
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fabrication of regenerated cellulose nanoparticles by mechanical disintegration of cellulose after dissolution and regeneration from a deep eutectic solvent†
Juho Antti
Sirviö
 *
*
Fibre and Particle Engineering Research Unit, University of Oulu, P.O. Box 4300, 90014 Oulu, Finland. E-mail: juho.sirvio@oulu.fi
First published on 3rd December 2018
Abstract
Cellulose nanoparticles are promising sustainable nanosized materials for use, for example, as nanofillers (such as reinforcement agents), green water purification chemicals, and oil dispersants. In this work, regenerated cellulose nanoparticles (RCNPs) were produced by mechanical disintegration of wood cellulose fibers and microcrystalline cellulose after dissolution and regeneration from a deep eutectic solvent (DES). An easily obtained DES based on guanidine hydrochloride and anhydrous phosphoric acid (molar ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) was used to dissolve cellulose at room temperature. The degree of polymerization (DP) of cellulose was observed to decrease during the dissolution, whereas the crystallinity changed from cellulose I to cellulose II. After precipitation and washing, the regenerated cellulose was easily disintegrated to evenly distributed nanosized (diameter around 6 nm) fibrous cellulose nanoparticles. RCNPs were investigated as fillers in poly(vinyl alcohol) (PVA) composite films. Due to their small, fibrous and flexible nature, at low concentrations (1–5 wt%) RCNPs improved the elongation of the PVA film without diminishing its strength properties. At higher concentrations, RCNPs from wood pulp improved the tensile strength, as well as the modulus of PVA, similar to commercial cellulose nanocrystals. Therefore, this method provides a feasible way to obtain a unique nanosized cellulose material with good uniform size distribution and adjustable reinforcement properties.
:2) was used to dissolve cellulose at room temperature. The degree of polymerization (DP) of cellulose was observed to decrease during the dissolution, whereas the crystallinity changed from cellulose I to cellulose II. After precipitation and washing, the regenerated cellulose was easily disintegrated to evenly distributed nanosized (diameter around 6 nm) fibrous cellulose nanoparticles. RCNPs were investigated as fillers in poly(vinyl alcohol) (PVA) composite films. Due to their small, fibrous and flexible nature, at low concentrations (1–5 wt%) RCNPs improved the elongation of the PVA film without diminishing its strength properties. At higher concentrations, RCNPs from wood pulp improved the tensile strength, as well as the modulus of PVA, similar to commercial cellulose nanocrystals. Therefore, this method provides a feasible way to obtain a unique nanosized cellulose material with good uniform size distribution and adjustable reinforcement properties.
Introduction
Nanomaterials are an emerging class of novel materials usually described as solid components that have at least one dimension at the nanometric scale (less than 1000 nm).1 Due to their nanometric dimensions, nanoparticles exhibit considerably different properties compared to their bulk counterparts. The vast variety of properties are related to high surface area, reactivity, and high strength. Nanoparticles can contain a single element (e.g., zero valent nanoparticles of metals2,3 and non-metals4), a composition of different metals and components (e.g., metal oxides5,6), and polymers.7,8 Nanoparticles are studied as drug carriers,9 anti-microbial materials,10 reinforcement agents,11 and optical materials, among other applications.12,13An important source of natural polymeric nanomaterials is cellulose.14 Cellulose is the most abundant renewable organic polymer on earth and is widely available as a plant component. Plants have nanosized cellulose fibrils (nanofibrils), which then form bundles called microfibrils and then cellulose fibers. Nanosized cellulose can be isolated from natural fibers, for example, using chemical, enzymatic, mechanical, or combined methods. In addition to these high aspect ratio particles, the fabrication of spherical cellulose nanoparticles (SCNPs) has been studied.15,16 Recently, TENCEL cellulose (obtained after the dissolution and regeneration of cellulose from N-methylmorpholine-N-oxide (NMMO)) has been studied as a source of nanosized cellulose (spherical nanostructured cellulose gels and carboxymethylated SCNPs).17,18 Compared to cellulose nanofibers (CNFs) and cellulose nanocrystals (CNCs) which mostly have a cellulose I crystalline structure, regenerated cellulose has the crystalline structure of cellulose II. In addition, when regenerated cellulose is used as a source, the destruction of the fiber structure leads to the production of nanoparticles with different morphologies (e.g., spherical) compared to those of nanocellulose obtained directly from a natural source.
Deep eutectic solvents (DESs) are a relatively novel class of solvents, catalysts, and reagents.19–21 DESs are related to more well-known ionic liquids, exhibiting similar properties such as good solvent capacity and low-vapor pressure. Compared to ionic liquids, DESs are generally easily obtained from cheap and widely available chemicals by simply mixing at elevated temperature. Many DES components can even be obtained from nature,22 and DESs are known to have low toxicity.23 DESs are many times more biodegradable than ionic liquids.24
DESs are recognized as promising media for nanoparticle fabrication, including metal nanoparticles.25,26 Recently, DESs have been utilized in the production of CNFs27–30 and CNCs.31,32 In this study, a DES based on guanidine hydrochloride and anhydrous phosphoric acid was used to dissolve cellulose at room temperature. Dissolving pulp and microcrystalline cellulose were studied as cellulose sources. Regenerated cellulose nanoparticles (RCNPs) were produced with mechanical disintegration of the regenerated cellulose. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were used to investigate the morphology of the regenerated cellulose and cellulose nanoparticles, respectively. Diffuse reflectance infrared Fourier transform (DRIFT) spectroscopy and X-ray photoelectron spectroscopy (XPS) were used for chemical analysis of the cellulose materials, and crystallinity changes were investigated with X-ray diffraction (XRD). To demonstrate the unique properties of RCNPs, they were utilized as nanosized fillers for poly(vinyl alcohol) (PVA) composite films and their tensile properties were compared to those of pure PVA films and commercial CNC containing PVA composite films.
