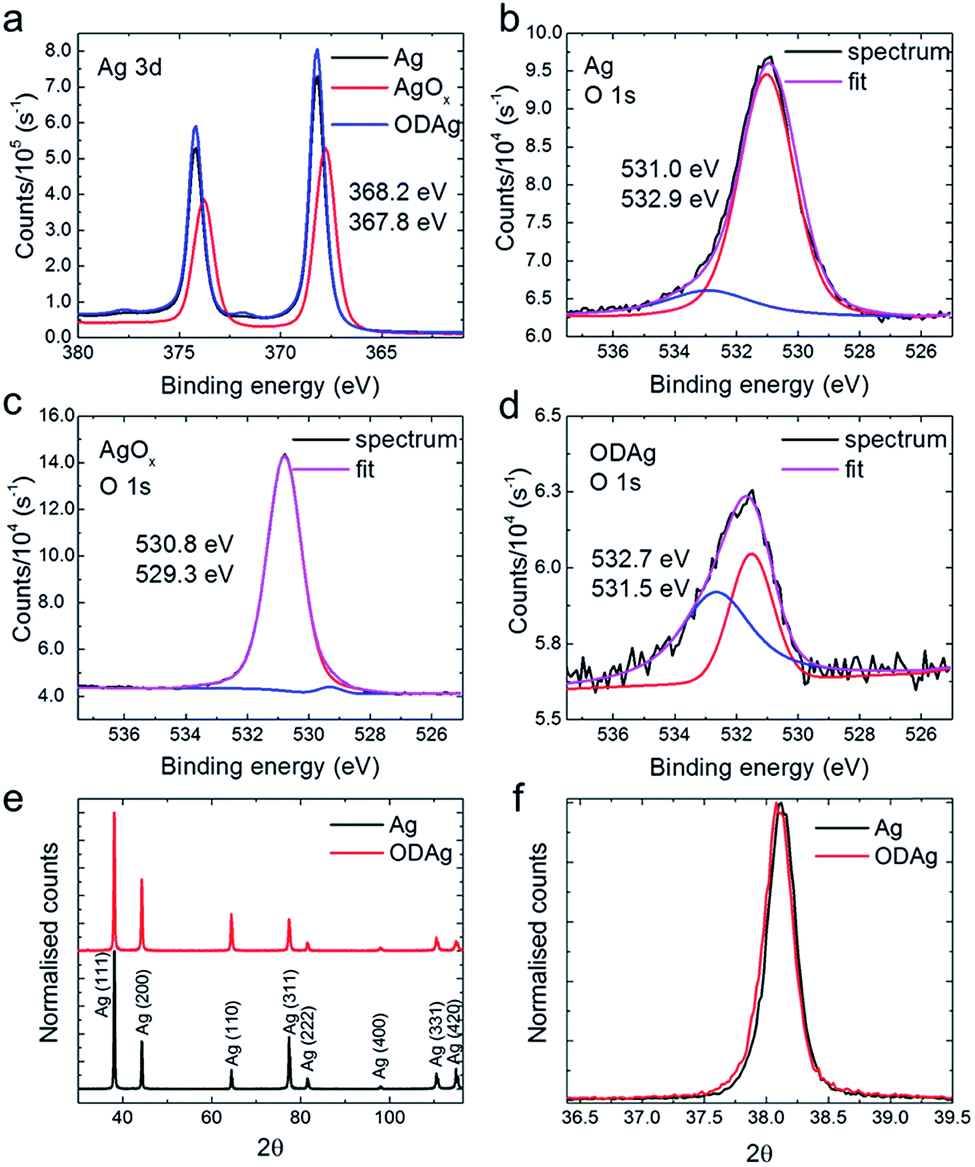 | ||
| Fig. 2 (a) Ag 3d XPS spectra of Ag, the silver oxide precursor and ODAg. The precursor is not metallic but an oxide, as can be concluded from the shift in the Ag 3d peak. The O 1s spectrum and its fit of (b) Ag, (c) AgOx and (d) ODAg, the red and blue lines indicate the deconvoluted peaks. (e) and (f) are GIXRD patterns of Ag and ODAg. (e) When the spectra are normalised it becomes apparent that Ag (220) grows relative to Ag (111). (f) There is no significant strain in the ODAg catalyst. | ||
To see whether there are any structural changes between Ag and ODAg, grazing incidence X-ray diffraction (GIXRD) patterns were measured and compared. To be more sensitive to the surface of the catalyst and to be able to measure all reflections (not just the reflections parallel to the surface as for the standard Bragg–Brentano XRD configuration), GIXRD was chosen as the most suitable XRD technique. A change in the ratio between (111) and (220) can be observed in the GIXRD data measured at a grazing incidence angle of 4° θ of oxide-derived silver compared to the pure silver foil (Fig. 2e). For the ODAg films, the ratio (220) over (111) is higher compared to that of untreated Ag. The presence of the (220) peak is an indirect proof of the presence of (110) which is not visible in the XRD pattern since it is a forbidden reflection for a fcc lattice. The increased (220) reflection is already observed in the silver oxide pattern, in addition to several smaller peaks representing both Ag2O and AgO structures (Fig. S3†).
The Ag (110) facet was shown by both experimental20 and theoretical5 work to be more active than the very stable (111) facet, thus the increased presence of the (220) reflection in the ODAg electrodes could potentially explain the improved electrocatalytic activity and selectivity. However, one should keep in mind that XRD, even when measured in grazing incidence mode, is still a bulk technique that will penetrate several hundreds of nanometers into the catalyst material. This means that even though the facets underlying the surface can be more (110) orientated in the case of ODAg, this does not prove that the actual surface facets are also (110). We can however say from the uniformity found in the peak shape (Fig. 2f) that the lattice is not strained in a higher or lower degree than the silver foil, nor that the crystallite size seems to have changed.
To better understand the cause of improved electronic, compositional or short-range structural difference between Ag and ODAg, a short-range order characterization technique that can be used under real operating conditions is required.
Operando X-ray absorption spectroscopy
X-ray absorption fine structure spectroscopy (XAFS) is widely used to study materials and systems operando or in situ due to the high energy X-rays that can be used; the X-rays can easily penetrate through air, water and various soft materials that are generally used to build an electrochemical cell.21 XAFS is divided into X-ray absorption near-edge spectroscopy (XANES) and extended X-ray absorption fine structure (EXAFS). The XANES spectra provide information on the oxidation state, geometry and electronic configuration of the absorber atom while EXAFS data fitting allows the ability to reconstruct its local environment to give the concentration of neighbouring atoms (N), the absorber atom's distance to the neighbouring atom (R), and disorder (Debye–Waller factor: σ) on a short length scale.X-ray absorption spectra were recorded at the Dutch-Belgian beamline (DUBBLE, 26A) of the European Synchrotron Radiation Facility (ESRF) in Grenoble, France at the silver K-edge (25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 515.59 eV). Fig. 3a shows the Fourier transforms of reference transmission EXAFS spectra for a 5 μm Ag thin film and pellets of silver(I) oxide and silver(II) oxide powder dispersed in a cellulose matrix. The EXAFS spectra are extracted and fitted according to the EXAFS equation (ESI, eqn (3)†); the data analysis details are reported in the ESI.† Due to the limited energy (k = 2.5–10) range achievable in the operando conditions, the data analysis has been mainly focused on the first two nearest neighbours (coordination shells) of the silver absorber atom: the Ag–O and Ag–Ag coordination shells. The fitting results of the reference metal and oxides in Fig. 3a are reported in Table 1.
