
Conjugated materials containing dithieno[3,2-b:2′,3′-d]pyrrole and its derivatives for organic and hybrid solar cell applications
Yanfang
Geng
 a,
Ailing
Tang
a,
Keisuke
Tajima
*c,
Qingdao
Zeng
a and
Erjun
Zhou
*ab
a,
Ailing
Tang
a,
Keisuke
Tajima
*c,
Qingdao
Zeng
a and
Erjun
Zhou
*ab
aCAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing 100190, China. E-mail: zhouej@nanoctr.cn
bHenan Institutes of Advanced Technology, Zhengzhou University, 97 Wenhua Road, Zhengzhou 450003, China
cEmergent Functional Polymers Research Team, RIKEN Center for Emergent Matter Science (CEMS), 2-1 Hirosawa, Wako 351-0198, Japan. E-mail: keisuke.tajima@riken.jp
First published on 14th November 2018
Abstract
The growing number of new π-conjugated polymers and small-molecules has promoted the rapid development of organic solar cells (OSCs) over the past two decades. Among the most widely used building blocks, dithieno[3,2-b:2′,3′-d]pyrrole (DTP) and its derivatives have occupied an important position in both electron-rich and electron-deficient semiconductors since their first introduction into photovoltaic materials in 2007. The incorporation of basic DTP units such as N-alkyl DTP, N-acyl DTP and pyrrolo[3,2-d:4,5-d′]bisthiazole (PBTz), and their derivatives including fused DTP, and the lactam and imide conjugated heterocycles into photovoltaic polymers and small-molecules largely enhanced the open circuit voltage (VOC), short-circuit current (JSC), fill factor (FF) and finally power conversion efficiency (PCE) of OSCs. In this review, we provide a comprehensive overview of the progress in DTP-based materials, summarize the current state of DTP research and describe the relationship between the molecular structure and device performance, which could pave a way for the further rational design of novel DTP-derived building blocks and the corresponding photovoltaic materials.
1. Introduction
Organic solar cells (OSCs) have received considerable attention due to their fascinating advantages of low-cost and large-area production through solution processes. The first OSC was fabricated with a bilayer structure by Tang et al. in 1986, which showed a low power conversion efficiency (PCE) owing to the poor exciton dissociation efficiency.1 In 1995, the bulk heterojunction (BHJ) OSC consisting of interpenetrating p-type organic donor materials (D) and n-type organic acceptor materials (A) solved this problem.2,3 After decades of extensive investigation, PCEs have exceeded the threshold of 10% and reached 17% approaching the requirement of industrial applications.4–7 The large improvement of the efficiency in OSCs can be mainly attributed to (1) the design and synthesis of novel organic photovoltaic materials, (2) optimization of fabrication conditions for the devices and (3) investigation on the mechanism of device operation.The molecular design is a fundamental and important factor to enhance the device performance. Usually, organic photovoltaic materials should meet several requirements such as broad and strong light absorption, suitable frontier molecular orbitals including the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) energy levels, high charge mobility and good solubility.8–13 To realize these parameters, the chemical structures of photovoltaic materials should be finely controlled. Over the past two decades, it had been clearly proven that the use of alternating donor–acceptor (D–A) copolymers, incorporating electron-donating and electron-accepting units into their backbones, is the most promising strategy to control the properties of the photovoltaic polymers.9 Therefore, the exploration of promising electron-donating and electron-accepting building blocks is always a hot topic.
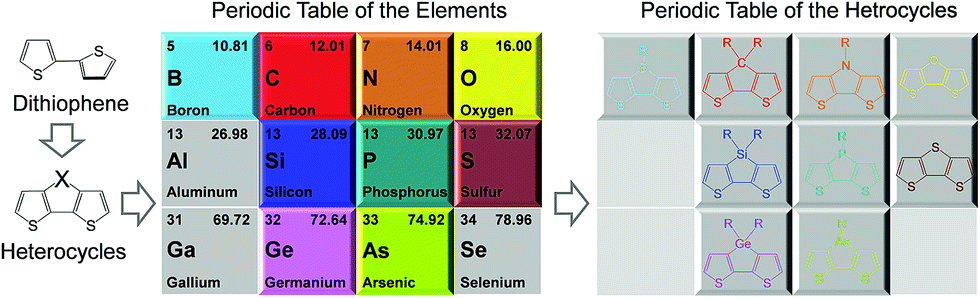
Thiophene is the simplest but the most important electron-donating building block to construct conjugated materials due to easily available methods and high performance.14–16 However, rotation around the inter-annular single bonds along the backbone leads to reduced planarity between the adjacent thiophene units (Fig. 1). It is desirable to lock the entire π-system into a coplanar conformation through a second strand of bonds, affording a fused-ring, ladder type molecular constitution. Strikingly, the introduction of heteroatoms at the bridge of neighboring thiophene rings to form various fused tricyclic systems can maximize planarity, which can enhance the overlap of p orbitals and provide more effective conjugation along the backbone.17
 | ||
| Fig. 1 Planar building blocks based on fused bithiophenes with various atoms. | ||
Typical fused bithiophenes are bridged by boron (B), carbon (C), nitrogen (N), oxygen (O), silicon (Si), phosphorus (P), sulfur (S), germanium (Ge), and arsenic (As), which are respectively named dithieno[3,2-b:2′,3′-d]borole (DTB),18,19 cyclopenta[2,1-b:3,4-b′]dithiophene (CPDT),20–22 dithieno[3,2-b:2′,3′-d]pyrrole (DTP),23–25 dithieno[3,2-b:2′,3′-d]furan (DTF),26 silolo[3,2-b:4,5-b']dithiophene (DTS),27,28 dithieno[3,2-b:2′,3′-d]phosphole (DTPh),29 dithieno[3,2-b:2′,3′-d]thiophene (DTT),30,31 germolo[3,2-b:4,5-b']dithiophene (DTG),32,33 and dithieno[3,2-b:2′,3′-d]arsole (DTA).34 Among them, DTB, DTF, and DTPh are not widely used in photovoltaic cells due to their air instability or improper energy levels and bandgaps. Although some studies proved that DTA has very good environmental stability and large potential to modulate the optoelectronic property, little research has been reported about its utilization in organic solar cells. CPDT, DTS, DTG, and DTT are the most widely investigated building blocks and have to some extent become indispensable units for designing a new generation of π-conjugated photovoltaic materials. Herein, it is necessary to illustrate the performance of typical polymers firstly.
These fused-bithiophenes have been incorporated with electron-accepting units, such as benzo[c][1,2,5]thiadiazole (BT), thieno[3,4-c]pyrrole-4,6-dione (TPD), and pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione (DPP), as presented in Fig. 2. A copolymer A1 with the combination of famous BT35–38 and CPDT exhibited strong light harvesting ability in the near-infrared (NIR) region,39 and an optical band gap of 1.46 eV is ideal for photovoltaic applications. Meanwhile, the strong intermolecular interaction leads to an ordered local structure in the BHJ active layer. These parameters contributed together to a significant enhancement of PCE up to 5.5%. In comparison with the A1 device, the deep HOMO level of the fluorinated polymer A2 leads to a higher open-circuit voltage (VOC) of 0.7 V.40 Moreover, the short-circuit current (JSC) and fill factor (FF) increased owing to the finely tuned morphology upon the fluorine substitution, which can reduce geminate and non-geminate recombination.41–43 A PCE as high as 6.1% for A2 was attained in combination with [6,6]-phenyl-C71-butyric acid methyl ester (PC71BM) as the acceptor. The copolymer A5 produced by combining the CPDT unit with another electron deficient TPD unit exhibited broad light absorption and good thermal stability.44 The relatively high PCE of 6.4% indicates that the fine tuning of the electron-accepting unit is critical to optimize the morphology of the active layer without changing other fabrication conditions.
 | ||
| Fig. 2 High performance photovoltaic polymers based on five rigidified bithiophene building blocks (EH: 2-ethylhexyl, BO: 2-butyloctyl, and OD: 2-octyldodecyl). | ||
When the bridged carbon atom was replaced by silicon, another rigidified building block DTS can be obtained. The study found that the hole mobility of a DTS-based polymer is three times higher than that of a CPDT-based analogue polymer, which can be attributed to the orderly structure in the solid state.38,45,46 When incorporating DTS and BT, the obtained low-bandgap copolymer A3 (Fig. 2) showed a blue-shifted λmax relative to A1.38 A PCE of 5.9% was attained by using A3 in combination with PC71BM. After the TPD unit is incorporated as the acceptor unit, an efficiency of 7.3% based on the polymer A6 and PC71BM was achieved.32
In addition to C and Si as bridged atoms, Ge can also be used to bridge bithiophene to form a DTG unit, in which the covalent radii of C–Ge are similar to those of C–Si bonds. The copolymer A4 based on DTG and BT exhibited a higher charge carrier mobility of up to 0.11 cm2 V−1 s−1 compared to analogous DTS-based copolymer A3.47 Although the PCE of just 4.5% is not the highest value in comparison with those of A1 and A3, A4-based BHJ solar cells achieved a relatively high JSC of 18.6 mA cm−2 because of the appropriate morphology.32 It is important to mention that the enhanced JSC is at the expense of decreased VOC, resulting in low PCE. When TPD was selected to copolymerize with DTG, another high-performance copolymer A7 was synthesized by Reynolds and co-workers.32,48 By using a nanocomposite film as the electron-transporting layer, inverted A7:PC71BM BHJ devices show a PCE in excess of 8% due to the improved charge collection efficiency.49
DTT with a highly extended heteroarene structure has also attracted considerable attention because DTT-based materials exhibited high crystallinity and charge carrier mobility. For example, a copolymer A8 containing DTT and DPP exhibited a low bandgap of 1.22 eV and high hole mobility of 0.60 cm2 V−1 s−1, which might be due to the strong intramolecular charge transfer as well as the strong intermolecular π–π stacking.50 As a result, a PCE of 6.05% with a JSC of 13.7 mA cm−2, a VOC of 0.66 V, and a FF of 66.1% was achieved. Very recently, it was found that a copolymer A5 in combination with DTT with dialkyloxy-BT showed a well-ordered molecular packing, high charge mobility and nanofibrillar morphology in the active film with PC71BM.51 As a result, A5:PC71BM gave a PCE as high as 9.02% with a JSC of 15.04 mA cm−2, VOC of 0.81 V, and FF of 0.74.
There are still a large number of materials exhibiting excellent photovoltaic properties in addition to the molecules discussed above. Recently, researchers have given some detailed overviews of their development.52 In particular, bridging fused thiophene with C, Si, Ge and S atoms results in promising building blocks as an electron-rich unit for high-performance donor materials. Besides these four kinds of electron-donating units, DTP units have also received great attention because the electron-rich N atom can increase the stability in the oxidized state and further decrease the band gap. Rasmussen et al. successively provided two full reviews of DTP and some fused bithiophene based materials, which focused on their photophysical properties and application in electronic devices.53,54 The reported DTP-based polymers and small molecules are mainly comprised of the original DTP unit or N-aryl DTP. However, there has been rapid progress in DTP-based building blocks in recent years.
In addition to the change of N-substituents, many researchers have reported novel DTP-based units through modification of the DTP backbone. So far, there have been numerous building blocks derived from DTP, as shown in Fig. 3. The family of DTP building blocks could be easily classified as four types: (1) basic building blocks, including N-alkyl DTP, N-acyl DTP and pyrrolo[3,2-d:4,5-d′]bisthiazole (PBTz); (2) fused DTP derivatives, including pyrrole-modified pentathiophene (NPTA), di(1-benzothieno)[3,2-b:2′,3′-d]pyrrole (DBTP), dithieno[2,3-d:2′,3′-d′]thieno[3,2-b:3′,2′-b′]dipyrrole (DTDP) or so-called S,N-heteropentacenes (SN5) and S,N-heterohexacenes (SN6); (3) DTP-based lactam building blocks, such as dithieno[3,2-b:2′,3′-d]pyridin-5(4H)-one (DTPO), [7,7′-bidithieno[3,2-b:2′,3′-d]pyridine]-5,5′(4H,4′H)-dione (BDTP) and [7,7′-bidithieno[3,2-b:2′,3′-d]pyridine]-5,5′(4H,4′H)-dione (TD); and (4) DTP-based imide building blocks, including N-alkyl-2,2′-bithiophene-3,3′-dicarboximide (BTI) and [2-(thiophen-2′-yl)-5-(thiophen-2′′-yl)thieno[3,2-b]-thiophene-3,3′:6,3′′-bis(dicarboximide)] (TBI).
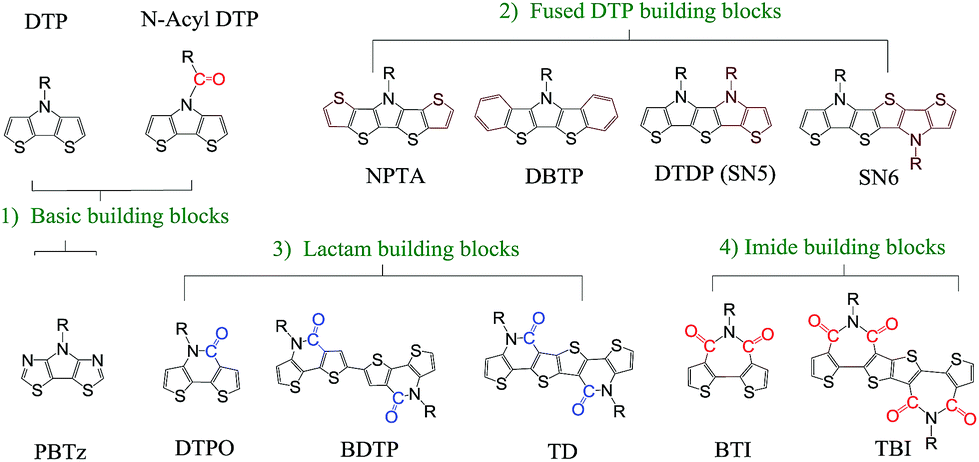 | ||
| Fig. 3 DTP-based units discussed in this review, including (1) basic, (2) fused DTP, (3) lactam and (4) imide building blocks. | ||
For instance, the instability of N-alkyl DTP can be overcome by decreasing the HOMO energy level after incorporating an N-acyl group.55 An electron-deficient PBTz unit can also lead to a smaller bandgap and lower-lying HOMO energy level compared to the original DTP due to the more electron-deficient flanking thiazole than thiophene.56 In addition, the conjugation of DTP can be extended through several fusion methods, such as incorporating thiophene or benzene on both sides of DTP affords NPTA or DBTP,57,58 combining three thiophene units with pyrrolidine offers DTDP,59 and fusing two DTP units gives SN6.60 In combination with the advantages that the strong electron-withdrawing acryl group can result in a low-lying HOMO energy level and the π-extending can enlarge the planar aromatic structure, lactam and imide building blocks were designed by incorporating one (DTPO) or two (BTI) N-acyl groups or changing the connection between two DTPO or BTI, such as BDTP, TD or TBI.61–64 The polymer based on a pentacyclic lactam acceptor unit exhibited outstanding photovoltaic properties.65 The PCE of DTP-based materials has improved recently, with a lab-scale efficiency of more than 10%, which brings great potential for commercialized devices in the near future. With the developments in processing conditions and device architectures, we believe that the efficiency will increase even more.
Therefore, it would be meaningful to make a summary of the recent advances in DTP-based materials, highlighting the structural features, which are the key points to provide high-efficiency and stable devices. In this account, we primarily summarize recent progress in novel DTP-based polymers and their device performances. Some considerations about the relationship between the structure and performance would be discussed from the point of view of molecular structures showing different photovoltaic properties. We hope this study could pave a way for the further design of novel DTP-related building blocks and the corresponding photovoltaic materials.
2. Synthesis of DTP-based building blocks
Since the first report on the synthesis of the original DTP by Zanirato and coworkers in 1983,23 so many DTP-based functional materials have been developed. Here, we introduce the synthesis method of the above-mentioned four classes of DTP derivatives. N-alkyl or N-aryl functionalized DTPs could be easily generated by amination of 3,3′-dibromo-2,2′-bithiophene with alkyl-amine or aryl-amine using Pd catalysis as depicted in Scheme 1a.66 It is also applicable to synthesizing fused DTPs (NPTA and DBTP) and PBTz materials from the corresponding brominated precursors in high yields.58 On the other hand, Pd-catalyzed tandem Buchwald–Hartwig coupling of the precursors with amine molecules, R-NH2, can afford other two ring-fused DTPs.59 For the production of N-acyl DTPs, Cu-catalyzed tandem C–N bond formation has been utilized (Scheme 1b).67,68 The synthesis of lactam structures can be achieved by the reaction of N-(2-alkyl)thiophene-3-amine with the corresponding dibromide acyl chlorides, which can afford bis-amide molecules followed by Pd-catalyzed intramolecular cyclization transformation reactions.69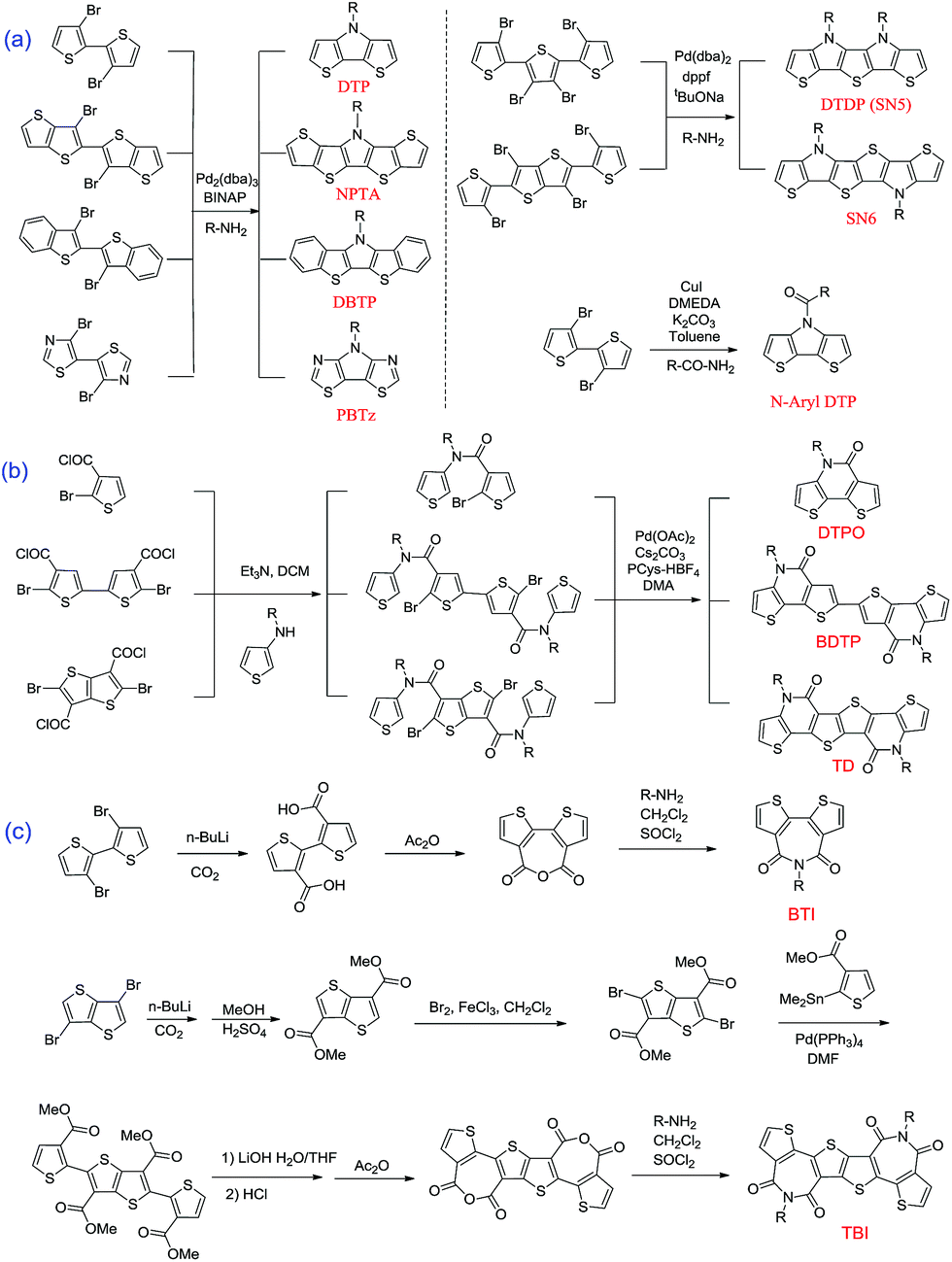 | ||
| Scheme 1 Common synthetic routes for the original DTP and derived segments: (a) basic and fused DTP; (b) lactam building blocks; (c) imide building blocks. | ||
Similar to the other DTP-based building blocks, two imide building blocks, BTI and TBI, were also synthesized from bromide precursors such as 3,3′-dibromo-2,2′-bithiophene and 3,6-dibromothieno[3,2-b]thiophene, respectively (Scheme 1c). The difference is that 3,3′-dicarboxylate-2,2′-bithiophene was directly cyclized in refluxing acetic anhydride (Ac2O) solution to yield the key 2,2′-bithiophene-3,3′-dicarboxylic anhydride intermediate. However, in the synthesis procedure of TBI, the thieno[3,2-b]thiophene-3,6-dicarboxylic acid was first esterified, and subsequently underwent bromination, Stille coupling, demethylation and finally dehydration by Ac2O to yield a dicarboxylic anhydride precursor. At the last step, the obtained anhydride intermediates underwent amination with alkyl amides to respectively produce BTI and TBI building blocks.
3. Conjugated photovoltaic materials based on the original DTP building block
The π-conjugated photovoltaic materials including polymers and small molecules based on the original DTP moiety have been systemically investigated from 2007.70 In particular, as one of the most strong donor units, DTP has been widely used to construct many low bandgap materials. We have paid attention earlier to DTP-based D–A polymers in combination with various acceptor units.71–75 Meanwhile, many other DTP-containing π-conjugated materials have also been studied by Wang,76,77 Janssen,78–81 and Yu.82,83 It has been proved that DTP-based polymers generally show a low bandgap and near-infrared photocurrent response, which strongly depend on the copolymerized units. In this section, the structural features of various photovoltaic materials containing the original DTP unit and different electron-deficient building blocks, the device performance as well as the their correlation are discussed.3.1 Photovoltaic polymers based on DTP and BT
A series of polymers based on DTP and BT are summarized and their chemical structures are displayed in Fig. 4 and the corresponding device performance is tabulated in Table 1. In 2008, we copolymerized DTP with a famous electron-withdraw building block, 4,7-di(2-thienyl)-2,1,3-benzothiadiazole (DTBT) and obtained several D–A type polymers.71 The copolymer B1 shows a low optical band gap (Eg = 1.46 eV) and broad absorption with peaks at 451 and 697 nm in the film. The best PCE of OSCs reached 2.18% in combination with [6,6]-phenyl-C61-butyric acid methyl ester (PC61BM) as the acceptor. After directly coupling DTP and BT units, copolymers B2a–B2c display strong absorption in the wavelength range of 600–900 nm.76 The enhanced absorption coefficients decreased as the length of alkyl chain in DTP increased. The low PCEs of 1.23%, 2.06% and 2.80% are mainly due to the poor film quality. An analogous polymer B2d bearing a branched side chain, 2-ethylhexyl (EH), showed an optical bandgap of 1.41 eV, and the best PCE of 1.64% was recorded in combination with PC71BM with an optimized mixed ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.84 After increasing the number of thiophene spacers, the blend active film B3:PC71BM showed a PCE of 3.12% with a largely increased JSC of up to 11.43 mA cm−2 because of the favorable interpenetrating network for charge transfer. The results indicate that the attached alkyl chain substituents can tune the solubility and the enlarged planarity of the polymeric backbone can strengthen π–π stacking. However, the low efficiency needs further adjusting of the energy level to improve VOC and optimizing of the morphology of the blend active layer to increase the JSC and FF.
:2.84 After increasing the number of thiophene spacers, the blend active film B3:PC71BM showed a PCE of 3.12% with a largely increased JSC of up to 11.43 mA cm−2 because of the favorable interpenetrating network for charge transfer. The results indicate that the attached alkyl chain substituents can tune the solubility and the enlarged planarity of the polymeric backbone can strengthen π–π stacking. However, the low efficiency needs further adjusting of the energy level to improve VOC and optimizing of the morphology of the blend active layer to increase the JSC and FF.
 | ||
| Fig. 4 Photovoltaic polymers based on DTP and BT (EH: 2-ethylhexyl). | ||
| λ max [nm] | HOMO/LUMOb [eV] | μ h [cm−2 V−1 s−1] | Active layer | V OC [V] | J SC [mA cm−2] | FF | PCE [%] | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| a In films on the plate. b Estimated from the onset oxidation and reduction potentials of the cyclic voltammetry profile. c Obtained from organic thin-film transistors. d “NA” represents no numerical values from the references. | |||||||||
| B1 | 697 | −5.00/−3.43 | NA |
B1:PC61BM = 1:1 |
0.52 | 9.47 | 0.44 | 2.18 | 71 |
|
B1:PC71BM = 1:2 |
0.50 | 9.90 | 0.48 | 2.37 | 84 | ||||
| B2a | 776 | −4.89/−3.08 | NA |
B2a:PC61BM = 1:3 |
0.43 | 6.19 | 0.46 | 1.23 | 76 |
| B2b | 777 | −4.86/−3.07 | NA |
B2b:PC61BM = 1:3 |
0.43 | 11.1 | 0.43 | 2.06 | 76 |
| B2c | 771 | −4.81/−3.08 | NA |
B2c:PC61BM = 1:3 |
0.54 | 11.9 | 0.44 | 2.80 | 76 |
| B2d | 723 | −4.98/−3.51 | NA |
B2d:PC71BM = 1:2 |
0.54 | 5.75 | 0.53 | 1.64 | 84 |
| B3 | 464, 611 | −4.99/−3.40 | NA |
B3:PC71BM = 1:2 |
0.48 | 11.43 | 0.57 | 3.12 | 84 |
| B4 | 698, 764 | −4.88/−3.47 | NA |
B4:PC71BM = 1:4 |
0.31 | 7.89 | 0.48 | 1.17 | 85 |
|
B4:ICBA = 1:3 |
0.54 | 7.06 | 0.47 | 1.71 | 85 | ||||
| B5 | 615 | −5.47/−3.07 | 2.0 × 10−4 |
B5:PC71BM = 1:2 |
0.84 | 9.78 | 0.50 | 4.2 | 49 |
| B6 | 615 | NA | NA |
B6:PC61BM = 1:3 |
0.72 | 4.97 | 0.39 | 1.40 | 78 |
After introducing alkoxyl side chains in the BT unit, D–A copolymer B4 showed semi-crystalline character because the non-covalent S⋯O interactions as well as the soluble alkoxyl groups maximize the planarity of the backbone and the intermolecular interactions.85 However, B4:PC71BM showed a low VOC of 0.31 V, which might be attributed to the high-lying HOMO (−4.88 eV) induced by the electron donating nature of the alkoxy chain. After selecting indene-C60 bisadduct (ICBA) as the acceptor, B4:ICBA OSCs exhibited a PCE of 1.71% with a JSC of 7.06 mA cm−2. The slightly lower JSC than the B4:PC71BM device can be attributed to the partially blocked electron transport by the intercalated structure of ICBA. The PCEs of B1–B4 polymers were limited by rather low VOC (0.43–0.52 V) due to their still relatively higher HOMO levels. Besides controlling the alkoxy substitution to a low-lying HOMO level for enabling intramolecular sulfur–oxygen interaction,86 the HOMO level can also be tuned by selecting appropriate units.87–89 For example, after coupling DTP with fluorene and BT, D–A polymer B5 exhibited a much lower HOMO level (−5.47 eV).49 The B5:PC71BM device showed a relatively high PCE of 4.2% with a high VOC of 0.84 V. After introducing ethynylene units into the polymeric backbone, B6 showed an increased oxidation potential and thereby a VOC as high as 0.72 V compared to its analogue.78
With PCBM (PC61BM or PC71BM) as the acceptor, these DTP–BT polymers show relatively high-lying LUMO and HOMO energy levels, leading to large LUMO(D)–LUMO(A) but small LUMO(A)–HOMO(D) energy offsets. As a result, the exciton dissociation is restrained, while the charge recombination is unbridled. Together with the bad phase separation in blend films, the performances of these devices have not been improved. It should be noted that the frontier molecular orbitals, crystallinity and solubility of BT-based materials have been finely tuned by different group attachment on the BT backbone.90,91 Therefore, further molecular modification on DTP–BT backbone would be one effective way to enhance the performance of the device. In addition, by selecting other electron-accepting units to copolymerize DTP, high performance can be obtained after reasonable design of photovoltaic polymers, which would be discussed as follows.