Materials and methods
Materials
Cellulose dissolving pulp (softwood) was obtained as dry sheets, whose properties are presented elsewhere.33 Before the dissolution experiment, the dry sheets were disintegrated in water followed by filtration, washing with technical ethanol (VWR) for 30 min, and filtration again before drying in an oven (60 °C). Microcrystalline cellulose (Avicel), guanidine hydrochloride (NH2C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH)NH2·HCl) and crystalline (anhydrous) phosphoric acid (H3PO4) (98%+, Acros Organics) were obtained from Sigma Aldrich and WVR, respectively. The commercial CNC suspensions (BGB ultra) were purchased from Blue Goose Biorefineries Inc., Canada.
NH)NH2·HCl) and crystalline (anhydrous) phosphoric acid (H3PO4) (98%+, Acros Organics) were obtained from Sigma Aldrich and WVR, respectively. The commercial CNC suspensions (BGB ultra) were purchased from Blue Goose Biorefineries Inc., Canada.
Cellulose dissolution and regeneration
At first, the DES was produced by mixing guanidine hydrochloride and anhydrous phosphoric acid in a beaker in a molar ratio of 1:2. After mixing, the beaker was placed in an oven at 80 °C until a clear, slightly viscous solution was obtained (the DES can be formed even at room temperature; however, an oven was used to limit the water content in the DES). The freshly prepared DES was cooled to room temperature in a desiccator. Then, dry cellulose was mixed with the DES at a mass ratio of 1:20 at room temperature for 24 h in a closed vessel. Dissolution was verified using an optical microscope. After the dissolution time, the mixture was poured into a beaker that contained ethanol. The regenerated cellulose was filtered and washed with ethanol and then with water until the filtrate became neutral. The product was stored at 4 °C in a non-dried state.
Disintegration of regenerated cellulose
The regenerated cellulose was diluted to a consistency of 1 wt% in deionized water. An Ultra Turrax treatment (10000 rpm) was then applied for 1 min. A microfluidizer (Microfluidics M-110EH-30, USA) was used for mechanical disintegration. The regenerated cellulose dispersion was passed twice through 400 and 200 μm chambers at a pressure of 1300 bar and four times through 200 and 87 μm chambers at a pressure of 1500 bar.
Cellulose nanoparticle reinforced poly(vinyl alcohol) composites
Prior to film fabrication, PVA was dissolved in deionized water at 90 °C to obtain 1% solution. After allowing the PVA solution to cool to room temperature, the desired amount of RCNPs or CNCs in water was added (1, 2.5, 5, and 10 wt% with respect to the mass of PVA). The dispersion was mixed for 5 minutes and sonicated in an ultrasound bath for 10 minutes to remove possible air bubbles and then cast on a polypropylene Petri disk. After around 5 days, the films were dry and they were peeled off from the Petri disk and stored at 50% humidity at 23 °C.Diffuse reflectance infrared Fourier transform spectroscopy
The chemical characterization of dissolving pulp and samples obtained after various dissolution times was performed using DRIFT. The spectra were collected with a Bruker Vertex 80v spectrometer (USA) from freeze-dried samples. The spectra were obtained in the 600–4000 cm−1 range, and 40 scans were taken at a resolution of 2 cm−1 from each sample.Scanning electron microscopy
SEM (Zeiss Zigma HD VP, Germany) images were obtained from the samples (freeze-dried via liquid nitrogen and vacuum drying) filtered on a polycarbonate membrane with a pore size of 0.2 μm. The accelerating voltage during imaging was 0.5 kV.Transmission electron microscopy
The morphological features of the fabricated RCNPs were analyzed with a JEOL JEM-2200FS transmission electron microscope (Japan). Each sample was prepared by dilution with ultrapure water. A carbon-coated copper grid was then prepared by coating it with polylysine. A small droplet of a 0.1% solution of polylysine was placed on top of the grid and allowed to stand for 3 min. The excess polylysine was removed from the grid by touching the droplet with one corner of a filter paper. A small droplet of the diluted RCNPs sample was then placed on top of the grid, and the excess sample was removed with the method described above. The samples were negatively stained with 2% uranyl acetate solution by placing a droplet on top of each specimen. The excess uranyl acetate was removed as described above. The grids were dried at room temperature and analyzed at 200 kV under standard conditions. The dimensions of the RCNPs from around 150 individual nanoparticles were measured using the ImageJ measuring program (1.50i).Optical transmittance
The transmittance of the 0.1% RCNP suspensions was measured at wavelengths of 200–800 nm using a Shimadzu UV-Vis spectrometer (Japan).X-ray diffraction
The crystalline structures of the original pulps and the CNCs were investigated using wide-angle X-ray diffraction. Measurements were conducted on a Rigaku SmartLab 9 kW rotating anode diffractometer (Japan) using Cu Kα radiation (40 kV, 135 mA; λ = 1.79030 Å). Samples were prepared by pressing tablets of freeze-dried celluloses to a thickness of 1 mm. Scans were taken over a 2θ (Bragg angle) range from 5° to 50° at a scanning speed of 10° s−1, using a 0.5° step.X-ray photoelectron spectroscopy
XPS of the original cellulose and the regenerated cellulose were obtained using a Thermo Fisher Scientific ESCALAB 250Xi (UK) XPS equipped with a monochromatic Al Kα X-ray source and operated at 300 W with a combination of electron flood gun and ion bombarding for charge compensation. The take-off angle was 45° in relation to the sample surface. Low-resolution survey scans were taken with a 1 eV step and 150 eV analyzer pass energy; high-resolution spectra were taken with a 0.1 eV step and 20 eV analyzer pass energy. All measurements were performed in an ultra-high vacuum chamber pressure (5 × 10−9 mbar). Before the measurement, the dry sample was pressed on the indium film.Tensile properties of cellulose nanoparticle reinforced poly(vinyl alcohol) composites
The tensile tests were performed with a universal material-testing machine (Zwick D0724587, Switzerland) equipped with a 100 N load cell. The films were cut into strips 5 mm wide, and their thicknesses were measured using a precision thickness gauge (Hanatek FT3, UK). Three different locations within the gauge length were measured to calculate the average thickness of each film. The thickness of the films varied from 45 to 58 μm (Fig. S3†) depending on the amount and type of added nanoparticles. For the tensile tests, a 40 mm gauge length was set under a strain rate of 4 mm min−1, and five strips in total were measured for each sample. The tests were conducted in a relative humidity of 50% at a temperature of 23 °C using a prestrain of ∼0.1 N. Prior to testing, each specimen was conditioned for at least two days in this environment.Results and discussion
Dissolution of cellulose was accomplished using the DES formed between crystalline (anhydrous) phosphoric acid and guanidine hydrochloride. When the two components were mixed together, a clear, slightly viscous liquid was formed after heating at 80 °C for 30 min. The DES was formed even at room temperature in a desiccator after several days without mixing. Phosphoric acid is highly hygroscopic, resulting in wetting and formation of a liquid if the chemical is left at room temperature in an open vessel. Therefore, before the cellulose was dissolved, the DES was dried at 80 °C for 30 min and stored in a desiccator.Cellulose was dissolved in the DES after mixing for 24 h at room temperature in a closed vessel to produce a clear viscous solution. Regeneration of cellulose could be accomplished using an anti-solvent, such as ethanol or water. The solution becomes green/blue after 24 h of dissolution, indicating that some chromophoric groups are formed, most likely from cellulose degradation products (Fig. 1). For example, it has been reported that coloration of the cellulose solution in N-methylmorpholine N-oxide (NMMO) is due to the formation of condensation products of carbohydrates.34 After regeneration and washing of cellulose, the final product appeared white, indicating that chromophoric groups were removed by water washing.