515.59 eV). Fig. 3a shows the Fourier transforms of reference transmission EXAFS spectra for a 5 μm Ag thin film and pellets of silver(I) oxide and silver(II) oxide powder dispersed in a cellulose matrix. The EXAFS spectra are extracted and fitted according to the EXAFS equation (ESI, eqn (3)†); the data analysis details are reported in the ESI.† Due to the limited energy (k = 2.5–10) range achievable in the operando conditions, the data analysis has been mainly focused on the first two nearest neighbours (coordination shells) of the silver absorber atom: the Ag–O and Ag–Ag coordination shells. The fitting results of the reference metal and oxides in Fig. 3a are reported in Table 1.
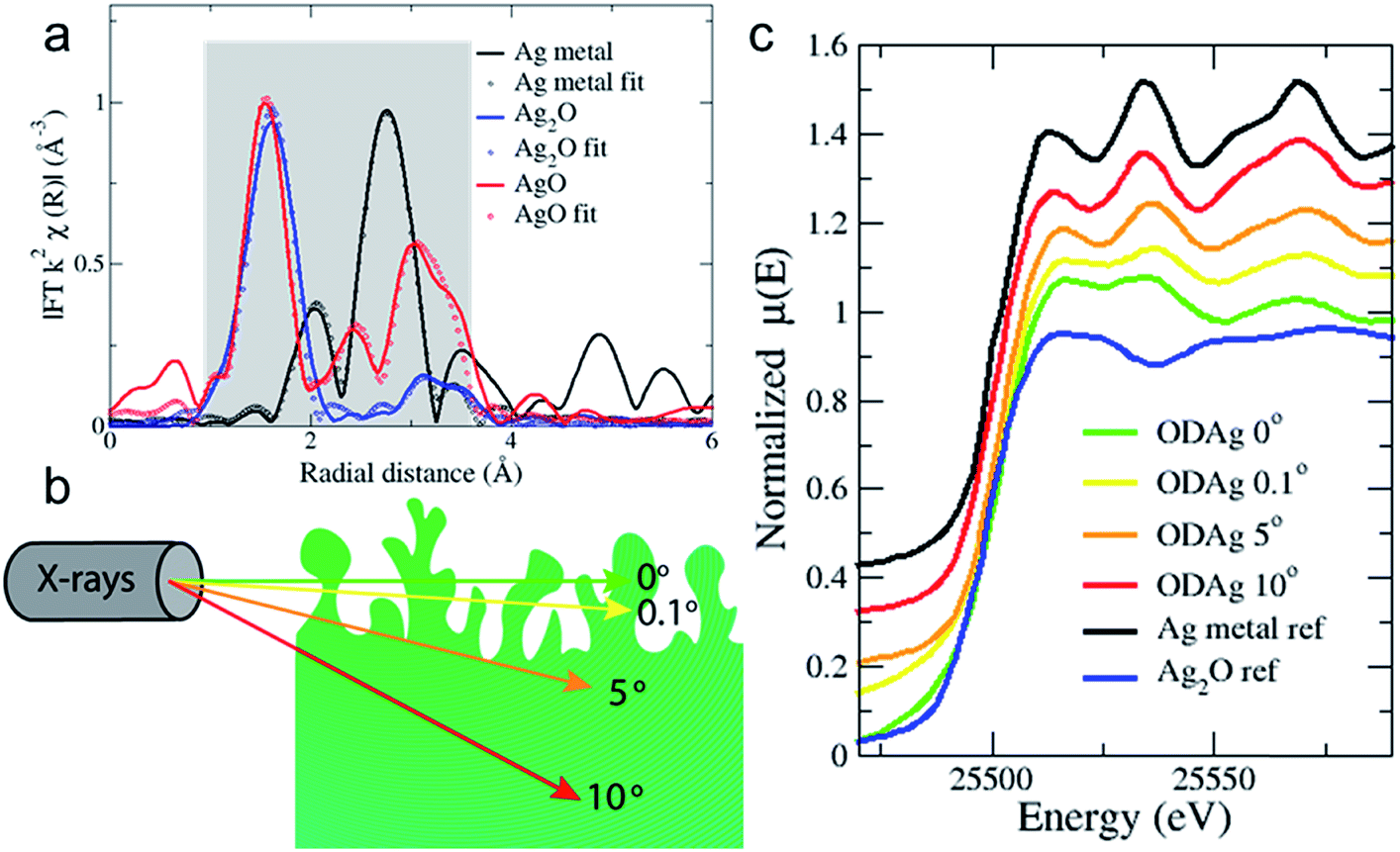 | ||
| Fig. 3 (a) k2-Weighted Fourier transformed EXAFS transmission spectra of silver and silver oxide reference samples. Grey shadow denotes fitted area. (b) A schematic showing how the grazing incidence X-rays penetrate the ODAg. (c) Normalised XANES spectra of the transmission metal and Ag2O references compared to ex situ oxide-derived silver samples, showing the difference in chemical composition of the film when the X-rays penetrate less deep into the material. | ||
| Sample | Bond | R 1 (error R1) | N 1 (error N1) | σ 1 (error σ1) |
|---|---|---|---|---|
| a Average multiple scattering factor for Ag–O. | ||||
| Ag metal | Ag–Ag | 2.86 (0.001) | 12.00 (0.2) | 0.007 (0.0003) |
| Ag2O | Ag–O | 2.06 (0.001) | 1.99 (0.4) | 0.004 (0.0001) |
| Ag–Ag | 3.34 (0.02) | 7.51 (0.7) | 0.036 (0.0009) | |
| AgO | Ag–O | 2.02 (0.02) | 1.99 (0.5) | 0.003 (0.008) |
| MSa | 2.66 (0.09) | 5.16 (0.9) | 0.04 (0.008) | |
| Ag–Ag | 3.29 (0.005) | 11.47 (2.3) | 0.02 (0.003) | |
The reference samples were measured in transmission mode to ensure optimal data quality and to avoid self-absorption processes.22 However, for samples measured in an electrochemical cell, the total sample + cell thickness would be too large for the X-rays to travel through, therefore all sample measurements were taken in fluorescence mode. It is common practice to do this at a configuration where the incident X-rays make a 45° degree angle to the sample. However, EXAFS is a bulk technique and high energy X-rays at 45° will penetrate into the sample >10 μm (Fig. 3b). In order to reduce the penetration depth and give more information from the near surface region, we again used a grazing incidence mode. Using this approach, the X-ray beam is only able to probe the surface of the catalyst and will provide surface-sensitive information.23
The probing depth into the sample is demonstrated in Fig. S4,† which shows the attenuation length of the X-ray beam into silver as a function of the incident angle of the X-rays. The attenuation length is defined as the depth into the material (along the surface normal) where the X-ray beam intensity drops to 1/e of its original value. When the sample is tilted only 0° or 0.1° with respect to the incoming X-rays, for instance, the beam is only expected to penetrate the first 2 or 3 nm of the sample. Fig. 3c shows the striking difference in the measured XANES spectra of an ex situ ODAg sample that was used in CO2 reduction before the EXAFS measurement as a function of varying angles of incidence. Interestingly, when the XANES spectrum is taken at 10° (red line, penetration depth of 3 μm) (Fig. 3c), the spectrum appears similar to pure metallic silver (black line). The XANES spectra of ODAg obtained at shallow angles (0° and 0.1°), however, are not reminiscent of either a pure metallic compound nor a pure silver oxide (blue line). Instead, the samples seem to be a mixture of both metallic and oxide composition, which can be seen from the second feature after the edge at 25534.1 eV; for metallic silver, this feature is 2 times higher than the first feature at 25512.8 eV, for the ODAg samples at 0° and 0.1° they are similar in size. This can be understood as a mixture of the metallic positive contribution and the oxide depression at the same energy. The spectrum measured at 5° (orange line, penetration depth of 1.7 μm) seems more metallic than the 0° and 0.1° spectra but not as strongly metallic as the 10° spectrum. This result highlights how important it is to measure in grazing incidence mode to ensure we observe mainly the surface of the silver electrocatalysts and to reduce bulk metallic signals as much as possible. These results also conclude that our oxide-derived fabrication method only modifies the first ∼2 μm of the Ag foil, while the rest of the 250 μm foil remains pure silver, as can be concluded from Fig. S4.†
While giving important information, XANES cannot, however, provide enough information to properly understand the electronic, structural or compositional properties of the catalysts. Instead, EXAFS spectra are recorded and fitted to provide this information. To ensure that the oxide-like structure found in ex situ spectra are not just the results of spontaneous self-oxidation in air, the spectra are acquired under electrochemical operating conditions.