3.2 Photovoltaic polymers based on DTP and DPP
Since Farnum et al. reported the synthesis of a DPP structure for the first time in 1974,92 DPP units have been widely utilized as important moieties in polymers and small molecules.93–95 Here, we describe several DTP–DPP-based photovoltaic polymers with various spacer units as shown in Fig. 5 and Table 2. A narrow bandgap copolymer C1a with the absorption band extending to the region of micrometer wavelength was first synthesized in 2009.73 The C1a:PC61BM device gave a PCE of 1.12%, which was one of the highest values for a micrometer-response polymer at that time. After replacing EH with butyl, D–A copolymer C1b showed a broad absorption in the range of 500–1100 nm with a tail extending to 1300 nm and a higher hole mobility of 0.05 cm2 V−1 s−1 measured by the field-effect transistor (FET) method.96 As a result, a high JSC of 14.87 mA cm−2 and PCE of 2.71% were achieved. The different photovoltaic characterization between two analogue polymers C1b and C1a implies that the side alkyl chain in the DPP segment exhibits a large effect not only on the solubility of polymers for solution-processed fabrication, but also on the electron affinity, molecular packing and thus the photovoltaic performance.97 | ||
| Fig. 5 Photovoltaic polymers based on DTP and DPP (EH: 2-ethylhexyl). | ||
| λ max [nm] | HOMO/LUMOb [eV] | μ h [cm−2 V−1 s−1] | Active layer | V OC [V] | J SC [mA cm−2] | FF | PCE [%] | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| a In films on the plate; the tildes indicate the values were estimated from the given profile. b Estimated from the onset oxidation and reduction potentials of the cyclic voltammetry profile. c Obtained from organic thin-film transistors. d “NA” represents no numerical values from the reference. | |||||||||
| C1a | 852 | −5.02/−3.64 | NA |
C1a:PC61BM = 1:3 |
0.44 | 4.47 | 0.57 | 1.12 | 73 |
| C1b | ∼800 | −4.90/−3.63 | 0.05 |
C1b:PC71BM = 1:2 |
0.38 | 14.87 | 0.48 | 2.71 | 96 |
| C2 | 800 | −4.86/−3.56 | 1.7 × 10−2 |
C2:PC71BM = 1:1.5 |
0.42 | 22.65 | 0.52 | 4.99 | 102 |
| C3 | ∼870 | −5.26/−3.68 | 8 × 10−2 |
C3:PC61BM = 1:2 |
0.43 | 20.5 | 0.54 | 4.8 | 79 |
| C4 | ∼920 | −5.17/−3.75 | 7 × 10−2 |
C4:PC61BM = 1:2 |
0.35 | 18.6 | 0.52 | 3.3 | 79 |
| C5 | ∼915 | −5.35/−3.92 | NA |
C5:PC71BM = 1:2 |
0.52 | 12.2 | 0.58 | 3.7 | 81 |
| C6 | ∼860 | −5.61/3.94 | NA |
C6:PC71BM = 1:2 |
0.69 | 14.9 | 0.54 | 5.6 | 80 |
| C7 | 593 | −5.19/−3.45 | 8.25 × 10−4 |
C7:PC71BM = 1:1 |
0.67 | 6.61 | 0.30 | 1.32 | 110 |
In addition to the alkyl chain, alkylthio, alkoxy, and conjugated groups should be key considerations to optimize the polymer structure in order to tune the optical and electrical properties.98,99 The conjugated side groups have been proved to be a promising strategy to pursue promising photovoltaic polymers with broad absorption and high charge mobility.100,101 We introduced a tris(thienylenevinylene) (TTV) conjugated group terminated with a hexyl alkyl chain into the 3-position of the thiophene unit and obtained polymer C2.102 The JSC was enhanced to 22.6 mA cm−2, which can be considered as the first device with a JSC over 20 mA cm−2 and is still one of the highest records for OSCs since then. The external quantum efficiency (EQE) profiles as shown in Fig. 6 reveal that the substituted TTV group largely improved the EQE, in particular in the region of 650–1100 nm to better match the solar spectrum compared to the famous P3HT polymer. Very recently, it was found that introducing a conjugated side chain can also enhance the hole mobility in the mixed film and facilitate the charge separation at the D/A interface than other analogues without conjugated side chains.103 Therefore engineering side chains is an easy and effective strategy to tune the properties of the polymer, which should be kept in mind while constructing photovoltaic materials.
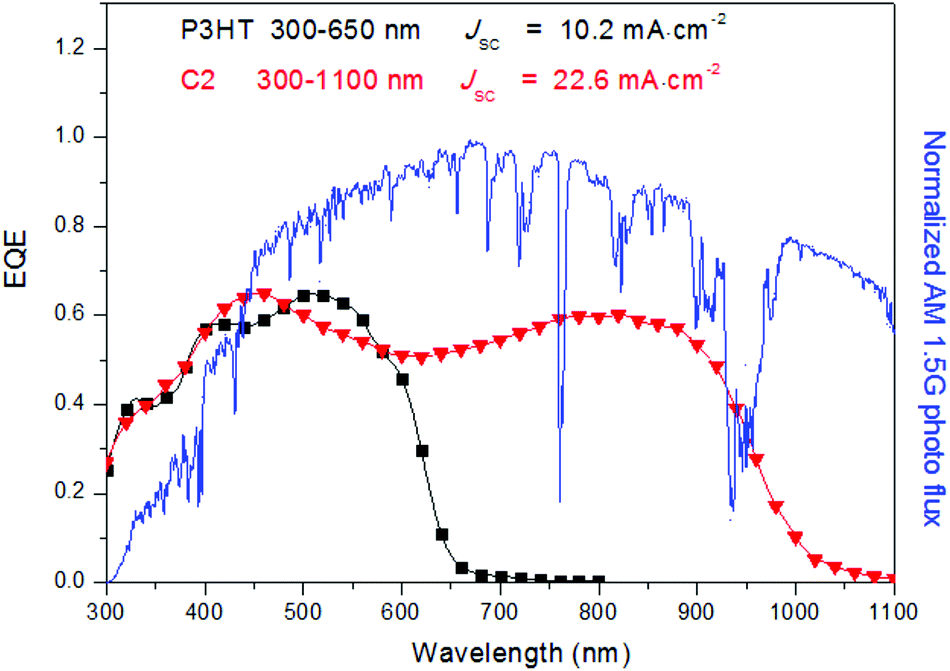 | ||
| Fig. 6 EQE curves of PSCs based on the P3HT:PC61BM and C2:PC71BM, the normalized AM1.5 photon flux spectrum is also shown for comparison. The figure was adopted from the reference with permission.102 | ||
It is well known that selenophene has stronger electron-donating properties than thiophene, which would largely affect the energy levels.104,105 Janssen and co-workers synthesized copolymers C3 and C4 with thiophene and selenophene adjacent to the DPP unit.79,81 Although both polymers exhibited a high NIR photo-response, a better PCE of 4.8% with a VOC of 0.43 V, a JSC of 20.5 mA cm−2 and a FF of 0.54 was recorded for C3 than C4 (PCE = 3.3%; VOC = 0.35 V; JSC = 18.6 mA cm−2; FF = 0.52). When retroreflective optical foil was used, the JSC can be further enhanced due to the improved light absorption. In order to enhance VOC with efficient charge generation, the other electron-deficient unit BT was introduced into the polymer C3, and a new polymer C5 with an ultra-low bandgap of 1.19 eV was obtained, resulting from the increased electron withdrawing properties by combination of two DPP and BT units. The photovoltaic device gave a JSC of 12.2 mA cm−2, a moderate VOC of 0.52 V and a PCE of 3.7%.106
Although the combined incorporation of several π-conjugated units into the backbone is a viable approach to finely tune the energy levels, the difficulty of syntheses has also increased. One of the critical issues that account for the low PCE is the significant loss in VOC, which largely depends on the energy difference between the ionization potential (IP) of the donor polymer and electron affinity (EA) of the acceptor.107,108 The IP of the polymer can be tuned by incorporating strong electron-rich and electron-deficient building blocks into the backbone. For example, electronegative atoms in the backbone can increase both the IP and EA of the polymer considerably. By replacing the thiophene unit with a thiazole unit, copolymer C6 showed an increased PCE of 5.6% with a VOC of 0.69 V, a JSC of 14.9 mA cm−2 and a FF of 0.54.80,81,109 A phenyl-flanked DPP-based polymer C7 showed a medium bandgap of 1.74 eV, which is larger by over 0.5 eV than that of the thiophene-flanked analogue C1. Although the obtained VOC of 0.67 V is comparable with that of thiazole-flanked analogue C6, a much lower PCE of 1.32% was observed.110 One of the main reasons that account for the low PCE of C6 might be the poor molecular packing because there are steric interactions between the DPP core and phenyl groups, leading to twisted backbone geometry.
3.3 Photovoltaic polymers based on DTP and other electron-deficient building blocks
The polymers discussed above showed relatively high JSC values; however the obtained VOC values are quite low. The high-lying HOMO energy level might be responsible for low VOC, and thus other electron-deficient building blocks as displayed in Fig. 7 should be considered to copolymerize with the DTP unit. A suitable building block can modulate the bandgap, energy level and device performance simultaneously as summarized in Table 3. As a planar fused building block, TPD has been widely used for constructing D–A type polymers in combination with diverse electron-donating units.48,111–115 After copolymerizing TPD with DTP, we obtained an alternating copolymer D1, in which the branched alkyl side chain EH attached to the TPD segment is beneficial to the solution processing.75 The device D1:PC71BM gave a PCE of 1.63% with a VOC of 0.66 V. In general, the properties of D–A type polymers can be straightforwardly tuned by introduction of thiophene spacers between D and A units.114,116–119 In particular, the planarity of the polymer backbone can be improved, leading to stronger and broader absorption. Keeping this principle in mind, we introduced thiophene spacers between DTP and TPD segments and obtained D2.120 Upon optimizing the film morphology by selecting different solvents or additives, a PCE of 2.11% with a VOC 0.58 V, a JSC of 7.03 mA cm−2, and a FF of 0.52 was observed. | ||
| Fig. 7 Photovoltaic polymers based on DTP and other electron-deficient building blocks (EH: 2-ethylhexyl). | ||
| λ max [nm] | HOMO/LUMOb [eV] | μ h [cm−2 V−1 s−1] | Active layer | V OC [V] | J SC [mA cm−2] | FF | PCE [%] | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| a In films on the plate. b Estimated from the onset oxidation and reduction potentials of the cyclic voltammetry profile. c Obtained from organic thin-film transistors. d HOMO and LUMO levels calculated at the B3LYP/6-31G(d,p) level. e Hole mobility determined by the space charge limited current (SCLC) method. f Annealing at 130 °C. g “NA” represents no numerical values from the reference. | |||||||||
| D1 | 598 | −5.11/−3.63 | NA |
D1:PC71BM = 1:3 |
0.66 | 4.98 | 0.50 | 1.63 | 75 |
| D2 | 650 | −5.15/−3.48 | NA |
D2:PC71BM = 1:2 |
0.58 | 7.03 | 0.52 | 2.11 | 120 |
| D3 | 333, 374, 436, 722 | −4.17/−2.31d | 2.94 × 10−4 |
D3:PC61BM = 1:3 |
0.63 | 2.69 | 0.61 | 1.40 | 126 |
| D4 | 624 | −5.04/−3.48 | NA |
D4:PC71BM = 1:2 |
0.46 | 7.78 | 0.49 | 1.77 | 74 |
| D5 | 453, 696 | −5.04/−3.62 | 8.3 × 10−5e |
D5:PC71BM = 1:3 |
0.51 | 7.16 | 0.42 | 1.55 | 135 |
| D6 | 681 | −4.81/−3.35 | NA |
D6:PC61BM = 1:3 |
0.74 | 5.87 | 0.40 | 1.74 | 78 |
| D7 | 530 | −5.23/−3.17 | NA |
D7:PC61BM = 1:2 |
0.68 | 2.70 | 0.31 | 0.58 | 82 |
| D8 | 571 | −4.99/−3.18 | NA |
D8:PC61BM = 1:2 |
0.46 | 2.54 | 0.33 | 0.39 | 82 |
| D9 | 562 | −4.79/−3.30 | 5.5 × 10−4 |
D9:PC71BM = 1:2 |
0.31 | 3.06 | 0.32 | 0.30 | 142 |
| D10 | 536 | −4.80/−2.69 | 2.11 × 10−5 |
D10:PC71BM = 1:1 |
0.69 | 2.80 | 0.31 | 0.62 | 143 |
| D11 | 521 | −5.00/−2.87 | 5.9 × 10−4e |
D11:PC71BM = 1:1.2 |
0.62 | 6.17 | 0.55 | 2.10 | 144 |
| D12 | 390, 790 | −4.77/−2.69 | NA |
D12:PC61BM = 1:2 |
0.46 | 5.25 | 0.41 | 0.99 | 83 |
| D13 | 570 | −4.93/−2.40 | 0.11f |
D13:PC61BM = 1:4 |
0.37 | 7.12 | 0.40 | 1.06 | 149 and 150 |
| D14 | 553 | −5.22/−3.44 | 5.11 × 10−5 |
D14:PC71BM = 1:2 |
0.73 | 6.68 | 0.37 | 1.85 | 151 |
| D15 | 723 | −5.2/−3.9 | NA |
D15:PC71BM = 1:3 |
0.64 | 5.37 | 0.36 | 1.24 | 152 |
Another electron-deficient segment, the quinoxaline (Qx) unit, was also very effective in the construction of D–A type conjugated polymers.121–125 We found that the combination of a 5,8-bis(2-thienyl)-2,3-diphenylquinoxaline (DTQx) segment with DTP gave a copolymer D3 with an optical bandgap of 1.61 eV.74 The D3:PC71BM device realized a PCE of 1.77% with a low VOC of 0.46 V. By copolymerizing N-aryl DTP and Qx derivative units, soluble copolymer D4 exhibited a PCE of 1.40% with a weight ratio of 1:3 because the domain size became large with increasing the PC61BM content in the blend film.126 In addition, thieno[3,4-b]pyrazine (TP), as the analogue of the BT unit and Qx unit, has also been used as an attractive acceptor unit because the TP moiety could not only make the polymeric backbone more planar, but also facilitate reduction of the bandgap of the resulting conjugated polymers.127–134 For example, TP-based copolymer D5 showed a PCE of 1.55% with a VOC of 0.51 V, a JSC of 7.16 mA cm−2, and a FF of 0.42.135 After introducing triple bonds between DTP and TP units, D6:PC61BM devices show a PCE up to 1.74% with a VOC of 0.74 V, a JSC of 5.87 mA cm−2, and a FF of 0.40.78 It is consistent with previous reports that the introduction of ethynylene units into the backbone can enhance the VOC through increasing the oxidation potential of the polymer.136–139
Additionally, the symmetric, compact and planar phthalimide (PI) moiety can also be considered as a promising electron-accepting building block. Two copolymer D7 and D8 are comprised of alternating DTP and PI units without and with a thiophene bridge between DTP and PI, respectively.82 The introduction of thiophene spacers induced a wider bandgap of 1.71 eV for D8 than 1.66 eV of D7. Although the incorporation of thiophene spacers broadened the absorption, a much higher PCE of 2.84% was obtained for D7 with a JSC of 12.6 mA cm−2, a VOC of 0.56 V, a FF of 0.40 in comparison with D8. The fused aromatic ring, benzobisthiazole (BBT) can increase the long-range order of polymers and small molecules due to the strong intermolecular interactions.140,141 Therefore, thermally robust BBT-based polymers are promising conjugated materials due to the high crystallinity and oxidative stability properties. Copolymer D9 combining DTP and 2,6-BBT exhibited a high-lying HOMO energy level of −4.79 eV and an optical bandgap of 1.85 eV.142 And, 4,8-BBT can also be explored to prepare polymer D10; however their performances are unsatisfactory so far.143
As discussed above, a series of D–A copolymers combining BT and DTP exhibit promising performance because of the attractive BT unit possessing good π–π stacking, strong intermolecular interactions, and simple synthesis procedure.36 After replacing of the S atom with a N atom, another electron-accepting moiety benzotriazole (BTZ) can be obtained. Copolymer D11 by using DTP as the donor block and BTZ as the acceptor block showed good solubility because of the substituted alkyl chain on BTZ and thiophene.144D11 also exhibited good ability of sunlight harvesting and a narrow bandgap; however D11:PC71BM devices exhibited low efficiency due to the large phase separated structure in the blend film.
It has been reported that isoindigo (iID)-based polymers showed a remarkable high mobility of up to 3.62 cm−2 V−1 s−1 as a result of the rigid and planar structure.145–147 The best PCE of up to 8.2% for an iID-based polymer was reported by Geng and co-workers in 2014.148 D–A copolymer D12 based on a DTP unit and an iID unit gave the best PCE of 1.73% when large-scale ITO-free devices were fabricated.83 When the DTP was combined with alkyl- or carboxylate-substituted thiophene, D–A copolymers D13 and D14 were synthesized.149,150 It was found that D13 exhibited a mobility of 0.11 cm2 V−1 s−1, and a PCE of 1.06% with a VOC only of 0.37 V. D14 shows a low-lying HOMO energy level, resulting in a larger VOC of 0.74 V.151 Bay-annulated indigo (BAI), which can be synthesized just in a single step from indigo dye, has also been considered as a potent electron deficient unit for the design of polymers.57 D–A polymer D15 incorporating DTP, thiophene and BAI exhibits a low bandgap of 1.4 eV, low-lying LUMO energy level of −3.9 eV and good solubility in common solvents. Unfortunately, the highest PCE of up to 1.24% was obtained especially owing to the unsatisfactory active layer morphology.152
Except for the acceptor moieties mentioned above, there would be many groups that are expected to copolymerize with DTP to produce new polymers. It should be pointed out that both the units and side chains should be considered as a priority because the former determine the optical and electrical properties, while the latter determine the planarity of the polymeric backbone, packing of molecules and the morphology of the blend film. Hence selecting suitable backbone units together with optimizing the side chains will greatly facilitate the development of polymer donors.
3.4 Photovoltaic materials based on DTP-containing small molecules
With the development of polymers possessing good light-harvesting and charge mobility, significant advances have been achieved for their devices in combination with fullerene derivatives as acceptors.153–155 The BHJ solar cells combining fullerene derivatives and DTP-based conjugated polymers have reached the best PCE of 5.6%.81 However, the polymeric donors still have some problems should be addressed such as difficult control of the structural regularity and molecular weight, resulting in poor reproducibility. It is also a long-standing challenging task for the fundamental study of polymers due to their naturally complex and disordered entanglement structures in the sold state. A solution to those problems is utilizing soluble small molecules as the donor.156,157The solution-processed OSCs with small donor molecules have realized a PCE of over 10%.158,159 These outstanding achievements in OSCs with small donors opened a new era of designing novel small molecules with excellent properties. Recently, numerous small molecules featuring D–A–D (or A–D–A) symmetrical or unsymmetrical structures have been used as donor materials.160–163 The typical alternating D and A architecture allows these small molecule donors to display strong absorption capability at long wavelengths as well as favorable charge transfer properties. In this section, recent progress in small-molecule donors is reviewed with respect to the fundamental understanding of the photophysical properties of DTP-based materials. The current state of their devices is summarized (Table 4) and the advantages of small molecules have been analyzed.
| λ max [nm] | HOMO/LUMOb [eV] | μ h [cm−2 V−1 s−1] | Active layer | V OC [V] | J SC [mA cm−2] | FF | PCE [%] | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| a Film on the plate. b Determined by cyclic voltammetry. c Organic FET devices, [h] and [e] indicate hole mobility and electron mobility, respectively. d Hole mobility evaluated using the SCLC method. e IP/EA values determined by ultraviolet photoelectron spectroscopy (UPS) and inverse photoemission spectroscopy (IPES), respectively. f Density functional theory (DFT)-calculated HOMO and LUMO energy levels. g TheHOMO level was estimated from the absorption onset in thin solid films, and LUMO level was calculated from the optical band gap. h “NA” represents no numerical value from the reference. | |||||||||
| E1 | 558 | NA | NA |
E1:C60 |
1.21 | 0.52 | 0.39 | 0.24 | 164 |
| E2a | 698 | −5.28/−3.68 | 0.9 × 10−4d |
E2a:PC61BM = 1:2 |
0.83 | 8.8 | 0.66 | 4.8 | 166 and 167 |
| E2b | 700 | −5.27/−3.67 | 1.1 × 10−4d |
E2b:PC61BM = 1:2 |
0.84 | 8.4 | 0.66 | 3.50 | 166 and 169 |
| E2c | 691 | −5.28/−3.64 | 0.6 × 10−4d |
E2c:PC61BM = 1:2 |
0.81 | 10.5 | 0.66 | 4.60 | 166 |
| E3a | 700 | −5.30/−3.75 | 1.2 × 10−4d |
E3a:PC61BM = 1:2 |
0.83 | 8.2 | 0.65 | 3.23 | 166 |
| E3b | 700 | −5.30/−3.73 | 1.6 × 10−4d |
E3b:PC71BM = 1:2 |
0.83 | 12.0 | 0.71 | 6.90 | 169 |
| E3c | 694 | −5.31/−3.75 | 1.1 × 10−4d |
E3c:PC61BM = 1:1 |
0.84 | 11.4 | 0.63 | 5.13 | 166 |
| E3d | 707 | −5.28/−3.70 | NA |
E3d:PC71BM = 1:2 |
0.79 | 12.46 | 0.69 | 6.82 | 170 |
| E3e | 720 | −5.28/−3.74 | NA |
E3e:PC71BM = 1:2 |
0.74 | 12.85 | 0.70 | 6.57 | 170 |
| E4 | NA | NA | 10−4 |
E4:PC61BM = 1:2 |
0.83 | 10.0 | 0.70 | 6.00 | 172 |
| E5 | 654 | −5.25/−3.78 | NA |
E5:PC71BM = 1:2 |
0.86 | 14.06 | 0.68 | 8.22 | 174 |
| E6a | 514 | −5.60/−3.47 | NA |
E6a:PC71BM = 2:3 |
1.10 | 3.2 | 0.35 | 1.2 | 176 |
| E6b | 532 | −5.33/−3.45 | NA |
E6b:PC71BM = 2:3 |
0.95 | 5.8 | 0.47 | 2.6 | 176 |
| E7a | 660 | −5.20/−3.37 | 1.72 × 10−4d |
E7a:PC70BM = 1:0.8 |
0.70 | 11.58 | 0.63 | 5.13 | 177 |
| E7b | 622 | −5.19/−3.36 | 3.10 × 10−6d |
E7b:PC70BM = 1:2 |
0.75 | 9.03 | 0.47 | 3.16 | 177 |
| E8 | 638 | −4.74/−3.26 | NA |
E8:PC71BM = 1:0.8 |
0.67 | 8.22 | 0.55 | 3.03 | 211 |
| E9 | 670 | −5.16/−3.39 | NA |
E9:PC71BM = 1:2 |
0.84 | 11.94 | 0.65 | 6.53 | 178 |
| E10a | 688 | −4.75/−2.87e | NA |
E10a:PC71BM = 1:1 |
0.74 | 8.8 | 0.39 | 2.55 | 179 |
| E10b | 616 | −4.72/−2.84e | NA |
E10b:PC71BM = 1:1 |
0.80 | 10.8 | 0.61 | 5.19 | 179 |
| E10c | 618 | −4.67/−2.89e | NA |
E10c:PC71BM = 1:1 |
0.67 | 12.8 | 0.62 | 5.72 | 179 |
| E11a | 604 | −5.39/−3.43 | NA |
E11a:PC71BM = 1:2 |
0.87 | 10.15 | 0.58 | 5.12 | 187 |
| E11b | 608 | −5.48/−3.46 | NA |
E11b:PC71BM = 1:2 |
0.92 | 10.68 | 0.60 | 5.90 | 187 |
| E12a | 652 | −5.55/−3.65g | 9.56 × 10−5d |
E12a:PC71BM = 1:2 |
0.91 | 10.52 | 0.57 | 5.46 | 191 |
| E12b | 636 | −5.66/−3.69g | 1.94 × 10−4d |
E12b:PC71BM = 1:2 |
0.98 | 12.23 | 0.66 | 7.91 | 191 |
| E13 | 566 | −4.99/−3.10 | NA |
E13:PC71BM = 2:3 |
0.70 | 6.03 | 0.31 | 1.3 | 194 |
| E14 | 618 | −5.78/−3.70 | 1.12 × 10−5d |
E14:PC61BM = 1:1 |
0.75 | 3.16 | 0.33 | 0.88 | 195 |
| E15 | 647 | −4.78/−2.68 | 7.6 × 10−6 |
E15:PC61BM = 1:2 |
0.51 | 1.2 | 0.27 | 0.19 | 197 |
| E16a | 586 | −5.4/−3.4f | NA |
E16a:C70 = 1:2 |
0.89 | 11.56 | 0.37 | 3.9 | 203 |
| E16b | 645 | −5.2/−3.4f | NA |
E16b:C70 = 1:2 |
0.76 | 11.63 | 0.46 | 4.1 | 203 |
| E16c | 660 | −5.0/−3.4f | NA |
E16c:C70 = 1:2 |
0.66 | 9.72 | 0.37 | 2.6 | 203 |
| E17a | 480 | −5.6/−3.2f | NA |
E17a:C70 = 1:2 |
0.98 | 7.97 | 0.33 | 2.5 | 203 |
| E17b | 529 | −5.4/−3.2f | NA |
E17b:C70 = 1:2 |
0.94 | 11.39 | 0.50 | 5.5 | 203 |
| E17c | 537 | −5.1/−2.9f | NA |
E17c:C70 = 1:2 |
0.85 | 10.43 | 0.41 | 3.8 | 203 |
| E18 | 834 | −5.40/−3.90g | NA |
E18:PC61BM = 1:1 |
0.81 | 7.33 | 0.36 | 2.14 | 204 |
Roncali and coworkers first evaluated small-molecule donor E1 (Fig. 8) with two dicyanovinyl substituents on the flanks of a rigid DTP block and found that the bilayer devices gave a comparable efficiency to that of P3HT.164 Compared with other small molecules with CPDT or carbazole as the donor core, E1 shows larger current, suggesting that the amino group in DTP contributes to hole-transport. Additionally, it has been observed that variations of the alkyl side chain have a pronounced impact on the morphology of the blend of small molecules, which in turn affects their photovoltaic properties.165 Depending on the outer or inner position of the hexyl chain on the thiophene unit, three A–D–A molecules (E2a, E2b, and E2c) and three isomeric molecules (E3a, E3b, and E3c) were obtained with three kinds of branched alkyl substituents (ethylhexyl, hexyldecyl, and octylnonyl) attached on DTP.166 A JSC of 8.8 mA cm−2, VOC of 0.83 V and FF of 0.66 resulted in a PCE of 4.8% for the E2a device; however E3a displays a rather low PCE of 0.8%.167 This startling difference can be probably ascribed to the different morphology of the blend active layer, resulting from the different position of the hexyl side chain.