 | ||
| Fig. 1 Dissolving pulp directly after addition in the DES (left) and after 24 hours at room temperature under mixing (right). | ||
Cellulose could not be dissolved in an open vessel under normal conditions or when 85% phosphoric acid in water is used instead of crystalline phosphoric acid. The poor dissolution of cellulose in DES in presence of water is contrary to the previous studies where 85% phosphoric acid in water has been shown to be efficient solvent for cellulose.35 Water is known to act as the competing hydrogen bond forming agent during the cellulose dissolution.36 For example, anhydrous NMMO and hydrated NMMO are efficient solvents for cellulose whereas when two or more water molecules are present, NMMO no longer acts as a solvent for cellulose.37 In addition, it is well known that the amount of water is a crucial parameter in the dissolution of cellulose in ionic liquids. Some ionic liquids can tolerate small quantities of water (a small amount of water can even act as the co-solvent for cellulose38), whereas anhydrous conditions are required with other ionic liquids.39 In DES systems, it has been shown that a small amount of water can be incorporated as part of the DES system, altering their hydrogen-bonding pattern.40 Therefore, it is likely that water in the phosphoric acid–guanidine hydrochloride DES prevents efficient interaction between cellulose and the DES, thus hindering the dissolution of cellulose.
The maximum dissolution of both dissolving pulp and MCC in the DES at room temperature was found to be 5%. At higher concentrations, a clear solution was obtained with both cellulose samples, however, a few small fiber fragments were observed in the optical microscopy images. When high molecular weight cellulose (bleached birch pulp33) was used, small fiber fragments were present even at a cellulose concentration of 5%. The 5% solution is not optimal for the production of traditional regenerated cellulose materials, such as fibers and films. However, 5% is similar to the cellulose fibers/liquid ratio used during the production of CNCs by sulfuric acid hydrolysis.41
High acid concentration in the DES can lead to the hydrolysis of cellulose even under mild conditions (room temperature). After regeneration, the yield of both samples was around 80%, indicating that some of the material dissolved during the washing step. The DP of both the samples decreased to around 80, which is significantly lower compared to the original DP of dissolving pulp (1800) and MCC (373). The similar DP of both regenerated samples shows that the severity of the hydrolysis of cellulose is independent of the DP of the starting material. The independence of the DP of regenerated cellulose on the DP of the starting material might indicate that during the dissolution, cellulose reached the leveling-off DP. It has been previously shown that the leveling-off DP depends on the properties of cellulose, for example on the crystalline structure.42 In the mild acid hydrolysis, the leveling-off DP of cellulose I is shown to be 170, whereas the leveling-off DP of cellulose II has been observed to be less (50).43 Therefore, it can be assumed that the DP of regenerated cellulose from the DES is close the leveling-off DP of cellulose II.
Although the DP of regenerated cellulose is most likely too low for bulk cellulose applications, like films and yarns, it is in line with the DP of some cellulosic nanomaterials reported in the literature.44,45 It can be assumed that the dissolution of cellulose is due to the degradation of cellulose. It is well known that the DP of cellulose has a high impact on the dissolution of cellulose, for example in aqueous NaOH/urea. High DP cellulose is insoluble in aqueous NaOH/urea, whereas lowering the DP results in efficient dissolution.46
The possible chemical modification of cellulose during the dissolution was investigated using XPS and DRIFT spectroscopy. Based on XPS, the elemental constitution of both dissolving pulp and MCC was only carbon and oxygen, which is in line with the chemical structure of cellulose. After dissolution and regeneration, small amounts of nitrogen and phosphorus were observed in both samples. Regenerated MCC and dissolving pulp contained 0.42 wt% and 0.82 wt% of nitrogen, corresponding to 0.1 mmol g−1 and 0.2 mmol g−1 of guanidine groups, respectively.
Based on the XPS analysis, the ratio between nitrogen and phosphorus in both samples was around 3:1. In addition, no chloride was observed. Therefore, it is possible that during the formation of DES, a guanidine salt of phosphoric acid is formed. Although phosphoric acid is a weaker acid than hydrochloric acid, it is possible that equilibrium exists between the formation of guanidine phosphonate and hydrochloride. Hydrochloric acid is a volatile component and therefore if a minor amount is formed during the DES preparation, it might be removed, especially at higher temperature, leading to the formation of guanidine phosphate.