A schematic of the custom-made cell used to acquire the operando EXAFS spectra is shown in Fig. 4a and a photograph of the cell is given in Fig. S5.† The design consists of a flow cell where the working electrode (WE) (the silver electrocatalyst) faces up. The WE was covered by a 0.5 mm layer of 0.1 M KHCO3 electrolyte which was kept saturated with CO2 in a reservoir outside of the cell. A 0.5 mm polypropylene (PP) window was clamped on top of the electrolyte allowing for transmission of the X-rays. The reference (RE) and counter electrode (CE) were positioned in the vertical parts of the electrolyte flow; with the RE placed just before and the CE just after the WE. The fluorescence detector was placed in close proximity to the WE. The cell was then mounted on a motorized stage that could rotate with respect to the incoming X-ray beam, to allow full control over the angle of incidence. While we were able to perform ex situ sample measurements at extremely low grazing angles, such low angles are not possible during in situ or operando measurements. At an angle of 0.1°, for example, the distance that the beam would have to travel through the polypropylene window and electrolyte layer becomes almost 60 cm, making cell design and alignment difficult, while resulting in large signal attenuation before the beam can reach the sample. Therefore the angle was increased to 6° for operando measurements, which results in a beam travelling distance through the electrolyte and polypropylene of roughly 1 cm. In this configuration, the polypropylene window and electrolyte transmit about 70% of the photons while the attenuation length of the X-rays into the silver was 1.9 μm, which closely resembles the previously observed thickness of the ODAg layer. It is quite remarkable that it is possible to measure operando EXAFS under a grazing incidence angle of only 6°, since it creates a beam path length through the electrolyte and polypropylene of 1 cm. The ability to gain meaningful information from this experiment is further made possible because silver is a heavy element: the high energy (25515.59 eV) is able to pass through 1 cm of material while losing only 30% of its photons.
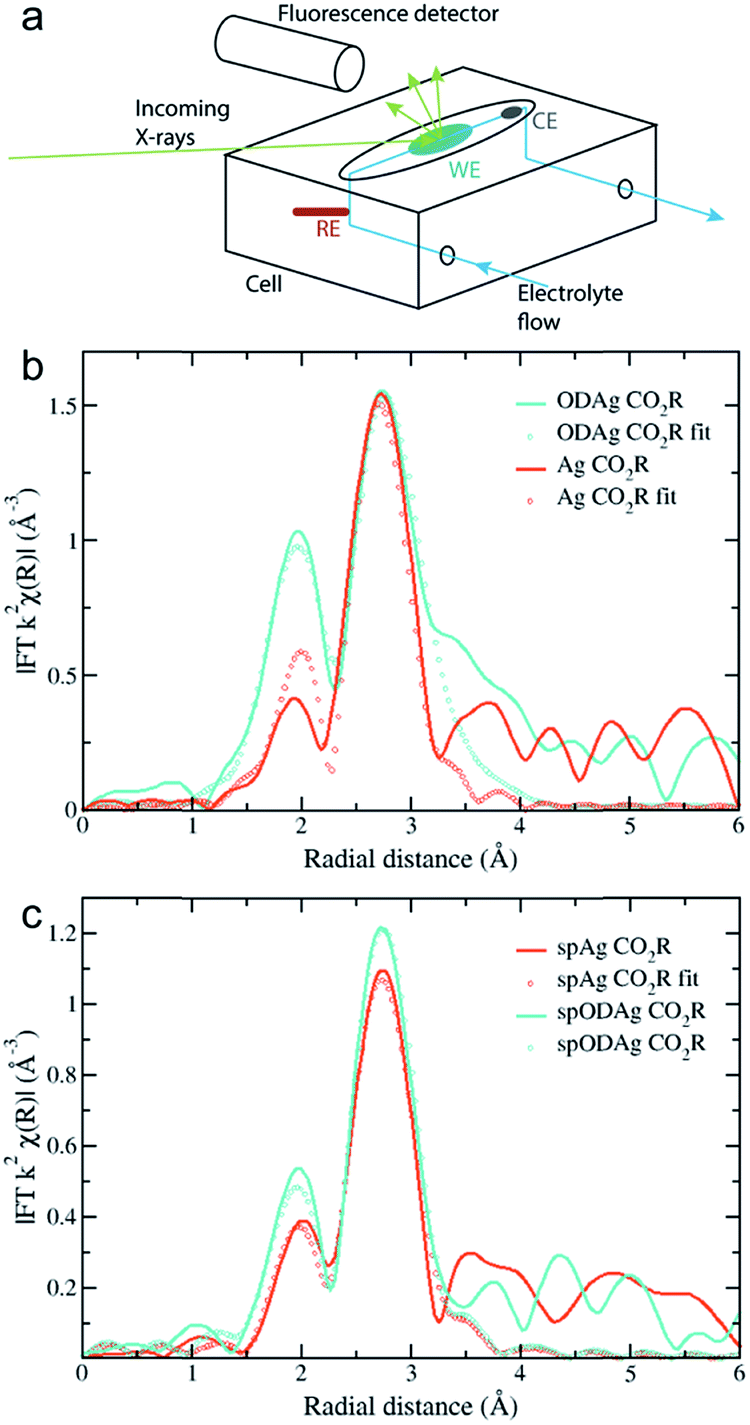 | ||
| Fig. 4 (a) A schematic of the home-made flow cell used for the operando EXAFS measurements. (b) k2-Weighted Fourier transformed EXAFS fluorescence spectra of (b) a Ag catalyst and an ODAg catalyst and of (c) a spAg catalyst and a spODAg catalyst during CO2 reduction. Fitted region is 1.5 to 3.2 Å. | ||
The results of these 6° measurements are shown in Fig. 4b where the Fourier transform of an ODAg sample is compared to that of a pure silver foil, both measured under operando conditions. Both spectra are corrected for self-absorption using the transmission spectrum of silver foil (Fig. 3a) as a reference to obtain reasonable coordination numbers. The EXAFS in K-space k2χ(k) and first derivative of μ(E) are presented in Fig. S6a and c.† Due to the electrolyte layer and the room temperature conditions, the longer range order of the samples could not be properly measured. The Fourier transformed spectra were fit for the first two shells: a Ag–O shell and the first Ag fcc Ag–Ag shell (Table 2). For both the transmission foil and the Ag foil measured during CO2 reduction, the Ag–O shell was not observed during the fitting process. This is expected since oxygen is normally not found in a silver fcc lattice. For the ODAg silver however, a coordination number of 0.69 for the Ag–O shell was obtained. Interestingly, the Ag–O distance in our ODAg is longer then it would be in Ag2O or AgO: 2.28 Å compared to 2.07 Å.24 Similar values for the Ag–O distance and coordination number have been shown before in literature where small Ag nanoparticles were deposited on a silica support.25 In this case, Balerna et al. explain the Ag–O bond as the bond between the oxygen of the silica and the Ag nanoparticles. Note that due to the relatively long interatomic distance, the binding energy is weaker than that of a pure silver oxide and thus the oxidation state of silver will be between Ag0 and Ag+.
