
Electrochemically activated cobalt nickel sulfide for an efficient oxygen evolution reaction: partial amorphization and phase control†

Abstract
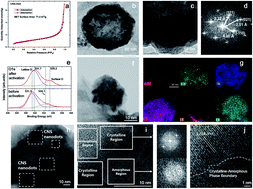
It has recently been demonstrated that the OER activity of transition metal sulfides (TMSs) could be enhanced by the introduction of a thin amorphous layer on a pristine surface. We report here a novel strategy to enhance the OER by developing cobalt nickel sulfide (CoxNi1−xS2, CNS) with a high density of crystalline and amorphous phase boundaries. Electrochemical activation (ECA) can partially amorphize hollow CNS nanoparticles derived from surface-selective sulfidation. The ECA-treated CNS (ECA-CNS) electrocatalyst, which is comprised of CNS nanodots separated by thin amorphous layers, shows high densities of crystalline and amorphous phase boundaries. This catalyst shows superior OER catalytic performance with a current density of 10 mA cm−2 at a small overpotential of 290 mV, a low Tafel slope of 46 mV dec−1, a high mass activity of 217 A g−1, a high turnover frequency of 0.21 s−1 at an overpotential of 340 mV, and excellent stability in alkaline media.
 Please wait while we load your content...
Please wait while we load your content...