Due to the minor chemical modification of cellulose, the dissolution of cellulose in the DES cannot be seen as a pure physical phenomenon, but at least partly derivatization dissolution. Derivatizing dissolution is well known for example when cellulose is dissolved in formic acid.47 In addition, it has been reported that the dissolution of cellulose in alkylimidazolium ionic liquids, like 1-ethyl-3-methylimidazolium acetate leads to a minor chemical modification of cellulose as the imidazolium group can be chemically attached to cellulose during the dissolution.48
Previously, the phosphorylation of cellulose has been suggested to be one of the main factors for the dissolution of cellulose in aqueous phosphoric acid.35 However, in the case of aqueous phosphoric acid, the phosphate groups are readily hydrolyzed back to phosphoric acid by addition of water as the anti-solvent and non-phosphorylated regenerated cellulose is obtained. The main difference between aqueous phosphoric acid and the DES used here is the presence of water. In the DES, the amount of water is limited (but not excluded due to the hygroscopicity of the used chemicals), whereas in aqueous phosphoric acid there is 15% of water in the solvent system. Due to the presence of a significant amount of water, the degree of esterification of cellulose in aqueous phosphoric acid is likely lower compared to esterification in the DES. Therefore, a small amount of phosphate ester groups remains after regeneration of cellulose from the DES, despite the washing of the product with an excess of water. Previously, highly phosphorylated cellulose was obtained using phosphoric acid together with ammonium polyphosphate49 and a small amount of phosphate ester groups was attached to CNCs when aqueous phosphoric acid was used as the hydrolytic medium.50 Although both of the reactions were done at elevated temperature, compared to room temperature used in the current study, it can be assumed that cellulose phosphate is formed during the cellulose dissolution in the DES and a small amount of phosphate groups remains in regenerated cellulose after washing with water.
The presence of the guanidine groups was observed in the DRIFT spectra of the regenerated cellulose (Fig. 2). After regeneration, a new peak appeared around 1660 cm−1. This peak is superimposed on the water vibration band at 1640 cm−1 (can be seen in the original dissolving pulp and MCC spectra) and the NH2 vibration of the guanidine group (at a wavenumber of 1670 cm−1).51 No direct observation of phosphate groups can be seen in the spectra due to small amounts of phosphate groups and the overlap with the original peak of the cellulose related to C–O–C from the glucosidic units.52
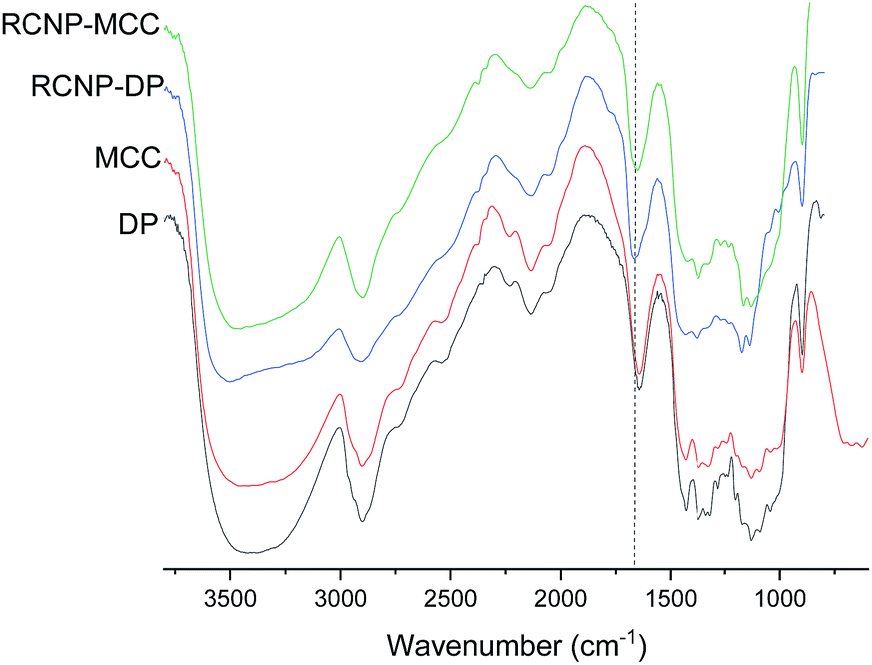 | ||
| Fig. 2 DRIFT spectra of the original cellulose and product obtained after different dissolution times. NH2 vibration of the guanidine group is indicated by a dashed line. | ||
The crystalline structure of cellulose was observed to change from cellulose I to cellulose II during the dissolution in the DES (Fig. 3). The XRD diffraction pattern of the original cellulose exhibited the characteristic cellulose I main diffraction angles around 22.5°, 15°, and 16.5° (appears as a broad signal centered close to 15°) corresponding to the primary (200) lattice plane peak and the overlapped (110) and (1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) peaks, respectively.53 The original CrI of both MCC and dissolving pulp was around 80%. After the dissolution and regeneration, characteristic cellulose I peaks disappeared and were replaced with the main peaks of cellulose II ((020), (110), and (10)) with the diffraction angles close to 22°, 20°, and 12°. These observations are in line with previous studies in which cellulose was dissolved in ionic liquids.54–56
0) peaks, respectively.53 The original CrI of both MCC and dissolving pulp was around 80%. After the dissolution and regeneration, characteristic cellulose I peaks disappeared and were replaced with the main peaks of cellulose II ((020), (110), and (10)) with the diffraction angles close to 22°, 20°, and 12°. These observations are in line with previous studies in which cellulose was dissolved in ionic liquids.54–56
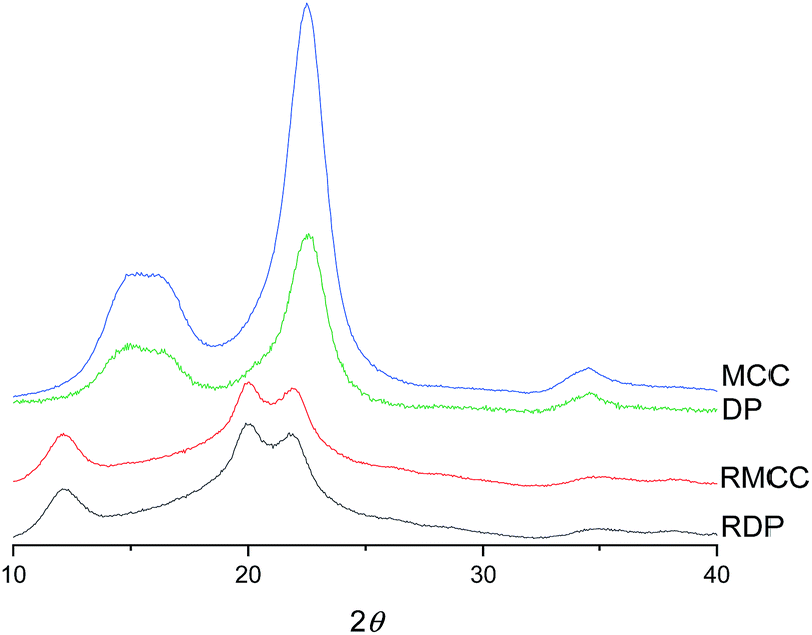 | ||
| Fig. 3 XRD diffraction patterns of original and regenerated microcrystalline cellulose (MCC and RMCC) and dissolving pulp (DP and RDP). | ||
RCNPs were produced from the regenerated cellulose using mechanical disintegration with a microfluidizer. After a 1% suspension of the regenerated cellulose was passed through the chambers of the microfluidizer, a highly transparent solution was obtained. At a wavelength of 600 nm, the transmittance of 0.1% MCC and dissolving pulp-based RCNP suspensions was 84% and 94%, respectively (Fig. 4c). The high transmittance of the RCNP suspensions indicates that the size of the nanocellulose particles was uniformly distributed, and bigger particles and agglomerates were present only in minor amounts. In addition, the RCNP dispersions remained stable at room temperature at least for several months, demonstrating their good colloidal stability.