| Sample | Bond | R 1 (error R1) | N 1 (error N1) | σ 1 (error σ1) |
|---|---|---|---|---|
| Ag CO2R | Ag–O | 0 | 0 | 0 |
| Ag–Ag | 2.86 (0.07) | 12.00 | 0.013 (0.01) | |
| OD-Ag CO2R | Ag–O | 2.28 (0.05) | 0.69 (0.2) | 0.001 (0.0006) |
| Ag–Ag | 2.88 (0.02) | 11.44 (2.6) | 0.02 (0.0004) |
These results are remarkable, especially when compared to the ex situ XPS spectra, which were not able to identify oxygen as part of the oxide-derived silver. The ultra-high vacuum conditions needed to acquire XPS data might make the ODAg self-reduce to Ag0. Considering the presence of oxygen that was observed in the EXAFS data which has a long bond distance, the electron sharing between the silver and the oxygen is smaller than in a pure oxide, which makes the weakly bound O quite susceptible to removal in vacuum conditions. This could explain why other reports on oxide-derived silver systems have not been able to identify oxygen as an active component in CO2 reduction.
Sputtered Ag and Ag foil
During previous experiments at −0.8 V vs. RHE, it was noticed that a sputtered silver layer on a Ti foil provided a higher selectivity towards CO of 51% than pure Ag foil with a faradaic efficiency of 25% (Fig. S2†). In order to check whether this difference could correlate with the long oxygen bond we found for ODAg, we fabricated sputtered ODAg (denoted as spODAg) and compared the EXAFS spectra of this sample and an otherwise untreated sputtered silver film (spAg) with the Ag and ODAg samples made from Ag foil. The spAg catalyst was made by sputtering a 600 nm layer of Ag on Ti foil (spAg, SEM image in Fig. S7a†) and the treated catalyst, spODAg, was fabricated through the same method as ODAg but using the sputtered Ag on Ti as the substrate (SEM image in Fig. S7c†). GIXRD and XPS data on these samples can be found in Fig. S8 and S9,† respectively. Because the total silver layer for the sputtered catalysts is much thinner, the self-absorption correction for the acquired EXAFS data was not necessary. Accordingly, the spectra were fitted without any self-absorption correction. The EXAFS in K-space k2χ(k) and first derivative of μ(E) are presented in Fig. S6b and d.† Interestingly, whereas the Ag and ODAg show a rather large difference in EXAFS spectra, the sputtered spAg and spODAg are quite similar to each other (Fig. 4c). They can both be fit by a Ag–O shell and a Ag–Ag shell (Table 3). The Ag–O shell gives a very low coordination number of 0.26 for spAg and 0.24 for spODAg. The Ag–O distances are 2.32 and 2.23 Å for spAg and spODAg, respectively. These oxygen species might originate from the sputtering treatment used to deposit the silver layer. The silver–silver shell of the spAg and the spODAg have coordination numbers of 8.60 and 9.77, respectively, indicating a small reactive silver cluster size.12,25 These clusters might spontaneously react to air, forming the Ag–O species.| Sample | Bond | R (error R) | N (error N) | σ (error σ) |
|---|---|---|---|---|
| spAg CO2R | Ag–O | 2.32 (0.03) | 0.24 (0.1) | 0.005 |
| Ag–Ag | 2.84 (0.02) | 8.60 (0.5) | 0.013 (0.0005) | |
| spODAg CO2R | Ag–O | 2.23 (0.03) | 0.26 (0.02) | 0.005 |
| Ag–Ag | 2.85 (0.006) | 9.77 (0.7) | 0.014 (0.0003) |
Model clusters
The absolute amount of oxygen given by the coordination number can be better understood when it is compared to model systems. Metallic silver clusters of different sizes, surrounded by oxygen atoms taken from a silver oxide shell were modelled, and shown in Fig. 5. These clusters gave an average Ag–O distance of 2.3 Å. The theoretical average coordination number of the Ag–O shell was calculated and can be compared to our experimental data. For a very small cluster consisting of only 2 Ag–Ag shells, the Ag–O coordination number is 1.97, roughly the same as for pure silver