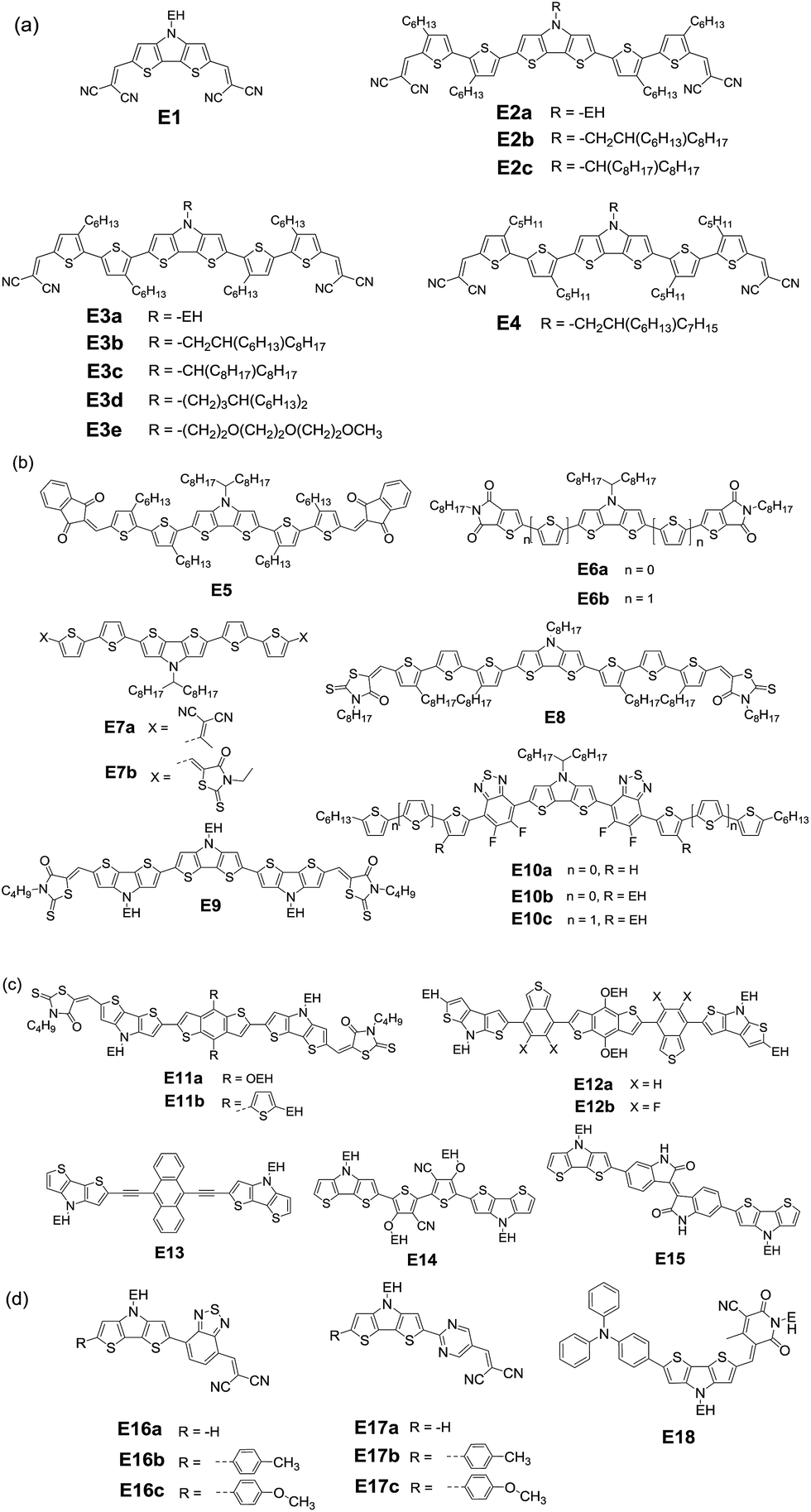 | ||
| Fig. 8 DTP-based small molecules (EH: 2-ethylhexyl): (a) molecules with CN as end-capped group; (b) molecules with other kinds of end-capped group; (c) molecules with other units as the central units; (d) molecules with unsymmetrical structures. | ||
However, it is difficult to evaluate the influence of side chains because there are still many factors affecting the final performance. For example, solvent vapor annealing (SVA) treatment dramatically improved the PCE of E3a:PC61BM up to 4.4% with a significantly enhanced JSC of 8.2 mA cm−2 and FF of 0.65 in virtue of the improved morphology of the active layer.166 After a series of SVA treatments, the PCE of the E3b:PC71BM device largely increased up to 7.1%.168–170 In a recent study, the nanoscale morphology of the E3b:PC71BM active layer strongly depended on the SVA duration time. By using a ZnO layer as the optical spacer to optimize the light absorption, an efficiency of 7.74% was achieved upon using the optimal D:A ratio and thicknesses of the active layer.171 The study found that the E4:PC61BM blend morphology was also a function of SVA time.172 With SVA time increasing the donor crystallinity is enhanced, and therefore, the charge transport is improved while the recombination is reduced. Unfortunately, further increase in SVA time resulted in recombination and low mobility because of too large donor crystallites. Besides the SVA time, the used solvent of SVA should also be a considerable factor to improve the performance.170
Branched alkyl chains are generally used in both small molecules and polymers in order to provide sufficient solubility and tune the film morphology. The wide-spread branching point is the C2-position such as EH, which can avoid steric hindrance to ensure sufficient π–π stacking of the backbone. A recent study reveals that the transition of C4-position branching (E3d) from the C2-position greatly increases the PCE up to 8.2% in comparison to E3b devices.173 A careful analysis indeed showed a significantly different active layer morphology, which strongly depends on the branched alkyl chains. When a triethylene oxide side chain is connected to the nitrogen of the DTP unit, E3e exhibited similar solubility in the same solution-process.170
In addition to the dicyanovinyl group, the unit 1,3-indanedione can also be introduced as a terminal group. By optimizing the device structure, such as employing a hole transport layer (HTL) and annealing treatment, a higher PCE of 7.54% is obtained for E5:PC71BM.174 A large enhancement of PCE up to 8.22% was realized when CuSCN was used as the HTL. Therefore, the interface modification of the HTL is fundamentally important to enhance the PCE through improving the charge selectivity at the electrodes and minimizing the energy barrier for charge extraction. When a weaker electron-accepting thieno[2,3-c]pyrrole-4,6-dione was utilized, the absorption of small molecules can be blue shifted.175 The conjugation length was extended for E6b by incorporating thiophene spacers compared with E6a.176 Using DIO as the solvent additive, solution-processed devices with PC71BM as the acceptor respectively gave PCEs of 1.2% and 2.6%. It should be noted that VOC values were up to 1.1 V, although low JSC values were obtained due to the wide bandgap. In order to enhance the device performance, it is important to further pursue other end-groups to tune the structures of DTP-based small molecules.
E7b has slightly lower decomposition temperature as compared to E7a because the aliphatic unit in rhodanine probably decomposes at high temperature in the presence of oxygen in air.177 And, rhodanine can largely improve the solubility as compared to the DCV analogue. However, the influence of terminal rhodanine and the 2,2-dicyanovinyl (DCV) group on the absorption, energy level and bandgap is not so pronounced. Unexpectedly, the E7a:PC70BM device showed a best PCE of 5.13%, which is much larger than that of E7b:PC70BM. The obvious difference can be ascribed to the response to TSA and TA treatment of blend films. The RMS roughness of the E7a:PC70BM film becomes higher after annealing treatments, whereas the surface of the E7b:PC70BM blend film was not influenced significantly by TA treatment. The results indicate that the terminal DCV groups effectively increase intermolecular interactions and thus broaden the optical absorption, enhance the charge carrier mobility and as expected lead to higher photovoltaic properties.
With octyl-rhodanine as the terminal acceptor unit, A–D–A small molecule E8 exhibits broad absorption with a low bandgap of 1.49 eV. The optimized E8:PC71BM device gave a PCE of 3.03% with a JSC of 8.22 mA cm−2, a VOC of 0.67 V and FF of 0.55. If the terthiophene unit was replaced with a DTP unit, E9 with three DTP units exhibited NIR absorption, excellent stability, good solubility for solution processing, and suitable energy levels compared to PC71BM.178 However, BHJ solar cell E9:PC71BM displayed a similar PCE of 3.04% (JSC = 8.22 mA cm−2, VOC = 0.86 V and FF = 0.43) to E8. After SVA treatment, a significantly enhanced PCE up to 6.53% with a largely improved JSC (11.94 mA cm−2) and FF (0.65) was achieved. In the same way as the other systems discussed above, SVA treatment improves the crystallinity and nanoscale morphology of the active layer, which might be the main driving force for enriching exciton dissociation and charge transport.
Small molecule E10b with branched alkyl chains showed a favorable active layer order, resulting in a higher PCE of 5.3% compared to E10a (PCE = 2.7%).179 After extending the main chain with a substituted thiophene ring, π-extended small molecule E10c showed a best PCE of ca. 6% with an enhanced JSC of 12.8 mA cm−2 and FF of 0.62. After SVA treatment, the higher-intensity lamellar reflection for the E10c:PC61BM blend film indicates the higher structural ordering. These results indicate that post-processing strongly depends on the molecular structure. Compared to their polymer counterparts, the particularly promising advantages of small molecules are the monodisperse properties and the wide range of bandgaps. However, small molecules tend to undergo morphological variations in BHJ thin films when subjected to processing additives and thermal annealing or SVA steps.180–183 Therefore, the optimization of their morphologies relying specifically on solution-processing approaches for ordered structures with adequate interpenetrating networks is still challenging.
It is noted that small molecules based on benzo[1,2-b:4,5-b′]dithiophene (BDT) possess excellent properties, especially large charge carrier mobility triggered by the efficient intermolecular interactions owing to the flat fused structure of BDT.184–186 With a butyl-rhodanine unit as the end acceptor, small molecules E11a and E11b exhibited good solution processability, high thermal annealing stability and broad and efficient light absorption.187 The devices of E11a:PC71BM and E11b:PC71BM showed PCEs of 5.12% and 5.90%, respectively, upon SVA treatment. The slightly higher PCE of the E11b device compared with that of E11a might be due to the efficient intermolecular packing of the E11b active layer resulting from the alkyl-thiophene substituent. If the thiol group is replaced with a dicyanomethylene group in rhodanine, another efficient electron-accepting group would lead to marked differences in the structure and performance.188
As discussed above, BT is one of the most important electron-deficient building blocks. In fact, small molecules combined with BT as the acceptor and CPDT or DTS as the donor showed a high mobility of up to 0.17 cm2 V−1 s−1.189,190 Recently, the influence of F substitution on the photovoltaic properties of small donors was first demonstrated by introducing fluorinated BT to DTP–BDT materials.191E12a and E12b exhibit excellent thermal stability, efficient light harvesting ability and appropriate energy levels, indicating that they are suitable donors when employing PC71BM as the acceptor. The E12b:PC71BM device showed a superior performance (PCE = 7.91%) to that of the E12a:PC71BM device (PCE = 5.46%). The slightly improved VOC from 0.91 V to 0.98 V is owing to the low-lying HOMO level induced by the fluorine substitution, and the increased JSC up to 12.23 mA cm−2 is attributed to the favorable morphology resulting in improved charge transport efficiency. Therefore, it is proved again that the attachment of F atoms can not only lead to the improvement of VOC but also enhance the JSC and FF when located at appropriate positions.
Except for the heteroaromatic unit as the acceptor, the incorporation of rigid and electron-rich anthracene in combination with ethynylene spacers could lead to a low bandgap and high carrier mobility.192,193 After coupling two DTP units with an anthracenyl core, one kind of D–A–D molecule E13 was obtained.194 The device E13:PC71BM showed a high PCE of 1.3% due to the inhomogeneous blend film. The result indicates that the planar conjugated molecular structures lead to an appropriate heterojunction with the fullerene acceptor although the performance of devices with PC61BM as the acceptor is very low.
When a 3-alkoxy-4-cyanothiophene (ACNT) acceptor unit was connected with two DTP donor units, a D–A–D small molecule E14 was obtained.195,196 Upon thermal annealing (TA), E14:PC61BM OSCs gave a PCE of 0.88% with a JSC of 3.16 mA cm−2, a VOC of 0.75 V, and a FF of 0.33. Additionally, iID has been integrated into small donor molecules as an efficient acceptor.145 When the NH group of iID is unsubstituted, the formed hydrogen-bonding interactions might enhance the intermolecular stacking. However, Roncali and colleagues found that the intermolecular hydrogen-bonding induced by the presence of a free NH group in E15 hindered the organization of molecules into a structure with favorable exciton diffusion and charge transport.197 As an interesting path to molecular organization, hydrogen-bonding could provide molecular design rules for further investigation of the structure–performance relationships of the devices.
Compared with the symmetrical structures A–D–A or D–A–D, the unsymmetrical structure D–π–A is suspected to have interesting properties due to its high molecular dipole.198,199 In particular, the unsymmetrical D–A–A architecture enables small molecules to exhibit smaller excitation energy and lower HOMO energy levels.200–202 In 2014, Wong et al. developed a series of D–A–A type small molecules E16 and E17, in which a rigid and coplanar DTP moiety served as the donor component while BT or pyrimidine was adopted as the central acceptor.203 These D–A–A molecules exhibited a good light harvesting properties except E16a and E17a, indicating that the terminally substituted aryl groups can extend the molecular conjugation and thus lead to better sunlight harvesting. E16 molecules show red-shifted absorption, thus showing higher JSC than pyrimidine-embedded E17 as shown in Fig. 9. E17b:PC71BM gave the best PCE up to 5.6% with a VOC of 0.94 V, a JSC of 11.34 mA cm−2, a FF of 0.52. Gupta et al. recently introduced a DTP unit between the triphenylamine donor part and cyanopyridone acceptor fragment and obtained the small molecule E18 with a bandgap of 1.49 eV.204 The BHJ solar cells displayed a PCE of 2.14% with a VOC of 0.81 V, JSC of 7.33 mA cm−2 and FF of 0.36. Therefore, further improvement of efficiency needs further exploitation of the bridged DTP unit.
 | ||
| Fig. 9 (a) Absorption spectra of DTP-based D–A–A donors in CH2Cl2. (b) Spectral mismatch-corrected J–V characteristics (under 1 sun, AM 1.5G illumination) (reprinted with permission from ref. 203). | ||
In this section, we mainly discussed DTP-based small molecules, which are formed by alternating donor and acceptor units with symmetric or asymmetric structures. The study found that these molecules possess planar structures, which tend to aggregate and self-assemble to form phase separation domains with a suitable length scale facilitating charge generation and transport. Although small molecule solar cells (SMSCs) showed superior performance compared to polymers, particularly the VOC values have been largely improved, many challenges remain to strive for commercial viability. It should be noted that one of the common limitations of small molecules is their low charge carrier mobility. At present, the study of other kinds of small molecules is also in full swing,205–207 in which impressive progress has been made and the best performance of small molecules has been over 10%.208,209 In particular as a hole-transport material (HTM), DTP-based small molecules possessing high conductivity and hole mobility exhibit high PCEs over 18%, which is comparable to the state-of-the-art, well-known, and relatively expensive spiro-OMeTAD.210 Therefore, it is expected to greatly improve the performance of DTP-based small molecules by introducing new building blocks because of a sensitive response of performance to a small change of chemical structure.
3.5 Applications of DTP-based polymers in hybrid solar cells
In fullerene-based OSCs, the absorption of sunlight mainly comes from organic donors because the absorption of fullerene is weak.212 The morphology of blend films easily began to deteriorate at high temperature because fullerene molecules tend to aggregate.213,214 In contrast to these addressed shortcomings, inorganic semiconductors exhibited excellent advantages such as high optical absorbance, tunable band gaps, high electron conductance, and easy solution processing with conjugated polymers.215–217 In addition, there are longer lived polarons during the electron transfer from the organic donor to the inorganic acceptor compared to the organic acceptor.218 Over the past few years, hybrid solar cells (HSCs) employing a polymer as the donor and an inorganic semiconductor as the acceptor have attracted considerable attention as new generation photovoltaic devices.219–221 Among a variety of inorganic semiconductors, the bandgap and charge mobility of lead sulfide (PbS) and lead selenide (PbSe) are easier to tune than those of cadmium selenide (CdSe).222–226 But the small HOMO energy difference between PbS (EHOMO = −5.0 to −5.2 eV) and donor P3HT (EHOMO = −5.1 eV) seriously hinders the hole transportation.227,228 Thus, it is crucial to design polymers with better energy-level alignment to further improve the performance of HSCs. So far, many polymers have been reported to be used in HSCs, in which DTP-based polymers as shown in Fig. 10 exhibited very good properties (Table 5). | ||
| Fig. 10 DTP-based polymers in hybrid solar cells (EH: 2-ethylhexyl). | ||
| λ max [nm] | HOMO/LUMOb [eV] | μ [cm−2 V−1 s−1] | Active layer | V OC [V] | J SC [mA cm−2] | FF | PCE [%] | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| a Films on the plate. b Estimated from the onset oxidation and reduction potentials of the cyclic voltammetry profile. c “NA” represents no numerical value from the reference. | |||||||||
| F1 | NA | −4.64/−2.61 | NA |
F1:PbS = 1:9 |
0.38 | 4.2 | 0.34 | 0.55 | 229 |
| NA | NA | NA |
F1:PbS = 1:9 |
0.40 | 2.1 | 0.33 | 0.25 | 218 | |
| F6 | 421, 767 | NA | NA |
F6:PbS = 1:9 |
0.57 | 13.06 | 0.51 | 3.78 | 234 |
| −4.95/−3.31 |
F6:PbS = 1:19 |
0.55 | 13.29 | 0.56 | 4.10 | 224 | |||
| −4.81/−3.38 |
F6:PbS0.4Se0.6 = 1:15 |
0.57 | 14.66 | 0.66 | 5.50 | 235 | |||
| F7 | 397, 662 | −5.00/−3.75 | NA |
F7:PbS = 1:19 |
0.58 | 11.06 | 0.56 | 3.60 | 224 |
| F8 | 413, 780 | −4.88/−3.52 | NA |
F8:PbS = 1:19 |
0.49 | 12.76 | 0.56 | 3.50 | 224 |
| F9 | 460, 650 | −5.05/−3.49 | NA |
F9:PbS = 1:19 |
0.44 | 12.75 | 0.51 | 2.86 | 224 |
| F10 | 422, 595 | −5.12/−3.47 | NA |
F10:PbS = 1:19 |
0.41 | 12.06 | 0.49 | 2.42 | 224 |
HSCs with blends of F1 and PbS exhibited a PCE of 0.55% with a JSC of 4.2 mA cm−2, a VOC of 0.38 V and a FF of 0.34, which is two orders of magnitude greater than that of P3HT.229 Although the PCE is still low, it is significantly higher than that of devices made from blends of polymers and low-bandgap quantum dots.230,231 No photoinduced absorption was detected on the F2:PbS or F3:PbS blend films, while long-lived photo-induced charge separation in the blends of F4:PbS and F5:PbS was confirmed.232,233 By employing low bandgap polymer F6 together with a PbS acceptor, a breakthrough PCE of 3.8% was achieved in virtue of the broad spectral response of a favorable heterojunction.234 In 2013, a record-high PCE of 5.5% with a FF of 67% was achieved for F6:PbSxSe1−x HSCs.235 Another noticeable feature is the spontaneously formed vertical phase segregation in the F6:PbSxSe1−x blend film, which facilitates charge dissociation and transport. A recent study on the charge transfer dynamics of a F6:PbS hybrid film found that over 80% electron transfer from the LUMO level of F6 to the conduction band of PbS quantum dots occurs within 1–5 ps.236 The emergence of a charge transfer state (CTs) at the interface might be the major factor to accelerate the charge separation and transfer. Moreover, the charge transfer rate and efficiency can be enhanced if the F6:PbS ratio is increased.
Ma and co-workers thoroughly investigated many HSCs containing DTP-based conjugated polymers (F6–F10) as the donor and PbS nanocrystals as the acceptor.224 Based on device optimization, a high PCE of 4.23% was achieved for the F6:PbS device. After introducing F atoms into the BT backbone, the resulting F7 showed a lower HOMO energy level, leading to a slightly increased VOC value. Compared with F6, the introduction of an electron-withdrawing DPP unit made the absorbance edge of F8 red shift. Nonetheless, the HSCs employing F8 deliver a lower VOC of 0.49 eV along with a decreased PCE of 3.5%. When a conjugated bridge such as thiophene and furan are introduced into the polymer backbone, the corresponding polymers F9 and F10 exhibited lower-lying HOMO energy levels of −5.05 eV and −5.12 eV, respectively. It was found that the aggregation of polymer F9 and F10 hindered the hole transport and resulted in low PCEs based on the morphology characterization.
From these results, it is easy to see that the efficiency of hybrid photovoltaics is mainly affected by the properties of both materials. The polymer contributes towards achieving flexible devices, and inorganic semiconductors increase the opto-electronic properties and the stability of the device. A large number of possible HSCs have been reported because of the large diversity of polymer donors and inorganic acceptors.237,238 However, the performance of HSCs is limited by the undesirable isolated domains in the blend film, which can serve as charge traps. To solve this problem, it is proposed to develop a processing method. Recently, the highest efficiency has reached up to 6.36% based on a P3HT:CdTe device after optimizing the preparation conditions.239 The present section is limited to the polymer donor to remain focused; therefore, the device efficiency can be further enhanced through optimizing the inorganic component, processing method and device structure.
3.6 Summary
In this section, we have focused on various DTP-based polymers and small molecules, including their chemical structures, photovoltaic properties and device performance. The C2-based device gave a high JSC of 22.65 mA cm−2, which is one of the highest values so far.102 The reason for the relatively low PCE of 4.99% might be the low charge generation efficiency induced by the relative high-lying HOMO energy level. Although the polymer B5-based device gave a high VOC of 0.84 V among these PSCs, it is regrettable that the high VOC is at the expense of JSC, and thus the PCE of 4.2% is still low.49 For DTP-based SMSCs, the PCEs have been improved up to 8.22% with a VOC of 0.86 eV, JSC of 14.06 mA cm−2, and FF of 0.68 for E5 molecules.174 The high performance is mainly attributed to the thermal optimization and utilization of CuSCN as a HTL.One of the main reasons that accounts for the low performance of PSCs has been considered to be the energy loss Eloss relative to VOC and Eg, which defined as Eloss = Eg − eVOC, where Eg is the lowest value of Eg(D) and Eg(A).107 According to the above function, we estimated the energy losses of the original DTP-based materials from the cited reference. Fig. 11 shows how PCE is related to Eloss for the above DTP-based polymers and small molecules. It can be seen that the lowest attainable energy loss is 0.6 eV.106 Efficient devices always have the energy losses in the range of 0.6–1.0 eV for SMSCs and 0.8–1.2 eV for PSCs. An energy loss of 0.61 eV was estimated for the most efficient E5 device. Recently, an energy loss as low as 0.51 eV has been obtained with a PCE higher than 6%.240 Although the reason for the low energy loss is not clear yet, these results indicate that there are many opportunities to further enhance the PCE of DTP-based polymers by reducing the energy loss. In particular, exploiting the aromatic molecular backbones with extended π orbital delocalization would be a reasonable molecular design concept.241
 | ||
| Fig. 11 The estimated maximum PCE vs. the estimated energy loss (Eloss) for the above referred DTP-based solar cells. | ||
4. Conjugated photovoltaic materials based on fused DTP
The optimization of molecular structures is still necessary for further progress, although the efficiency has been considerably improved to realize commercial applications. In addition to incorporating different electron-donating and electron-accepting units into the polymer or small molecules, the fused aromatic heterocyclic unit has been proved to be a promising building block because the shared C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C not only can extend the conjugation resulting in broad sunlight absorption but also strengthen the intermolecular π–π stacking facilitating charge transport.242,243 For instance, a family of N-heteropentacenes shows excellent stability, solubility and electronic properties after introducing a varied number of N atoms at different backbone positions of pentacene.244,245 Large heteroacenes are believed to show advantageous opto-electronic properties for high-performance devices. In fact, in addition to the number and position of heteroatoms, the type of atom such as O or S would offer more chances to tune the stability and energy level. Among various heteroacenes containing more than one atom, S,N-heteroacenes have been considered as excellent building blocks to prepare promising conjugated materials.246
C not only can extend the conjugation resulting in broad sunlight absorption but also strengthen the intermolecular π–π stacking facilitating charge transport.242,243 For instance, a family of N-heteropentacenes shows excellent stability, solubility and electronic properties after introducing a varied number of N atoms at different backbone positions of pentacene.244,245 Large heteroacenes are believed to show advantageous opto-electronic properties for high-performance devices. In fact, in addition to the number and position of heteroatoms, the type of atom such as O or S would offer more chances to tune the stability and energy level. Among various heteroacenes containing more than one atom, S,N-heteroacenes have been considered as excellent building blocks to prepare promising conjugated materials.246
DTP can be regarded as one of the simplest heteroacenes containing fused two thiophenes bridged with a N atom. The great interest in DTP has extended to a family of planar and rigidified S,N-heteroacenes with the promise of pursuing high-performance materials. According to the position of fused groups relative to the central DTP, there are main two kinds of S,N-heteroacenes as presented in Fig. 12. When three thiophene units are combined together with a N bridge, a typical fused DTDP is formed. If the middle thiophene ring in DTDP is replaced with a thieno[3,2-b]thiophene (TT) unit, a SN6 unit can be obtained. In contrast to DTDP and SN6, in which different units are fused at one side of DTP, NPTA and DBTP contain thiophene and benzene rings attached on both sides of DTP, respectively. In this section, conjugated materials containing these heteroacenes as well as their application in solar cells will be introduced.