 | ||
| Fig. 4 TEM image of MCC (a) and dissolving pulp (b) RCNPs and (c) the transmittance of 0.1% RCNP dispersions. | ||
In the TEM images, the RCNPs appeared as coarse, fibrous nanoparticles and the diameters of MCC and dissolving pulp RCNPs were 5.8 ± 1.4 and 5.6 ± 1.4 nm, respectively (Fig. 4a and b). The length of the individual fibers was in tens of nanometers. It was also observed that at higher concentrations the particles formed a network-like structure, similar to the CNFs (see ESI† for more TEM images). Compared to the CNFs and CNCs, the RCNPs appeared more flexible, as the typical long and strain regions of the CNFs and CNCs could not be observed. This was likely due to the different formation process and crystalline structure of the RCNPs compared to those of the CNFs and CNCs. CNFs and CNCs are produced from natural fibers, which are formed during biosynthesis as rod-like regions already exist in the natural fibers and are liberated during the production of CNFs and CNCs. However, in the case of RCNPs, the fiber structure is completely disintegrated after dissolution, and the cellulose molecules are more freely oriented during regeneration.
The formation of fiber-like regenerated nanoparticles after the disintegration of cellulose regenerated from the DES is in contrast to a previous study where circular nanoparticles were obtained after homogenization of sugarcane bagasse cellulose regenerated from an ionic liquid.57 In addition, spherical nanosized cellulose particles were previously produced after cellulose regeneration from phosphoric acid58 and thermally induced gelation from an aqueous NaOH/thiourea solution.59 Therefore, previous regenerated nanoparticles have a significantly different morphology compared to the current study. As stated before, the use of phosphoric acid-guanidine hydrochloride DES results in a severe decrease in the DP of cellulose. Therefore, it can be assumed that the formation of thin, fiber-like regenerated cellulose nanoparticles is due to the hydrolysis of cellulose during the dissolution. It is possible that shorter cellulose polymer chains are easier to disintegrate into small nanoparticles using mechanical treatment.
The RCNPs were investigated as a nanosized additive for PVA. Different cellulose nanoparticles are known to improve the tensile strength and modulus of many polymers, like PVA.60 The use of CNCs or CNFs generally leads to a stiffer material, resulting in a decrease in the elongation. It can be seen from Fig. 5a that addition of commercial CNCs to PVA resulted in a composite with slightly lower elongation compared to pristine PVA. On the other hand, when RCNPs were used at low concentrations (1–5 wt%), the ductility of the composites was significantly improved compared to that of pristine PVA and CNC reinforced composites. The highest elongation at break, 104%, was found for the composite containing 2.5 wt% of MCC-based RCNPs, being around 2.8 times higher compared to that of the pure PVA film. The elongation of composites containing dissolving pulp-based RCNPs was slightly lower compared to that of MCC-based RCNPs, but still over two times higher compared to that of PVA and CNC reinforced PVA films. At a high concentration (10 wt%), the elongation of RCNP composites was similar or even slightly lower compared to that of PVA.
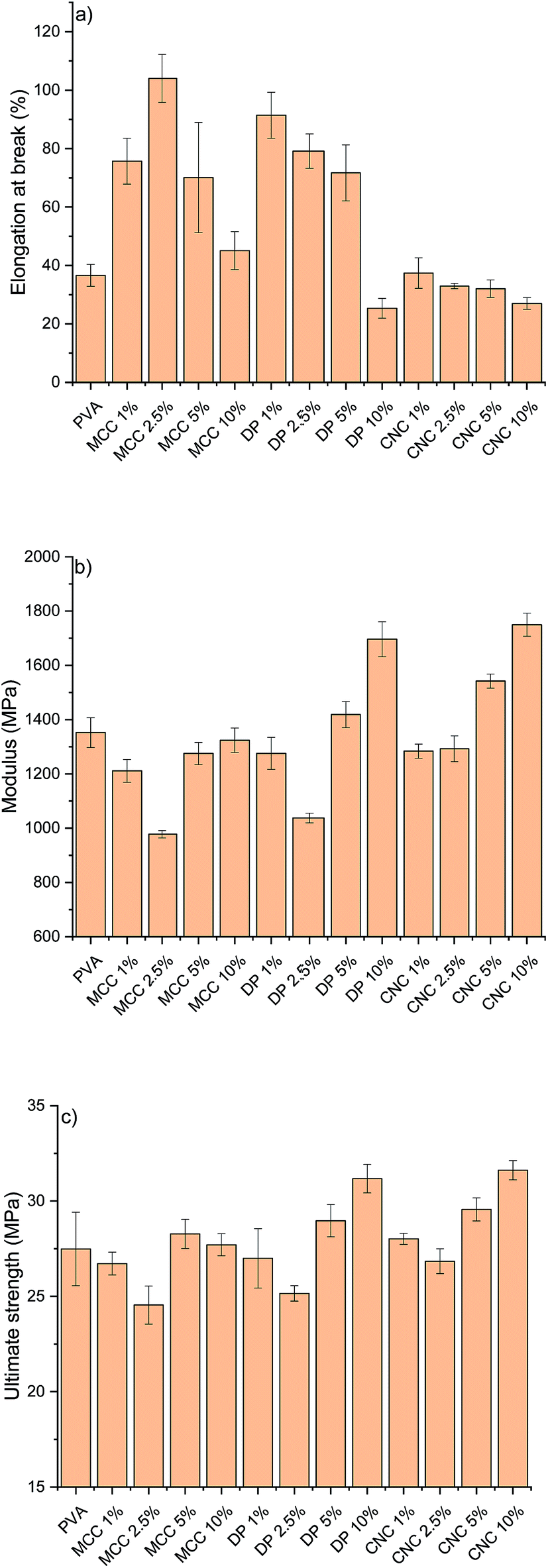 | ||
| Fig. 5 (a) Elongation at break, (b) modulus, and (c) tensile strength of pristine PVA films and composites with various amounts of different nanoparticles (MCC = MCC based regenerated cellulose nanoparticles; DP = dissolving pulp based regenerated cellulose nanoparticles; CNC = commercial cellulose nanocrystals). | ||
It appears that at low concentrations, RCNPs have plasticizer-like behavior. In contrast to the small molecular plasticizer, which typically increases the elongation of polymers, but decreases the modulus and strength,61,62 the use of RCNPs does not decrease the strength of the PVA (Fig. 5c). The ultimate tensile strength of PVA composites containing 1–5 wt% of RCNPs was in line with that of pure PVA. It should be also noted that the ultimate tensile strength of PVA films reinforced with 1–5 wt% CNCs did not significantly differ from that of pure PVA. At low RCNP content, the modulus decreased slightly compared to that of pure PVA and CNC reinforced films. Especially at 2.5 wt%, the modulus of PVA containing both MCC and dissolving pulp based RCNPs decreased around 30%.