oxide. However, when we increase the cluster size to 3 or 4 Ag–Ag shells, the Ag–O coordination numbers drop to 0.37 and 0.21 respectively. The large decrease in coordination number for these very small clusters shows how quickly the Ag–O component drops to 0 and thus below the EXAFS detection limit. Since our experimental data shows non-zero coordination numbers, we can conclude that we have a polydisperse silver with very small single crystal clusters that contain oxygen. When we compare this to our experimental data, the amount of oxygen we find by fitting is certainly not negligible. The 2-shell cluster contains 48% of oxygen, the 3- and 4-shell clusters contain 37% and 36% oxygen, respectively. Since many of the silver atoms are so far away from the oxygen atoms, the averaged coordination number still decreases quickly with cluster size.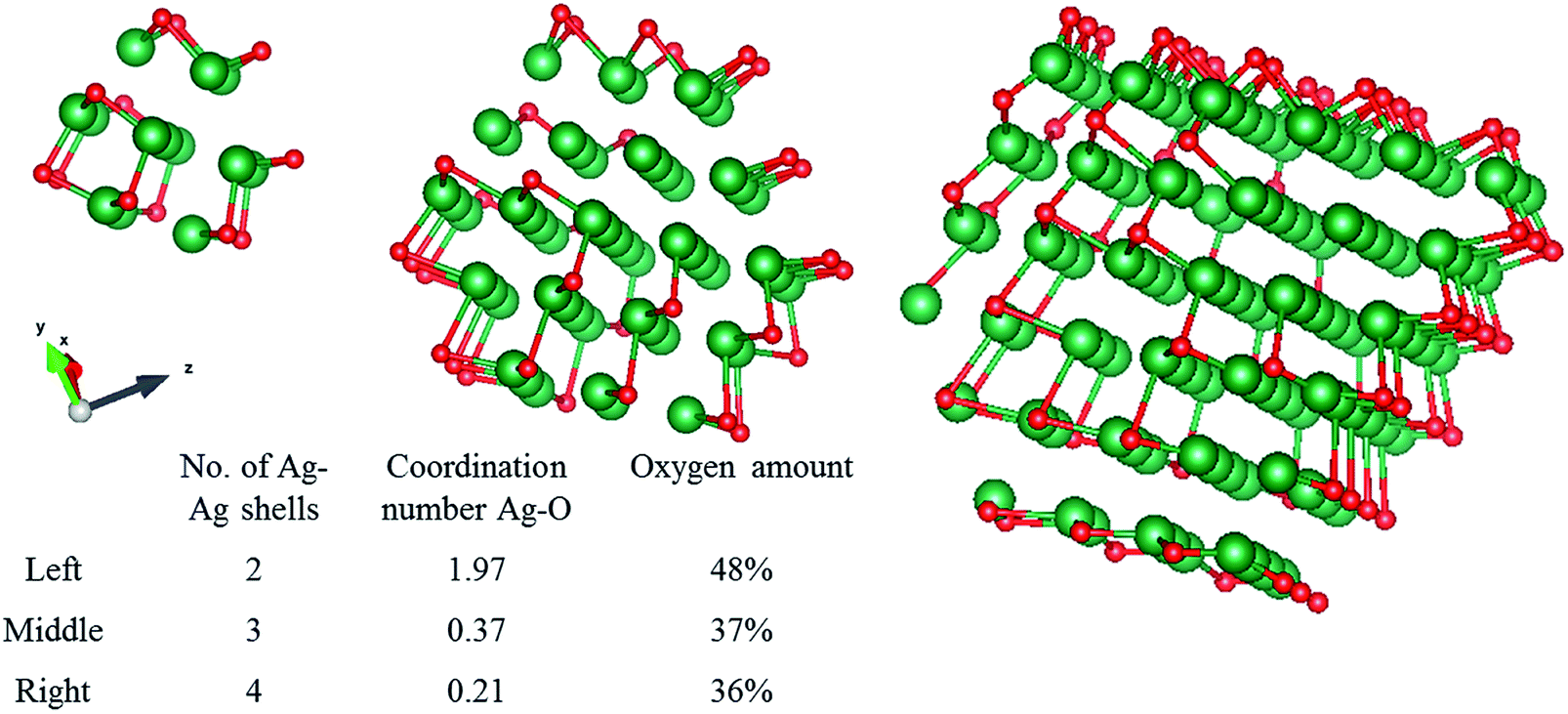 | ||
| Fig. 5 Silver clusters consisting of 2, 3 and 4 Ag–Ag shells were modelled with one Ag–O shell as the outer shell. Note that in this 2D image, the oxygen appears to be dispersed throughout the cluster, in reality it is only present at the outer edge of the cluster. Red is oxygen, green is silver. | ||
CO2 electrolysis
All catalysts examined above were also tested for CO2 reduction, focusing on their activity and selectivity at −0.8 V vs. RHE. The trend in faradaic efficiency for CO is as follows: Ag < spAg < spODAg < ODAg (Fig. S2†). Since ODAg has a seven times larger electrochemical surface area (ECSA) than Ag (Fig. 6a), the difference between their CO selectivity can possibly be explained with the local pH-effect: the local pH at the silver surface is increased by the formation of CO and H2 (since these half reactions consume water molecules and produce hydroxide ions (OH−)), as shown in reaction (1) and (2).| CO2 + H2O + 2e− → CO + 2OH− | (1) |
| 2H2O + 2e− → H2 + 2OH− | (2) |
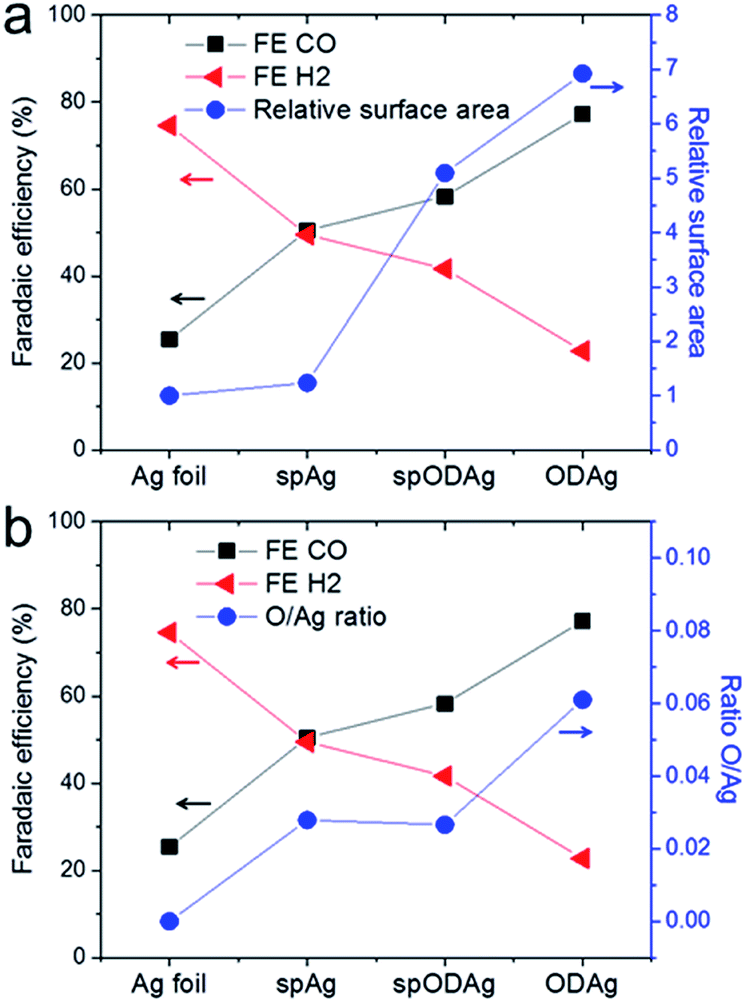 | ||
| Fig. 6 (a) The relation between the selectivity of the catalysts and the relative surface area (as determined by the double layer capacitance, where the relative surface area of the Ag foil is taken as 1). (b) The relation between the catalyst selectivity and the O concentration found in the catalyst as calculated by the ratio between the coordination numbers of the Ag–O and Ag–Ag shell found by EXAFS fitting of the operando measurements. | ||
The hydroxide ions have to travel to the anode to act as reactants in the oxygen producing half reaction. The catalyst's surface porosity inhibits fast OH− exchange between the reaction site and the bulk electrolyte, thus creating a higher pH at the nanostructured silver surface which favours CO production while suppressing H2 evolution.
However, the pure Ag foil and the spAg have quite a selectivity discrepancy between them, and the local pH effect alone cannot account for this since they are both relatively flat enough to not have any mass transfer limitations based on porosity, in fact their ECSAs are almost identical (Fig. 6a). Interestingly, the spAg and spODAg have a factor 4 difference in surface area, but only a small difference in FECO. Whereas some of the differences in CO selectivity can be explained by the local pH effect due to the nanostructured morphology of the samples, there seems to be another factor that also benefits the improvement in CO2 reduction selectivity. When the CO2 selectivity is compared with the concentration of Ag–O bonds found by EXAFS fitting, we find similar trends. The O/Ag ratio (defined as the ratio between the Ag–O coordination number and the Ag–Ag coordination number) follows roughly the same trend as the FE of CO (Fig. 6b) and may therefore play a critical role in the improved CO selectivity of both sputtered silver and oxide-derived silver as compared to silver foil.