 | ||
| Fig. 12 Chemical structures of four fused DTP building blocks. | ||
4.1 Small molecules based on SN5 and SN6
A new generation of A–D–A small donor molecules capped with DCV emerged (Fig. 13).247 The presence of different substituents on the N atom largely affected the solubility, stability and charge transport ability.248 Note that a best PCE up to 6.5% for G1 with a propyl substituent was achieved in combination with C60 as the acceptor.247 The high PCE can be attributed to the enhanced JSC of 10.20 mA cm−2 and FF of 0.69 (Table 6), which resulted from the favorable blend morphology and phase separation as shown in Fig. 14. In contrast, G2 (JSC = 7.97 mA cm−2, FF = 0.49) with a hexyl substituent and G3 (JSC = 1.94 mA cm−2, FF = 0.40) with a tolyl substituent showed low PCE because of the decreased JSC and FF. The improvement of JSC for G1 compared to G2 and G3 is in good agreement with the EQE measurement, in which the spectra cover the range of 300–700 nm with a maximum EQE close to 70% at 600 nm for G1. Another A–D–A donor molecule G4 bearing an EH side chain shows a PCE of 3.1% with a JSC of 6.4 mA cm−2 and FF of 0.43.248 It is worth mentioning that the VOC was largely enhanced up to 1.13 V from 0.95 V. Very recently, the PCE was improved up to 5.64% with a JSC of 10.9 mA cm−2, a VOC of 0.98 V, and a FF of 0.52 for G4 molecules by optimization of the device.249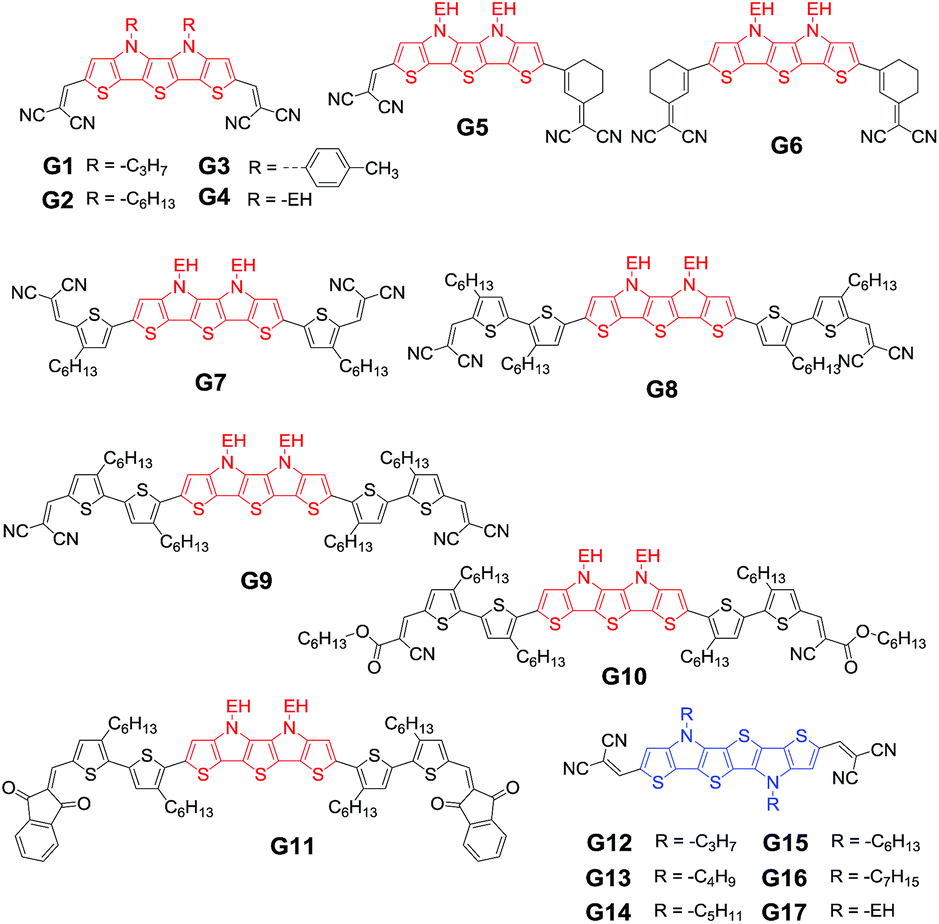 | ||
| Fig. 13 Small molecules based on DTDP and SN6 (EH: 2-ethylhexyl). | ||
| λ max [nm] | HOMO/LUMOb [eV] | μ h [cm−2 V−1 s−1] | Active layer | V OC [V] | J SC [mA cm−2] | FF | PCE [%] | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| a Films. b Estimated from the onset of the respective redox waves. c Measured using an organic FET device. d Hole mobility determined by the SCLC method. e Device characteristics of planar-mixed heterojunction (PMHJ) solar cells. f Could not be measured due to the limited solubility. g “NA” represents no data from the reference. | |||||||||
| G1 | 595, 637 | −5.64/−3.78 | NA |
G1:C60 = 1:1 |
0.92 | 10.20 | 0.69 | 6.5 | 247 |
| G2 | 594, 630 | −5.65/−3.79 | NA |
G2:C60 = 1:1 |
0.95 | 7.97 | 0.49 | 3.7 | 247 |
| G3 | 579, 624 | −5.70/−3.79 | NA |
G3:C60 = 1:1 |
0.95 | 1.94 | 0.40 | 0.7 | 247 |
| G4 | 592, 630 | −5.67/−3.85 | NA |
G4:PC61BM = 2:3 |
1.13 | 6.4 | 0.43 | 3.1 | 248 |
| 570 | −5.43/−3.53 |
G4:C70 = 1:1e |
0.98 | 10.90 | 0.52 | 5.58 | 249 | ||
| G5 | 625, 680 | −5.48/−3.74 | NA |
G5:PC61BM = 2:3 |
0.95 | 9.4 | 0.47 | 4.2 | 248 |
| G6 | 638, 718 | −5.35/−3.68 | NA |
G6:PC61BM = 1:1 |
0.88 | 10.8 | 0.51 | 4.9 | 248 |
| G7 | 725 | −5.26/−3.77 | 7.3 × 10−5d | CH3NH3PbI3 | 0.99 | 16.4 | 0.65 | 10.5 | 251 |
| G8 | 675 | −5.10/−3.74 | 6.6 × 10−5d | CH3NH3PbI3 | 0.90 | 15.2 | 0.68 | 9.5 | 251 |
| G9 | 642 | −5.16/−3.45 | 9.98 × 10−6 |
G9:PC71BM = 1:2 |
0.82 | 11.48 | 0.60 | 5.64 | 252 |
| G10 | 604 | −5.15/−3.34 | 8.38 × 10−6 |
G10:PC71BM = 1:2 |
0.82 | 9.24 | 0.56 | 4.24 | 252 |
| G11 | 648 | −5.25/−3.61 | 1.38 × 10−5 |
G11:PC71BM = 1:2 |
0.92 | 11.64 | 0.62 | 6.64 | 252 |
| G12 | 647, 600 | NAf | NA |
G12:C60 = 2:1 |
0.90 | 10.7 | 0.61 | 5.9 | 60 |
| G13 | 647, 598 | NAf | NA |
G13:C60 = 2:1 |
0.95 | 10.0 | 0.62 | 5.9 | 60 |
| G14 | 643, 596 | −5.58/−3.83 | NA |
G14:C60 = 2:1 |
0.96 | 12.2 | 0.61 | 7.1 | 60 |
| G15 | 638, 591 | −5.56/−3.74 | 0.021 |
G15:C60 = 2:1 |
0.96 | 10.3 | 0.60 | 5.9 | 60 and 253 |
| G16 | 640, 594 | −5.58/−3.77 | NA |
G16:C60 = 2:1 |
0.97 | 11.0 | 0.62 | 6.6 | 60 |
| G17 | 589 | −5.41/−3.55 |
G17:C70 = 1:1.5 |
0.84 | 9.51 | 0.38 | 3.00 | 249 | |
 | ||
| Fig. 14 (a) J–V characteristics and (b) EQE spectra of m-i-p bulk-heterojunction solar cells prepared with G1–G3 as donors and C60 as the acceptor (active area 6.4 mm2). (c) Tapping mode AFM topography images (1 μm × 1 μm) of blend films of G1:C60, G2:C60 and G3:C60 (reprinted with permission from ref. 247). | ||
If one 1-(1,1-dicyanomethylene)-cyclohex-2-ene (DCC) moiety was added between the DTDP skeleton and DCV terminal group, the backbone conjugation of molecules can be increased because of the additional double bonds.250 Bäuerle and co-workers designed two small molecules G5 with DCV and DCC as terminal units, and G6 with two DCC as terminal units.248 As a result, the DCC unit not only induced red-shifted and broadened absorption, but also lowered the bandgap by raising the HOMO energy level. With PC61BM as the acceptor, a highest PCE of 4.9% was achieved for G6 molecules. By comparison of G4, G5 and G6, the VOC values sequentially decreased from 1.13 V to 0.95 V and 0.88 V, while the JSC sequentially increased from 6.4 mA cm−2 to 9.4 mA cm−2 and 10.8 mA cm−2. The obvious trade-off between VOC and JSC can be attributed to the influence of terminal acceptor units.
In addition to being used as the donor, these molecules can also be utilized as hole-transport materials. After introducing alkyl-substituted thiophene into the backbone, two novel A–D–A oligomers G7 and G8 showed excellent PCEs of 10.5% and 9.5% in perovskite-based solar cells.251 The study on G9 indicates that the position of the alkyl chain on thiophene has a great impact on the performance.252 Using cyanoacetate and indenedione instead of DCV, two molecules G10 and G11 are respectively obtained. It is found that the red-shifted charge transfer band of G11 promoted the photocurrent, while the lower HOMO of G11 gives a relatively high VOC. In addition, the long alkoxy chain attached to the terminal part of the molecular scaffold seems to force the G10 molecules to pack on top of each other, which might be one reason for the low photocurrent.
In order to obtain thermally stable molecules with stronger absorption and better charge transport, one TT unit was used to replace the middle thiophene core of DTDP. Recently, a series of SN6-based molecules G12–G17 were obtained by varying the length of alkyl side chains substituted on the N atoms.60,249 Owing to the similar conjugated backbone, these molecules showed similar absorption properties and energy levels. Among them, G15 gave good optoelectronic properties and a high charge carrier mobility of 0.021 cm2 V−1 s−1.253 When an EH side chain was attached on N atoms, the vacuum-processed solar cells with G17 as the donor and C70 as the acceptor showed a PCE of 3.02%. By device optimization, an excellent PCE of up to 7.1% was achieved for G14-based vacuum-processed BHJ solar cells. In comparison with their DTP analogue E1, DTDP-based G4 and SN6-based G17 gave a largely improved JSC up to 6.4 mA cm−2 and 9.51 mA cm−2, respectively; however, a highest VOC of 1.21 V was observed for E1. These results indicate that fused S,N-heteroacene structures are promising building blocks for photovoltaic applications, although there are still many paths to further improve the PCE through molecular design.
4.2 Small molecules and polymers based on NPTA or DBTP
NPTA has symmetrical π-conjugated systems; therefore, it is expected to help in obtaining high performance small molecules as shown in Fig. 15. With DPP as the terminal acceptor unit and NPTA as the donor unit, a solution-processable small molecule H1 was designed.57 Compared to the analogue pentathiophene, the electron-rich pyrrole in the NPTA unit brought a red-shifted absorption. The substituted dodecyl chain on the N atom improved the crystallinity of H1 and miscibility with PC71BM. A PCE as high as 3.83% with a JSC of 10.69 mA cm−2, a VOC of 0.71 V and a FF of 48.3% was achieved based on the H1:PC71BM device with a 1.5:1 weight ratio (Table 7).
 | ||
| Fig. 15 Small molecules and polymers based on NPTA and DBTP (EH: 2-ethylhexyl). | ||
| λ max [nm] | HOMO/LUMOb [eV] | μ h [cm−2 V−1 s−1] | Active layer | V OC [V] | J SC [mA cm−2] | FF | PCE [%] | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| a Absorption maximum of films. b Estimated from the onset of the respective redox waves. c Hole mobility estimated by the SCLC method. d Calculated by the DFT method using B3LYP/6-31G(d). e “NA” represents no data from the reference. | |||||||||
| H1 | 648 | −4.96/−3.67 | 1.62 × 10−4 |
H1:PC71BM = 1.5:1 |
0.71 | 10.69 | 0.48 | 3.69 | 57 |
| H2 | 413, 549 | −4.83/−2.57d | 4.80 × 10−4 |
H2:PC71BM = 1:3 |
0.81 | 9.03 | 0.51 | 3.73 | 58 |
| H3 | 388, 536 | −4.93/−2.70d | 8.52 × 10−5 |
H3:PC71BM = 1:2 |
0.81 | 8.26 | 0.34 | 2.27 | 58 |
| H4 | 396, 552 | −4.74/−2.52d | 1.07 × 10−3 |
H4:PC71BM = 1:3 |
0.65 | 11.10 | 0.54 | 3.90 | 58 |
| H5 | 406, 653, 714 | −4.71/−2.57d | 1.53 × 10−3 |
H5:PC71BM = 1:2 |
0.65 | 15.54 | 0.64 | 6.46 | 58 |
| H6 | 421,568 | −4.60/−2.19d | 3.55 × 10−5 |
H6:PC71BM = 1:3 |
0.59 | 7.06 | 0.54 | 2.25 | 58 |
| H7 | 376, 605 | −5.24/−3.55 | NA |
H7:PC71BM = 1:2 |
0.82 | 12.62 | 0.69 | 7.04 | 254 |
After introducing benzene rings at both sides of the DTP unit, the obtained DBTP showed poor electron-donating ability, resulting in a low-lying HOMO energy level. A series of novel DBTP-based polymers H2–H6 gave effectively enhanced VOC and similar JSC and FFs compared to their DTP-based analogues (Table 7).58 As mentioned above, halogenation is an effective way to tune the energy levels of organic semiconducting materials. After introducing a fluorine atom into the BT unit, an approximate 0.33 eV increase in the HOMO energy level was observed for H3 compared to non-fluorinated H2. The H3:PC71BM device yielded a PCE of 4.16% (VOC = 0.65 V, JSC = 11.10 mA cm−2, and FF = 0.54), while H2:PC71BM devices yielded the best PCE of 4.02% (VOC = 0.81 V, JSC = 9.03 mA cm−2, and FF = 0.51). Upon replacing the alkyl side chain with an alkoxyl group substituted onto the BT unit, the H4 device shows a similar VOC to that of the H2 device; however, the FF value largely decreased. Therefore, the substituents on the bridged thiophene unit might be better for a good morphology than that on the BT unit.
If the substituted BT unit was replaced with a DPP unit, polymer H5 exhibited a broad absorption from 550 nm to 850 nm, resulting in the highest PCE of 6.80% with an improved JSC of 15.54 mA cm−2. In comparison with the non-fused DTP analogue C3, the extension of π-conjugation through the benzene ring fused with the DTP moiety is successfully used in polymers to increase the VOC value. A copolymer H6 combining DBTP with DTQx units showed much lower hole mobility than H1–H5 copolymers. Due to the moderate bandgap and slightly high-lying HOMO level, a better performance (PCE = 2.41%) than the DTP analogue D4 was obtained.74 After coupling DBTP and iID, polymer H7 showed a surprisingly high PCE of 7.04% with a VOC of 0.818 V, a JSC of 12.62 mA cm−2, and a FF of 68.6% based on H7:PC71BM solar cells. From the above discussion we know that the introduction of an iID group can greatly increase the mobility.
4.3 Summary
This section reviews some polymers and small molecules based on four typical fused-DTP units and found that the enlarged π-conjugated system can efficiently facilitate π-electron delocalization and intermolecular π–π stacking, which can directly enhance the charge carrier transport. Note that, among these fused-DTP-based small molecules and polymers, a VOC as high as 1.13 V was obtained by G4, a JSC as high as 15.54 mA cm−2 was observed in the H5-based device, a FF as high as 0.69 was recorded for the G1, and the highest PCE was obtained using G11. From the performance of isonomic A–D–A molecules E1, G4 and G14, it can be seen that the “fused” strategy reliably affects and improves the light-electricity conversion efficiency. In particular, the highest VOC of E1 resulted in a large loss in the charge separation efficiency. The final enhanced PCE of G11 might result from the fact that the increased JSC partly compensates for the loss of VOC. To our knowledge, this trade-off between JSC and VOC can be suppressed through manipulating the molecular structure and optimizing device parameters.255For OSC applications strongly involving charge transport through the active layer, an important aspect of heteroacenes is their molecular ordering in the solid state, which is the privilege of planar cores substituted with long side chains. It is generally known that molecules with face-to-face packing exhibited hole mobility superior to those with edge-to-face or even edge-to-edge packing. The position and length of the alkyl side chain strongly affect the molecular packing and should be carefully considered for molecular design. In addition, the fused-DTP units discussed above are five-numbered or six-numbered S,N-heteroacenes. With increasing the number of fused rings, the molecules can be easily modified with tunable optical and electronic properties and multi-level structures.256 Therefore, opportunities lie in the design of larger heteroacenes with the attachment of different substituents, and also the development of novel processed methods to achieve appropriate blend films. Many research studies are beginning to focus on other parameters such as stability, solubility and interfacial issues to maximize the performance. In the following sections, we will continue to adjust the structure of DTP to yield functional building blocks to pursue high-performance DTP-based solar cells.
5. Conjugated photovoltaic materials based on N-acyl DTP and PBTz
Although a diversity of N-alkyl DTP units have recently been considered as interesting and promising electron-deficient building blocks for high-performance OSCs, one limitation of such N-alkyl DTP is their high HOMO energy level decreasing stability under environmental conditions.55 There are two effective solutions to this limitation, such as incorporating N-acyl groups to produce N-acyl DTP and replacing thiophene with thiazole in the DTP heterocycle to produce PBTz, as presented in Fig. 16.56,67 Since 2010, a series of N-acyl DTP based polymers and small molecules (Fig. 17) have been synthesized by combining different acceptor derivatives. It was found that the N-acyl DTP polymers have relatively stabilized HOMO and LUMO energy levels in comparison with N-alkyl-DTP based analogues. The replacement of thiophene with thiazole in the fused heterocycles can improve the oxidative stability. The main challenge of the application of N-acyl DTP and PBTz based polymers would be tuning the molecular structure and optimizing the device conditions. This section will mainly discuss typical polymers and small molecules, and take a close look at the relationship between chemical structures and electronic performance (Table 8). | ||
| Fig. 16 Two building blocks N-acyl DTP and PBTz derived from DTP. | ||
 | ||
| Fig. 17 Photovoltaic polymers and small molecules based on N-acyl DTP and PBTz (EH: 2-ethylhexyl). | ||
| λ max [nm] | HOMO/LUMOb [eV] | μ h [cm−2 V−1 s−1] | Active layer | V OC [V] | J SC [mA cm−2] | FF | PCE [%] | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| a Absorption maximum of films. b Estimated from the onset of the respective redox waves. c Hole mobility estimated by the SCLC method. d TheHOMO level was measured with photoelectron spectroscopy in air (PESA), and LUMO level was estimated from the optical bandgap. e Could not be observed due to difficulties in the deposition on the substrate. f HOMO levels were estimated by UPS and the LUMO levels were estimated from the optical bandgap. g Field effect hole mobility. h “NA” represents no data from the reference. | |||||||||
| I1 | ∼660 | −5.00/−3.56d | 9.01 × 10−5c |
I1:PCBDAN = 1:1 |
0.52 | 17.1 | 0.44 | 3.95 | 258 |
| I2 | 580 | −5.27/−3.08 | NA |
I2:PC71BM = 1:3 |
0.66 | 8.89 | 0.56 | 3.29 | 259 |
| I3 | 685 | −5.39/−3.31 | NA |
I3:PC71BM = 1:3 |
0.82 | 13.30 | 0.53 | 5.82 | 259 |
| I4 | 703 | −5.18/−3.34 | NA |
I4:PC71BM = 1:3 |
0.67 | 12.57 | 0.54 | 4.53 | 263 |
| I5 | 666 | −5.31/−3.40 | NA |
I5:PC71BM = 1:3 |
0.76 | 6.66 | 0.47 | 2.39 | 263 |
| I6 | 650 | −5.50/−3.45 | NA |
I6:PC71BM = 1:3 |
0.64 | 7.19 | 0.51 | 2.34 | 263 |
| I7 | 681 | −5.35/−3.43 | NAe |
I7:PC71BM = 1:2 |
0.67 | 8.37 | 0.44 | 2.56 | 264 |
| I8 | 680 | −5.3/−3.9 | NA |
I8:PC71BM = 1:4 |
0.82 | 6.67 | 0.44 | 2.41 | 152 |
| I9 | 677 | −5.3/−3.8 | NA |
I9:PC71BM = 1:4 |
0.80 | 6.83 | 0.41 | 2.22 | 152 |
| I10 | 620 | −5.30/−3.67f | 7.5 × 10−5g |
I10:PC71BM = 1:3.5 |
0.64 | 2.8 | 0.44 | 0.8 | 265 |
| I11 | 653 | −5.25/−3.69f | 5.5 × 10−5g |
I11:PC71BM = 1:3.5 |
0.63 | 4.7 | 0.60 | 1.7 | 265 |
| I12 | 620 | −5.15/−3.45f | 1.1 × 10−3g |
I12:PC71BM = 1:3.5 |
0.61 | 9.4 | 0.53 | 3.0 | 265 |
| I13 | 560 | −5.20/−3.55f | 6.7 × 10−5g |
I13:PC71BM = 1:3.5 |
0.66 | 6.0 | 0.51 | 2.0 | 265 |
5.1 Polymers and small molecules based on N-acyl DTP
It has been found that the N-functionalized side chain in DTP largely affects the absorption and HOMO energy.257 The absorption red-shift and stabilization increase were observed in the same order alkyl < aryl < acyl < benzoyl. Based on this consideration, a polymer I1 composed of N-benzoyl DTP with BDT was designed.258 By combination with a fullerene derivative PCBDAN as the acceptor, a PCE of 3.95% was achieved with a high JSC of 17.1 mA cm−2. Meanwhile, two N-acyl substituted DTP polymers I2 and I3 were synthesized, and the I3:PC71BM device achieved a high PCE of 4.0% with a VOC of 0.80 V and a JSC of 9.88 mA cm−2.259 In comparison with the analogue polymer D1, the introduction of an acyl unit at the N atom significantly enhanced the JSC up to 13.30 mA cm−2 after applying a conjugated cathode interlayer. Overall, the relatively high efficiency of these three polymers (I1–I3) can be mainly attributed to the increased VOC, resulting from the lower HOMO energy level by changing the N-alkyl to N-acyl.Qxs are promising electron-deficient units due to their high EA.260 It has been reported that Qx-containing copolymers could afford high performance with a high VOC exceeding 0.9 V.261,262 For further improving the performance of N-acyl DTP derivatives, a new polymer I4 combining N-acyl-DTP with Qx was reported.263 The fabricated devices showed a best PCE of 4.81% with an enhanced JSC of 12.57 mA cm−2 but a relatively low VOC of 0.67 V. In order to improve VOC, fluorinated polymers I5 and I6 were designed; however the PCE dramatically decreased, which might be attributed to the unfavorable interpenetrating blend network. In order to study the impact of molecular structure, very recently a similar small molecule I7 was obtained after replacing the central DTS unit with an N-acyl DTP unit.264 As a result, the variation of donor unit largely affects the properties, and particularly the crystallinity was greatly modified. In addition, two kinds of BAI-based polymer I8 and I9 containing N-acyl DTP and BAI were synthesized recently.152 As a result, the polymer I8 afforded a slightly better efficiency of 2.41% than I9 (PCE = 2.22%), which is one of the highest PCEs among reported BAI-based devices. All the devices seem to be limited by the active layer morphology because of the low solubility, and they need high-boiling solvents to completely dissolve the polymers. Therefore, much effort on molecular engineering is required to increase the miscibility without changing the low-lying LUMO levels and NIR absorption spectra.
5.2 Polymers based on PBTz
A series of PBTz based polymers I10–I13 coupled with different acceptor units have been studied.265 It was found that these polymers possessed good solubility and bandgaps in the range of 1.56–1.7 eV. A highest PCE of 3.0% was obtained for I12:PC71BM because of stronger crystallization of I12 resulting in longer length scale phase segregation in the blend film. Recently, a kind of small molecule N-alkyl-PBTz-bridged bis(naphthalene diimide) was studied.266 It was found that its electron mobility as high as 0.19 cm2 V−1 s−1 strongly depended on the side chain substituted on the N atoms of pyrrole and NDI. In addition, after coupling with electron-donating thienylenevinylene (TV), copolymer PTBz–TV substituted with an n-hexadecyl side chain on pyrrole exhibited a high hole mobility of 0.062 cm2 V−1 s−1.267 These results indicate that this tricycle PBTz has both weak donor and acceptor properties, which will be a promising building block for developing high-performance p-type or n-type conjugated materials although there are very few reports on this kind of material at present.5.3 Summary
In this section, a series of polymers and small molecules based on N-acyl DTP have been simply discussed. It was found that in this specific class of DTP groups, the incorporated N-acyl group yields more stable polymers with deep HOMO levels and low bandgaps. In comparison to N-alkyl DTP polymers, the appropriate LUMO level results in facile electron injection. In addition, the film morphology can also be optimized leading to an efficient charge transport. Hence, use of N-acyl DTP is a promising approach to devices with increased performance. Continuous structure modification should enable the N-acyl functional group to play a greater role.6. Conjugated photovoltaic materials based on lactam or imide building blocks derived from DTP
After introducing an electron withdrawing carbonyl unit, N-acyl-substituted DTP materials usually showed lower-lying HOMO energy levels and consequently higher VOC in comparison with DTP-based materials as mentioned above. Thus, the HOMO energy level can be further reduced by the carbonyl group. In addition, a more π-extended ring with an enlarged planar aromatic structure would be more favorable for the π-electron delocalization, resulting in a deep HOMO energy level.268 Combining the advantages of the carbonyl group and polycyclic structure, a novel kind of DTPO unit with the carbonyl migrating into the DTP heterocycle was designed. When two acyl groups were integrated into the heterocycle, a seven-membered heterocycle BTI can be obtained. A lactam TD unit containing six fused aromatic rings with two directly connecting DTPO units was designed. Similar to the design concept of TD, a more π-extended aromatic structure TBI comprises two fused BTI units. If starting with the structure of SN6, TD and TBI also can be considered as SN6 derivatives, in which two or four carbonyl units were incorporated into the DTP heterocycle. In comparison with DTPO and BTI, the superior features such as more electron-withdrawing groups, extended conjugation and enlarged planarity in TD and TBI are expected to enhance the light absorbance, reducing the bandgap, and improve the charge mobility. Recently, several BTI-based polymers have been summarized by Guo and co-workers.62In this section, a series of semiconducting polymers containing various structures such as DTPO, TD, BTI and TBI (Fig. 18) with their photovoltaic properties and their outstanding performances in OSC applications will be discussed. We would focus on the evolution of molecular structures and their impact on the photovoltaic properties including light absorption capability, energy level structure, and charge carrier mobility. The purpose of our review is to elaborate the relationship between the molecular structure and photovoltaic properties through molecular design and give some guidance for future molecular design.