At the filler content of 10 wt%, RCNPs had a similar reinforcement effect to CNCs, i.e. the elongation of PVA decreased along with improvement in strength properties. Especially, dissolving pulp-based RCNPs exhibited the potential ability to reinforce the PVA film. The modulus and ultimate tensile strength of the PVA film containing 10 wt% of dissolving pulp-based RCNPs significantly improved compared to those of pure PVA and the values were in line with those of the film containing 10 wt% of CNCs.
A possible explanation for the different behavior of RCNPs compared to the CNCs might be in their morphology. CNCs are stiff, rod-like materials, which provide reinforcement to the polymer matrix, but do not allow polymer chains to slide when the tension is increased. On the other hand, from the TEM images it is apparent that RCNPs could be randomly oriented from kinks. In addition, their diameters were in tens of nanometers, so they might work similarly to bearing balls, i.e. their small size allows them to rotate along with sliding PVA chains, while constantly breaking and newly forming hydrogen bonds provide strength to the composite structure. Due to their nanometric size (i.e. large surface area) and similar chemical nature (i.e. the presence of a high amount of hydroxyl groups), RCNPs have good interaction between PVA molecules, and their flexibility allows polymer chains to move more freely during the tension at lower nanoparticle loading. Unlike the molecular plasticizers, which interfere with internal bonding of the polymer matrix, RCNPs also provide reinforcement as they also contain some stiffer, crystalline regions (Fig. 3).
On the other hand, at higher nanoparticle loading, RCNPs could form a more network-like structure similar to CNFs (see TEM images in the ESI†). The formed network then provides a structure that is more rigid and the sliding of PVA chains is limited. This can be seen as the improved tensile strength and modulus, but also in the decrease of elongation-at-break.
In the literature, several different cellulose-based nanomaterials have been used as reinforcement agents for PVA and other polymers. For example, CNFs and bacterial cellulose were used to increase the modulus of PVA gradually from 126 MPa to 200 MPa and 260 MPa, respectively. However, a gradual decrease in the elongation was observed when the amount of CNF and BC was increased from 0 to 10%. Decrease in the elongation was especially prominent in the case of CNFs.63 Strong PVA nanocomposites have been obtained using cross-linking CNC13,64 and sodium tetraborate decahydrate cross-linking PVA with CNCs.65 However, decrease in the elongation is severe when nanoparticles are cross-linked with PVA. Spherical nanocellulose from linen has been shown to increase the tensile strength of PVA at a concentration of 20%.66 When the amount of nanoparticles was increased, the elongation was observed to increase from 26.8 to 108.5%, which is similar to the results obtained with RCNPs. However, as high as 60% nanoparticle loading was required to increase the elongation at break of the PVA.
Recently, chemically modified UV-blocking CNFs have been produced, and were used to significantly increase the properties of PVA. At a CNF content of 5%, the tensile strength of PVA was increased from around 30 MPa to over 90 MPa.67 A similar increase in the modulus was also observed. Interestingly, when the amount of CNFs was increased from 0 to 10%, elongation at break gradually increased from around 150% to over 300%. This 2.3 times increase in elongation was slightly lower than what was obtained with 2.5% of MCC-based RCNPs (2.8 times increase). However, it should be noted that the use of UV-blocking CNFs showed a simultaneous increase in the tensile strength and modulus along with improved elongation. The increase in the elongation of PVA with UV-blocking CNFs was supposed to be due to the presence of the lubricating epoxidized soybean oil moiety in the modified CNFs structure, whereas in the case of RCNPs, the increase in elongation is likely due to the morphology of nanoparticles. It should be noted that although UV-blocking CNFs were readily produced from well-known (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) oxidized CNFs, the production requires several solvent exchange steps, use of organic solvents and protection gas, and high temperature.