It is necessary to put these results in the context of previous findings in order to extrapolate deeper meaning. Over the past years, a number of reports have been published about silver-based nanostructured electrocatalysts that all have a precursor containing a form of silver oxide.10–12,19,26 During CO2 reduction these catalysts reduce back to a nanostructured form of metallic silver. All of these catalysts can thus fall within the class of oxide-derived silver catalysts, although they have different fabrication methods and starting morphologies/compositions. Different possible explanations were given in these reports that try to elucidate the active component within these catalysts. It seems quite likely that there are a number of factors that together create such active and selective catalysts; thus it is fair that the effects of local pH, higher surface areas and residual oxygen species are in essence, not mutually-exclusive. Strikingly, whereas some of these previous works claim to have found traces of oxygen in their catalyst, to which they attribute to the improved catalytic performance, others claim to be certain oxygen has completely disappeared from the catalysts' surface and can therefore not contribute to the reaction. A range of different characterization techniques were employed to try and find the presence of oxygen. The difficulty with many of these measurement techniques is that they are often recorded ex situ, so the composition of the catalyst may be different during electrolysis than under vacuum/air characterization. Some of the characterization techniques give indirect information about the catalyst structure: a change in work function as measured by Kelvin Probe Force Microscopy (KPFM) or Ultraviolet Photoelectron Spectroscopy (UPS) could indicate increased oxygen levels but it could also mean higher impurity levels or a different crystal facet exposure. Another difficulty lies in the bulk sensitivity of some techniques: both X-ray diffraction (XRD) and X-ray absorption spectroscopy (XAS) probe several micrometres far into the bulk, thus the contributions from the surface of the catalyst can be masked by signals from the bulk. An extra complication with XRD is that the presence of certain facets in the bulk has no real link to the presence of that same facet on the surface; often surfaces will rearrange to the most stable formation possible, regardless of their bulk structure. The last issue is the use of ultra-high vacuum characterization techniques: silver oxides are not very stable, and they can reduce to metallic silver under ultra-high vacuum conditions.14 This means that a lack of oxygen in an XPS or Auger spectrum is not representative of the state of the same catalyst under ambient or electrochemical conditions. Besides this it can be difficult to obtain conclusive results from XPS spectra. A Ag 3d peak will not shift far from metallic silver when weakly bound to an oxygen atom. As discussed above, the O 1s spectrum has many contributions from both silver and contamination that are hard to assign.
The discrepancy in these previous studies then highlight the importance of techniques that can measure a catalyst's surface under actual reaction environments, either in ambient or under operating conditions. These efforts are important to gain information about the catalyst in its working state.
Based upon the findings of our work, the presence of O in the ODAg catalyst seems to be acting as a dopant, similar to other non-metallic p-type dopants such chloride,27 iodide4 and even amine or thiol groups.28 For these doped silver catalysts, it has been shown that trace amounts of non-metallic species such as I or Cl can have a significant effect on the selectivity and activity of CO2 electroreduction, and therefore most likely impact the reaction mechanism. Therefore, in addition to the impact of the nanostructured morphology on the local reaction environment, the electronic effect of oxygen should also be taken into account. A similar conclusion has been recently found by Zhou et al., who used EXAFS to show how partial positive charges induced by boron doping on copper can stabilize CO2 reduction intermediates.29
Bond length calculations
To strengthen the understanding of the presence of oxygen in a Ag catalyst for improved CO2 electroreduction, DFT calculations and a Bader charge analysis were performed to determine the bond length and partial charge of the long Ag–O bond, presented in Fig. 7a. In a low-coverage case on Ag(110), the two most favourable sites for an oxygen atom to sit on are the hollow and the long-bridge site. Of these two different sites, the hollow site provides the most stable configuration. The bond lengths found for these scenarios closely resemble the Ag–O distances that were found in EXAFS fitting. Therefore we assume that the long Ag–O bond is situated in the ODAg in a similar manner to what is calculated here and has a partial charge of O0.95− as calculated from the Bader analysis. The positive charge on the silver is shared by many atoms and is thus small: Ag0.17+ for atoms close to the oxygen.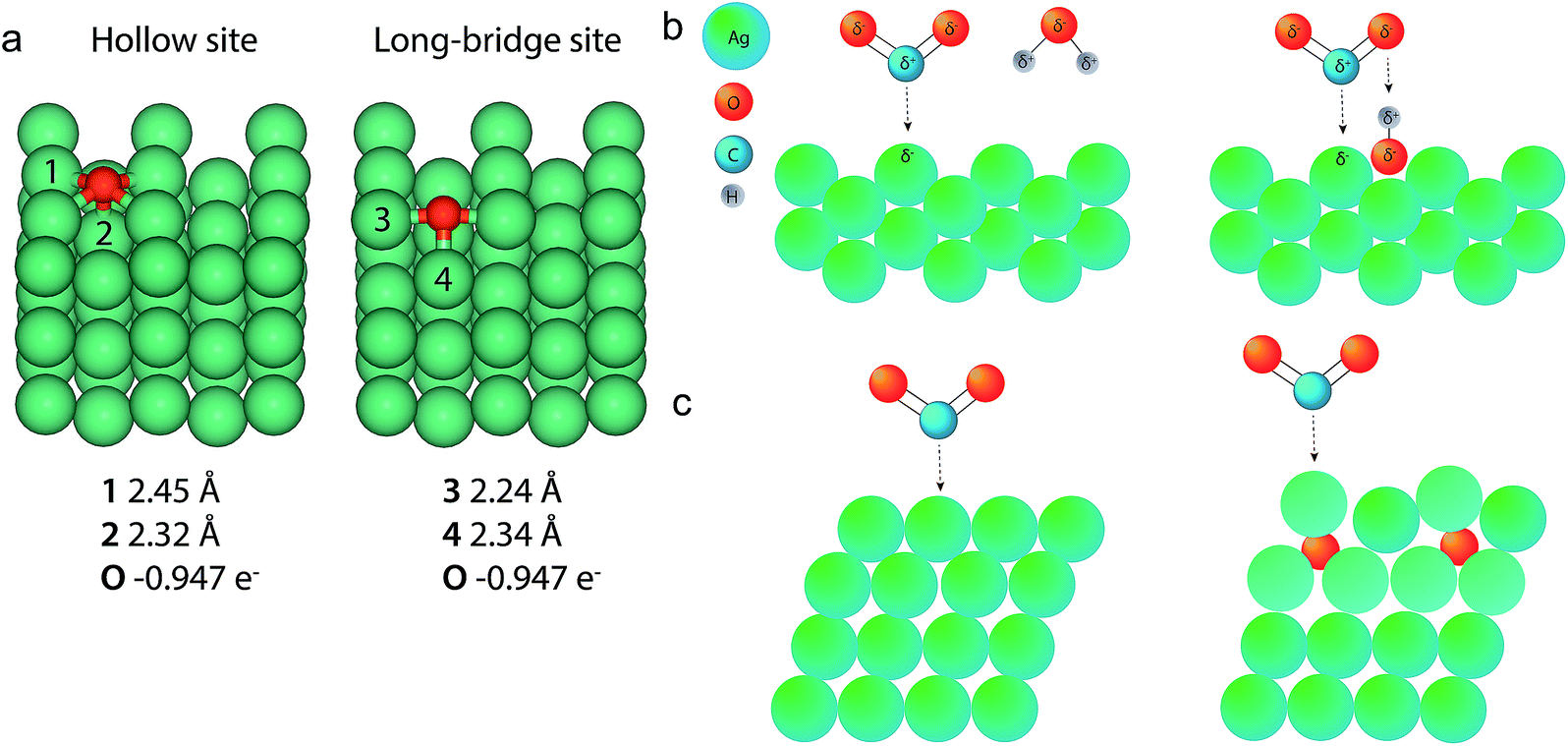 | ||
| Fig. 7 (a) DFT calculations and Bader analysis for O on a Ag (110) slab give Ag–O bond lengths very similar to those found in the EXAFS data fitting. (b) Left: a CO2 molecule binds to a Ag(110) facet in the presence of a water molecule. Right: the presence of a water molecule. Right: the presence of oxygen in the cavities of a silver (110) facet can facilitate the binding of CO2 to the silver through the presence of a shared proton. (c) Left: a CO2 molecule binding to a Ag(111) surface. Right: subsurface O below Ag (111) facets shift the d-band of silver toward the Fermi level (indicated by a change in color of the affected Ag atoms), increasing the reactivity of the subsurface O–Ag(111) facet compared to Ag(111). | ||
Potential mechanisms as a result of the presence of oxygen