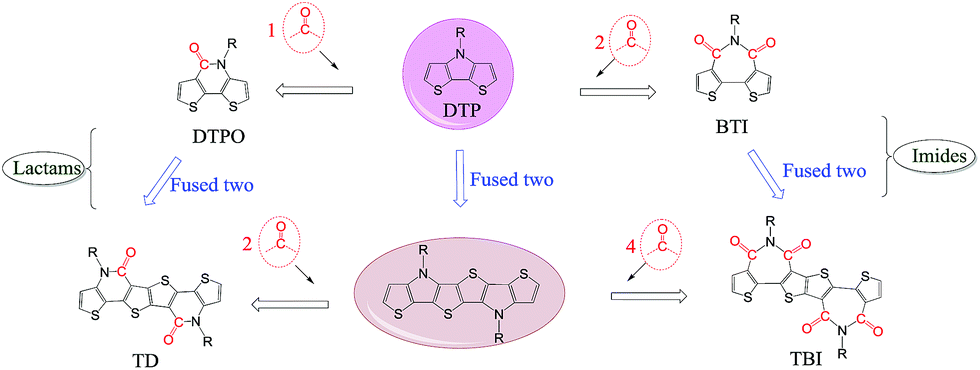 | ||
| Fig. 18 The evolution of the original DTP unit to fused DTP, lactam and imide building blocks. | ||
6.1 Polymers based on DTPO and TD
Comprising an N-acyl DTP building block, a DTPO unit with a more π-extended six-membered lactam ring and a larger planar aromatic structure is expected to show excellent photovoltaic properties. By combining with the famous BDT acceptor unit, a series of D–A copolymers J1–J3 as shown in Fig. 19 have shown significantly improved photovoltaic performance (Table 9).61 There are two side-chain substituted positions, one is the N atom of DTPO and the other is the benzene ring of BDT. When butyloctyl (BO) was attached on DTPO, the optical bandgaps are in the order J1a > J2 > J3a depending on the substituents on BDT. Note that J3a showed the lowest LUMO energy level, while J2 showed the highest HOMO energy level. Under the same device conditions, they showed little difference in VOC and JSC values; however, the FF of J3a:PC71BM is much higher than those of the other two systems because of the long pathway favorable for charge transport and separation, resulting from the large crystalline domain in the pristine J3a film. With variation of the side chains on the N atom of DTPO, J1b and J3b with longer OD alkyl chains showed slightly wider optical bandgaps and better solubility than their BO-substituted analogues J1a and J3a. The as-cast or inverted devices of J1a and J3a showed a similar high PCE up to 6.84% with VOC in the range of 0.93–0.96 V. | ||
| Fig. 19 Photovoltaic polymers based on DTPO and TD lactams (EH: 2-ethylhexyl). | ||
| λ max [nm] | HOMO/LUMOb [eV] | μ h [cm−2 V−1 s−1] | Active layer | V OC [V] | J SC [mA cm−2] | FF | PCE [%] | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| a Absorption maximum of films. b Estimated from the onset of the respective redox waves. c Measured using the SCLC method. d Field effect hole mobility. e “NA” represents no data from the reference. | |||||||||
| J1a | 570, 527 | −5.27/−2.68 | 2.83 × 10−4 |
J1a:PC71BM = 1:1.5 |
0.87 | 8.30 | 0.45 | 3.28 | 61 and 69 |
| J1b | 530, 571 | −5.38/−3.37 | 1.25 × 10−4d |
J1b:PC71BM = 1:2 |
0.93 | 10.4 | 0.71 | 6.84 | 61 |
| J2 | 573, 530 | −5.47/−2.68 | 4.91 × 10−4 |
J2:PC71BM = 1:1.5 |
0.85 | 9.26 | 0.52 | 4.08 | 69 |
| J3a | 579, 536 | −5.36/−2.83 | 4.42 × 10−4 |
J3b:PC71BM = 1:1.5 |
0.89 | 8.50 | 0.71 | 5.33 | 69 |
| J3b | 541, 582 | −5.44/−3.35 | 1.41 × 10−4d |
J3a:PC71BM = 1:2 |
0.96 | 10.5 | 0.71 | 6.84 | 61 |
| J4 | 500 | −5.59/−2.77 | 2.09 × 10−4 |
J4:PC71BM = 1:1.5 |
0.86 | 3.50 | 0.32 | 0.95 | 69 |
| J5 | 574 | −5.39/−2.87 | 4.40 × 10−4 |
J5:PC71BM = 1:1.5 |
0.75 | 8.47 | 0.41 | 2.57 | 69 |
| J6 | 630, 668 | −5.26/−3.21 | 3.60 × 10−4 |
J6:PC71BM:ITIC = 1:1.2:0.3 |
0.84 | 15.82 | 0.69 | 9.20 | 269 |
| J7 | 531, 566 | −5.31/−3.27 | 1.0 × 10−3 |
J7:PC71BM = 1:2 |
0.97 | 10.55 | 0.72 | 7.33 | 272 |
| J8 | 532, 569 | −5.31/−3.27 | 1.0 × 10−3 |
J8:PC71BM = 1:2 |
0.94 | 9.14 | 0.64 | 5.47 | 272 |
| J9 | 620 | −5.52/−2.74 | NA |
J9:PC71BM = 1:1.2 |
0.98 | 11.56 | 0.70 | 7.94 | 273 |
| Inverted J9:PC71BM = 1:1.2 |
0.96 | 12.59 | 0.76 | 9.13 | 273 | ||||
| 0.98 | 13.65 | 0.76 | 10.15 | 278 | |||||
| Tandem solar cells | 1.67 | 9.85 | 0.60 | 11.35 | 275 | ||||
| J10 | 640 | −5.51/−2.80 | 1.69 × 10−3 |
J10:PC71BM = 1:1.2 |
0.91 | 9.96 | 0.75 | 6.76 | 63 |
| J11 | 590 | −5.53/−2.68 | 3.00 × 10−4 |
J11:PC71BM = 1:1.2 |
0.89 | 10.37 | 0.55 | 5.05 | 63 |
| J12a | 578, 629 | −5.30/−2.63 | 5.98 × 10−4 |
J12a:PC71BM = 1:0.8 |
0.85 | 11.95 | 0.68 | 6.90 | 64 |
| J12b | 579, 630 | −5.36/−2.56 | 4.06 × 10−4 |
J12b:PC71BM = 1:0.8 |
0.87 | 9.67 | 0.72 | 6.02 | 64 |
| J13a | 602, 656 | −5.31/−2.74 | 2.53 × 10−4 |
J13a:PC71BM = 1:0.8 |
0.85 | 13.55 | 0.71 | 8.18 | 64 |
| J13b | 597, 651 | −5.34/−2.70 | 9.79 × 10−4 |
J13b:PC71BM = 1:0.8 |
0.83 | 10.92 | 0.69 | 6.22 | 64 |
After replacing the BDT unit with bis(2-hexylthiophene), polymer J4 showed a dramatically decreased PCE of only 0.95% (VOC = 0.86 V, JSC = 3.50 mA cm−2 and FF = 0.32), which might be attributed to the narrow UV-vis absorption and low hole mobility of 2.09 × 10−5 cm2 V−1 s−1. After selecting rigid DTS as the acceptor unit to copolymerize with DTPO, polymer J5 showed a smaller bandgap of 2.52 eV than J4 (2.82 eV). The J5:PC71BM device exhibited an extended EQE at 700 nm with a PCE of 2.57%. When a difluorobenzothiadiazole (FBT) unit is introduced to couple with the DTPO unit, a low bandgap D–A copolymer J6 (1.62 eV) was developed.269 With a famous small molecule 3,9-bis(2-methylene-(3-(1,1-dicyanomethylene-indanone))-5,5,11,11-tetrakis(4-hexylphenyl)-dithieno[2,3-d:2′,3′-d′]-s-indaceno[1,2-b:5,6-b′]dithiophene) (ITIC) as the acceptor, J6:ITIC exhibited a much higher PCE of 7.58% than J6:PC71BM (PCE = 2.91%). Upon addition of DIO as the solvent additive, the performance of J6:ITIC devices dramatically decreased down to 2.58%; however, J6:PC71BM exhibited a largely increased PCE up to 8.75%. The morphology characterization indicates that DIO might inhibit J6 and PC71BM from aggregation, while promoting the J6:ITIC blend film to form large domains. Combined with the advantages of these two systems, a ternary device using the blend active layer J6:PC71BM:ITIC with a ratio of 1:1.2:0.3 showed a decent PCE of 9.20% due to the complementary absorption and stepwise-aligned energy level.
Since the advent of the popular ladder-type unit indacenodithiophene (IDT),270 and its derivative indacenodithieno[3,2-b]thiophene (IDTT) by substituting two outward thiophenes on IDT,271 photovoltaic materials based on them have been widely developed. By coupling an IDT or an IDTT unit with DTPO, two wide bandgap polymers J7 and J8 displayed a planar polymer backbone and strong intermolecular π–π stacking, and thus large charge carrier transport due to the ladder-type π-extended structures of IDT and IDTT.272 As a result, J7 devices showed a higher PCE of 7.33% than J8 devices, which can be attributed to the different morphologies of their blend films with PC71BM. Ladder-type units comprising of linearly fused aromatic or heteroaromatic structures not only can extend the conjugation length of the backbone but also restrain the rotational disorder, which benefits the charge carrier mobility.
In order to bring out the advantages of the DTPO structure, a BDTP unit was designed by directly connecting two DTPO units. BDTP-based materials are expected to show broadened and red-shifted light absorption, tunable energy levels, and appropriate lamellar arrangement due to the enlarged π-conjugation length, two electron-withdrawing carboxyl groups and a single bond between two fused DTPO units. There have been reported three typical polymers J9, J10 and J11 as shown in Fig. 19, in which thiophene, selenophene, and the BDT donor unit were coupled with BDTP, respectively. It was found that J9 showed a narrow optical bandgap of 1.86 eV, and J9:PC71BM gave a PCE of 7.94% (VOC = 0.98 V, JSC = 11.56 mA cm−2, and FF = 61.0%).273 Note that the PCE of J9:PC71BM was improved up to 9.13% with an inverted device structure, and was enhanced to 10.15% by using poly(9,9-bis(N,N-dimethylamino)) (PFN) as a buffer layer.274 The JSC was up to 13.65 mA cm−2, while the absorption wavelength in the range of 300–800 nm results in insufficient utilization of sunlight. A tandem solar cell based on J9:PC71BM and DPPEZnP-TEH:PC61BM active layers achieved a higher PCE of 11.35% by utilizing the wide range of the solar spectrum.275 However, the J10:PC71BM inverted solar cells showed a lower PCE of 6.76% compared to the J9:PC61BM inverted device.63 With BDT as the donor unit, J11 showed a broader bandgap of 1.98 eV, and the J11:PC61BM inverted solar cell gave a low PCE of 5.05%, which might affect by the lower hole mobility than J10. In contrast to the series of DTPO-based polymers mentioned above, the coupling of BDTP-based polymers with BDT did not exhibit good photovoltaic properties. In addition to the light absorption and energy level, it is important to note that the introduction of BDT disrupted the π–π stacking of large BDTP units, resulting in a low charge mobility. From this point of view, a reasonable choice of the D unit and A unit is critical in the D–A type polymers. So far there have been reported a wide variety of D and A units, and thus high-efficiency photovoltaic materials can be designed in consideration of basic requirements and scientific issues.8
Among the current high-performance materials containing IDT or IDTT ladder-type units,276,277 ladder-type building blocks would be components in the next generation of photovoltaic materials. By fusing two DTPO units together, a symmetrical hexacyclic lactam TD unit was developed.64 Compared with the BDTP unit, the rigid and coplanar TT unit at the center of the TD unit would increase the conjugation planarity of TD. J12a and J12b showed an optical bandgap of 1.82 eV just slightly lower than that of analogue J9 because of the different conjugation lengths between BDTP and TD. Devices J12a:PC71BM and J12b:PC71BM exhibited PCEs of 6.90% and 6.02%, respectively. The shorter 2-hexyldecyl side chain resulted in much more ordered molecular packing of J12a than J12b, which is beneficial to higher photocurrent in J12a:PC71BM. On replacing the thiophene donor unit with selenophene, two J10 analogues J13a and J13b with a low bandgap of 1.75 eV showed high PCEs up to 8.18% and 6.22% with PC71BM as the acceptor. Thus it can be seen that a highly planar unit along the backbone would efficiently enhance the performance by increasing the JSC and FF values and maintaining VOC.
From the consideration of solubility and packing to the variation of branched side chains attached on the N atom, from the engineering of the polymeric backbone to the combination of diversified acceptor units with different π-conjugations, from the regulation of the energy level to the introduction of two DTPO units, and from the influence of backbone curvature to the comparison of unfused and fused DTPO units, we outline molecular design strategies and recent performance progress in these pyridone-based polymers. A synergy of the engineering of the side chain and backbone not only improves the performance but also broadens the application of DTP-based polymers. However, some challenges persist, especially in enhancing charge mobility, controlling the molecular packing, and improving the device stability, which require a collective effort of integrated consideration of both molecular engineering and process optimizing.
6.2 Polymers based on BTI and TBI
As a new kind of building block, BTI, exhibited bithiophene planarity and antiparallel molecular packing with close π–π stacking.279 The homopolymer is highly crystalline showing a high electron mobility of 0.04 cm2 V−1 s−1, which can be up to 0.19 cm2 V−1 s−1 when the molecular weight was increased two-fold. Although a HOMO energy level as low as −6.43 eV might prevent the charge transport in the device, other advantages such as good solubility, favorable geometry, and promising charge mobility would serve as the foundation for BTI-based polymers as effective donor materials. In order to solve the problems of poor solubility and unsuitable energy levels, it is necessary to further develop BTI-based polymers by selecting electron-donating units. Note that the central imide group favors the BTI unit to copolymerize with various monomers to realize a variety of BTI-based polymers as shown in Fig. 20 and Table 10. There is no doubt that BTI-based polymers would promote the development of OSCs through further structure modification and device optimization. | ||
| Fig. 20 Photovoltaic polymers based on BTI and TBI lactams (EH: 2-ethylhexyl). | ||
| λ max [nm] | HOMO/LUMOb [eV] | μ [cm−2 V−1 s−1] | Active layer | V OC [V] | J SC [mA cm−2] | FF | PCE [%] | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| a Films on the plate; the wave line indicates the estimated value from the given profile. b Calculated from the onset of oxidation and reduction waves in CV. c Field effect hole mobility. d Measured by the SCLC method. e LUMO levels were calculated according to the optical bandgap. f HOMO levels are determined from UPS, and LUMO values are determined from the optical bandgap. g HOMO levels evaluated by photoemission yield spectroscopy (PYS), and LUMO levels evaluated by low-energy inverse photoemission spectroscopy (LEIPS). h “NA” represents no data from the reference. | |||||||||
| K1 | 644 | −5.39/−2.54 | 1.9 × 10−4d |
K1:PC71BM = 1:1 |
0.92 | 9.62 | 0.62 | 5.5 | 282 |
| K2 | 592 | −5.43/−3.68e | 1.08 × 10−4 |
K2:PC71BM = 1:1 |
0.803 | 12.81 | 0.62 | 6.41 | 285 |
| K3 | 593 | −5.39/−3.64e | 1.66 × 10−4 |
K3:PC71BM = 1:1 |
0.774 | 12.30 | 0.50 | 4.77 | 285 |
| K4 | 602 | −5.72/−3.85f | 7.1 × 10−5 |
K4:PC71BM = 1:2 |
0.75 | 0.71 | 0.31 | 0.16 | 287 |
| K5 | 573 | −5.57/−3.76f | 1.1 × 10−3 |
K5:PC71BM = 1:2 |
0.95 | 8.15 | 0.69 | 5.37 | 287 |
| K6 | ∼625 | −5.58/−3.77 | 2.74 × 10−3 |
K6:PC71BM = 1:2 |
0.86 | 12.9 | 77.8 | 8.66 | 288 |
| K7 | 628 | −5.25/3.41f | 7.1 × 10−2 |
K7:PC71BM = 1:2 |
0.75 | 9.30 | 0.68 | 4.74 | 287 |
| K8 | 641 | −5.70/−3.22g | 1.4 × 10−4d |
K8:PC61BM = 1:2 |
1.05 | 8.2 | 0.66 | 5.6 | 289 |
| K9 | 660 | −5.58/−3.31g | 6.4 × 10−4d |
K9:PC61BM = 1:2 |
1.00 | 10.9 | 0.65 | 7.1 | 289 |
|
K9:PC71BM = 1:2 |
0.99 | 12.5 | 0.64 | 8.0 | 289 | ||||
| K10 | 654 | −5.85/−3.40g | 2.8 × 10−4d |
K10:PC61BM = 1:2 |
1.07 | 4.2 | 0.53 | 2.4 | 289 |
| K11 | 602 | −6.12/−3.56g | 1.2 × 10−4d |
K11:PC61BM = 1:2 |
0.40 | 0.34 | 0.32 | 0.05 | 289 |
The BDT unit, in particular that with two thienyl conjugated side chains, has been considered a highly promising unit for constructing high-efficiency donor materials.280,281 Polymer K1 coupling BTI with BDT showed a HOMO energy level of −5.39 eV, and a PCE as high as 5.5% was achieved with high a VOC of more than 0.9 V.282 Such fabricated devices also showed promising air and thermal stability in virtue of the intrinsic low-lying HOMO level and good device structure. The LUMO level of the donor should be in the range of −3.0 to −4.0 eV ensuring that the energy difference LUMOD–LUMOA is about 0.3 eV for efficient charge separation.283 Therefore, optimizing the LUMO level (−2.54 eV) and reducing the bandgap (∼1.95 eV) of K1 are prerequisites to achieve higher performance.284 In addition, the electron and hole mobilities in the K1:PC71BM blend should also be improved to be high enough for efficient charge extraction.
After coupling BTI with an electron-rich DTS or DTG building block, two BTI-based polymers K2 and K3 were obtained.285 Note that the HOMO energy levels of K2 (−5.43 eV) and K3 (−5.39 eV) are similar to that of K1, while the LUMO levels of K2 and K3 are −3.68 eV and −3.64 eV, respectively. The little different HOMO levels indicate that the strong electron-withdrawing imide group dominates the HOMO energy of BTI-based polymers. Therefore, the lower-lying LUMO energy level makes a small LUMO–LUMO offset of 0.3 eV with the PC71BM acceptor. The inverted device K2:PC71BM showed a best PCE of 6.41%, while the K3:PC71BM inverted device exhibited a slightly decreased PCE of 4.77%, due principally to the largely depressed FF over that of analogous K2. We supposed that the low device performance is possibly related to not only the molecular weight but also the molecular packing.
A series of BTI–oligothiophene polymers K4–K7 with sequentially varying conjugation lengths of oligothiophene subunits exhibited similar band gaps but greatly different charge-transport mobilities.286 The polymer with one thiophene exhibited an ambipolar behavior with a balanced electron and hole mobility of ∼10−4 cm2 V−1 s−1, while other polymers with two or four thiophene subunits showed greatly enhanced hole mobility of up to 10−3 and 10−2 cm2 V−1 s−1, respectively. By optimizing the device architecture, the highest hole mobility of these polymers can approach 0.1 cm2 V−1 s−1 comparable to that of P3HT. These results indicate that oligothiophenes are effective building blocks for high-mobility polymers, which can suppress the charge recombination. Recently, it was found that HOMO energy levels were raised with increasing the number of thiophene units, and the position of the alkyl chain on oligothiophene greatly affected the film morphology.287 Even under the same device conditions, their performance varied greatly in the order of K4 (0.16%) < K7 (4.74%) < K5 (5.37%) < K6 (7.70%). Therefore, the optimal number of thiophene units along the backbone of the polymer might be three.
As a promising material, K6 has been the focus of research recently. A high PCE up to 8.7% (JSC = 12.9 mA cm,2VOC = 0.859 V, and FF = 78%) was achieved upon addition of the DIO solvent additive.288 The JSC value being comparable to the theoretical one might be consistent with the efficient charge transport due to a highly ordered π–π stacking structure. In addition, the elaborately controlled bicontinuous network with vertical phase gradation in the blend film is a critical factor for high-performance devices as shown in Fig. 21. The charge pair dissociation at the face-on interface was more efficient and resulted in smaller geminate recombination loss. Therefore, a high PCE over 10% can be realized by further optimizing the architecture to suppress the carrier recombination and reduce the energy loss to the maximum.
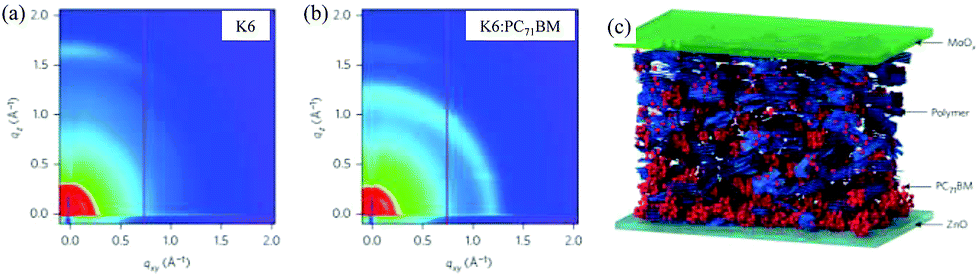 | ||
| Fig. 21 2D GIWAXS images of a neat K6 film (a) and K6:PC71BM blend film (b). (c) Schematic of the K6:PC71BM blend film showing vertical phase gradation with a polymer-rich layer near the MoOx/blend interface and a PC71BM-rich layer near the blend/ZnO interface (reprinted with permission from ref. 288). | ||
In the exploration of high-performance polymers, a deep frontier molecular orbital energy level is a key parameter for the oxidative stability of materials, high VOC and ambient electron transport in devices. One of the most efficient strategies is to develop new electron-withdrawing units. Similar to the evolution of TD from DTPO, a fascinating acceptor is the heterocycle unit TBI containing two fused BTI units. On introducing two thiophene units along the backbone, K8 shows a HOMO of −5.70 eV and LUMO of −3.22 eV, which are just deeper than those of the BTI analogue by 0.1 eV. When an electron-deficient bithiazole unit was coupled with TBI, K11 shows a much deeper HOMO of −6.12 eV and LUMO of −3.56 eV. Note that the energy level decreased with increasing the electron-withdrawing capability of the coupling units. In addition, the absorption maximum of K11 showed a blue-shift relative to the other three polymers. A common feature is that these polymers K8–K11 displayed good molecular ordering due to the strong intermolecular interactions. But K8 and K11 formed an edge-on orientation, while K9 and K10 formed a face-on orientation.
With PC61BM as the acceptor, K9:PC71BM exhibited a highest PCE up to 8.0% with a high JSC of 10.9 mA cm−2. K8, K9 and K10 devices gave a remarkably high VOC of more than 1.0 eV, which can be attributed to the deep HOMO energy levels. The different JSC values agreed with the polymer orientation in the thin film. The face-on orientation is favorable for charge transport and thus better charge collection for K9, whereas the edge-on orientation and the amorphous structure are unfavorable for the charge transport in the case of K8 and K10. It should be noted that K11 did not show any photovoltaic effect because the electron transfer might be prevented by the smaller LUMO–LUMO offset between K11 and PC61BM. Instead, K11 would be promising for use as an acceptor material, which will be discussed in the following section. These multi-functional applications demonstrate that the TBI building block is a promising acceptor group for constructing semiconducting materials.
6.3 Summary
In this section, we have discussed recent advances in the research of four novel building blocks including DTPO, TD, BTI and TBI by incorporating either one or two carbonyl groups into the pyrrole ring of DTP. It was found that the energy levels of BTI- and TBI-based polymers were much deeper than those of analogues containing one-carbonyl-incorporated DTPO and TD building block. In addition, the bandgaps of DTPO- and TD-based polymers are in a large range of 2.01–2.85 eV, while the bandgaps were narrowed down to 1.75 eV for BTI-based polymers. Note that the TBI-based polymer showed large bandgaps in the range of 2.27–2.56 eV. This might be due to the inappropriate selection of copolymerized units because a lower degree of backbone planarity usually leads to a larger bandgap. In addition, the backbone planarity also affects the absorption capability and charge mobility. A higher degree of backbone planarity generally affords a better absorption in the red region and a higher charge transport due to a higher degree of molecular lamellar ordering.Similar to the other polymers mentioned above, the charge mobility of most polymers is still in the order of 10−3 cm2 V−1 s−1. As a matter of fact, great progress in polymeric semiconducting materials with high mobility has been made in recent years. For example, semiconducting polymers based on thienoisoindigo and DPP moieties show a hole mobility (μh) over 14 cm2 V−1 s−1290,291 and an ultrahigh hole mobility up to 36 cm2 V−1 s−1 through a nanotemplating technique.292 Through optimization of the molecular structure and preparation methods, the improved charge mobility can be attributed to the enlarged π-conjugated backbone and enhanced intermolecular π-orbital overlap.293 Together with the characterization of solution-processability and high stability, we believe that this kind of polymer can meet the requirements for commercial application in OSCs although there are still a few challenges, including the design concept, synthesis, fabrication of devices, and investigation of their electronic properties.