Conclusions
A new type of fibrous cellulose nanomaterial was produced using mechanical disintegration of dissolved and regenerated wood cellulose. Cellulose was dissolved in a DES based on guanidine hydrochloride and anhydrous phosphoric acid at room temperature. The driving force toward the dissolution was attributed to the decrease in the DP of cellulose due to the highly acidic medium. Uniformly sized, fibrous RCNPs with a diameter around 6 nm were produced from regenerated cellulose with mechanical disintegration. Due to the flexible nature and short diameters of RCNPs, at lower concentrations they could be used to significantly improve the ductility of PVA, with minimal decrease in the strength and modulus. At higher concentrations, the elongation of PVA films decreased while the tensile strength and modulus increased in a similar manner to those of commercial CNCs. Due to the use of easily available chemicals at room temperature, the method introduced here could open a sustainable way to produce cellulosic nanomaterials with interesting morphologies and properties for composite applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Matias Nissinen is acknowledged for his contribution to the experimental part of the study. Elisa Wirkkala, Marcin Selent, Ilkka Miinalainen, and Sami Saukko are gratefully acknowledged for their contribution to the elemental and limiting viscosity analysis, XRD and XPS, SEM, and TEM measurements, respectively. Ass. Prof. Henrikki Liimatainen is gratefully acknowledged for his help with the manuscript writing and fruitful discussion.References
- W. H. De Jong and P. J. Borm, Int. J. Nanomed., 2008, 3, 133–149 CrossRef CAS.
- X. Chen, D. Ji, X. Wang and L. Zang, IOP Conf. Ser. Earth Environ. Sci., 2017, 51, 012004 CrossRef.
- Y.-C. Yeh, B. Creran and V. M. Rotello, Nanoscale, 2012, 4, 1871–1880 RSC.
- M. Rai, A. P. Ingle and P. Paralikar, Expert Rev. Anti-Infect. Ther., 2016, 14, 969–978 CrossRef CAS PubMed.
- J. Pal and T. Pal, Nanoscale, 2015, 7, 14159–14190 RSC.
- A. B. Djurišić, Y. H. Leung, A. M. C. Ng, X. Y. Xu, P. K. H. Lee, N. Degger and R. S. S. Wu, Small, 2015, 11, 26–44 CrossRef PubMed.
- A. Nasir, A. Kausar and A. Younus, Polym.-Plast. Technol. Eng., 2015, 54, 325–341 CrossRef CAS.
- A. Reisch and A. S. Klymchenko, Small, 2016, 12, 1968–1992 CrossRef CAS.
- A. Puri, K. Loomis, B. Smith, J.-H. Lee, A. Yavlovich, E. Heldman and R. Blumenthal, Crit. Rev. Ther. Drug Carrier Syst., 2009, 26, 523–580 CrossRef CAS.
- A. P. Richter, J. S. Brown, B. Bharti, A. Wang, S. Gangwal, K. Houck, E. A. Cohen Hubal, V. N. Paunov, S. D. Stoyanov and O. D. Velev, Nat. Nanotechnol., 2015, 10, 817–823 CrossRef CAS PubMed.
- H. Liu and L. C. Brinson, Compos. Sci. Technol., 2008, 68, 1502–1512 CrossRef CAS.
- N. G. Khlebtsov and L. A. Dykman, J. Quant. Spectrosc. Radiat. Transfer, 2010, 111, 1–35 CrossRef CAS.
- J. A. Sirviö, M. Visanko, J. P. Heiskanen and H. Liimatainen, J. Mater. Chem. A, 2016, 4, 6368–6375 RSC.
- D. Klemm, F. Kramer, S. Moritz, T. Lindström, M. Ankerfors, D. Gray and A. Dorris, Angew. Chem., Int. Ed., 2011, 50, 5438–5466 CrossRef CAS PubMed.
- J. Zhang, T. J. Elder, Y. Pu and A. J. Ragauskas, Carbohydr. Polym., 2007, 69, 607–611 CrossRef CAS.
- T. Fattahi Meyabadi, F. Dadashian, G. Mir Mohamad Sadeghi and H. Ebrahimi Zanjani Asl, Powder Technol., 2014, 261, 232–240 CrossRef CAS.
- M. Beaumont, T. Nypelö, J. König, R. Zirbs, M. Opietnik, A. Potthast and T. Rosenau, Green Chem., 2016, 18, 1465–1468 RSC.
- M. Beaumont, H. Rennhofer, M. Opietnik, H. C. Lichtenegger, A. Potthast and T. Rosenau, ACS Sustainable Chem. Eng., 2016, 4, 4424–4432 CrossRef CAS.
- J. A. Sirviö and J. P. Heiskanen, ChemSusChem, 2017, 10, 455–460 CrossRef PubMed.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- A. Paiva, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, ACS Sustainable Chem. Eng., 2014, 2, 1063–1071 CrossRef CAS.
- Y. Dai, J. van Spronsen, G.-J. Witkamp, R. Verpoorte and Y. H. Choi, Anal. Chim. Acta, 2013, 766, 61–68 CrossRef CAS PubMed.
- B.-Y. Zhao, P. Xu, F.-X. Yang, H. Wu, M.-H. Zong and W.-Y. Lou, ACS Sustainable Chem. Eng., 2015, 3, 2746–2755 CrossRef CAS.
- K. Radošević, M. Cvjetko Bubalo, V. Gaurina Srček, D. Grgas, T. Landeka Dragičević and I. Radojčić Redovniković, Ecotoxicol. Environ. Saf., 2015, 112, 46–53 CrossRef PubMed.
- A. Abo-Hamad, M. Hayyan, M. A. AlSaadi and M. A. Hashim, Chem. Eng. J., 2015, 273, 551–567 CrossRef CAS.
- O. S. Hammond, K. J. Edler, D. T. Bowron and L. Torrente-Murciano, Nat. Commun., 2017, 8, 14150 CrossRef CAS PubMed.
- P. Li, J. A. Sirviö, A. Haapala and H. Liimatainen, ACS Appl. Mater. Interfaces, 2017, 9, 2846–2855 CrossRef CAS PubMed.
- J. A. Sirviö, M. Visanko and H. Liimatainen, Green Chem., 2015, 17, 3401–3406 RSC.
- T. Selkälä, J. A. Sirviö, G. S. Lorite and H. Liimatainen, ChemSusChem, 2016, 9, 3074–3083 CrossRef PubMed.
- J. A. Sirviö, Carbohydr. Polym., 2018, 198, 34–40 CrossRef PubMed.
- J. A. Sirviö, M. Visanko and H. Liimatainen, Biomacromolecules, 2016, 17, 3025–3032 CrossRef PubMed.
- Y. Liu, B. Guo, Q. Xia, J. Meng, W. Chen, S. Liu, Q. Wang, Y. Liu, J. Li and H. Yu, ACS Sustainable Chem. Eng., 2017, 5, 7623–7631 CrossRef CAS.
