
Adapting FeS2 micron particles as an electrode material for lithium-ion batteries via simultaneous construction of CNT internal networks and external cages†
 a
Fang
Lian,
*a
a
Fang
Lian,
*a
Abstract
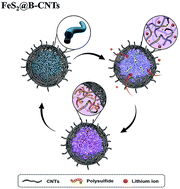
Volume changes, polysulfide shuttle effects, and low Li-ion/electronic conductivity of sulfide electrodes limit their application in Li-ion batteries with high-energy density. Here, micron FeS2 particles with bifunctional carbon nanotubes (FeS2@B–CNTs) including CNT internal conductive networks and external protective cages were prepared by a one-step solvothermal method. The internal CNTs anchor FeS2via chemical binding, which much more effectively inhibited the shuttle effect than physical confinement alone. Meanwhile, reticular CNT cages were generated on the surface of the composite FeS2 micron particles, buffering volumetric changes during cycling and further restraining the shuttle of polysulfides. Additionally, a three-dimensional CNT framework offered a primarily continuous charge transfer pathway. The FeS2@B–CNTs electrode had a high initial coulombic efficiency of 91.3% and delivered a capacity of 698 mA h g−1 over 500 cycles at 1000 mA g−1. The Li-ion diffusion coefficient is two orders of magnitude higher than those of previous reports, which improved the rate performance of the FeS2@B–CNTs electrode. These studies on micron FeS2 spheres shed light on the design strategies for the application of sulfide electrodes.
 Please wait while we load your content...
Please wait while we load your content...