7. Conjugated photovoltaic materials based on DTP and its derivatives as electron acceptors
With development of new electron-donating small molecules and polymers in the past few decades, the PCE of OSCs has reached over 11% for fullerene-based BHJ devices.294–296 Despite widespread use of fullerenes as electron acceptors, it is difficult to further tune their absorption and electronic structures over a wide range by generally molecular modification. The development of nonfullerene acceptor molecules has become an important approach to overcome the shortcomings of fullerenes and to further improve the performance. Since a polymer was first used as the electron acceptor in all-polymer solar cells (all-PSCs) in 1995,2 the best PCE has extended up to 8%.297 Recently, non-fullerene small acceptor molecules have realized the highest PCE of over 12%.298 Compared with the remarkable success in p-type polymers and small molecules, DTP and its derivatives have been rarely used to construct n-type photovoltaic materials. The reason might be the relative high-lying energy levels, which need strong electron-withdrawing units to combine with DTP derivatives. So far, only a few DTP-based n-type polymers and small molecules have been reported mainly based on the original DTP or TBI building blocks. Due to the relatively high energy levels of DTP-based materials, the choice of p-type materials is limited. Six types of donor materials as shown in Fig. 22 including two kinds of polythiophenes, and one DTP-based and three BDT-based polymers have been used to fabricate devices in combination with these DTP-based acceptors.299,300 In this section, the focus of our discussion is on the correlation between the molecular structure and device performance, which would play a vital role in guiding molecular design in organic electron materials (Table 11).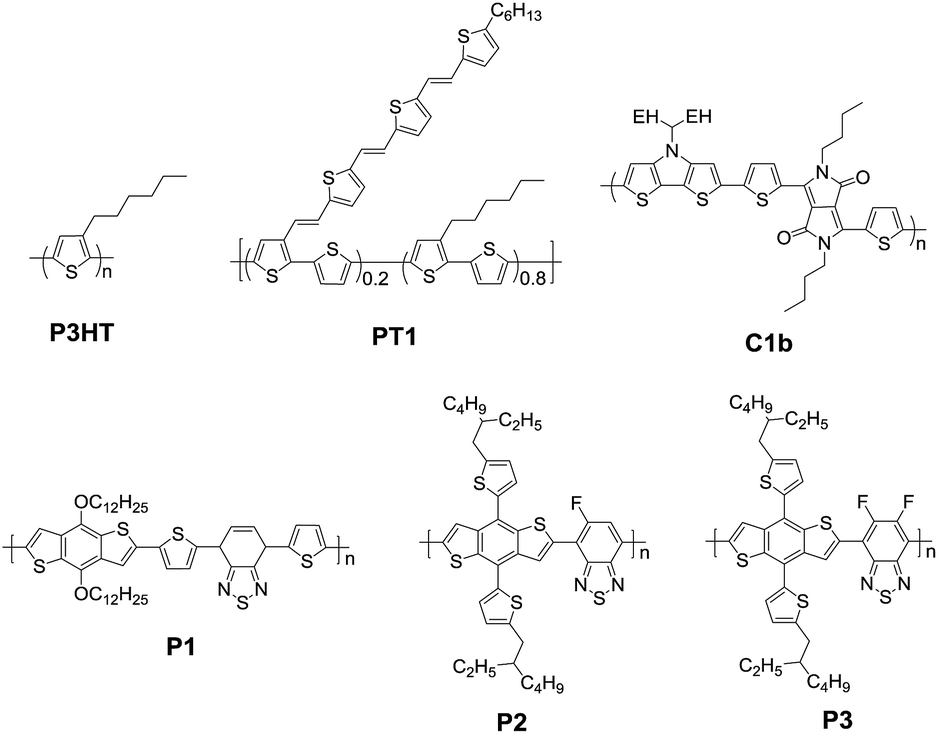 | ||
| Fig. 22 p-Type polymers combined with DTP-based n-type materials (EH: 2-ethylhexyl). | ||
| λ max [nm] | HOMO/LUMOb [eV] | μ e [cm−2 V−1 s−1] | Active layer | V OC [V] | J SC [mA cm−2] | FF | PCE [%] | Ref. | |
|---|---|---|---|---|---|---|---|---|---|
| a Film on plate. b Calculated from the onset of oxidation and reduction waves in CV. c Measured according to the SCLC method. d Obtained from organic FET devices. e “NA” represents no data from the reference. | |||||||||
| L1 | NA | NA | NA |
P2:L1 = 1:1 |
0.62 | 8.6 | 0.50 | 2.68 | 304 |
|
P3:L1 = 1:1 |
0.72 | 9.4 | 0.52 | 3.53 | 304 | ||||
| L2 | NA | NA | NA |
P2:L2 = 1:1 |
0.61 | 8.56 | 0.54 | 2.82 | 304 |
|
P3:L2 = 1:1 |
0.76 | 9.3 | 0.57 | 4.05 | 304 | ||||
| L3 | 361, 484, 715 | −5.49/−3.83 | NA |
C1b:L3 = 1:1 |
0.42 | 1.86 | 0.53 | 0.41 | 299 |
| P3HT:L3 = 2:1 |
0.46 | 0.76 | 0.50 | 0.17 | 320 | ||||
| PT1:L3 = 2:1 |
0.66 | 3.05 | 0.46 | 0.93 | 320 | ||||
| L4 | 441, 746 | −5.27/−4.00 | NA | PT1:L4 = 2:1 |
0.60 | 0.68 | 0.31 | 0.13 | 299 |
|
C1b:L4 = 1:1 |
0.28 | 0.24 | 0.38 | 0.02 | 299 | ||||
| L5 | NA | −5.61/3.94 | NA | P3HT:L5 = 2:1 |
0.72 | 4 | 0.51 | 1.5 | 80 |
| L6 | 654 | −5.85/−3.40 | 0.02 | P3HT:L6 = 2:1 |
0.64 | 2.5 | 0.46 | 0.73 | 289 |
| L7 | 602 | −6.12/−3.56 | 0.05 | P3HT:L7 = 2:1 |
0.59 | 3.5 | 0.48 | 1.0 | 289 |
| M1 | 532 | −5.14/−3.74 | 5.39 × 10−5d | P3HT:M1 = 1:1.5 |
0.57 | 3.28 | 0.63 | 1.18 | 312 |
| M2 | 631, 687 | −5.55/−3.77 | 6.0 × 10−5 | P3HT:M2 = 1:1 |
0.78 | 2.60 | 0.32 | 0.65 | 313 |
| M3 | 750 | −5.27/−4.00 | 1.06 × 10−4 |
P1:M3 = 1:2 |
0.83 | 10.53 | 0.56 | 4.89 | 315 |
| M4 | 888 | −5.24/−4.05 | 2.21 × 10−4 |
P1:M4 = 1:2 |
0.80 | 13.77 | 0.63 | 6.94 | 315 |
| M5 | ∼720 | −6.42/−2.34 | 2.42 × 10−4 |
PBDB-T![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :M5a = 1:1 :M5a = 1:1
|
0.88 | 16.5 | 0.66 | 9.6 | 316 |
| M6 | ∼850 | −6.58/−2.52 | 3.94 × 10−4 |
PBDB-T:M5b = 1:1
|
0.78 | 23.2 | 0.73 | 13.2 | 316 |
| M7 | 779 | −5.36/−3.82 | 1.97 × 10−4 |
PBDB-T:M5a = 1:1
|
0.96 | 8.55 | 0.52 | 4.31 | 317 |
| M8 | 821 | −5.42/−3.94 | 1.28 × 10−3 |
PBDB-T:M5b = 1:1
|
0.85 | 21.61 | 0.72 | 13.13 | 317 |
7.1 DTP-based polymers as electron acceptors
Since the first investigation of PDI-based polymers used in all-PSCs started in 2007,301 many small molecules and polymers containing PDI units have been used in OSCs up to now. PDI-based small molecules offered a potential replacement for fullerenes as acceptors,302 and the highest PCE has recently reached to 8.47%.303 Although naphthalene imide (NDI) has a similar structure to PDI, NDI-based materials show wide bandgaps, low electron affinity and weak electron capture ability. With two-fluorinated polymer P3 as the donor and L1 (Fig. 23) as the acceptor, the best devices show a slightly higher PCE than one-fluorinated polymer P2.304 Recently, the PDI-DTP donor polymer L2 displayed higher performance with PCE increased up to 4.05%.304 In 2010, we first copolymerized DTP and PDI to obtain polymer L3.299 Through selecting donor polymers, the highest PCE of 0.93% was obtained for the PT1:L3 system.48 It should be noted that the strong aggregation behaviour of PDI and NDI impedes the improvement of solar cell performance. By introducing thiophene as a spacer to decrease the steric hindrance, the polymer L4 film shows extended absorption over 1 μm due to the strong intermolecular interaction. However L4 showed very low PCE resulting from, at least in part, the low solubility of L4. | ||
| Fig. 23 The reported DTP-based polymers used as electron acceptors (EH: 2-ethylhexyl). | ||
Except for NDI and PDI units, there are a series of units that have been used to construct acceptor materials by copolymerizing with DTP derivatives.305 Recently, an n-type polymer based on DPP and thiazole has been designed, which not only features broad absorption, low-lying energy levels and high mobility but also gave high performance.108 In consideration of increasing the mobility while retaining efficient NIR absorption, L5 was designed in combination with electron-deficient DPP substituted with thiazole units and DTP. With P3HT as the electron donor, P3HT:L5 devices gave a PCE of 1.5% with a JSC of 4.0 mA cm−2, a VOC of 0.72 V, and a FF of 0.51 highly depending on the processing solvent, TA treatments as well as the thickness of the active layer.80 The result indicates that the performance can be further improved after morphology optimization by balancing the pure crystalline and amorphous mixed regions.
There is no defined boundary between p-type and n-type materials because p-type polymers could perform electron transport by using an appropriate device structure. For instance, K8 and K9 respectively have hole mobilities of 0.06 and 0.05 cm2 V−1 s−1, K10 shows a balanced hole mobility of 0.03 cm2 V−1 s−1 and electron mobility of 0.02 cm2 V−1 s−1, and the electron mobility of K11 is 0.05 cm2 V−1 s−1. In other words, K10 exhibits both p-type and n-type properties, while K11 can function as an n-type polymer. By using P3HT as the donor and K8 and K9 as acceptors, no photovoltaic effect can be observed; however, L6 (K10) and L7 (K11) respectively showed PCEs of 0.73% and 1.0%. These results indicate that TBI is a versatile unit in both p-type and n-type materials, which would afford highly efficient devices through molecular design.
7.2 DTP-based small molecules as electron acceptors
Compared with polymeric acceptors, small molecule acceptors (SMAs) exhibit superior properties for OSC applications because of several key factors including good processability, high electron mobility and excellent stability. Driven by the demand for non-fullerene SMAs,306–309 significant progress in the field of small molecule acceptors have been made since the first observation of bilayer OSCs by using a perylene tetracarboxylic derivative as the acceptor in 1986.310 Among various fused heterocycle π-systems, DTP-based small molecules exhibited electron mobilities up to 1.5 cm2 V−1 s−1, which are much higher than that of TT and DTT analogues.311 Although fewer studies have been reported on DTP-based SMAs in comparison with donors, their superior properties as acceptors have attracted much attention.Poe et al. recently synthesized a DTP-based n-type molecule (M1) utilizing 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) as an end-capped acceptor unit as shown in Fig. 24.312 P3HT:M1 inverted BHJ devices afforded an efficiency of 1.18%, which might be attributed to the strong absorption resulting from the BODIPY groups. When two DCV moieties were end-capped to the central DTP donor unit with bridged mono-thiophene, a new acceptor M2 with broad absorption and a low-lying LUMO energy level was obtained.313 The P3HT:M2 device achieved a PCE of 0.65% with a low JSC of 2.60 mA cm−2 and a FF of 0.32. The inferior performance might be ascribed to the low electron mobility, and thus the conjugation of the backbone should be extended in order to increase the intermolecular π–π stacking.314
 | ||
| Fig. 24 The reported DTP-based small molecules used as electron acceptors (EH: 2-ethylhexyl). | ||
1-Butyl-4-methyl-2,6-dioxopyridine-3-carbonitrile (PY) and 3-dicyanomethyleneindan-1-one (DCI) are two kinds of moieties with stronger electron-accepting ability than DCV. By replacing DCV with PY and DCI, the initial donor molecules can be switched into acceptor molecules such as M3 and M4.315 Besides the type conversion, the stronger electron-accepting terminal units can extend the absorption wavelength up to 1000 nm and lower the frontier molecular orbitals similar to the fullerene derivatives. In order to make them solution-processable, the branched octylnonyl is substituted at the central DTP-nitrogen and the hexyl chains are attached at the thiophene unit. With a simple polymer P1 as the donor, the optimized P1:M3 and P1:M4 devices exhibited PCEs of 4.89% and 6.94%, respectively. Their electron mobilities were improved by one order of magnitude compared with M1 and M2. The higher efficiency of P1:M4 devices can be confirmed by the 69% EQE at 880 nm generating a significantly improved JSC and FF as well as high VOC resulting in low energy losses.
Very recently, Chen and co-workers designed two new SMAs (M5 and M6) with SN6 as the central rigid backbone, inspired by the advantages of SN6 such as strong intramolecular charge transfer (ICT), tunable energy levels, and efficient electron transfer.316 They found that M5 and M6 assuredly exhibited red-shift absorption, better crystallinity and higher charge mobility compared with their carbon analogues. As a result, a very high PCE of 13.2% was achieved with PBDB-T as the polymer donor. At the same time, Tang et al. realized another excellent study.317 A new derivative of IDT, 5,5,12,12-tetrakis(4-hexylphenyl)-indacenobis(dithieno[3,2-b:2′,3′-d]pyrrol) (INP) was designed, which can also be considered as a derivative of SN6 after incorporating the central indacene unit between two flanked DTP units. Two SMAs (M7 and M8) end-capped with 3-(dicyanomethylidene)indan-1-one (IC) exhibited strong electron-donating, good processability, broad absorption and efficient charge transport compared with IDT analogues. With the same polymer donor PBDB-T, OSC devices afforded an equivalent PCE of 13.13% to that of M6.
Their photovoltaic parameters shown in Table 11 reveal that the PCEs of M6 and M8-based devices are much higher than those of the M5 and M7 devices, which indicates that fluorine-containing end-capped building blocks play an important role. In addition, M7 and M8 possess large rigid fused-rings, which easily induce negative aggregation in blend films. Therefore, the SN6 building block may be suitable for constructing various SMAs from the view of extending the conjugation. More than this, two nitrogen atoms in SN6 provide great properties to modify the molecular backbone for tuning molecular packing and further the active-layer morphology. With our recent research focusing on designing SMAs in mind,318,319 we speculate that the emergence of many end-capped electron-withdrawing units would bring about the development of SMAs based on these fused-DTP building blocks.
7.3 Summary
In short summary, typical n-type materials based on DTP and its derivatives have been discussed including their photo-physical properties and applications in OSCs. As far as we know, non-fullerene devices have achieved a PCE over 6% in polymer/polymer systems,321–323 and a PCE over 10% in polymer/small molecule systems,324,325 with the development of non-fullerene acceptors over the past few decades. Therefore, the performance of DTP-based polymer acceptors lags far behind those of other polymers. One of the critical reasons for the poor efficiency of DTP-based polymer acceptors is their non-ideal large-scale domain size, which reduces the miscibility with the donor polymer. Thus, the enhancement of crystallinity and electron mobility is an urgent and important issue for their development. As discussed above, modification of the alkyl chain could modulate both intramolecular and intermolecular interactions, influencing the solubility, crystallinity, capability of absorption and charge mobility. Despite the selection of the side chain, most efforts should also be devoted to manipulating acceptor units for optimizing the D–A polymer backbones. We believe that further improvements are possible if additional efforts would be directed toward optimizing the structure, energy level and processing conditions.Compared with the variability of DTP-based polymers, DTP-based SMAs often offer a well-defined molecular structure and readily tunable light absorption and energy levels. In particular the recent achievements offering PCEs over 13% confirm the great potential applications of DTP-based SMAs in OSC devices. Due to the tendency of crystallinity, they are expected to form well-ordered domains on the same order as exciton diffusion length by tuning the molecular planarity. In addition, we found that one limiting parameter to further improve the PCE of SN6-based SMA systems may be the relatively small VOC values. With the recent developments of SMAs, this factor would be enhanced by modifying the molecular structure or selecting a suitable donor in the not too distant future. Although the molecular properties of DTP-based acceptors can be reasonably regulated through diversiform chemical structure modification, there is both a great space and large challenge to realize promising DTP-based SMAs.
8. Conclusion and outlook
Due to worldwide research efforts, numerous DTP-based materials including polymers and small molecules have been developed and played a significant role in achieving high-efficiency organic and hybrid solar cells. In this current review, we have focused on the recent advances of conjugated materials containing original or derivative DTP groups essentially used as electron-donating and electron-accepting motifs. Since the application of DTP-based polymers in OSCs in 2007, DTP-based materials have sprung up like mushrooms and increased year by year. As discussed above, DTP-based devices appear to be more promising due to their rapidly and steadily improved PCE. Although the investigation of DTP-based SMSCs started relatively late in 2014, a PCE exceeding 10% for DTDP-based small molecule G7 has been reported. Very recently, a tandem solar cell based on the DTPO polymer J9 exhibited a high PCE of 11.35%. Among these single-junction devices, the highest JSC (23.2 mA cm−2), VOC (1.21 V) and FF (0.76) were obtained for DTP-based small molecular acceptor M6, small molecule E1 and BTI-based polymer K6, respectively. However, these largest JSC, VOC and FF values do not belong to the same material. To realize commercial applications, it is necessary to improve the PCE of both p-type and n-type semiconducting materials simultaneously.As is known to all, the PCE of devices strongly depends on the molecular structures and device fabrication technology. Herein, the strategy to further pursue novel conjugated materials containing DTP-based building blocks can satisfy the basic requirements for most photovoltaic materials involving as low as possible optical bandgaps to broaden the absorption and capture more photons, as long as possible exciton diffusion distances to achieve the charge separation states, as fast as possible charge mobility to reduce charge recombination, as suitable as possible HOMO and LUMO energy levels to ensure large VOC and decrease energy loss, and so on. However, it is still a big challenge to comprehensively understand the relationship between the intrinsic properties of materials and performance. For further improving the efficiency, it is necessary and important to understand and then resolve these issues through molecular engineering and device technology. We found that several types of trade-offs between these photovoltaic properties blocked the further improvement of the efficiency. Hereinafter, we summarize the trade-off effects as well as the corresponding strategies.
(1) In order to harvest as many photons as possible in the range of the solar spectrum, low bandgap materials are preferred for high JSC. However the low bandgap original DTP-based polymers usually have high-lying HOMO energy levels resulting in low VOC values. In addition to selecting appropriate units to couple with the original DTP unit, the DTP-based derivative building blocks have low-lying HOMO levels and finally give higher VOC values. Although the low-lying HOMO level induced a larger bandgap for lactam- or imide-based polymers, much higher performances have been obtained compared with the original DTP polymers due to the enhanced charge carrier mobility. Therefore, the HOMO energy level of the donor should be optimum with respect to that of the acceptor through introducing functional carbonyl groups to achieve an efficient charge separation with a minimal concomitant energy loss.
(2) The LUMO energy level of the donor should be aligned with that of the acceptor to keep adequate driving force for the charge generation. However, with increasing the energy gap between the donor and acceptor, charge carrier dissociation is eventually replaced by recombination at some point. By using PC71BM as the acceptor, polymers based on a lactam building block not only have low-lying HOMO energy levels but also have higher-lying LUMO energy levels, which facilitate exciton dissociation at the D/A interface. The enhanced JSC values indicate that efficient charge generation and transport make up for a loss induced by the large bandgap. Therefore, much more research effort should be focused on the design of DTP-based derivatives with a hetero-ring structure.
(3) The crystallinity is critical for efficient exciton diffusion and charge transport; the over-improved crystallinity often is at the expense of large domain sizes, which reduces the charge separation efficiency. Longer or larger side chains improve the solubility but decrease the crystallinity. On the one hand, fused DTP small molecules can resolve the trade-off between solubility and crystallinity. On the other hand, suitable lengths of side chains such as, in particular, the branched side chains such as hexyldecyl, and decyldodecyl should be used to ensure the phase-separation thin-film morphology with ordered molecular packing. At the D/A interface, the local crystalline structure favored the charge transport. Therefore, the intermolecular interaction should be optimized by tuning the backbones, side chains, and functional substitutions.
(4) In order to harvest as many photons as possible, the active layer should be as thick as possible. However the largely enhanced absorption tends to be at the cost of dramatic charge recombination. Tandem solar cells can efficiently utilize the photons, and meanwhile, reduce recombination loss. In addition, TA treatments can not only optimize the crystallinity of the active layer but also bring segregation resulting in interfacial recombination, therefore both TA and SVA treatments should be used to optimize the performance.
In order to minimize these trade-offs, in particular that between JSC and VOC, much effort should be devoted to suitable molecular design to reduce various losses. Fine-tuning the absorption, energy level alignment, charge mobility, phase separation and so on is critical to balance the JSC and VOC values. All these trade-offs are ultimately linked to the molecule and device design. Therefore, the efficiency of devices containing DTP-based units would increase and the future of this unit looks bright in organic photovoltaic devices by changing the donor parts or acceptor parts as well as the device structures. The relationship between the structure and performance of organic solar cells needs to be further understood clearly.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support from the National Key Research and Development Program of China (2017YFA0206600), the Key Research Program of Frontier Sciences, Chinese Academy of Sciences (Grant No. QYZDB-SSW-SLH033), and the National Natural Science Foundation of China (NSFC, No. 51473040, 51673048, 21875052, 21602040, and 51873044).Notes and references
- C. W. Tang, Appl. Phys. Lett., 1986, 48, 183–185 CrossRef CAS.
- J. J. M. Halls, C. A. Walsh, N. C. Greenham, E. A. Marseglia, R. H. Friend, S. C. Moratti and A. B. Holmes, Nature, 1995, 376, 498–500 CrossRef CAS.
- G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789–1791 CrossRef CAS.
- C. C. Chen, W. H. Chang, K. Yoshimura, K. Ohya, J. B. You, J. Gao, Z. R. Hong and Y. Yang, Adv. Mater., 2014, 26, 5670–5677 CrossRef CAS PubMed.
- H. Q. Zhou, Y. Zhang, C. K. Mai, S. D. Collins, G. C. Bazan, T. Q. Nguyen and A. J. Heeger, Adv. Mater., 2015, 27, 1767–1773 CrossRef CAS PubMed.
- A. B. Yusoff, D. Kim, H. P. Kim, F. K. Shneider, W. J. da Silva and J. Jang, Energy Environ. Sci., 2015, 8, 303–316 RSC.
- L. Meng, Y. Zhang, X. Wan, C. Li, X. Zhang, Y. Wang, X. Ke, Z. Xiao, L. Ding, R. Xia, H.-L. Yip, Y. Cao and Y. Chen, Science, 2018, 361, 1094–1098 CrossRef CAS PubMed.
- Y. F. Li, Acc. Chem. Res., 2012, 45, 723–733 CrossRef CAS PubMed.
- T. Xu and L. P. Yu, Mater. Today, 2014, 17, 11–15 CrossRef CAS.
- I. Etxebarria, J. Ajuria and R. Pacios, Org. Electron., 2015, 19, 34–60 CrossRef CAS.
- H. Zhang, L. Ye and J. H. Hou, Polym. Int., 2015, 64, 957–962 CrossRef CAS.
- Z. B. Henson, K. Mullen and G. C. Bazan, Nat. Chem., 2012, 4, 699–704 CrossRef CAS PubMed.
- P. M. Beaujuge and J. M. J. Frechet, J. Am. Chem. Soc., 2011, 133, 20009–20029 CrossRef CAS PubMed.
- J. W. Chen and Y. Cao, Acc. Chem. Res., 2009, 42, 1709–1718 CrossRef CAS PubMed.
- F. P. V. Koch, M. Heeney and P. Smith, J. Am. Chem. Soc., 2013, 135, 13699–13709 CrossRef CAS PubMed.
- L. Zhang, N. S. Colella, F. Liu, S. Trahan, J. K. Baral, H. H. Winter, S. C. B. Mannsfeld and A. L. Briseno, J. Am. Chem. Soc., 2013, 135, 844–854 CrossRef CAS PubMed.
- E. Zhou, K. Hashimoto and K. Tajima, Polymer, 2013, 54, 6501–6509 CrossRef CAS.
- S. Kim, K.-h. Song, S. O. Kang and J. Ko, Chem. Commun., 2004, 68–69 RSC.
- Y. Ren and F. Jakle, Dalton Trans., 2016, 45, 13996–14007 RSC.
- G. Zotti, G. Schiavon, A. Berlin, G. Fontana and G. Pagani, Macromolecules, 1994, 27, 1938–1942 CrossRef CAS.
- C. Soci, I.-W. Hwang, D. Moses, Z. Zhu, D. Waller, R. Gaudiana, C. J. Brabec and A. J. Heeger, Adv. Funct. Mater., 2007, 17, 632–636 CrossRef CAS.
- Y. Bai, J. Zhang, D. Zhou, Y. Wang, M. Zhang and P. Wang, J. Am. Chem. Soc., 2011, 133, 11442–11445 CrossRef CAS PubMed.
- P. Zanirato, P. Spagnolo and G. Zanardi, J. Chem. Soc., Perkin Trans. 1, 1983, 2551–2554 RSC.
- J. Liu, R. Zhang, G. Sauve, T. Kowalewski and R. D. McCullough, J. Am. Chem. Soc., 2008, 130, 13167–13176 CrossRef CAS PubMed.
- E. Zhou, M. Nakamura, T. Nishizawa, Y. Zhang, Q. Wei, K. Tajima, C. Yang and K. Hashimoto, Macromolecules, 2008, 41, 8302–8305 CrossRef CAS.
- Y. X. Zhang, X. Cai, Y. Z. Bian, X. Y. Li and J. Z. Jiang, J. Phys. Chem. C, 2008, 112, 5148–5159 CrossRef CAS.
- K. L. Chan, M. J. McKiernan, C. R. Towns and A. B. Holmes, J. Am. Chem. Soc., 2005, 127, 7662–7663 CrossRef CAS PubMed.
- B. McDearmon, Z. A. Page, M. L. Chabinyc and C. J. Hawker, J. Mater. Chem. C, 2018, 6, 3564–3572 RSC.
- T. Baumgartner, T. Neumann and B. Wirges, Angew. Chem., Int. Ed., 2004, 43, 6197–6201 CrossRef CAS PubMed.
- A. Bolognesi, M. Catellani, S. Destri, R. Zamboni and C. Taliani, J. Chem. Soc., Chem. Commun., 1988, 246–247 RSC.
- M. E. Cinar and T. Ozturk, Chem. Rev., 2015, 115, 3036–3140 CrossRef CAS PubMed.
- C. M. Amb, S. Chen, K. R. Graham, J. Subbiah, C. E. Small, F. So and J. R. Reynolds, J. Am. Chem. Soc., 2011, 133, 10062–10065 CrossRef CAS PubMed.
- C. E. Small, S. Chen, J. Subbiah, C. M. Amb, S. W. Tsang, T. H. Lai, J. R. Reynolds and F. So, Nat. Photonics, 2012, 6, 115–120 CrossRef CAS.
- J. P. Green, Y. Han, R. Kilmurray, M. A. McLachlan, T. D. Anthopoulos and M. Heeney, Angew. Chem., Int. Ed., 2016, 55, 7148–7151 CrossRef CAS PubMed.
- S. H. Park, A. Roy, S. Beaupre, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297–303 CrossRef CAS.
- H. X. Zhou, L. Q. Yang, A. C. Stuart, S. C. Price, S. B. Liu and W. You, Angew. Chem., Int. Ed., 2011, 50, 2995–2998 CrossRef CAS PubMed.
- N. Blouin, A. Michaud and M. Leclerc, Adv. Mater., 2007, 19, 2295–2300 CrossRef CAS.
- J. Hou, H.-Y. Chen, S. Zhang, G. Li and Y. Yang, J. Am. Chem. Soc., 2008, 130, 16144–16145 CrossRef CAS PubMed.
- J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497–500 CrossRef CAS PubMed.
- S. Albrecht, S. Janietz, W. Schindler, J. Frisch, J. Kurpiers, J. Kniepert, S. Inal, P. Pingel, K. Fostiropoulos, N. Koch and D. Neher, J. Am. Chem. Soc., 2012, 134, 14932–14944 CrossRef CAS PubMed.
- T. Imae, Curr. Opin. Colloid Interface Sci., 2003, 8, 307–314 CrossRef CAS.
- Y. Liang, D. Feng, Y. Wu, S.-T. Tsai, G. Li, C. Ray and L. Yu, J. Am. Chem. Soc., 2009, 131, 7792–7799 CrossRef CAS PubMed.
- H.-Y. Chen, J. Hou, S. Zhang, Y. Liang, G. Yang, Y. Yang, L. Yu, Y. Wu and G. Li, Nat. Photonics, 2009, 3, 649–653 CrossRef CAS.
- C. Gong, H. Bin Yang, Q. L. Song, Z. S. Lu and C. M. Li, Sol. Energy Mater. Sol. Cells, 2011, 95, 969–973 CrossRef CAS.
- R. Kroon, M. Lenes, J. C. Hummelen, P. W. M. Blom and B. de Boer, Polym. Rev., 2008, 48, 531–582 CrossRef CAS.
- E. G. Wang, L. Wang, L. F. Lan, C. Luo, W. L. Zhuang, J. B. Peng and Y. Cao, Appl. Phys. Lett., 2008, 92, 033307 CrossRef.
- Z. P. Fei, J. S. Kim, J. Smith, E. B. Domingo, T. D. Anthopoulos, N. Stingelin, S. E. Watkins, J. S. Kim and M. Heeney, J. Mater. Chem., 2011, 21, 16257–16263 RSC.
- T. Y. Chu, J. Lu, S. Beaupre, Y. Zhang, J. R. Pouliot, S. Wakim, J. Zhou, M. Leclerc, Z. Li, J. Ding and Y. Tao, J. Am. Chem. Soc., 2011, 133, 4250–4253 CrossRef CAS PubMed.
- C. E. Small, S. Chen, J. Subbiah, C. M. Amb, S.-W. Tsang, T.-H. Lai, J. R. Reynolds and F. So, Nat. Photonics, 2012, 6, 115–120 CrossRef CAS.
- J. W. Jung, F. Liu, T. P. Russell and W. H. Jo, Energy Environ. Sci., 2012, 5, 6857 RSC.
- G. P. Kini, S. Oh, Z. Abbas, S. Rasool, M. Jahandar, C. E. Song, S. K. Lee, W. S. Shin, W. W. So and J. C. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 12617–12628 CrossRef CAS PubMed.
- R. S. Kularatne, H. D. Magurudeniya, P. Sista, M. C. Biewer and M. C. Stefan, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 743–768 CrossRef CAS.
- S. C. Rasmussen and S. J. Evenson, Prog. Polym. Sci., 2013, 38, 1773–1804 CrossRef CAS.
- S. C. Rasmussen, S. J. Evenson and C. B. McCausland, Chem. Commun., 2015, 51, 4528–4543 RSC.
- K. Ogawa and S. C. Rasmussen, J. Org. Chem., 2003, 68, 2921–2928 CrossRef CAS PubMed.
- M. Al-Hashimi, J. G. Labram, S. Watkins, M. Motevalli, T. D. Anthopoulos and M. Heeney, Org. Lett., 2010, 12, 5478–5481 CrossRef CAS PubMed.
- Q. C. Yu, W. F. Fu, J. H. Wan, X. F. Wu, M. M. Shi and H. Z. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 5798–5809 CrossRef CAS PubMed.
- I. H. Jung, J. H. Kim, S. Y. Nam, C. Lee, D. H. Hwang and S. C. Yoon, Macromolecules, 2015, 48, 5213–5221 CrossRef CAS.
- K. Mitsudo, S. Shimohara, J. Mizoguchi, H. Mandai and S. Suga, Org. Lett., 2012, 14, 2702–2705 CrossRef CAS PubMed.
- C. Wetzel, A. Mishra, E. Mena-Osteritz, K. Walzer, M. Pfeiffer and P. Baeuerle, J. Mater. Chem. C, 2016, 4, 3715–3725 RSC.