- J. Sirvio, U. Hyvakko, H. Liimatainen, J. Niinimaki and O. Hormi, Carbohydr. Polym., 2011, 83, 1293–1297 CrossRef CAS.
- T. Rosenau, A. Potthast, W. Milacher, I. Adorjan, A. Hofinger and P. Kosma, Cellulose, 2005, 12, 197–208 CrossRef CAS.
- Y.-H. P. Zhang, J. Cui, L. R. Lynd and L. R. Kuang, Biomacromolecules, 2006, 7, 644–648 CrossRef CAS PubMed.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974–4975 CrossRef CAS PubMed.
- E. Maia, A. Peguy and S. Pérez, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1981, 37, 1858–1862 CrossRef.
- R. Parthasarathi, K. Balamurugan, J. Shi, V. Subramanian, B. A. Simmons and S. Singh, J. Phys. Chem. B, 2015, 119, 14339–14349 CrossRef CAS PubMed.
- M. Mazza, D.-A. Catana, C. Vaca-Garcia and C. Cecutti, Cellulose, 2009, 16, 207–215 CrossRef CAS.
- O. S. Hammond, D. T. Bowron and K. J. Edler, Angew. Chem., Int. Ed., 2017, 56, 9782–9785 CrossRef CAS PubMed.
- E. Mascheroni, R. Rampazzo, M. A. Ortenzi, G. Piva, S. Bonetti and L. Piergiovanni, Cellulose, 2016, 23, 779–793 CrossRef CAS.
- O. A. Battista, S. Coppick, J. A. Howsmon, F. F. Morehead and W. A. Sisson, Ind. Eng. Chem., 1956, 48, 333–335 CrossRef CAS.
- E. Niinivaara, S. A. A. Arshath, K. Nieminen, A. Bismarck and E. Kontturi, Adv. Sustainable Syst., 2018, 2, 1800026 CrossRef.
- B. Sun, M. Zhang, Q. Hou, R. Liu, T. Wu and C. Si, Cellulose, 2015, 23, 439–450 CrossRef.
- M. Lee, M. H. Heo, H. Lee, H.-H. Lee, H. Jeong, Y.-W. Kim and J. Shin, Green Chem., 2018, 20, 2596–2610 RSC.
- H. Qi, C. Chang and L. Zhang, Cellulose, 2008, 15, 779–787 CrossRef CAS.
- T. Heinze and T. Liebert, Prog. Polym. Sci., 2001, 26, 1689–1762 CrossRef CAS.
- G. Ebner, S. Schiehser, A. Potthast and T. Rosenau, Tetrahedron Lett., 2008, 49, 7322–7324 CrossRef CAS.
- N. K. Luneva and T. I. Ezovitova, Russ. J. Appl. Chem., 2014, 87, 1558–1565 CrossRef CAS.
- S. Camarero Espinosa, T. Kuhnt, E. J. Foster and C. Weder, Biomacromolecules, 2013, 14, 1223–1230 CrossRef CAS PubMed.
- W. J. Jones, Trans. Faraday Soc., 1959, 55, 524–531 RSC.
- D. M. Suflet, G. C. Chitanu and V. I. Popa, React. Funct. Polym., 2006, 66, 1240–1249 CrossRef CAS.
- A. D. French, Cellulose, 2014, 21, 885–896 CrossRef CAS.
- H. Zhang, J. Wu, J. Zhang and J. He, Macromolecules, 2005, 38, 8272–8277 CrossRef CAS.
- S. Tang, G. A. Baker, S. Ravula, J. E. Jones and H. Zhao, Green Chem., 2012, 14, 2922–2932 RSC.
- F. Boissou, A. Mühlbauer, K. D. O. Vigier, L. Leclercq, W. Kunz, S. Marinkovic, B. Estrine, V. Nardello-Rataj and F. Jérôme, Green Chem., 2014, 16, 2463–2471 RSC.
- J. Li, X. Wei, Q. Wang, J. Chen, G. Chang, L. Kong, J. Su and Y. Liu, Carbohydr. Polym., 2012, 90, 1609–1613 CrossRef CAS PubMed.
- X. Hao, W. Shen, Z. Chen, J. Zhu, L. Feng, Z. Wu, P. Wang, X. Zeng and T. Wu, Carbohydr. Polym., 2015, 123, 297–304 CrossRef CAS PubMed.
- L. Weng, L. Zhang, D. Ruan, L. Shi and J. Xu, Langmuir, 2004, 20, 2086–2093 CrossRef CAS.
- B. Tan, Y. Ching, S. Poh, L. Abdullah and S. Gan, Polymers, 2015, 7, 2205–2222 CrossRef CAS.
- M. Mohsin, A. Hossin and Y. Haik, J. Appl. Polym. Sci., 2011, 122, 3102–3109 CrossRef CAS.
- H. Tian, D. Liu, Y. Yao, S. Ma, X. Zhang and A. Xiang, J. Food Sci., 2017, 82, 2926–2932 CrossRef CAS PubMed.
- K. Yuwawech, J. Wootthikanokkhan and S. Tanpichai, Effects of Two Different Cellulose Nanofiber Types on Properties of Poly(vinyl alcohol) Composite Films, https://www.hindawi.com/journals/jnm/2015/908689/, accessed November 20, 2018 Search PubMed.
- J. A. Sirviö, S. Honkaniemi, M. Visanko and H. Liimatainen, ACS Appl. Mater. Interfaces, 2015, 7, 19691–19699 CrossRef PubMed.
- S. Tanpichai and K. Oksman, J. Appl. Polym. Sci., 2018, 135, 45710 CrossRef.
- M. M. Ibrahim, W. K. El-Zawawy and M. A. Nassar, Carbohydr. Polym., 2010, 79, 694–699 CrossRef CAS.
- X. Niu, Y. Liu, G. Fang, C. Huang, O. J. Rojas and H. Pan, Biomacromolecules, 2018, 19, 4565–4575 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: High resolution TEM images of RCNPs. See DOI: 10.1039/c8ta09959f |
| This journal is © The Royal Society of Chemistry 2019 |