The role of oxygen in the silver lattice can now be explained by several hypotheses. The first hypothesis would be that the observed oxygen is part of a reaction intermediate of the CO2 reduction. This hypothesis seems unlikely since the Ag–O is also observed in the ex situ samples (Fig. 7b). In addition, if oxygen were part of a reaction intermediate, this would mean that we are able to observe a single monolayer of material using a bulk technique (EXAFS), which seems unlikely.Another hypothesis relates to the bond lengths found by the DFT calculations. The hollow or long-bridge site in the silver (110) facet if exposed to the electrolyte can host oxygen atoms, as is shown in Fig. 6a. If these oxygen atoms bound to CO2 directly, a stable and thus unreactive carbonate species would formed. If instead a proton would bind to the oxygen on the silver surface first, the combination of a negatively charged silver atom next to a positively charged proton would present the perfect substrate for a CO2 molecule to bind to. This is schematically presented in Fig. 6b: the proton can initially be shared between the silver–oxygen and the CO2–oxygen, but once CO2 is properly adsorbed on the surface the proton can become exclusively bound to CO2, forming the well-known COOH intermediate.30 This hypothesis relies on the presence of oxygen in Ag(110) facets. However, the fact that our GIXRD studies show an increased concentration of silver (110), does not necessarily prove that (110) is present in higher concentrations at the surface of ODAg. Nevertheless, due to the increased roughness on the nanoscale (Fig. 1d and S6c†), we can say with high confidence that the exposed facets will contain many defects, kinks and other types of stepped sites. These sites can also facilitate the presence of oxygen atoms that bind to protons and follow a similar reaction mechanism as explained for the (110) facet. The low coordination number of spAg (Table 3) also indicates many such defects exist on this catalyst. Once the atomic oxygen sits at such a site, it also becomes more difficult for the Ag to rearrange to a more stable surface configuration, meaning the presence of the oxygen actually stabilizes the defects and the activity of the ODAg can be maintained more easily.
Another hypothesis stems from heterogeneous catalysis
In the field of ethylene epoxidation catalysed by silver, subsurface oxygen is found to have a very important role in increasing the reactivity of the catalyst.31,32 It was found that the presence of subsurface oxygen shifts the d-band towards the Fermi level, increasing the reactivity of silver as schematically depicted in Fig. 7c.31 It is known that for d-band metals, a valence band (i.e. metal d-band centre) closer to the Fermi level increases reactivity for the metal. This effect on CO2 reduction was demonstrated for gold–copper nanoparticles.33 Since the (111) facet is the most energetically stable surface facet for silver, it is imaginable that it is still present in large quantities at the rough ODAg surface. When the mixed silver oxide quickly reduces back to form ODAg (Fig. 1a), some oxygen atoms can become stuck in a potential well, meaning that the energy barrier to reduce the last remaining oxygen atoms can be quite high when there is no other oxygen around to form molecular O2. Below the surface of the catalyst this seems like a plausible scenario. This creates silver atoms in close proximity to the subsurface O that are more reactive than before the oxide-derived treatment and can more easily bind CO2 to form COOH.Conclusions
We have shown how the use of operando EXAFS has identified the presence of a long Ag–O bond where the oxygen may be a key participant in the reduction of CO2 to CO on oxide-derived silver catalysts. The silver needs a long and thus atomic oxygen bond in order to alter its reactivity and improve its susceptibility to CO2 binding. Both the presence of oxygen and nanostructuring of the catalyst to increase the local pH aid the activity and selectivity for CO production on silver electrocatalysts and are therefore vital components of an active and selective CO2 reduction catalyst. The knowledge gained from this research can hopefully help to design future silver-based CO2 reduction catalysts.In recent years, many groups have looked into the possible presence of oxygen species remaining after CO2 reduction and competing conclusions were drawn from these studies. We hope to positively contribute to the scientific discussion by adding operando results that are surface sensitive enough to measure the catalytically active surface layer.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financed by the Netherlands Organisation for Scientific Research (NWO, project number FOM-13CO19) and Shell. We thank DUBBLE for funding and gratefully acknowledge its staff for assisting in our synchrotron experiments. The authors would like to thank Bernard Dam for fruitful discussions, Joost Middelkoop for cell design and Bartek Trześniewski, Ming Ma, Marco Valenti and Kai Liu for preliminary XAS measurements.References
- Y. Hori, Electrochemical CO2 Reduction on Metal Electrodes, in Modern Aspects of Electrochemistry, ed. C. G. Vayenas, M. E. Gamboa-Aldeco and R. E. White, Springer, New York, 2008, vol. 42, pp. 89–189 Search PubMed.
- K. P. Kuhl, T. Hatsukade, E. R. Cave, D. N. Abram, J. Kibsgaard and T. F. Jaramillo, Electrocatalytic Conversion of Carbon Dioxide to Methane and Methanol on Transition Metal Surfaces, J. Am. Chem. Soc., 2014, 136, 14107–14113 CrossRef CAS PubMed.
- G. O. Larrazábal, A. J. Martín, S. Mitchell, R. Hauert and J. Pérez-Ramírez, Synergistic Effects in Silver–Indium Electrocatalysts for Carbon Dioxide Reduction, J. Catal., 2016, 343, 266–277 CrossRef.
- Y. Zhang, L. Ji, W. Qiu, X. Shi, A. M. Asiri and X. Sun, Iodide-Derived Nanostructured Silver Promotes Selective and Efficient Carbon Dioxide Conversion into Carbon Monoxide, Chem. Commun., 2018, 54, 2666–2669 RSC.
- J. Rosen, G. S. Hutchings, Q. Lu, S. Rivera, Y. Zhou, D. G. Vlachos and F. Jiao, Mechanistic Insights into the Electrochemical Reduction of CO2 to CO on Nanostructured Ag Surfaces, ACS Catal., 2015, 5, 4293–4299 CrossRef CAS.
- M. Ma, B. J. Trzesniewski, J. Xie and W. A. Smith, Selective and Efficient Reduction of Carbon Dioxide to Carbon Monoxide on Oxide-Derived Nanostructured Silver Electrocatalysts, Angew. Chem., Int. Ed., 2016, 55, 9748–9752 CrossRef CAS PubMed.
- Y. Chen, C. W. Li and M. W. Kanan, Aqueous CO2 Reduction at Very Low Overpotential on Oxide-Derived Au Nanoparticles, J. Am. Chem. Soc., 2012, 134, 19969–19972 CrossRef CAS PubMed.
- A. Verdaguer-Casadevall, C. W. Li, T. P. Johansson, S. B. Scott, J. T. McKeown, M. Kumar, I. E. L. Stephens, M. W. Kanan and I. Chorkendorff, Probing the Active Surface Sites for CO Reduction on Oxide-Derived Copper Electrocatalysts, J. Am. Chem. Soc., 2015, 137, 9808–9811 CrossRef CAS PubMed.
- C. H. Lee and M. W. Kanan, Controlling H+vs. CO2 Reduction Selectivity on Pb Electrodes, ACS Catal., 2015, 5, 465–469 CrossRef CAS.
- K. Jiang, P. Kharel, Y. Peng, M. K. Gangishetty, H.-Y. G. Lin, E. Stavitski, K. Attenkofer and H. Wang, Silver Nanoparticles with Surface-Bonded Oxygen for Highly Selective CO2 Reduction, ACS Sustainable Chem. Eng., 2017, 5, 8529–8534 CrossRef CAS.
- M. S. Jee, H. Kim, H. S. Jeon, B. K. Min, Y. J. Hwang, B. K. Min, Y. J. Hwang, K. H. Chae and B. K. Min, Stable Surface Oxygen on Nanostructured Silver for Efficient CO2 electroreduction, Catal. Today, 2017, 288, 48–53 CrossRef CAS.
- H. Mistry, Y.-W. Choi, A. Bagger, F. Scholten, C. Bonifacio, I. Sinev, N. J. Divins, I. Zegkinoglou, H. S. Jeon and K. Kisslinger, et al., Enhanced Carbon Dioxide Electroreduction to Carbon Monoxide over Defect Rich Plasma-Activated Silver Catalysts, Angew. Chem., Int. Ed., 2017, 56, 1–6 CrossRef PubMed.
- Y. Yoon, A. S. Hall and Y. Surendranath, Tuning of Silver Catalyst Mesostructure Promotes Selective Carbon Dioxide Conversion into Fuels, Angew. Chem., Int. Ed., 2016, 128, 15508–15512 CrossRef.
- T. C. Kaspar, T. Droubay, S. A. Chambers and P. S. Bagus, Spectroscopic Evidence for Ag(III) in Highly Oxidized Silver Films by X-Ray Photoelectron Spectroscopy, J. Phys. Chem. C, 2010, 114, 21562–21571 CrossRef CAS.
- D. Lützenkirchen-Hecht and H. H. Strehblow, Anodic Silver(II) Oxides Investigated by Combined Electrochemistry, Ex Situ XPS and In Situ X-Ray Absorption Spectroscopy, Surf. Interface Anal., 2009, 41, 820–829 CrossRef.
- A. I. Boronin, V. I. Bukhityarov, A. L. Vishnevskii, G. K. Boreskov and V. I. Savchenko, XPS and UPS Studies of Oxygen Adsorption over Clean and Carbon-Modified Silver Surfaces, Surf. Sci., 1988, 201, 195–210 CrossRef CAS.
- A. A. El Mel, N. Stephant, J. Hamon, D. Thiry, A. Chauvin, M. Chettab, E. Gautron, S. Konstantinidis, A. Granier and P. Y. Tessier, Creating Nanoporosity in Silver Nanocolumns by Direct Exposure to Radio-Frequency Air Plasma, Nanoscale, 2016, 8, 141–148 RSC.
- W. Wei, X. Mao, L. A. Ortiz and D. R. Sadoway, Oriented Silver Oxide Nanostructures Synthesized through a Template-Free Electrochemical Route, J. Mater. Chem., 2011, 21, 432–438 RSC.
- L. Q. Zhou, C. Ling, M. Jones and H. Jia, Selective CO2 Reduction on a Polycrystalline Ag Electrode Enhanced by Anodization Treatment, Chem. Commun., 2015, 51, 17704–17707 RSC.
- N. Hoshi, M. Kato and Y. Hori, Electrochemical Reduction of CO2 on Single Crystal Electrodes of Silver, J. Electroanal. Chem., 1997, 440, 283–286 CrossRef CAS.
- H. S. Jeon, I. Sinev, F. Scholten, N. J. Divins, I. Zegkinoglou, L. Pielsticker and B. Roldan Cuenya, Operando Evolution of the Structure and Oxidation State of Size-Controlled Zn Nanoparticles during CO2 Electroreduction, J. Am. Chem. Soc., 2018, 140, 9383–9386 CrossRef CAS PubMed.
- L. Tröger, D. Arvanitis, K. Baberschke, H. Michaelis, U. Grimm and E. Zschech, Full Correction of the Self-Absorption in Soft-Fluorescence Extended X-Ray-Absorption Fine Structure, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 3283–3289 CrossRef.
- C. Maurizio, M. Rovezzi, F. Bardelli, H. G. Pais and F. D'Acapito, Setup for Optimized Grazing Incidence X-Ray Absorption Experiments on Thin Films on Substrates, Rev. Sci. Instrum., 2009, 80, 63904 CrossRef CAS PubMed.
- J. P. Allen, D. O. Scanlon and G. W. Watson, Electronic Structures of Silver Oxides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 115141 CrossRef.
- A. Balerna, L. Liotta, A. Longo, A. Martorana, C. Meneghini, S. Mobilio and G. Pipitone, Structural Characterization of Pumice-Supported Silver-Palladium Metal Clusters by Means of XAFS and AWAXS, Eur. Phys. J. D, 1999, 7, 89–97 CrossRef CAS.
- X. Peng, S. G. Karakalos and W. E. Mustain, Preferentially Oriented Ag Nanocrystals with Extremely High Activity and Faradaic Efficiency for CO2 Electrochemical Reduction to CO, ACS Appl. Mater. Interfaces, 2018, 10, 1734–1742 CrossRef CAS PubMed.
- Y.-C. Hsieh, S. D. Senanayake, Y. Zhang, W. Xu and D. E. Polyansky, Effect of Chloride Anions on the Synthesis and Enhanced Catalytic Activity of Silver Nanocoral Electrodes for CO2 Electroreduction, ACS Catal., 2015, 5, 5349–5356 CrossRef CAS.
- C. Kim, T. Eom, M. S. Jee, H. Jung, H. Kim, B. K. Min and Y. J. Hwang, Insight into Electrochemical CO2 Reduction on Surface-Molecule-Mediated Ag Nanoparticles, ACS Catal., 2016, 779–785 Search PubMed.
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, H. Yuan, J. Li, Z. Wang, P. De Luna and H. Yuan, et al., Dopant-Induced Electron Localization Drives CO2 Reduction to C2 Hydrocarbons, Nat. Chem., 2018, 1–20 Search PubMed.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrocatalytic Process of CO Selectivity in Electrochemical Reduction of CO2 at Metal Electrodes in Aqueous Media, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- Y. Xu, J. Greeley and M. Mavrikakis, Effect of Subsurface Oxygen on the Reactivity of the Ag(111) Surface, J. Am. Chem. Soc., 2005, 127, 12823–12827 CrossRef CAS PubMed.
- J. G. Serafin, A. C. Liu and S. R. Seyedmonir, Surface Science and the Silver-Catalyzed Epoxidation of Ethylene: An Industrial Perspective, J. Mol. Catal. A: Chem., 1998, 131, 157–168 CrossRef CAS.
- D. Kim, J. Resasco, Y. Yu, A. M. Asiri and P. Yang, Synergistic Geometric and Electronic Effects for Electrochemical Reduction of Carbon Dioxide Using Gold–Copper Bimetallic Nanoparticles, Nat. Commun., 2014, 5, 5948 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ta10412c |
| This journal is © The Royal Society of Chemistry 2019 |