- M. Hao, G. Luo, K. Shi, G. Xie, K. Wu, H. Wu, G. Yu, Y. Cao and C. Yang, J. Mater. Chem. A, 2015, 3, 20516–20526 RSC.
- X. G. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed.
- F. Lu, L. Qian, J. Cao, Y. Feng, B. Du and L. Ding, Polym. Chem., 2015, 6, 7373–7376 RSC.
- J. Cao, C. Zuo, B. Du, X. Qiu and L. Ding, Chem. Commun., 2015, 51, 12122–12125 RSC.
- J. Cao, Q. Liao, X. Du, J. Chen, Z. Xiao, Q. Zuo and L. Ding, Energy Environ. Sci., 2013, 6, 3224–3228 RSC.
- G. Koeckelberghs, L. De Cremer, W. Vanormelingen, W. Dehaen, T. Verbiest, A. Persoons and C. Samyn, Tetrahedron, 2005, 61, 687–691 CrossRef CAS.
- S. J. Evenson and S. C. Rasmussen, Org. Lett., 2010, 12, 4054–4057 CrossRef CAS PubMed.
- R. Martín, C. H. Larsen, A. Cuenca and S. L. Buchwald, Org. Lett., 2007, 9, 3379–3382 CrossRef PubMed.
- A. M. Schneider, L. Lu, E. F. Manley, T. Zheng, V. Sharapov, T. Xu, T. J. Marks, L. X. Chen and L. Yu, Chem. Sci., 2015, 6, 4860–4866 RSC.
- W. J. Belcher, S. C. Rasmussen and P. C. Dastoor, Polym. Prepr., 2007, 48, 11–12 CAS.
- E. J. Zhou, M. Nakamura, T. Nishizawa, Y. Zhang, Q. S. Wei, K. Tajima, C. H. Yang and K. Hashimoto, Macromolecules, 2008, 41, 8302–8305 CrossRef CAS.
- E. J. Zhou, S. Yamakawa, Y. Zhang, K. Tajima, C. H. Yang and K. Hashimoto, J. Mater. Chem., 2009, 19, 7730–7737 RSC.
- E. J. Zhou, S. P. Yamakawa, K. Tajima, C. H. Yang and K. Hashimoto, Chem. Mater., 2009, 21, 4055–4061 CrossRef CAS.
- E. Zhou, J. Cong, K. Tajima and K. Hashimoto, Chem. Mater., 2010, 22, 4890–4895 CrossRef CAS.
- E. J. Zhou, J. Z. Cong, K. Tajima, C. H. Yang and K. Hashimoto, Macromol. Chem. Phys., 2011, 212, 305–310 CrossRef CAS.
- W. Yue, Y. Zhao, S. Y. Shao, H. K. Tian, Z. Y. Xie, Y. H. Geng and F. S. Wang, J. Mater. Chem., 2009, 19, 2199–2206 RSC.
- H. Song, H. Tong, Z. Xie, L. Wang and F. Wang, Polymer, 2012, 53, 5103–5108 CrossRef CAS.
- R. S. Ashraf, J. Gilot and R. A. J. Janssen, Sol. Energy Mater. Sol. Cells, 2010, 94, 1759–1766 CrossRef CAS.
- K. H. Hendriks, W. Li, M. M. Wienk and R. A. Janssen, J. Am. Chem. Soc., 2014, 136, 12130–12136 CrossRef CAS PubMed.
- W. Li, K. H. Hendriks, A. Furlan, M. M. Wienk and R. A. Janssen, J. Am. Chem. Soc., 2015, 137, 2231–2234 CrossRef CAS PubMed.
- W. Li, K. H. Hendriks, A. Furlan, A. Zhang, M. M. Wienk and R. A. Janssen, Chem. Commun., 2015, 51, 4290–4293 RSC.
- W. Yue, T. T. Larsen-Olsen, X. Hu, M. Shi, H. Chen, M. Hinge, P. Fojan, F. C. Krebs and D. Yu, J. Mater. Chem. A, 2013, 1, 1785–1793 RSC.
- R. G. Brandt, W. Yue, T. R. Andersen, T. T. Larsen-Olsen, M. Hinge, E. Bundgaard, F. C. Krebs and D. Yu, J. Mater. Chem. C, 2015, 3, 1633–1639 RSC.
- Y. Geng, J. Cong, K. Tajima, Q. Zeng and E. Zhou, Polym. Chem., 2014, 5, 6797–6803 RSC.
- W. Lee, G. H. Kim, E. Jeong, X. Wang, S. Yum, S. J. Ko, S. Hwang, J. Y. Kim and H. Y. Woo, Macromol. Chem. Phys., 2013, 214, 2083–2090 CrossRef CAS.
- X. J. Guo, Q. G. Liao, E. F. Manley, Z. S. Wu, Y. L. Wang, W. D. Wang, T. B. Yang, Y. E. Shin, X. Cheng, Y. Y. Liang, L. X. Chen, K. J. Baeg, T. J. Marks and X. G. Guo, Chem. Mater., 2016, 28, 2449–2460 CrossRef CAS.
- M. Svensson, F. Zhang, S. C. Veenstra, W. J. H. Verhees, J. C. Hummelen, J. M. Kroon, O. Inganäs and M. R. Andersson, Adv. Mater., 2003, 15, 988–991 CrossRef CAS.
- F. Zhang, W. Mammo, L. M. Andersson, S. Admassie, M. R. Andersson and O. Inganäs, Adv. Mater., 2006, 18, 2169–2173 CrossRef CAS.
- O. InganÄs, F. Zhang and M. R. Andersson, Acc. Chem. Res., 2009, 42, 1731–1739 CrossRef PubMed.
- J. Du, M. C. Biewer and M. C. Stefan, J. Mater. Chem. A, 2016, 4, 15771–15787 RSC.
- T. C. Parker, D. G. Patel, K. Moudgil, S. Barlow, C. Risko, J.-L. Bredas, J. R. Reynolds and S. R. Marder, Mater. Horiz., 2015, 2, 22–36 RSC.
- D. G. Farnum, G. Mehta, G. G. I. Moore and F. P. Siegal, Tetrahedron Lett., 1974, 15, 2549–2552 CrossRef.
- B. Tieke, A. R. Rabindranath, K. Zhang and Y. Zhu, Beilstein J. Org. Chem., 2010, 6, 830–845 CrossRef PubMed.
- D. Chandran and K.-S. Lee, Macromol. Res., 2013, 21, 272–283 CrossRef CAS.
- M. Grzybowski and D. T. Gryko, Adv. Opt. Mater., 2015, 3, 280–320 CrossRef CAS.
- E. Zhou, Q. Wei, S. Yamakawa, Y. Zhang, K. Tajima, C. Yang and K. Hashimoto, Macromolecules, 2010, 43, 821–826 CrossRef CAS.
- R. C. Coffin, J. Peet, J. Rogers and G. C. Bazan, Nat. Chem., 2009, 1, 657–661 CrossRef CAS PubMed.
- W. Huag, M. Li, L. Zhang, T. Yang, Z. Zhang, H. Zeng, X. Zhang, L. Dang and Y. Liang, Chem. Mater., 2016, 28, 5887–5895 CrossRef.
- C. Cui and W.-Y. Wong, Macromol. Rapid Commun., 2016, 37, 287–302 CrossRef CAS PubMed.
- J. Hou, Z. a. Tan, Y. Yan, Y. He, C. Yang and Y. Li, J. Am. Chem. Soc., 2006, 128, 4911–4916 CrossRef CAS PubMed.
- J. Hou, L. Huo, C. He, C. Yang and Y. Li, Macromolecules, 2006, 39, 594–603 CrossRef CAS.
- E. J. Zhou, J. Z. Cong, K. Hashimoto and K. Tajima, Energy Environ. Sci., 2012, 5, 9756–9759 RSC.
- J. Huang, K. Nakano, K. Suzuki, Y. Chen, F. Wang, T. Koganezawa and K. Tajima, Macromolecules, 2017, 50, 3557–3564 CrossRef CAS.
- L. Dou, W.-H. Chang, J. Gao, C.-C. Chen, J. You and Y. Yang, Adv. Mater., 2013, 25, 825–831 CrossRef CAS PubMed.
- W. Li, K. H. Hendriks, M. M. Wienk and R. A. J. Janssen, Acc. Chem. Res., 2016, 49, 78–85 CrossRef CAS PubMed.
- D. Veldman, S. C. J. Meskers and R. A. J. Janssen, Adv. Funct. Mater., 2009, 19, 1939–1948 CrossRef CAS.
- R. A. J. Janssen and J. Nelson, Adv. Mater., 2013, 25, 1847–1858 CrossRef CAS PubMed.
- W. W. Li, W. S. C. Roelofs, M. Turbiez, M. M. Wienk and R. A. J. Janssen, Adv. Mater., 2014, 26, 3304–3309 CrossRef CAS PubMed.
- W. Li, Y. An, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem. A, 2015, 3, 6756–6760 RSC.
- L. Chen, D. Deng, Y. Nan, M. Shi, P. K. L. Chan and H. Chen, J. Phys. Chem. C, 2011, 115, 11282–11292 CrossRef CAS.
- M.-C. Yuan, M.-Y. Chiu, S.-P. Liu, C.-M. Chen and K.-H. Wei, Macromolecules, 2010, 43, 6936–6938 CrossRef CAS.
- Y. Zou, A. Najari, P. Berrouard, S. Beaupré, B. Réda Aïch, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2010, 132, 5330–5331 CrossRef CAS PubMed.
- D. Gendron and M. Leclerc, Energy Environ. Sci., 2011, 4, 1225–1237 RSC.
- A. Najari, S. Beaupre, P. Berrouard, Y. Zou, J.-R. Pouliot, C. Lepage-Perusse and M. Leclerc, Adv. Funct. Mater., 2011, 21, 718–728 CrossRef CAS.
- C. Cabanetos, A. El Labban, J. A. Bartelt, J. D. Douglas, W. R. Mateker, J. M. J. Frechet, M. D. McGehee and P. M. Beaujuge, J. Am. Chem. Soc., 2013, 135, 4656–4659 CrossRef CAS PubMed.
- Y. Sun, S. C. Chien, H. L. Yip, Y. Zhang, K. S. Chen, D. F. Zeigler, F. C. Chen, B. P. Lin and A. K. Y. Jen, J. Mater. Chem., 2011, 21, 13247–13255 RSC.
- P. M. Beaujuge, H. N. Tsao, M. R. Hansen, C. M. Amb, C. Risko, J. Subbiah, K. R. Choudhury, A. Mavrinskiy, W. Pisula, J. L. Bredas, F. So, K. Mullen and J. R. Reynods, J. Am. Chem. Soc., 2012, 134, 8944–8957 CrossRef CAS PubMed.
- S. C. Lan, P. A. Yang, M. J. Zhu, C. M. Yu, J. M. Jiang and K. H. Wei, Polym. Chem., 2013, 4, 1132–1140 RSC.
- M. Wang, X. W. Hu, L. Q. Liu, C. H. Duan, P. Liu, L. Ying, F. Huang and Y. Cao, Macromolecules, 2013, 46, 3950–3958 CrossRef CAS.
- E. J. Zhou, J. Z. Cong, K. Tajima, C. H. Yang and K. Hashimoto, J. Phys. Chem. C, 2012, 116, 2608–2614 CrossRef CAS.
- T. Yamamoto, K. Sugiyama, T. Kushida, T. Inoue and T. Kanbara, J. Am. Chem. Soc., 1996, 118, 3930–3937 CrossRef CAS.
- A. Gadisa, W. Mammo, L. M. Andersson, S. Admassie, F. Zhang, M. R. Andersson and O. Inganäs, Adv. Funct. Mater., 2007, 17, 3836–3842 CrossRef CAS.
- N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. Neagu-Plesu, M. Belletête, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732–742 CrossRef CAS PubMed.
- D. Ouyang, M. J. Xiao, D. Q. Zhu, W. G. Zhu, Z. K. Du, N. Wang, Y. H. Zhou, X. C. Bao and R. Q. Yang, Polym. Chem., 2015, 6, 55–63 RSC.
- A. Wickham, D. Sjolander, G. Bergstrom, E. G. Wang, V. Rajendran, C. Hildesjo, K. Skoglund, K. P. R. Nilsson and D. Aili, Adv. Funct. Mater., 2015, 25, 4274–4281 CrossRef CAS.
- K. M. Noone, S. Subramaniyan, Q. F. Zhang, G. Z. Cao, S. A. Jenekhe and D. S. Ginger, J. Phys. Chem. C, 2011, 115, 24403–24410 CrossRef CAS.
- L. M. Campos, A. Tontcheva, S. Gunes, G. Sonmez, H. Neugebauer, N. S. Sariciftci and F. Wudl, Chem. Mater., 2005, 17, 4031–4033 CrossRef CAS.
- F. L. Zhang, E. Perzon, X. J. Wang, W. Mammo, M. R. Andersson and O. Inganas, Adv. Funct. Mater., 2005, 15, 745–750 CrossRef CAS.
- Y. Zhu, R. D. Champion and S. A. Jenekhe, Macromolecules, 2006, 39, 8712–8719 CrossRef CAS.
- J. H. Hou, M. H. Park, S. Q. Zhang, Y. Yao, L. M. Chen, J. H. Li and Y. Yang, Macromolecules, 2008, 41, 6012–6018 CrossRef CAS.
- A. P. Zoombelt, J. Gilot, M. A. Wienk and R. A. J. Janssen, Chem. Mater., 2009, 21, 1663–1669 CrossRef CAS.
- E. J. Zhou, J. Z. Cong, S. Yamakawa, Q. S. Wei, M. Nakamura, K. Tajima, C. H. Yang and K. Hashimoto, Macromolecules, 2010, 43, 2873–2879 CrossRef CAS.
- H. M. Qin, L. S. Li, Y. Li, X. B. Peng, J. B. Peng, Y. Cao, N. Ismayil and W. Shi, Eur. Polym. J., 2012, 48, 2076–2084 CrossRef CAS.
- G. Saranya, P. Kolandaivel and K. Senthilkumar, Mol. Phys., 2013, 111, 3036–3046 CrossRef CAS.
- Q. Peng, X. Liu, Y. Qin, D. Zhou and J. Xu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4458–4467 CrossRef CAS.
- J. Cremer, P. Bäuerle, M. M. Wienk and R. A. J. Janssen, Chem. Mater., 2006, 18, 5832–5834 CrossRef CAS.
- F. Silvestri, A. Marrocchi, M. Seri, C. Kim, T. J. Marks, A. Facchetti and A. Taticchi, J. Am. Chem. Soc., 2010, 132, 6108–6123 CrossRef CAS PubMed.
- W. A. Braunecker, S. D. Oosterhout, Z. R. Owczarczyk, R. E. Larsen, B. W. Larson, D. S. Ginley, O. V. Boltalina, S. H. Strauss, N. Kopidakis and D. C. Olson, Macromolecules, 2013, 46, 3367–3375 CrossRef CAS.
- N. Leclerc, P. Chávez, O. Ibraikulov, T. Heiser and P. Lévêque, Polymers, 2016, 8, 11 CrossRef.
- S. A. Jenekhe and J. A. Osaheni, Science, 1994, 265, 765–768 CrossRef CAS PubMed.
- J. A. Osaheni and S. A. Jenekhe, J. Am. Chem. Soc., 1995, 117, 7389–7398 CrossRef CAS.
- E. Ahmed, S. Subramaniyan, F. S. Kim, H. Xin and S. A. Jenekhe, Macromolecules, 2011, 44, 7207–7219 CrossRef CAS.
- G. Conboy, R. G. D. Taylor, N. J. Findlay, A. L. Kanibolotsky, A. R. Inigo, S. S. Ghosh, B. Ebenhoch, L. K. Jagadamma, G. Thalluri, M. T. Sajjad, I. D. W. Samuel and P. J. Skabara, J. Mater. Chem. C, 2017, 5, 11927–11936 RSC.
- Z. Li, D. Zhou, L. Li, Y. Li, Y. He, J. Liu and Q. Peng, Polym. Chem., 2013, 4, 2496 RSC.
- J. G. Mei, K. R. Graham, R. Stalder and J. R. Reynolds, Org. Lett., 2010, 12, 660–663 CrossRef CAS PubMed.
- R. Stalder, J. G. Mei and J. R. Reynolds, Macromolecules, 2010, 43, 8348–8352 CrossRef CAS.
- T. Lei, J.-H. Dou and J. Pei, Adv. Mater., 2012, 24, 6457–6461 CrossRef CAS PubMed.
- Y. Deng, J. Liu, J. Wang, L. Liu, W. Li, H. Tian, X. Zhang, Z. Xie, Y. Geng and F. Wang, Adv. Mater., 2014, 26, 471–476 CrossRef CAS PubMed.
- W. Zhang, J. Li, L. Zou, B. Zhang, J. G. Qin, Z. S. Lu, Y. F. Poon, M. B. Chan-Park and C. M. Li, Macromolecules, 2008, 41, 8953–8955 CrossRef CAS.
- C. Gong, H. Bin Yang, Q. L. Song, Z. S. Lu and C. M. Li, Sol. Energy Mater. Sol. Cells, 2011, 95, 969–973 CrossRef CAS.
- M. J. Cho, J. Seo, S. H. Yoon, D. H. Choi and P. N. Prasad, Synth. Met., 2013, 182, 22–27 CrossRef CAS.
- J. Brebels, K. C. C. W. S. Klider, M. Kelchtermans, P. Verstappen, M. Van Landeghem, S. Van Doorslaer, E. Goovaerts, J. R. Garcia, J. Manca, L. Lutsen, D. Vanderzande and W. Maes, Org. Electron., 2017, 50, 264–272 CrossRef CAS.
- L. Dou, J. You, J. Yang, C.-C. Chen, Y. He, S. Murase, T. Moriarty, K. Emery, G. Li and Y. Yang, Nat. Photonics, 2012, 6, 180–185 CrossRef CAS.
- Z. He, C. Zhong, S. Su, M. Xu, H. Wu and Y. Cao, Nat. Photonics, 2012, 6, 591–595 CrossRef.
- Y. Zhong, M. T. Trinh, R. S. Chen, G. E. Purdum, P. P. Khlyabich, M. Sezen, S. Oh, H. M. Zhu, B. Fowler, B. Y. Zhang, W. Wang, C. Y. Nam, M. Y. Sfeir, C. T. Black, M. L. Steigerwald, Y. L. Loo, F. Ng, X. Y. Zhu and C. Nuckolls, Nat. Commun., 2015, 6, 8242 CrossRef CAS PubMed.
- S. Roquet, A. Cravino, P. Leriche, O. Alévêque, P. Frère and J. Roncali, J. Am. Chem. Soc., 2006, 128, 3459–3466 CrossRef CAS PubMed.
- J. Roncali, Acc. Chem. Res., 2009, 42, 1719–1730 CrossRef CAS PubMed.
- T. Liu, Y. Guo, Y. Yi, L. Huo, X. Xue, X. Sun, H. Fu, W. Xiong, D. Meng, Z. Wang, F. Liu, T. P. Russell and Y. Sun, Adv. Mater., 2016, 28, 10008–10015 CrossRef CAS PubMed.
- Y. Lin and X. Zhan, Acc. Chem. Res., 2016, 49, 175–183 CrossRef CAS PubMed.
- F. Lincker, B. Heinrich, R. De Bettignies, P. Rannou, J. Pecaut, B. Grevin, A. Pron, B. Donnio and R. Demadrille, J. Mater. Chem., 2011, 21, 5238–5247 RSC.
- H. X. Shang, H. J. Fan, Y. Liu, W. P. Hu, Y. F. Li and X. W. Zhan, Adv. Mater., 2011, 23, 1554–1557 CrossRef CAS PubMed.
- Q. Q. Shi, P. Cheng, Y. F. Li and X. W. Zhan, Adv. Energy Mater., 2012, 2, 63–67 CrossRef CAS.
- S. L. Shen, P. Jiang, C. He, J. Zhang, P. Shen, Y. Zhang, Y. P. Yi, Z. J. Zhang, Z. B. Li and Y. F. Li, Chem. Mater., 2013, 25, 2274–2281 CrossRef CAS.
- A. Yassin, T. Rousseau, P. Leriche, A. Cravino and J. Roncali, Sol. Energy Mater. Sol. Cells, 2011, 95, 462–468 CrossRef CAS.
- J. Min, Y. N. Luponosov, A. Gerl, M. S. Polinskaya, S. M. Peregudova, P. V. Dmitryakov, A. V. Bakirov, M. A. Shcherbina, S. N. Chvalun, S. Grigorian, N. Kaush-Busies, S. A. Ponomarenko, T. Ameri and C. J. Brabec, Adv. Energy Mater., 2014, 4, 1301234 CrossRef.
- C. D. Wessendorf, G. L. Schulz, A. Mishra, P. Kar, I. Ata, M. Weidelener, M. Urdanpilleta, J. Hanisch, E. Mena-Osteritz, M. Lindén, E. Ahlswede and P. Bäuerle, Adv. Energy Mater., 2014, 4, 1400266 CrossRef.
- M. Weidelener, C. D. Wessendorf, J. Hanisch, E. Ahlswede, G. Götz, M. Linden, G. Schulz, E. Mena-Osteritz, A. Mishra and P. Bäuerle, Chem. Commun., 2013, 49, 10865–10867 RSC.
- G. L. Schulz, M. Löbert, I. Ata, M. Urdanpilleta, M. Lindén, A. Mishra and P. Bäuerle, J. Mater. Chem. A, 2015, 3, 13738–13748 RSC.
- C. D. Wessendorf, A. Perez-Rodriguez, J. Hanisch, A. P. Arndt, I. Ata, G. L. Schulz, A. Quintilla, P. Bäuerle, U. Lemmer, P. Wochner, E. Ahlswede and E. Barrena, J. Mater. Chem. A, 2016, 4, 2571–2580 RSC.
- D. Popovic, I. Ata, J. Krantz, S. Lucas, M. Linden, E. Mena-Osteritz and P. Baeuerle, J. Mater. Chem. C, 2017, 5, 9920–9928 RSC.
- S. Ben Dkhil, M. Pfannmöller, I. Ata, D. Duche, M. Gaceur, T. Koganezawa, N. Yoshimoto, J.-J. Simon, L. Escoubas, C. Videlot-Ackermann, O. Margeat, S. Bals, P. Bäuerle and J. Ackermann, J. Mater. Chem. A, 2017, 5, 1005–1013 RSC.
- J. Min, X. C. Jiao, I. Ata, A. Osvet, T. Ameri, P. Bäuerle, H. Ade and C. J. Brabec, Adv. Energy Mater., 2016, 6, 1502579 CrossRef.
- I. Ata, S. B. Dkhil, M. Pfannmöller, S. Bals, D. Duché, J.-J. Simon, T. Koganezawa, N. Yoshimoto, C. Videlot-Ackermann, O. Margeat, J. Ackermann and P. Bäuerle, Org. Chem. Front., 2017, 4, 1561–1573 RSC.
- A. Mishra, T. Rana, A. Looser, M. Stolte, F. Würthner, P. Bäuerle and G. D. Sharma, J. Mater. Chem. A, 2016, 4, 17344–17353 RSC.
- M. Durso, C. Bettini, A. Zanelli, M. Gazzano, M. G. Lobello, F. De Angelis, V. Biondo, D. Gentili, R. Capelli, M. Cavallini, S. Toffanin, M. Muccini and M. Melucci, Org. Electron., 2013, 14, 3089–3097 CrossRef CAS.
- L. G. Mercier, A. Mishra, Y. Ishigaki, F. Henne, G. Schulz and P. Bäuerle, Org. Lett., 2014, 16, 2642–2645 CrossRef CAS PubMed.
- J. Min, Y. N. Luponosov, D. A. Khanin, P. V. Dmitryakov, E. A. Svidchenko, S. M. Peregudova, L. Grodd, S. Grigorian, S. N. Chvalun, S. A. Ponomarenko and C. J. Brabec, Org. Electron., 2018, 55, 42–49 CrossRef CAS.
- M. R. Busireddy, V. N. R. Mantena, N. R. Chereddy, B. Shanigaram, B. Kotamarthi, S. Biswas, G. D. Sharma and J. R. Vaidya, Phys. Chem. Chem. Phys., 2016, 18, 32096–32106 RSC.
- K. Wang, M. Azouz, M. Babics, F. Cruciani, T. Marszalek, Q. Saleem, W. Pisula and P. M. Beaujuge, Chem. Mater., 2016, 28, 5415–5425 CrossRef CAS.
- J. Y. Zhou, Y. Zuo, X. J. Wan, G. K. Long, Q. Zhang, W. Ni, Y. S. Liu, Z. Li, G. R. He, C. X. Li, B. Kan, M. M. Li and Y. S. Chen, J. Am. Chem. Soc., 2013, 135, 8484–8487 CrossRef CAS PubMed.
- Y. S. Chen, X. J. Wan and G. K. Long, Acc. Chem. Res., 2013, 46, 2645–2655 CrossRef CAS PubMed.
- Y. S. Liu, C. C. Chen, Z. R. Hong, J. Gao, Y. Yang, H. P. Zhou, L. T. Dou, G. Li and Y. Yang, Sci. Rep., 2013, 3, 3356 CrossRef PubMed.
- M. M. Li, F. Liu, X. J. Wan, W. Ni, B. Kan, H. R. Feng, Q. Zhang, X. Yang, Y. C. Wang, Y. M. Zhang, Y. Shen, T. P. Russell and Y. S. Chen, Adv. Mater., 2015, 27, 6296–6302 CrossRef CAS PubMed.
- Y. S. Liu, X. J. Wan, F. Wang, J. Y. Zhou, G. K. Long, J. G. Tian and Y. S. Chen, Adv. Mater., 2011, 23, 5387–5391 CrossRef CAS PubMed.
- P. Gautam, R. Misra, S. A. Siddiqui and G. D. Sharma, ACS Appl. Mater. Interfaces, 2015, 7, 10283–10292 CrossRef CAS PubMed.
- N. Lim, N. Cho, S. Paek, C. Kim, J. K. Lee and J. Ko, Chem. Mater., 2014, 26, 2283–2288 CrossRef CAS.
- M. R. Busireddy, V. N. R. Mantena, N. R. Chereddy, B. Shanigaram, B. Kotamarthi, S. Biswas, G. D. Sharma and J. R. Vaidya, Org. Electron., 2016, 37, 312–325 CrossRef CAS.
- Q. Zhang, B. Kan, F. Liu, G. K. Long, X. J. Wan, X. Q. Chen, Y. Zuo, W. Ni, H. J. Zhang, M. M. Li, Z. C. Hu, F. Huang, Y. Cao, Z. Q. Liang, M. T. Zhang, T. P. Russell and Y. S. Chen, Nat. Photonics, 2015, 9, 35–41 CrossRef CAS.
- P. Sonar, S. P. Singh, P. Leclere, M. Surin, R. Lazzaroni, T. T. Lin, A. Dodabalapur and A. Sellinger, J. Mater. Chem., 2009, 19, 3228–3237 RSC.
- P. Sonar, S. P. Singh, S. Sudhakar, A. Dodabalapur and A. Sellinger, Chem. Mater., 2008, 20, 3184–3190 CrossRef CAS.
- M. R. Busireddy, N. R. Chereddy, B. Shanigaram, B. Kotamarthi, S. Biswas, G. D. Sharma and J. R. Vaidya, Phys. Chem. Chem. Phys., 2017, 19, 20513–20522 RSC.
- A. Marrocchi, F. Silvestri, M. Seri, A. Facchetti, A. Taticchia and T. J. Marks, Chem. Commun., 2009, 1380–1382 RSC.
- M. Seri, A. Marrocchi, D. Bagnis, R. Ponce, A. Taticchi, T. J. Marks and A. Facchetti, Adv. Mater., 2011, 23, 3827–3831 CAS.
- R. Grisorio, G. Allegretta, G. P. Suranna, P. Mastrorilli, A. Loiudice, A. Rizzo, M. Mazzeo and G. Gigli, J. Mater. Chem., 2012, 22, 19752–19760 RSC.
- A. Yassin, G. Savitha, P. Leriche, P. Frère and J. Roncali, New J. Chem., 2012, 36, 2412–2416 RSC.
- N. Hergué, C. Mallet, P. Frère, M. Allain and J. Roncali, Macromolecules, 2009, 42, 5593–5599 CrossRef.
- A. Yassin, P. Leriche, M. Allain and J. Roncali, New J. Chem., 2013, 37, 502–507 RSC.
- H. Bürckstümmer, E. V. Tulyakova, M. Deppisch, M. R. Lenze, N. M. Kronenberg, M. Gsänger, M. Stolte, K. Meerholz and F. Würthner, Angew. Chem., Int. Ed., 2011, 50, 11628–11632 CrossRef PubMed.
- V. Steinmann, N. M. Kronenberg, M. R. Lenze, S. M. Graf, D. Hertel, K. Meerholz, H. Bürckstümmer, E. V. Tulyakova and F. Würthner, Adv. Energy Mater., 2011, 1, 888–893 CrossRef CAS.
- L. Y. Lin, Y. H. Chen, Z. Y. Huang, H. W. Lin, S. H. Chou, F. Lin, C. W. Chen, Y. H. Liu and K. T. Wong, J. Am. Chem. Soc., 2011, 133, 15822–15825 CrossRef CAS PubMed.
- S. W. Chiu, L. Y. Lin, H. W. Lin, Y. H. Chen, Z. Y. Huang, Y. T. Lin, F. Lin, Y. H. Liu and K. T. Wong, Chem. Commun., 2012, 48, 1857–1859 RSC.
- Y. H. Chen, L. Y. Lin, C. W. Lu, F. Lin, Z. Y. Huang, H. W. Lin, P. H. Wang, Y. H. Liu, K. T. Wong, J. G. Wen, D. J. Miller and S. B. Darling, J. Am. Chem. Soc., 2012, 134, 13616–13623 CrossRef CAS PubMed.
- H. I. Lu, C. W. Lu, Y. C. Lee, H. W. Lin, L. Y. Lin, F. Lin, J. H. Chang, C. I. Wu and K. T. Wong, Chem. Mater., 2014, 26, 4361–4367 CrossRef CAS.
- A. Gupta, A. Ali, M. Gao, T. B. Singh, A. Bilic, S. E. Watkins, U. Bach and R. A. Evans, Dyes Pigm., 2015, 119, 122–132 CrossRef CAS.
- W. N. Li, H. F. Yao, H. Zhang, S. S. Li and J. H. Hou, Chem. - Asian J., 2017, 12, 2160–2171 CrossRef CAS PubMed.
- S. D. Collins, N. A. Ran, M. C. Heiber and T. Q. Nguyen, Adv. Energy Mater., 2017, 7, 1602242 CrossRef.
- W. Q. Chen and Q. C. Zhang, J. Mater. Chem. C, 2017, 5, 1275–1302 RSC.
- B. Qiu, L. Xue, Y. Yang, H. Bin, Y. Zhang, C. Zhang, M. Xiao, K. Park, W. Morrison, Z.-G. Zhang and Y. Li, Chem. Mater., 2017, 29, 7543–7553 CrossRef CAS.
- B. Kan, M. Li, Q. Zhang, F. Liu, X. Wan, Y. Wang, W. Ni, G. Long, X. Yang, H. Feng, Y. Zuo, M. Zhang, F. Huang, Y. Cao, T. P. Russell and Y. Chen, J. Am. Chem. Soc., 2015, 137, 3886–3893 CrossRef PubMed.
- S. Mabrouk, M. Zhang, Z. Wang, M. Liang, B. Bahrami, Y. Wu, J. Wu, Q. Qiao and S. Yang, J. Mater. Chem. A, 2018, 6, 7950–7958 RSC.
- M. M. Li, W. Ni, H. R. Feng, B. Kan, X. J. Wan, Y. M. Zhang, X. Yang and Y. S. Chen, Chin. J. Chem., 2015, 33, 852–858 CrossRef.
- Y. J. He, H. Y. Chen, J. H. Hou and Y. F. Li, J. Am. Chem. Soc., 2010, 132, 1377–1382 CrossRef PubMed.
- J. M. Warman, M. P. de Haas, T. D. Anthopoulos and D. M. de Leeuw, Adv. Mater., 2006, 18, 2294–2298 CrossRef.
- J. Y. Yuan, H. L. Dong, M. Li, X. D. Huang, J. Zhong, Y. Y. Li and W. L. Ma, Adv. Mater., 2014, 26, 3624–3630 CrossRef PubMed.
- I. Gur, Science, 2005, 310, 1618 CrossRef PubMed.
- V. Sukhovatkin, S. Hinds, L. Brzozowski and E. H. Sargent, Science, 2009, 324, 1542–1544 CrossRef PubMed.
- J. Tang, X. H. Wang, L. Brzozowski, D. A. R. Barkhouse, R. Debnath, L. Levina and E. H. Sargent, Adv. Mater., 2010, 22, 1398–1402 CrossRef PubMed.
- K. M. Noone, S. Subramaniyan, Q. F. Zhang, G. Z. Cao, S. A. Jenekhe and D. S. Ginger, J. Phys. Chem. C, 2011, 115, 24403–24410 CrossRef.
- W. U. Huynh, J. J. Dittmer and A. P. Alivisatos, Science, 2002, 295, 2425–2427 CrossRef PubMed.
- Y. Y. Liang, Z. Xu, J. B. Xia, S. T. Tsai, Y. Wu, G. Li, C. Ray and L. P. Yu, Adv. Mater., 2010, 22, E135–E138 CrossRef PubMed.
- I. J. Kramer and E. H. Sargent, Chem. Rev., 2014, 114, 863–882 CrossRef PubMed.
- S. Dayal, N. Kopidakis, D. C. Olson, D. S. Ginley and G. Rumbles, Nano Lett., 2010, 10, 239–242 CrossRef PubMed.
- R. J. Zhou, R. Stalder, D. P. Xie, W. R. Cao, Y. Zheng, Y. X. Yang, M. Plaisant, P. H. Holloway, K. S. Schanze, J. R. Reynolds and J. G. Xue, ACS Nano, 2013, 7, 4846–4854 CrossRef PubMed.
- J. Yuan, A. Gallagher, Z. Liu, Y. Sun and W. Ma, J. Mater. Chem. A, 2015, 3, 2572–2579 RSC.
- F. W. Wise, Acc. Chem. Res., 2000, 33, 773–780 CrossRef PubMed.
- D. Yu, C. Wang and P. Guyot-Sionnest, Science, 2003, 300, 1277–1280 CrossRef PubMed.
- K. M. Noone, N. C. Anderson, N. E. Horwitz, A. M. Munro, A. P. Kulkarni and D. S. Ginger, ACS Nano, 2009, 3, 1345–1352 CrossRef PubMed.
- A. Guchhait, A. K. Rath and A. J. Pal, Appl. Phys. Lett., 2010, 96, 073505 CrossRef.
- K. M. Noone, E. Strein, N. C. Anderson, P. T. Wu, S. A. Jenekhe and D. S. Ginger, Nano Lett., 2010, 10, 2635–2639 CrossRef PubMed.
- E. H. Sargent, Nat. Photonics, 2009, 3, 325–331 CrossRef.
- M. D. Heinemann, K. von Maydell, F. Zutz, J. Kolny-Olesiak, H. Borchert, I. Riedel and J. Parisi, Adv. Funct. Mater., 2009, 19, 3788–3795 CrossRef.
- A. E. Colbert, E. M. Janke, S. T. Hsieh, S. Subramaniyan, C. W. Schlenker, S. A. Jenekhe and D. S. Ginger, J. Phys. Chem. Lett., 2013, 4, 280–284 CrossRef PubMed.
- E. Strein, A. Colbert, S. Subramaniyan, H. Nagaoka, C. W. Schlenker, E. Janke, S. A. Jenekhe and D. S. Ginger, Energy Environ. Sci., 2013, 6, 769–775 RSC.
- J. Seo, M. J. Cho, D. Lee, A. N. Cartwright and P. N. Prasad, Adv. Mater., 2011, 23, 3984–3988 CrossRef PubMed.
- Z. K. Liu, Y. X. Sun, J. Y. Yuan, H. X. Wei, X. D. Huang, L. Han, W. W. Wang, H. Q. Wang and W. L. Ma, Adv. Mater., 2013, 25, 5772–5778 CrossRef PubMed.
- W. P. Guo, J. Y. Yuan, H. C. Yuan, F. Jin, L. Han, C. X. Sheng, W. L. Ma and H. B. Zhao, Adv. Funct. Mater., 2016, 26, 713–721 CrossRef.
- P. Mueller-Buschbaum, M. Thelakkat, T. F. Faessler and M. Stutzmann, Adv. Energy Mater., 2017, 7, 1700248 CrossRef.
- X. Du, Q. Zeng, H. Zhang and B. Yang, Chin. J. Chem., 2017, 35, 551–561 CrossRef.
- X. Du, Q. Zeng, G. Jin, F. Liu, T. Ji, Y. Yue, Y. Yang, H. Zhang and B. Yang, Small, 2017, 13, 1603771 CrossRef PubMed.
- X. J. Long, Z. C. Ding, C. D. Dou, J. D. Zhang, J. Liu and L. X. Wang, Adv. Mater., 2016, 28, 6504–6508 CrossRef PubMed.
- C. B. Nielsen, S. Holliday, H. Y. Chen, S. J. Cryer and I. McCulloch, Acc. Chem. Res., 2015, 48, 2803–2812 CrossRef PubMed.
- M. D. Watson, A. Fechtenkotter and K. Mullen, Chem. Rev., 2001, 101, 1267–1300 CrossRef PubMed.
- H. J. Son, L. Lu, W. Chen, T. Xu, T. Zheng, B. Carsten, J. Strzalka, S. B. Darling, L. X. Chen and L. Yu, Adv. Mater., 2013, 25, 838–843 CrossRef PubMed.
- Z. Liang, Q. Tang, R. Mao, D. Liu, J. Xu and Q. Miao, Adv. Mater., 2011, 23, 5514–5518 CrossRef PubMed.
- Z. Liang, Q. Tang, J. Xu and Q. Miao, Adv. Mater., 2011, 23, 1535–1539 CrossRef PubMed.
- J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef PubMed.
- A. Mishra, D. Popovic, A. Vogt, H. Kast, T. Leitner, K. Walzer, M. Pfeiffer, E. Mena-Osteritz and P. Bäuerle, Adv. Mater., 2014, 26, 7217–7223 CrossRef PubMed.
- H. Kast, A. Mishra, G. L. Schulz, M. Urdanpilleta, E. Mena-Osteritz and P. Bäuerle, Adv. Funct. Mater., 2015, 25, 3414–3424 CrossRef.
- C.-L. Chung, C.-Y. Chen, H.-W. Kang, H.-W. Lin, W.-L. Tsai, C.-C. Hsu and K.-T. Wong, Org. Lett., 2016, 28, 229–238 Search PubMed.
- R. Fitzner, E. Mena-Osteritz, K. Walzer, M. Pfeiffer and P. Bäuerle, Adv. Funct. Mater., 2015, 25, 1845–1856 CrossRef.
- P. Qin, H. Kast, M. K. Nazeeruddin, S. M. Zakeeruddin, A. Mishra, P. Bäuerle and M. Grätzel, Energy Environ. Sci., 2014, 7, 2981–2985 RSC.
- H. Tee, H. Jo, D. Kim, S. Biswas, G. D. Sharma and J. Ko, Dyes Pigm., 2016, 129, 209–219 CrossRef.
- C. Wetzel, A. Mishra, E. Mena-Osteritz, A. Liess, M. Stolte, F. Wurthner and P. Bäuerle, Org. Lett., 2014, 16, 362–365 CrossRef CAS PubMed.
- I. H. Jung, J. H. Kim, S. Y. Nam, C. Lee, D. H. Hwang and S. C. Yoon, J. Mater. Chem. C, 2016, 4, 663–667 RSC.
- W. Ma, J. R. Tumbleston, M. Wang, E. Gann, F. Huang and H. Ade, Adv. Energy Mater., 2013, 3, 864–872 CrossRef CAS.
- W. Jiang, Y. Li and Z. Wang, Chem. Soc. Rev., 2013, 42, 6113–6127 RSC.
- S. J. Evenson, T. M. Pappenfus, M. C. R. Delgado, K. R. Radke-Wohlers, J. T. L. Navarrete and S. C. Rasmussen, Phys. Chem. Chem. Phys., 2012, 14, 6101–6111 RSC.
- D. Hong, M. Lv, M. Lei, Y. Chen, P. Lu, Y. Wang, J. Zhu, H. Wang, M. Gao, S. E. Watkins and X. Chen, ACS Appl. Mater. Interfaces, 2013, 5, 10995–11003 CrossRef CAS PubMed.
- W. Vanormelingen, J. Kesters, P. Verstappen, J. Drijkoningen, J. Kudrjasova, S. Koudjina, V. Liegeois, B. Champagne, J. Manca, L. Lutsen, D. Vanderzande and W. Maes, J. Mater. Chem. A, 2014, 2, 7535–7545 RSC.
- M. Thelakkat and H. W. Schmidt, Polym. Adv. Technol., 1998, 9, 429–442 CrossRef CAS.
- E. G. Wang, L. T. Hou, Z. Q. Wang, S. Hellstrom, F. L. Zhang, O. Inganas and M. R. Andersson, Adv. Mater., 2010, 22, 5240–5244 CrossRef CAS PubMed.
- Y. Kim, H. R. Yeom, J. Y. Kim and C. Yang, Energy Environ. Sci., 2013, 6, 1909–1916 RSC.
- J. Kesters, P. Verstappen, W. Vanormelingen, J. Drijkoningen, T. Vangerven, D. Devisscher, L. Marin, B. Champagne, J. Manca, L. Lutsen, D. Vanderzande and W. Maes, Sol. Energy Mater. Sol. Cells, 2015, 136, 70–77 CrossRef CAS.
- P. Verstappen, I. Cardinaletti, T. Vangerven, W. Vanormelingen, F. Verstraeten, L. Lutsen, D. Vanderzande, J. Manca and W. Maes, RSC Adv., 2016, 6, 32298–32307 RSC.
- R. Xia, M. Al-Hashimi, W. C. Tsoi, M. Heeney, D. D. C. Bradley and J. Nelson, Sol. Energy Mater. Sol. Cells, 2012, 96, 112–116 CrossRef CAS.
- Y. A. Getmanenko, S. Singh, B. Sandhu, C. Y. Wang, T. Timofeeva, B. Kippelen and S. R. Marder, J. Mater. Chem. C, 2014, 2, 124–131 RSC.
- D. Patra, J. Lee, J. Lee, D. N. Sredojevic, A. J. P. White, H. S. Bazzi, E. N. Brothers, M. Heeney, L. Fang, M. H. Yoon and M. Al-Hashimi, J. Mater. Chem. C, 2017, 5, 2247–2258 RSC.
- I. Osaka, M. Shimawaki, H. Mori, I. Doi, E. Miyazaki, T. Koganezawa and K. Takimiya, J. Am. Chem. Soc., 2012, 134, 3498–3507 CrossRef CAS PubMed.
- M. An, F. Xie, X. Geng, J. Zhang, J. Jiang, Z. Lei, D. He, Z. Xiao and L. Ding, Adv. Energy Mater., 2017, 7, 1602509 CrossRef.
- W. M. Zhang, J. Smith, S. E. Watkins, R. Gysel, M. McGehee, A. Salleo, J. Kirkpatrick, S. Ashraf, T. Anthopoulos, M. Heeney and I. McCulloch, J. Am. Chem. Soc., 2010, 132, 11437–11439 CrossRef CAS PubMed.
- Y.-X. Xu, C.-C. Chueh, H.-L. Yip, F.-Z. Ding, Y.-X. Li, C.-Z. Li, X. Li, W.-C. Chen and A. K. Y. Jen, Adv. Mater., 2012, 24, 6356–6361 CrossRef CAS PubMed.
- W. Gao, T. Liu, M. H. Hao, K. L. Wu, C. Zhang, Y. M. Sun and C. L. Yang, Chem. Sci., 2016, 7, 6167–6175 RSC.
- J. Cao, L. Qian, F. Lu, J. Zhang, Y. Feng, X. Qiu, H. L. Yip and L. Ding, Chem. Commun., 2015, 51, 11830–11833 RSC.
- H. Li, D. He, P. Mao, Y. Wei, L. Ding and J. Wang, Adv. Energy Mater., 2017, 1602663 CrossRef.
- K. Zhang, K. Gao, R. Xia, Z. Wu, C. Sun, J. Cao, L. Qian, W. Li, S. Liu, F. Huang, X. Peng, L. Ding, H.-L. Yip and Y. Cao, Adv. Mater., 2016, 28, 4817–4823 CrossRef CAS PubMed.
- W. Zhang, Y. Han, X. Zhu, Z. Fei, Y. Feng, N. D. Treat, H. Faber, N. Stingelin, I. McCulloch, T. D. Anthopoulos and M. Heeney, Adv. Mater., 2016, 28, 3922–3927 CrossRef CAS PubMed.
- H. Chen, M. Hurhangee, M. Nikolka, W. Zhang, M. Kirkus, M. Neophytou, S. J. Cryer, D. Harkin, P. Hayoz, M. Abdi-Jalebi, C. R. McNeill, H. Sirringhaus and I. McCulloch, Adv. Mater., 2017, 29, 1702523 CrossRef PubMed.
- H. Li, D. He, P. Mao, Y. Wei, L. Ding and J. Wang, Adv. Energy Mater., 2017, 1602663 CrossRef.
- J. A. Letizia, M. R. Salata, C. M. Tribout, A. Facchetti, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2008, 130, 9679–9694 CrossRef CAS PubMed.
- H. F. Yao, L. Ye, H. Zhang, S. S. Li, S. Q. Zhang and J. H. Hou, Chem. Rev., 2016, 116, 7397–7457 CrossRef CAS PubMed.
- L. J. Huo and J. H. Hou, Polym. Chem., 2011, 2, 2453–2461 RSC.
- N. J. Zhou, X. G. Guo, R. P. Ortiz, S. Q. Li, S. M. Zhang, R. P. H. Chang, A. Facchetti and T. J. Marks, Adv. Mater., 2012, 24, 2242–2248 CrossRef CAS PubMed.
- J. L. Bredas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971–5003 CrossRef CAS PubMed.
- P. Schilinsky, C. Waldauf, J. Hauch and C. J. Brabec, J. Appl. Phys., 2004, 95, 2816–2819 CrossRef CAS.
- X. G. Guo, N. J. Zhou, S. J. Lou, J. W. Hennek, R. P. Ortiz, M. R. Butler, P. L. T. Boudreault, J. Strzalka, P. O. Morin, M. Leclerc, J. T. L. Navarrete, M. A. Ratner, L. X. Chen, R. P. H. Chang, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2012, 134, 18427–18439 CrossRef CAS PubMed.
- X. G. Guo, R. P. Ortiz, Y. Zheng, Y. Hu, Y. Y. Noh, K. J. Baeg, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2011, 133, 1405–1418 CrossRef CAS PubMed.
- N. J. Zhou, X. G. Guo, R. P. Ortiz, T. Harschneck, E. F. Manley, S. J. Lou, P. E. Hartnett, X. G. Yu, N. E. Horwitz, P. M. Burrezo, T. J. Aldrich, J. T. L. Navarrete, M. R. Wasielewski, L. X. Chen, R. P. H. Chang, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2015, 137, 12565–12579 CrossRef CAS PubMed.
- X. G. Guo, N. J. Zhou, S. J. Lou, J. Smith, D. B. Tice, J. W. Hennek, R. P. Ortiz, J. T. L. Navarrete, S. Y. Li, J. Strzalka, L. X. Chen, R. P. H. Chang, A. Facchetti and T. J. Marks, Nat. Photonics, 2013, 7, 825–833 CrossRef CAS.
- M. Saito, I. Osaka, Y. Suda, H. Yoshida and K. Takimiya, Adv. Mater., 2016, 28, 6921–6925 CrossRef CAS PubMed.
- G. Kim, S.-J. Kang, G. K. Dutta, Y.-K. Han, T. J. Shin, Y.-Y. Noh and C. Yang, J. Am. Chem. Soc., 2014, 136, 9477–9483 CrossRef CAS PubMed.
- J. Y. Back, H. Yu, I. Song, I. Kang, H. Ahn, T. J. Shin, S.-K. Kwon, J. H. Oh and Y.-H. Kim, Chem. Mater., 2015, 27, 1732–1739 CrossRef CAS.
- C. Luo, A. K. K. Kyaw, L. A. Perez, S. Patel, M. Wang, B. Grimm, G. C. Bazan, E. J. Kramer and A. J. Heeger, Nano Lett., 2014, 14, 2764–2771 CrossRef CAS PubMed.
- V. Coropceanu, J. Cornil, D. A. da Silva, Y. Olivier, R. Silbey and J. L. Bredas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed.
- D. Deng, Y. J. Zhang, J. Q. Zhang, Z. Y. Wang, L. Y. Zhu, J. Fang, B. Z. Xia, Z. Wang, K. Lu, W. Ma and Z. X. Wei, Nat. Commun., 2016, 7, 13740 CrossRef CAS PubMed.
- G. C. Zhang, K. Zhang, Q. W. Yin, X. F. Jiang, Z. Y. Wang, J. M. Xin, W. Ma, H. Yan, F. Huang and Y. Cao, J. Am. Chem. Soc., 2017, 139, 2387–2395 CrossRef CAS PubMed.
- J. B. Zhao, Y. K. Li, G. F. Yang, K. Jiang, H. R. Lin, H. Ade, W. Ma and H. Yan, Nat. Energy, 2016, 1, 15027 CrossRef CAS.
- L. Gao, Z. G. Zhang, L. Xue, J. Min, J. Zhang, Z. Wei and Y. Li, Adv. Mater., 2016, 28, 1884–1890 CrossRef CAS PubMed.
- G. B. Lin, Y. K. Qin, Y. S. Guan, H. Xu, W. Xu and D. B. Zhu, RSC Adv., 2016, 6, 4872–4876 RSC.
- E. Zhou, K. Tajima, C. Yang and K. Hashimoto, J. Mater. Chem., 2010, 20, 2362–2368 RSC.
- E. J. Zhou, J. Z. Cong, Q. S. Wei, K. Tajima, C. H. Yang and K. Hashimoto, Angew. Chem., Int. Ed., 2011, 50, 2799–2803 CrossRef CAS PubMed.
- X. W. Zhan, Z. A. Tan, B. Domercq, Z. S. An, X. Zhang, S. Barlow, Y. F. Li, D. B. Zhu, B. Kippelen and S. R. Marder, J. Am. Chem. Soc., 2007, 129, 7246–7247 CrossRef CAS PubMed.
- P. E. Hartnett, A. Timalsina, H. Matte, N. J. Zhou, X. G. Guo, W. Zhao, A. Facchetti, R. P. H. Chang, M. C. Hersam, M. R. Wasielewski and T. J. Marks, J. Am. Chem. Soc., 2014, 136, 16345–16356 CrossRef CAS PubMed.
- Q. Wu, D. Zhao, A. M. Schneider, W. Chen and L. Yu, J. Am. Chem. Soc., 2016, 138, 7248–7251 CrossRef CAS PubMed.
- M. Li, Y. Qin, W. Dai and X. Luo, RSC Adv., 2018, 8, 3809–3815 RSC.
- X. Gao and Y. Hu, J. Mater. Chem. C, 2014, 2, 3099–3117 RSC.
- A. a. F. Eftaiha, J.-P. Sun, I. G. Hill and G. C. Welch, J. Mater. Chem. A, 2014, 2, 1201–1213 RSC.
- Y. Lin and X. Zhan, Adv. Energy Mater., 2015, 5, 1501063 CrossRef.
- C. Song, E. Wang, B. Dong and S. Wang, Prog. Chem., 2015, 27, 1754–1763 Search PubMed.
- C. Zhan, X. Zhang and J. Yao, RSC Adv., 2015, 5, 93002–93026 RSC.
- C. W. Tang, Appl. Phys. Lett., 1986, 48, 183–185 CrossRef CAS.
- L. E. Polander, S. P. Tiwari, L. Pandey, B. M. Seifried, Q. Zhang, S. Barlow, C. Risko, J. L. Bredas, B. Kippelen and S. R. Marder, Chem. Mater., 2011, 23, 3408–3410 CrossRef CAS.
- A. M. Poe, A. M. Della Pelle, A. V. Subrahmanyam, W. White, G. Wantz and S. Thayumanavan, Chem. Commun., 2014, 50, 2913–2915 RSC.
- J. Wu, Y. Ma, N. Wu, Y. Lin, J. Lin, L. Wang and C.-Q. Ma, Org. Lett., 2015, 23, 28–38 CAS.
- J. Song, C. Li, L. Ye, C. Koh, Y. Cai, D. Wei, H. Y. Woo and Y. Sun, J. Mater. Chem. A, 2018, 6, 18847–18852 RSC.
- A. Mishra, M. L. Keshtov, A. Looser, R. Singhal, M. Stolte, F. Wuerthner, P. Baeuerle and G. D. Sharma, J. Mater. Chem. A, 2017, 5, 14887–14897 RSC.
- C. Y. Huang, X. F. Liao, K. Gao, L. J. Zuo, F. Lin, X. L. Shi, C. Z. Li, H. B. Liu, X. S. Li, F. Liu, Y. W. Chen, H. Z. Chen and A. K. Y. Jen, Chem. Mater., 2018, 30, 5429–5434 CrossRef CAS.
- J. Sun, X. L. Ma, Z. H. Zhang, J. S. Yu, J. Zhou, X. X. Yin, L. Q. Yang, R. Y. Geng, R. H. Zhu, F. J. Zhang and W. H. Tang, Adv. Mater., 2018, 30, 1707150 CrossRef PubMed.
- B. Xiao, A. L. Tang, J. Q. Zhang, A. Mahmood, Z. X. Wei and E. J. Zhou, Adv. Energy Mater., 2017, 7, 1602269 CrossRef.
- A. Tang, B. Xiao, F. Chen, J. Zhang, Z. Wei and E. Zhou, Adv. Energy Mater., 2018, 8, 1801582 CrossRef.
- Z. Li, Y. Zhang, S.-W. Tsang, X. Du, J. Zhou, Y. Tao and J. Ding, J. Phys. Chem. C, 2011, 115, 18002–18009 CrossRef CAS.
- W. Lee, S. Jeong, C. Lee, G. Han, C. Cho, J.-Y. Lee and B. J. Kim, Adv. Energy Mater., 2017, 7, 1602812 CrossRef.
- K. Kranthiraja, S. Kim, C. Lee, K. Gunasekar, V. G. Sree, B. Gautam, K. Gundogdu, S.-H. Jin and B. J. Kim, Adv. Funct. Mater., 2017, 27, 1701256 CrossRef.
- J. W. Jo, J. W. Jung, H. Ahn, M. J. Ko, A. K. Y. Jen and H. J. Son, Adv. Energy Mater., 2017, 7, 1601365 CrossRef.
- Z. Luo, Y. Zhao, Z.-G. Zhang, G. Li, K. Wu, D. Xie, W. Gao, Y. Li and C. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 34146–34152 CrossRef CAS PubMed.
- W. T. Hadmojo, F. T. A. Wibowo, D. Y. Ryu, I. H. Jung and S.-Y. Jang, ACS Appl. Mater. Interfaces, 2017, 9, 32939–32945 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |