
Ordered intracrystalline pores in planar molybdenum oxide for enhanced alkaline hydrogen evolution†
Farjana
Haque
 a,
Ali
Zavabeti
a,
Bao Yue
Zhang
a,
Robi S.
Datta
a,
Yuefeng
Yin
bg,
Zhifeng
Yi
c,
Yichao
Wang
d,
Nasir
Mahmood
a,
Naresh
Pillai
a,
Nitu
Syed
a,
Hareem
Khan
a,
Azmira
Jannat
a,
Ning
Wang
*e,
Nikhil
Medhekar
*bg,
Kourosh
Kalantar-zadeh
*af and
Jian Zhen
Ou
*a
a,
Ali
Zavabeti
a,
Bao Yue
Zhang
a,
Robi S.
Datta
a,
Yuefeng
Yin
bg,
Zhifeng
Yi
c,
Yichao
Wang
d,
Nasir
Mahmood
a,
Naresh
Pillai
a,
Nitu
Syed
a,
Hareem
Khan
a,
Azmira
Jannat
a,
Ning
Wang
*e,
Nikhil
Medhekar
*bg,
Kourosh
Kalantar-zadeh
*af and
Jian Zhen
Ou
*a
aSchool of Engineering, RMIT University Melbourne, Victoria, Australia. E-mail: jianzhen.ou@rmit.edu.au
bSchool of Materials Science and Engineering, Monash University, Clayton, Victoria, Australia. E-mail: nikhil.medhekar@monash.edu
cInstitute for Frontier Materials, Deakin University, Geelong, Victoria, Australia
dSchool of Life and Environmental Sciences, Deakin University, Geelong, Victoria, Australia
eState Key Laboratory of Marine Resource Utilization in South China Sea, Hainan University, Haikou, Hainan, China. E-mail: wangn02@foxmail.com
fSchool of Chemical Engineering, University of New South Wales, Sydney, New South Wales, Australia. E-mail: k.kalantar-zadeh@unsw.edu.au
gSchool of Physics and Astronomy, Monash University, Clayton, Victoria, Australia
First published on 23rd November 2018
Abstract
Molybdenum based compounds are an emerging class of non-metallic catalytic materials for the hydrogen evolution reaction (HER) in acidic media. However, most of them lose considerable catalytic performance and exhibit poor long-term stability in alkaline media. Here, planar molybdenum oxide, with high alkaline stability and ordered intracrystalline pores, is developed as the HER candidate. The pores with diameters in the order of ∼5–7 Å are HER-active, and appear after an NH4+ doping-driven phase transition from the orthorhombic to hexagonal phase. Such a unique structure facilitates the diffusion of ionic entities and water molecules to the HER sites and helps in the removal of gaseous products, therefore improving the surface active area and reaction kinetics. These intracrystalline pores also reduce the long term stress on electrodes. The corresponding HER activity is extremely stable for >40 h in an alkaline medium at an overpotential of 138 mV with a Tafel slope of 50 mV dec−1. Such properties offer a superior combination compared to those of other reported molybdenum based nanostructures, hence providing a great opportunity for developing high-performance alkaline non-metal HER catalysts.
Introduction
Due to its environmental benignity and recyclability, hydrogen gas is one of the most attractive alternatives to the rapidly depleting fossil fuels.1–5 However, the sustainable and efficient production of hydrogen gas remains as the main challenge.6 Room-temperature water electrolysis is considered as one of the most promising production pathways, and is generally performed under either acidic or alkaline conditions.7–10 Acidic medium catalysis is usually expensive as the highly corrosive environment limits the choice of catalysts to noble metals, which hinders their wide implementation in many industries.2,11–15 In contrast, alkaline medium electrolysis is inherently low-cost but is offset by the high energy requirement.11,12 Therefore, searching for an economical catalyst with low required energy and long term stability in an alkaline medium remains an on-going challenge for the hydrogen production industry.16–19Molybdenum based compounds have been recognized as a class of non-metallic catalytic materials for the hydrogen evolution reaction (HER) of water electrolysis.3,20 In particular, molybdenum disulphide (MoS2) with a two-dimensional (2D) configuration has been investigated.20–26 In addition to the large surface-to-volume ratio, the unique 2D geometry results in a large quantity of unsaturated sulphur atoms on the edge and kink sites as well as basal steps distributed across the MoS2 planes, providing active sites for the HER.22 However, single MoS2 normally has poor long term stability in an alkaline medium and requires a modest to high overpotential.20 This is possibly due to the high activation energy of the initial Volmer step of water dissociation and the strong adsorption of hydroxyl anions on the surface.20,21 Other low-dimensional molybdenum based compounds, such as molybdenum carbides, borides, phosphides and nitrides, have also been explored as possible alkaline HER catalysts.27–31 However, such compounds cannot offer alkaline medium stability and low overpotentials simultaneously for long term stable HER performance.27–31
Molybdenum trioxide (MoO3) is known to be chemically inert in alkaline solutions at room temperature.3 It has a thermodynamically stable orthorhombic phase with a well-known layered crystal structure (so called ‘α-MoO3’), in which MoO6 octahedra establish planes that are held together by weak van der Waals forces (Fig. 1a).32 Due to its low conductivity and unfavourable H adsorption energy on O atoms, fully stoichiometric MoO3 is not considered as an efficient catalyst.33 The introduction of oxygen vacancies in the α-MoO3 crystal structure results in the generation of Mo dangling bonds and therefore defect states close to the conduction band minimum, which significantly improves the charge conductivity.34 In addition, the oxygen deficient structure favours the adsorption of water molecules in an alkaline medium, which serve as an electron acceptor and hence lower the HER energy barrier.35 However, the HER performance of sub-stoichiometric MoO3 decreases gradually during long term stability tests, possibly due to the reduction of catalytic reaction sites through the adsorption of OH− groups, saturating the coordination of the Mo6+ element.3,35,36
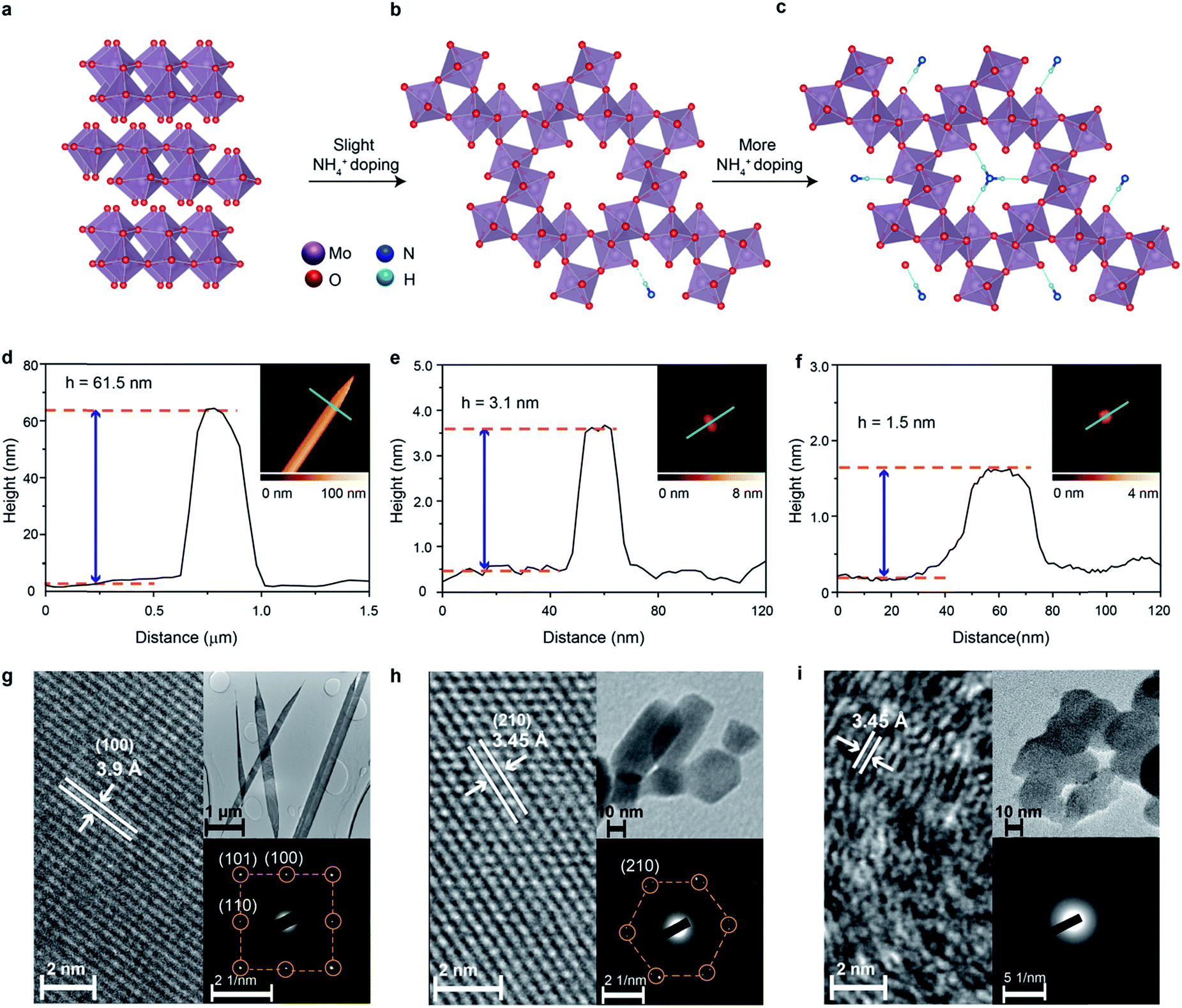 | ||
| Fig. 1 Schematics of the crystal structures of (a) α-MoO3, (b) Crys-AMO, and (c) Polycrys-AMO, AFM images of (d) α-MoO3, (e) Crys-AMO, and (f) Polycrys-AMO and HRTEM images of (g) α-MoO3, (h) Crys-AMO, and (i) Polycrys-AMO. The insets show the corresponding low resolution TEM images. | ||
The thermodynamically stable orthorhombic phase of MoO3 can be transformed to a crystal phase with hexagonal crystal coordination upon doping using small ions such as Na+, K+ and NH4+ under specific conditions.37,38 This hexagonal molybdenum oxide is constructed using zigzag MoO6 octahedral chains as the building blocks, which are interlinked through configuration isotherm positions. Therefore, the hexagonal crystal coordination results in the formation of highly-ordered one-dimensional (1D) tunnel-like pores with a calculated diameter of 5.7 Å across the planes (Fig. 1b).39 Small ions can reside in the centre of the pores (Fig. 1b and c) to further stabilize the hexagonal ring-like structure.40
The tunnelled structure of hexagonal molybdenum oxide, either with or without the residual ions, can potentially allow the facile diffusion of molecules and ions, and also offers more reaction sites, which results in improved HER performance.39 The presence of small ions in the crystal structure may facilitate the ion transport of hexagonal molybdenum oxide as it makes the structure more polar. At the same time, the concurrently injected electrons during ion doping have Drude-model-like behaviour and also improve the electron conductivity.41 More importantly, the hexagonal coordination of the MoO6 octahedra could result in the formation of non-periodic free energy barriers, or shallow potential barriers of low free energy, therefore facilitating a bypass channel for high charge transfer kinetics and possibly lowering the energy required for catalytic activity.38,41,42 One extra advantage of the intracrystalline pores is the reduction of stress caused by gaseous products over time that increases the lifetime of electrodes.
In this work, highly crystalline 2D molybdenum oxide with ordered hexagonal pores is obtained through the doping of an optimum level of NH4+ dopants in the MoO3 crystal structure using a hydrothermal synthesis method. The hypothesis of being a suitable HER catalyst in an alkaline medium is tested particularly in terms of overpotential, charge transfer kinetics, durability and long-term stability. The influence of the unique hexagonal intracrystalline pores and the NH4+ dopants in the crystal structure on the electronic band structure, surface area, charge conductivity and hydrogen adsorption energy is comprehensively investigated in conjunction with the study of HER performance. Additionally, we show that the electrocatalytic activity is higher than that of any other previously reported single Mo compounds, with an exceptional stability that exceeds 40 h of non-deteriorated operation.
Experimental
Synthesis of α-MoO3, Crys-AMO and Polycrys-AMO
All the samples were synthesized from peroxomolybdic solution. The peroxomolybdic solution was first prepared by adding 5 mL hydrogen peroxide (50 wt%, Sigma-Aldrich) into a 20 mL glass vial containing 0.5 g of Mo powder (99.9% purity, Aldrich). Afterward, 0, 150 and 350 mg of HMTA (hexamethylenetetramine, 99% purity, Sigma-Aldrich) dissolved in 10 mL DI water were slowly added into the peroxomolybdic solution and left stirring for 30 min. The solutions were then transferred into Teflon-lined stainless-steel autoclaves and the hydrothermal reaction was carried out at 200 °C for 24 h. The autoclaves were then naturally cooled down to room temperature after the experiments. All three samples were washed with DI water, centrifuged and dried in a natural environment.Structural and morphological characterization
AFM images were recorded using a Bruker Dimension Icon AFM with scan-assist software. Low and high resolution TEM measurements were conducted using JEOL 1010 and JEOL 2100F instruments with 80 and 100 keV acceleration voltages. XRD patterns were obtained using a Bruker D8 microdiffractometer equipped with a 0.5 mm collimator and a Vantec 500 detector. The experiment was conducted using a copper source with a potential of 40 kV and a current of 40 mA. Raman spectra were collected using a LabRAM HR Evolution Raman spectrometer (Horiba Scientific) under the excitation of a 532 nm laser with a power of 4.5 mW. FTIR spectroscopic measurements were performed on a PerkinElmer ATR FTIR spectrometer. XPS was performed using a Thermo Scientific K-alpha system. The samples were scanned under an Al Kα monochromated X-ray source with a pass energy of 50 kV and a dwell time of 50 ms. TGA was carried out using a Perkin-Elmer Pyris 1 instrument in N2 over a temperature range of 30 to 600 °C with a heating rate of 10 °C min−1. PESA spectra were collected using a Riken Keiki Model AC-2 PESA spectrometer with the probe light energy set to 50 nW and a power law of 1/3. UV-vis-NIR spectroscopy was carried out using a Cary 500 spectrophotometer. CO2 adsorption isotherms were measured at 273 K on an Autosorb iQ3 from Quantachrome Instruments, United States. The isotherm fitting and pore size distributions were calculated using the NLDFT model provided by Micromeritics.Electrochemical studies
HER catalytic activity tests were carried out in a standard three-electrode setup using a CHI 760D electrochemical workstation (CH Instruments). The cell setup contained a working electrode (nickel foam – 99.9% purity, MTI Corporation) coated with the samples, a Ag/AgCl reference electrode (in 1 M KCl aqueous solution), a graphite rod as the counter electrode and a 0.1 M KOH solution as the electrolyte. The reference electrode was calibrated against and converted to a reversible hydrogen electrode (RHE). Linear sweep voltammetry was performed at a 5 mV s−1 scan rate to obtain the polarization curves. All polarization curves were iR-corrected. The Ni foam was cleaned using a standard acidic solution (0.1 M HCl, 37%, Sigma-Aldrich). The Ni foam based working electrode was prepared as follows: 5 mg of each sample was mixed with a 2.5 mL DI water–isopropanol mixture (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and sonicated for 5 min, followed by the addition of 10 μL of polytetrafluoroethylene solution (60% in H2O, Sigma) and sonication for another 5 min. Finally, 100 μL of the suspension was drop-cast onto a Ni foam substrate with an area of 0.25 cm2. Electrochemical cyclic voltammetry measurements for determining EDLC were performed over a range of scan rates (20, 30, 40, 50 and 60 mV s−1) at 0.65 V vs. RHE in 0.1 M KOH.
:1) and sonicated for 5 min, followed by the addition of 10 μL of polytetrafluoroethylene solution (60% in H2O, Sigma) and sonication for another 5 min. Finally, 100 μL of the suspension was drop-cast onto a Ni foam substrate with an area of 0.25 cm2. Electrochemical cyclic voltammetry measurements for determining EDLC were performed over a range of scan rates (20, 30, 40, 50 and 60 mV s−1) at 0.65 V vs. RHE in 0.1 M KOH.
Theoretical calculation of hydrogen adsorption energy of α-MoO3, Crys-AMO and Polycrys-AMO
We used DFT calculations as implemented in the Vienna ab initio simulation package (VASP)43 to analyze the adsorption energy of the hydrogen of α-MoO3, Crys-AMO and Polycrys-AMO. The Perdew–Burke–Ernzerhof (PBE) form of the generalized gradient approximation (GGA) was used for describing electron exchange and correlation.44 All structures were relaxed until the ionic forces were smaller than 0.01 eV Å−1. A semiempirical functional developed by Grimme (DFT-D2) was employed to described dispersion forces. We used 5 × 5 × 15 Γ-centered grids for sampling the Brillouin zone. The plane-wave kinetic energy cut-off was set to 400 eV.Results and discussion
For producing hexagonal molybdenum oxide through NH4+ doping, peroxomolybdic acid solution is first prepared by dissolving Mo powder into a concentrated H2O2 solution. The post treatment of peroxomolybdic acid at elevated temperatures forms α-MoO3.45 Hexamethylenetetramine (HMTA) is employed as the source of the NH4+ dopant.42 NH4+ doped molybdenum oxide with a hexagonal crystal phase is expected to be obtained by reacting HMTA with the peroxomolybdic acidic solution in a pressurized autoclave at elevated temperatures.We also hypothesize that the added amount of HMTA will influence the degree of crystallinity and electronic band structure of the doped compound. In particular, the increase of the NH4+ dopant concentration may result in their placement on the top of the hexagonal intracrystalline pores (Fig. 1c), causing further exfoliation of the sample and reduction of the crystallinity.46 In addition, the concurrently injected electrons during the NH4+ doping process will enter into the electronic band structure and are expected to have Drude-like behavior, leading to the semiconductor-to-metal transition when the dopant concentration is significant.47 Therefore in this experiment, 0, 150 and 350 mg of HMTA are added into the peroxomolybdic acid solution, which is expected to produce undoped, low-level and high-level NH4+ doped molybdenum oxides, respectively. The morphological characterization of the samples is carried out utilizing atomic force microscopy (AFM). In the typical AFM representative image in Fig. 1d, it can be seen that the un-doped sample is belt-shaped with a thickness of ∼62 nm and a length of ∼8 μm. The thickness and length distributions of the belts are in the range of 55 to 65 nm and 8 to 14 μm, respectively, based on a large number of samples as shown in Fig. S1a and b in the ESI.† Low and high-resolution transmission electron microscopy (TEM) is also utilized to assess the dimensions and crystal structures of the samples. The length of the samples is found to be within 8–14 μm (upper inset in Fig. 1g). The high resolution TEM (HRTEM) image in Fig. 1g shows a belt with a lattice spacing of 3.9 Å, which corresponds to the (100) plane of α-MoO3.46 The corresponding selected area electron diffraction (SAED) pattern presented in the lower inset also shows the (100), (101) and (110) planes of α-MoO3 with an orthorhombic structure.48 Upon low-level doping with NH4+ ions, the sample is transformed into 2D nanoflake morphologies with a lateral dimension of ∼30 nm and a thickness of ∼3.1 nm, which corresponds to ∼2 monolayers of molybdenum oxide (Fig. 1e).38 The corresponding thickness distribution of the nanoflakes is in the range of 3 to 5 nm and lateral dimensions are within 20–50 nm (Fig. S1c and d, ESI†), which are also confirmed by the TEM image in the upper inset of Fig. 1h.
In view of the crystal structure, NH4+ doping results in the complete transformation of the sample from the orthorhombic to hexagonal system as evident by both the HRTEM (Fig. 1h) and SAED images (the lower inset of Fig. 1h). The lattice spacing of 3.45 Å in the HRTEM image represents the (210) plane of hexagonal molybdenum oxide,49 while the SAED pattern also indicates the hexagonal rings in the crystal structure. Therefore, we denote this sample as ‘crystalline ammonium-doped molybdenum oxide’ (Crys-AMO). For the high-level doped sample, while the 2D nature is preserved with a similar lateral dimension, the representative AFM image in Fig. 1f shows that the sample thickness is reduced to 1.5 nm.46 This trend is also confirmed by thickness and lateral dimension distribution charts (Fig. S1e and f, ESI†) and the corresponding TEM image (the upper inset of Fig. 1i). For the crystal structure, the lattice spacing of 3.45 Å in the HRTEM image in Fig. 1i is still observed. However, a large part of the sample becomes disordered, which is a typical observation for high-level ion doping of MoO3.47 This phenomenon can also be confirmed from the SAED pattern in the lower inset in Fig. 1i, in which a polycrystalline ring structure is observed. In this case, we denote this sample as ‘polycrystalline ammonium-doped molybdenum oxide’ (Polycrys-AMO).
X-ray diffraction (XRD) is also utilized to assess the crystal structure. For the un-doped sample, the dominant peaks at 12.6, 25.7, 38.92 and 67.4° correspond to the (020), (040), (060) and (0100) crystal planes of α-MoO3 as shown in Fig. 2a.50 For Crys-AMO, the dominant peaks at 16.8, 19.48, 25.7, 45.4 and 49° correspond to the planes of (110), (200), (210), (410) and (002) of hexagonal molybdenum oxide, respectively.42 For Polycrys-AMO, no obvious peaks appear except for a very weak peak at 26.5°, which is slightly right-shifted with reference to Crys-AMO, possibly due to the high-level NH4+ doping.51
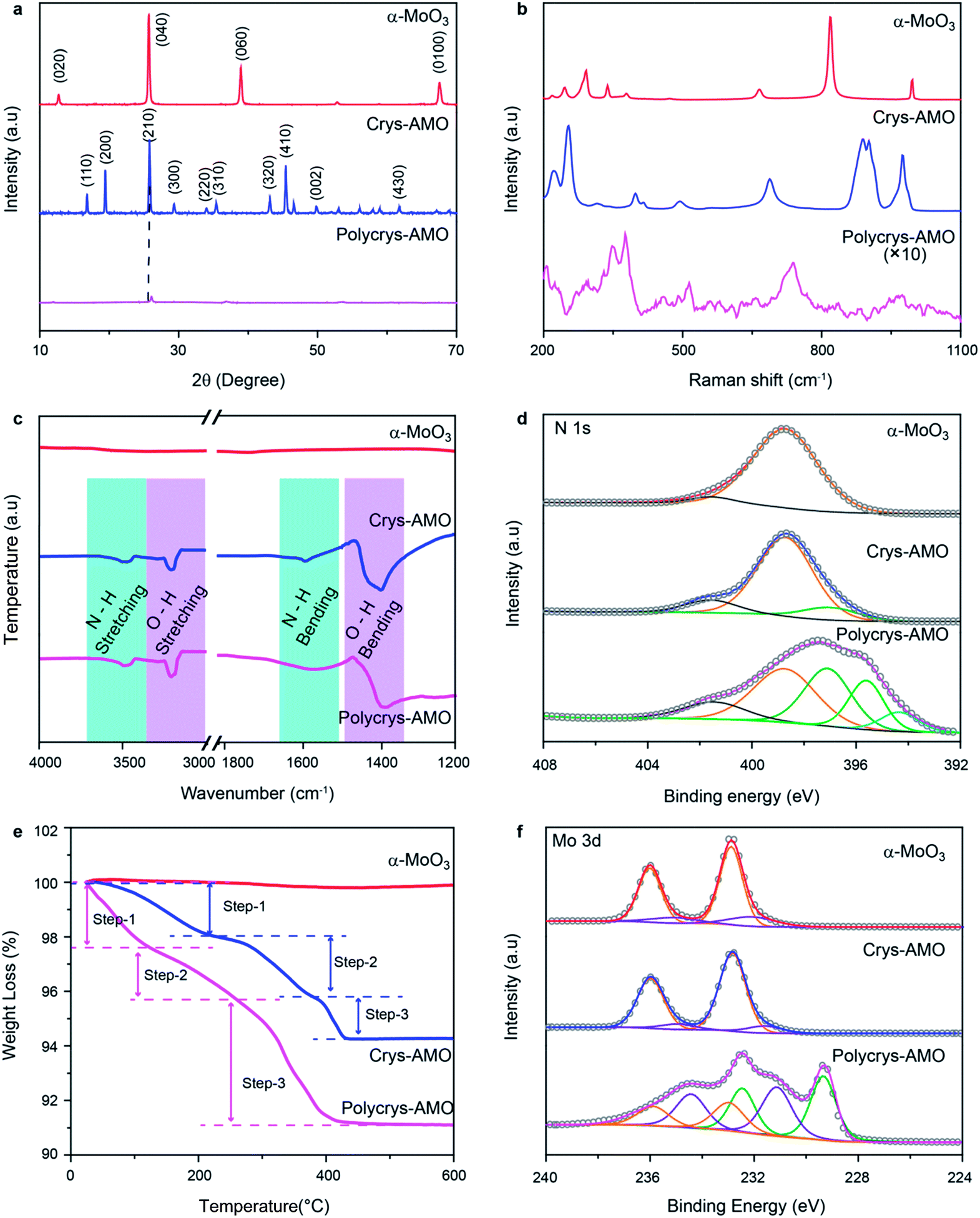 | ||
| Fig. 2 (a) XRD patterns, (b) Raman spectra, (c) FTIR spectra, (d) XPS N 1s spectra, (e) TGA patterns and (f) XPS Mo 3d spectra of the α-MoO3, Crys-AMO and Polycrys-AMO samples. | ||
Raman spectroscopy is used for investigating the influence of NH4+ dopants on the crystal structure and chemical bonding (Fig. 2b). For α-MoO3, strong peaks appear at 246, 284, 338, 379, 667, 818 and 995 cm−1, which are all ascribed to the orthorhombic structure (peak assignment in ESI Note 1). In the case of Crys-AMO, the distinguished peaks at 900, 906 and 976 cm−1 are due to the stretching vibrations of Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds from the MoO6 octahedra positioned in the hexagonal ring structure. Obvious peaks are also found at 320, 401, 420, 502 and 698 cm−1, which can be assigned to the O–Mo–O bending vibrations of the MoO6 octahedra.52 For Polycrys-AMO, the intensity of the Raman signal is greatly reduced possibly due to the reduction of crystallinity47 and the metallic nature of the sample.53 The broad peaks at 740 and 976 cm−1 are ascribed to the Mo5+/4+–O stretching vibration, which is possibly caused by NH4+ doping.50,53 A distinguished peak at ∼380 cm−1 and a weak broad peak at ∼450 cm−1 are also observed, which can be assigned to the symmetric coordinated oxygen (Mo–O) stretching vibration and the asymmetric coordinated oxygen (Mo–O) stretching vibration of sub-stoichiometric molybdenum oxides, respectively.46,50
O bonds from the MoO6 octahedra positioned in the hexagonal ring structure. Obvious peaks are also found at 320, 401, 420, 502 and 698 cm−1, which can be assigned to the O–Mo–O bending vibrations of the MoO6 octahedra.52 For Polycrys-AMO, the intensity of the Raman signal is greatly reduced possibly due to the reduction of crystallinity47 and the metallic nature of the sample.53 The broad peaks at 740 and 976 cm−1 are ascribed to the Mo5+/4+–O stretching vibration, which is possibly caused by NH4+ doping.50,53 A distinguished peak at ∼380 cm−1 and a weak broad peak at ∼450 cm−1 are also observed, which can be assigned to the symmetric coordinated oxygen (Mo–O) stretching vibration and the asymmetric coordinated oxygen (Mo–O) stretching vibration of sub-stoichiometric molybdenum oxides, respectively.46,50
As both XRD and Raman spectroscopy do not provide direct evidence of NH4+ doping, attenuated total reflection Fourier transform infrared (ATR-FTIR) spectroscopic measurements are conducted in the 1200–4000 cm−1 region. In Fig. 2c, it can be observed that there are no peaks for α-MoO3, confirming that there is no trace of NH4+ in the sample. For both Crys-AMO and Polycrys-AMO, the peaks at 3219 and 1600 cm−1 are attributed to the stretching and bending vibrations of the N–H bond, respectively, confirming the presence of NH4+ dopants in the crystal structure.41,49 In addition, the peaks at 3445 and 1560 cm−1 are ascribed to the stretching and bending vibrations of the O–H bond,41,49 respectively, which possibly originate from the hydrogen bonds between NH4+ ions and the MoO6 octahedra as well as crystalline water. X-Ray photoelectron spectroscopy (XPS) is employed to further confirm the presence of doped NH4+ ions and investigate the stoichiometry of the samples. Fig. 2d depicts the N 1s spectra of all three samples. It is known that the spectra of N 1s typically overlap with those of Mo 3p in molybdenum oxide.54 In order to clarify, we use α-MoO3 with no trace of NH4+ as the background reference and the peak at 398.8 eV is ascribed to the Mo6+ contribution.54,55 For Crys-AMO, a new peak centered at 397.1 eV represents the trace of N originating from NH4+ doping.55,56 For Polycrys-AMO, two additional peaks appear at 395.5 and 394.3 eV, which are ascribed to the Mo4+ and Moδ+ (0 < δ < 4) oxidation states, respectively.55,57 A shake-up satellite peak for all three samples is identified at ∼401.5 eV, which can be attributed to the surface adsorbed nitrogen.42,56
The NH4+ doping concentration of Crys-AMO and Polycrys-AMO can be determined by thermogravimetric analysis (TGA) as shown in Fig. 2e.42 For α-MoO3, there is almost no observable weight loss up to 600 °C.58 In contrast, three steps can be clearly observed in the weight loss chart for Crys-AMO at 40–200 °C, 200–390 °C and 390–450 °C, which correspond to the evaporation of surface adsorbed water molecules, the desorption of crystal water molecules and the release of NH4+ dopants, respectively.42,59 Therefore, the amounts of crystal water and NH4+ dopants in Crys-AMO are calculated to be 2.4% and 1.4%, respectively. As Crys-AMO can be formally described as (NH4)xMoO3−x(H2O)y,42x and y are estimated to be 0.11 and 0.2, respectively. Interestingly, the three above-mentioned steps in Polycrys-AMO occur at slightly lower temperatures, which are 30–150 °C, 150–290 °C and 290–450 °C, respectively.
The corresponding crystal water and NH4+ dopant contents are hence 1.5% and 4.6%, respectively, which indicates that the NH4+ dopant concentration in Polycrys-AMO is almost three times larger than that in Crys-AMO. The actual chemical composition of Polycrys-AMO is estimated to be (NH4)0.45MoO2.6 (H2O)0.15. XPS O 1s spectra of three samples are shown in Fig. S2 in the ESI.† For both α-MoO3 and Crys-AMO, the peak at 530.8 eV can be attributed to the Mo6+–O bond.47,60 An additional weak peak at 531.9 eV appears, which is ascribed to the adsorbed OH groups and the Mo5+–O bond.35,47,61 For Polycrys-AMO, another peak at 532.8 eV is observed, which is attributed to the Mo4+–O bond and indicates the existence of water molecules in the crystal structure.35,61
As the reduction of Mo oxidation states is mainly the result of NH4+ doping for both Crys-AMO and Polycrys-AMO, we also use the ratio of Mo6+:Mo5+:Mo4+ extracted from the Mo 3d spectra to further validate the estimation of the NH4+ doping content from the TGA patterns. For α-MoO3, the peaks at 232.6 and 236.1 eV both represent the Mo6+ oxidation state, while there is a small amount of Mo5+ present as is evident from the 231.5 and 234.8 eV peaks (Fig. 2f).62 Such sub-stoichiometry can also be observed in other reported low-dimensional α-MoO3 possibly due to the presence of defects in the sharp edges of the nanostructure.59 For Crys-AMO, there is no trace of Mo4+ and the ratio of Mo6+:Mo5+ is calculated to be 84:16, implying that x = 0.09 for (NH4)xMoO3−x which is close to the TGA result (x = 0.11). In the case of Polycrys-AMO, the 229.9 and 232.9 eV peaks indicate the Mo4+ oxidation state62,63 and the Mo6+:Mo5+:Mo4+ ratio is hence calculated to be 19:58:23, suggesting that x = 0.48, which is also similar to the TGA result (x = 0.45).
Fig. 3 shows the electronic band structures of all three samples. α-MoO3 (Fig. 3a) is a typical n-type semiconductor with a band gap energy of ∼3.05 eV, which is within our expectation.64 For Crys-AMO (Fig. 3b), the n-type semiconducting feature is preserved with a slightly narrower band gap energy of ∼2.85 eV, possibly due to the formation of localized states within the bandgap induced by the NH4+ dopants.65 On the other hand, Polycrys-AMO shows a metallic band structure as the Fermi level is moved above the conduction band minimum (CBM) (Fig. 3c). Such a semiconductor-to-metal transition can be due to the overlap of the electron wave functions associated with the occupation of the Mo 4d states by the free electrons introduced by the NH4+ dopants, which can be evidenced by the XPS valence spectrum (Fig. S4c, ESI†). As shown in Fig. S4c in the ESI,† two strong peaks located at the binding energies of ∼0.9 and ∼1.9 eV appear in the region between the Fermi level and VBM. The 0.9 eV peak represents the singly filled Mo 4d band originating from Mo5+, while the ∼1.9 eV peak results from the doubly occupied Mo4+ cations.66
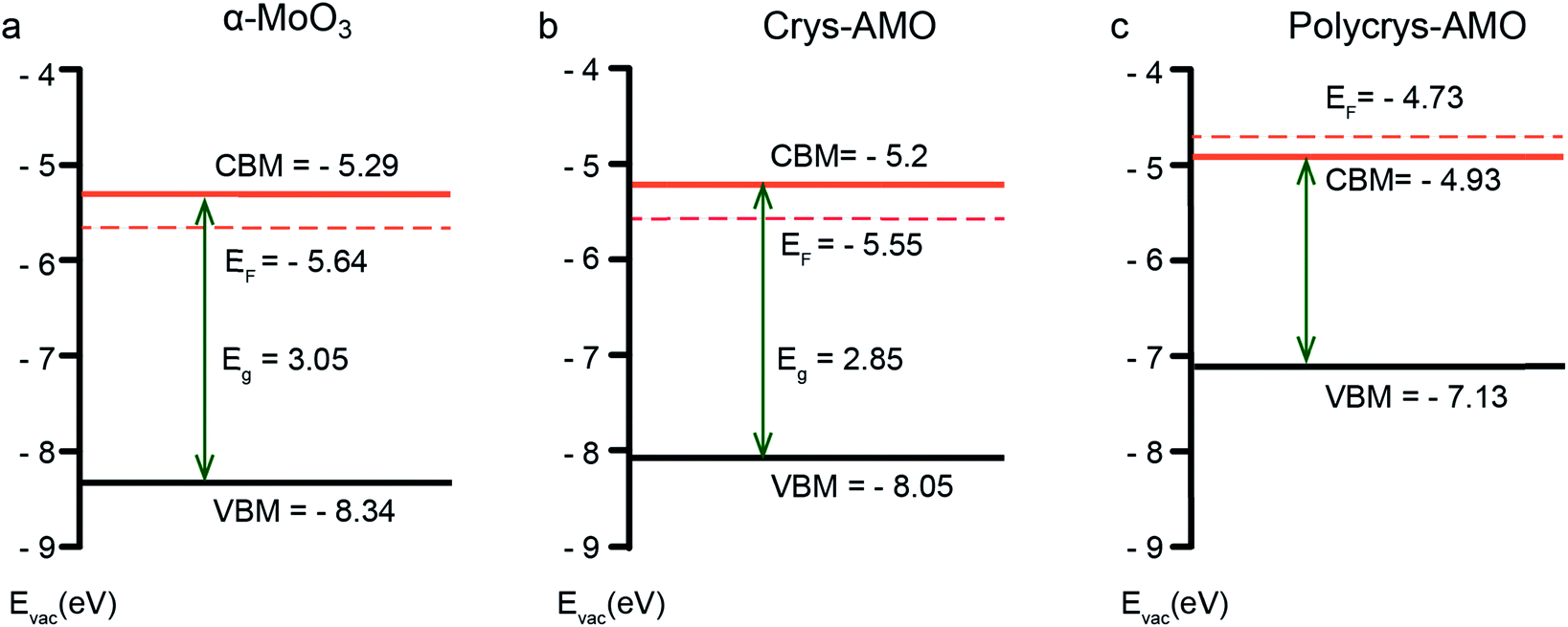 | ||
| Fig. 3 The electronic band structure of (a) α-MoO3, (b) Crys-AMO and (c) Polycrys-AMO based on the analysis of the UV-vis-NIR spectra, XPS valence spectra and photoelectron spectroscopy in air (PESA) results. In particular, the UV-vis-NIR spectroscopic results are used for estimating the optical bandgap of the material (Fig. S3, ESI†), while XPS valence spectroscopy (Fig. S4, ESI†) and PESA analysis (Fig. S5, ESI†) are carried out to determine the position of the valence band maximum (VBM) and Fermi level, respectively. | ||
The HER activities of α-MoO3, Crys-AMO and Polycrys-AMO are investigated in the alkaline medium. Fig. 4a shows the HER polarization curves of various electrocatalysts. For reference, commercial α-MoO3 (denoted as “com-MoO3”) has an overpotential of 220 mV to achieve the standard 10 mA cm−2 HER current density, indicating its low HER reactivity as expected from the literature.3 In comparison, there is a 10 mV improvement in the overpotential of our α-MoO3 belt sample, which is due to the presence of oxygen vacancy driven HER active sites.35 For both the Crys-AMO and Polycrys-AMO samples, the overpotentials are dramatically reduced to 138 and 160 mV, respectively.
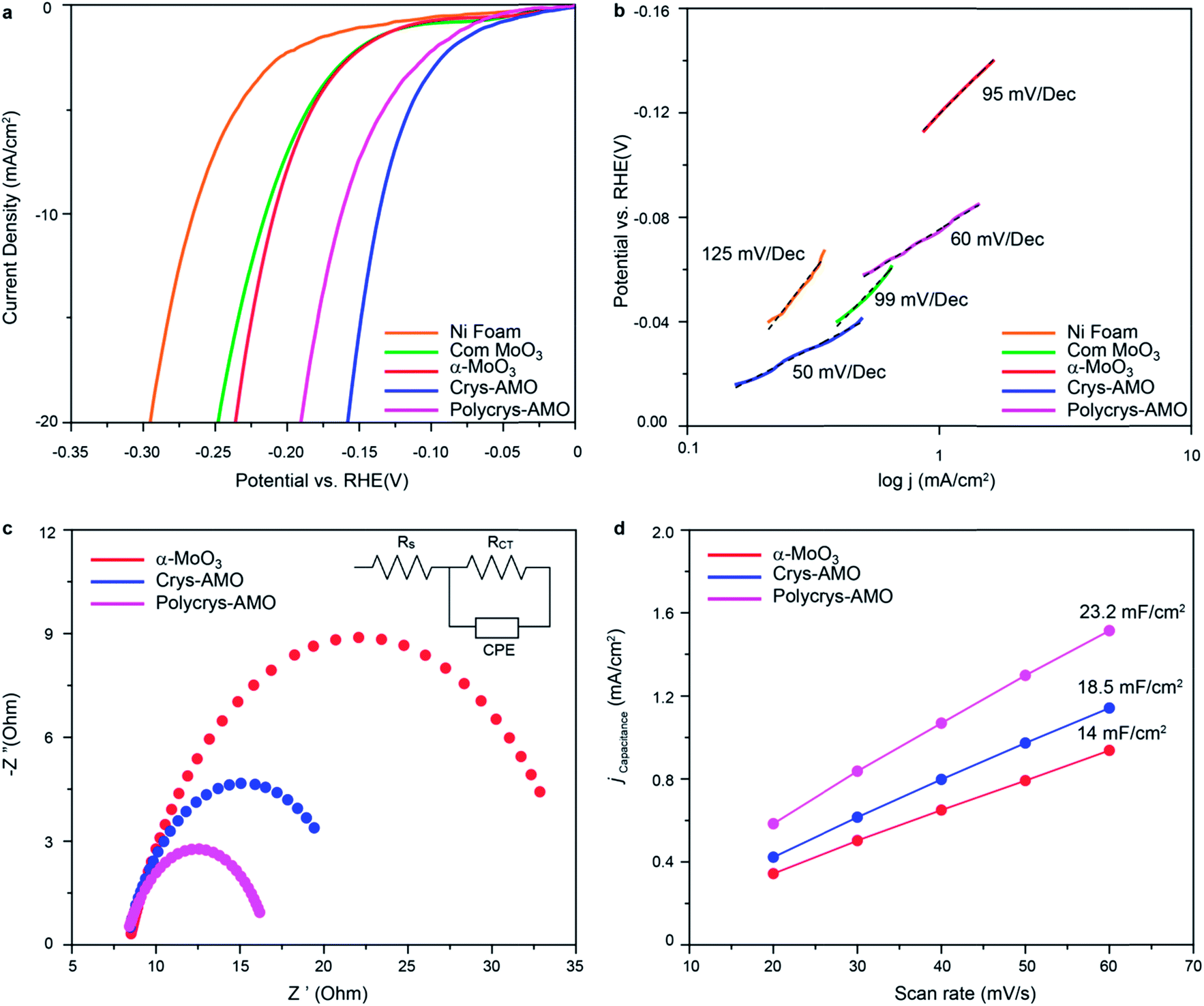 | ||
| Fig. 4 (a) Polarization curves of α-MoO3 (red curve), Crys-AMO (blue curve) and Polycrys-AMO (magenta curve) on the Ni foam electrode in 0.1 M potassium hydroxide (KOH), along with that of bare Ni foam (orange curve) and com MoO3 (green curve) for comparison and (b) their corresponding Tafel slopes; (c) EIS spectra of α-MoO3, Crys-AMO and Polycrys-AMO; the inset represents an electrochemical equivalent circuit model. (d) Electrochemically active surface area of α-MoO3, Crys-AMO and Polycrys-AMO. | ||
To determine the HER reaction kinetics, the Tafel slopes of all samples are extracted from their HER polarization curves (Fig. 4b). Large Tafel slopes of 125, 99 and 95 mV dec−1 are found in the bare Ni foam, com-MoO3 and our α-MoO3 sample, respectively. These observations suggest that the above-mentioned materials exhibit slow reaction rate for HER via the Volmer–Heyrovsky process.67 During this process, the adsorbed water molecules on the material surface are first broken into H+ and OH− ions (Volmer reaction). Then the H+ ions are transformed into H2 gas molecules by accepting electrons and combining with other H2O molecules (Heyrovsky reaction).2 In comparison, the Tafel slope values of Crys-AMO and Polycrys-AMO are greatly reduced to 50 and 60 mV dec−1, respectively, indicating a much faster HER rate during the Volmer–Heyrovsky process.
It is hypothesized that the orthorhombic-to-hexagonal crystal structure transformation by NH4+ doping activates the HER catalytic properties of molybdenum oxide with respect to the improvement of the charge transfer kinetics and the increased number of surface active sites with significantly reduced hydrogen adsorption energy. The charge transfer kinetics is directly related to the charge transfer resistance (Rct) of the material, which can be measured using electrochemical impedance spectroscopy (EIS).3 In Fig. 4c, the high-frequency intersection with the x-axis represents the uncompensated solution resistance (Rs), which is almost the same for all the samples (Table S1, ESI†). In addition, the diameter of the semi-circle in the EIS spectra of both Crys-AMO and Polycrys-AMO is reduced compared to that of α-MoO3.
By using the electrochemical equivalent circuit model shown in the inset of Fig. 4c, Rct is determined to be 13.9 and 11.7 Ω for Crys-AMO and Polycrys-AMO, respectively, which are much lower than that of α-MoO3 (26.5 Ω), indicating better conductivities and enhanced HER kinetics (Table S1, ESI†). It has been reported that the easy migration and diffusion characteristics of NH4+ dopants are mainly responsible for the dramatic increase of ion conductivity in both Crys-AMO and Polycrys-AMO, while the concurrently injected electrons during the ion doping process have a Drude-model-like behaviour and therefore contribute to the improvement of the electron conductivity.41
In addition, the number of surface active sites also immensely influences the HER performance.68 The electrochemically active surface area (ECSA) is used here for quantifying the surface active sites68 through the measurement of double layer capacitance (Cdl) (Fig. S6, ESI†).69 A specified voltage range of 0.6–0.7 V (Fig. S6 and S7, ESI†) was selected to obtain a rectangular shaped CV curve, ensuring that only the surface reaction without any contribution from the Faraday process is considered to determine the pure EDLC values.70 From Fig. 4d, the Cdl of α-MoO3, Crys-AMO and Polycrys-AMO is measured to be 14, 18.5, and 23.2 mF cm−2, respectively. This indicates that both Crys-AMO and Polycrys-AMO have much higher ECSAs than α-MoO3, which can be attributed to the higher number of surface active sites of the samples and hence larger HER current density (Fig. 4a).
Low-pressure CO2 adsorption isotherms are utilized regardless of N2 adsorption (due to the smaller size of pores, (Fig. S8, ESI†)) to further verify the surface active area (Fig. 5a). It can be observed that the surface adsorption values of both Crys-AMO and Polycrys-AMO are much higher than that of α-MoO3. By using the nonlocal density functional theory (NLDFT) model, the actual surface area of α-MoO3 is calculated to be 21.4 m2 g−1, while Crys-AMO and Polycrys-AMO have areas of 40.23 and 48.68 m2 g−1, respectively. Both BET and ECSA analyses show higher surface area of Polycrys-AMO compared to that of Crys-AMO due to the smaller thickness of 2D Polycrys-AMO sheets given that their lateral dimensions are similar.
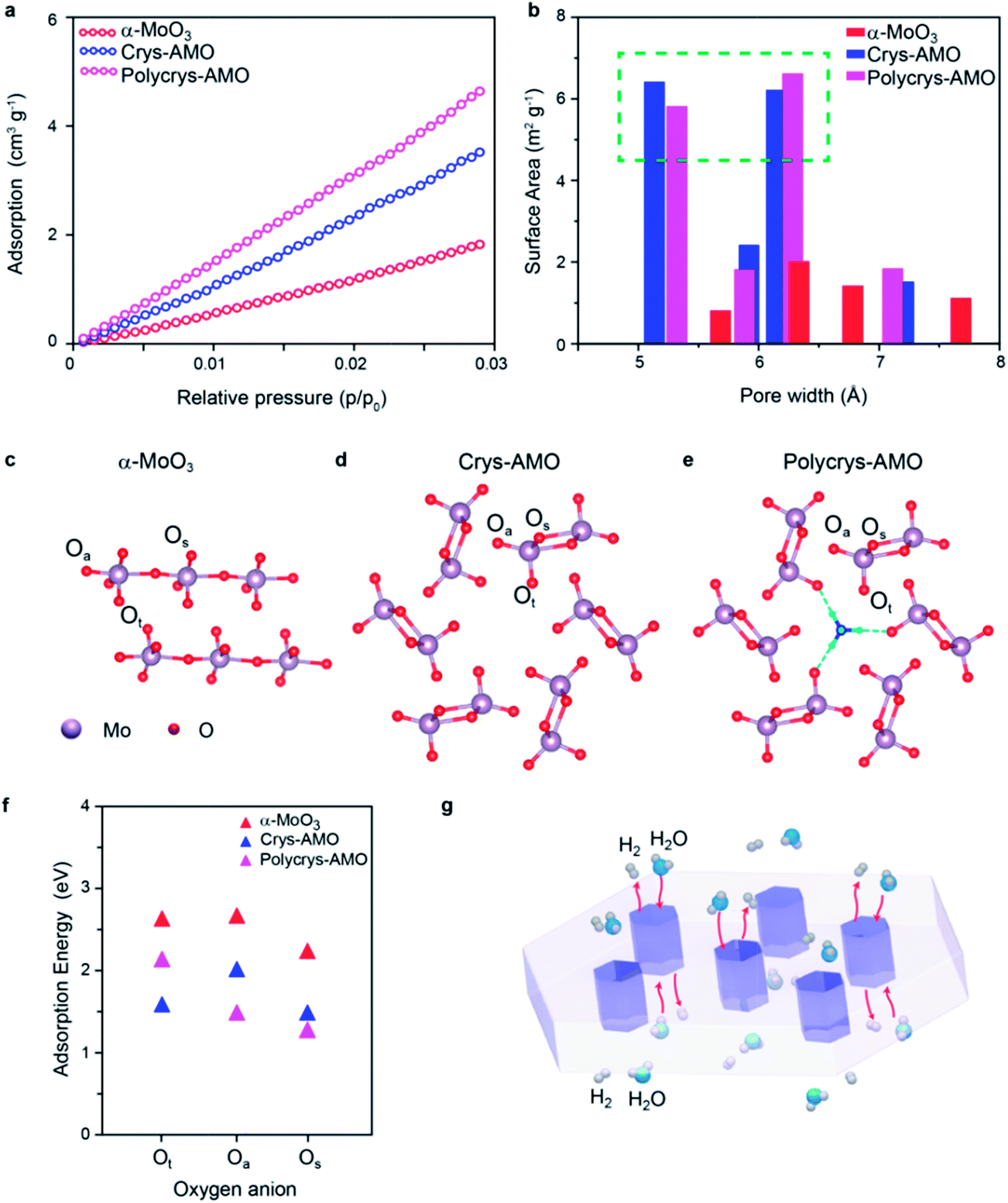 | ||
| Fig. 5 (a) Low pressure CO2 adsorption isotherms at 273 K of α-MoO3, Crys-AMO and Polycrys-AMO; (b) the corresponding pore size distributions calculated using the NLDFT method; simulated crystal structure of (c) α-MoO3, (d) Crys-AMO and (e) Polycrys-AMO; (f) hydrogen adsorption energy of α-MoO3, Crys-AMO and Polycrys-AMO; and (g) schematic of ion and molecule diffusion through the 1D hexagonal intracrystalline pores in the 2D lateral domain. | ||
The pore size distribution of each material calculated using the NLDFT model is shown in Fig. 5b. For α-MoO3, the concentration of pores with diameters less than 10 Å is very small, indicating that the pores are mostly inter-crystalline rather than intra-crystalline. In contrast, both Crys-AMO and Polycrys-AMO have distinct maxima at pore diameters of ∼5.3 and ∼6.4 Å. These diameters agree well with those of hexagonal rings (∼5.7 Å) from the simulated crystal structure (Fig. 5d), confirming that such an impressive improvement of the surface active sites is due to the hexagonal phase transformation.
Furthermore, density functional theory (DFT) calculations are performed to theoretically predict the surface active sites and estimate the energy needed for hydrogen adsorption for all three samples (Fig. 5c–e). Here, Ot, Os and Oa specify the terminal, symmetric bridging, and asymmetric bridging oxygen sites,33 respectively (Fig. 5c–e). According to Fig. 5f, the adsorption energies of hydrogen on the oxygen atom sites of α-MoO3 are all above 2 eV, confirming the low HER catalytic activity. The active sites for Crys-AMO are identified in both Ot and Os sites with significantly lower adsorption energies of 1.5 and 1.35 eV, respectively. In comparison, the active sites for Polycrys-AMO are changed to Oa and Os with similar adsorption energies of 1.4 and 1.25 eV, respectively.
As the Crys-AMO and Polycrys-AMO samples are mostly mono-layered or bi-layered, the pores act like 1D hexagonal tunnels across the whole sample as illustrated in Fig. 5g. These highly ordered intracrystalline pores are particularly useful in Crys-AMO and allow easy migration of water molecules and gaseous species. Although the hydrogen adsorption energies of the active sites for Crys-AMO (Ot) are slightly higher than those for Polycrys-AMO (Oa and Os), the higher HER performance of Crys-AMO is based on the reversible availability of pores for the next water molecule due to the existence of active sites in the form of Ot which quickly break it down into gaseous products. As the result, the effective utilization of every unit cell for the HER catalytic process can be realized for Crys-AMO and its charge transfer kinetics is significantly enhanced.41 In comparison, the Polycrys-AMO pores are partially blocked by NH4+ ionic dopants (Fig. 5e) and its Ot sites have a higher hydrogen binding energy, resulting in the unavailability of the pores for the HER. This may cause the degradation of the performance of Polycrys-AMO compared to that of Crys-AMO although it has a higher ECSA and BET surface area and a smaller charge transfer resistance.
Stability and durability are significant criteria to evaluate a HER catalyst.22 The long term HER stabilities of all the three samples are tested at the overpotentials determined in Fig. 4a for 40 h. Fig. 6a shows that the current density of non-doped α-MoO3 dropped by almost 80% in the first one hour during the stability test. This is due to the significant reduction of the HER active sites by the adsorption of OH− groups on the oxygen vacancies.36
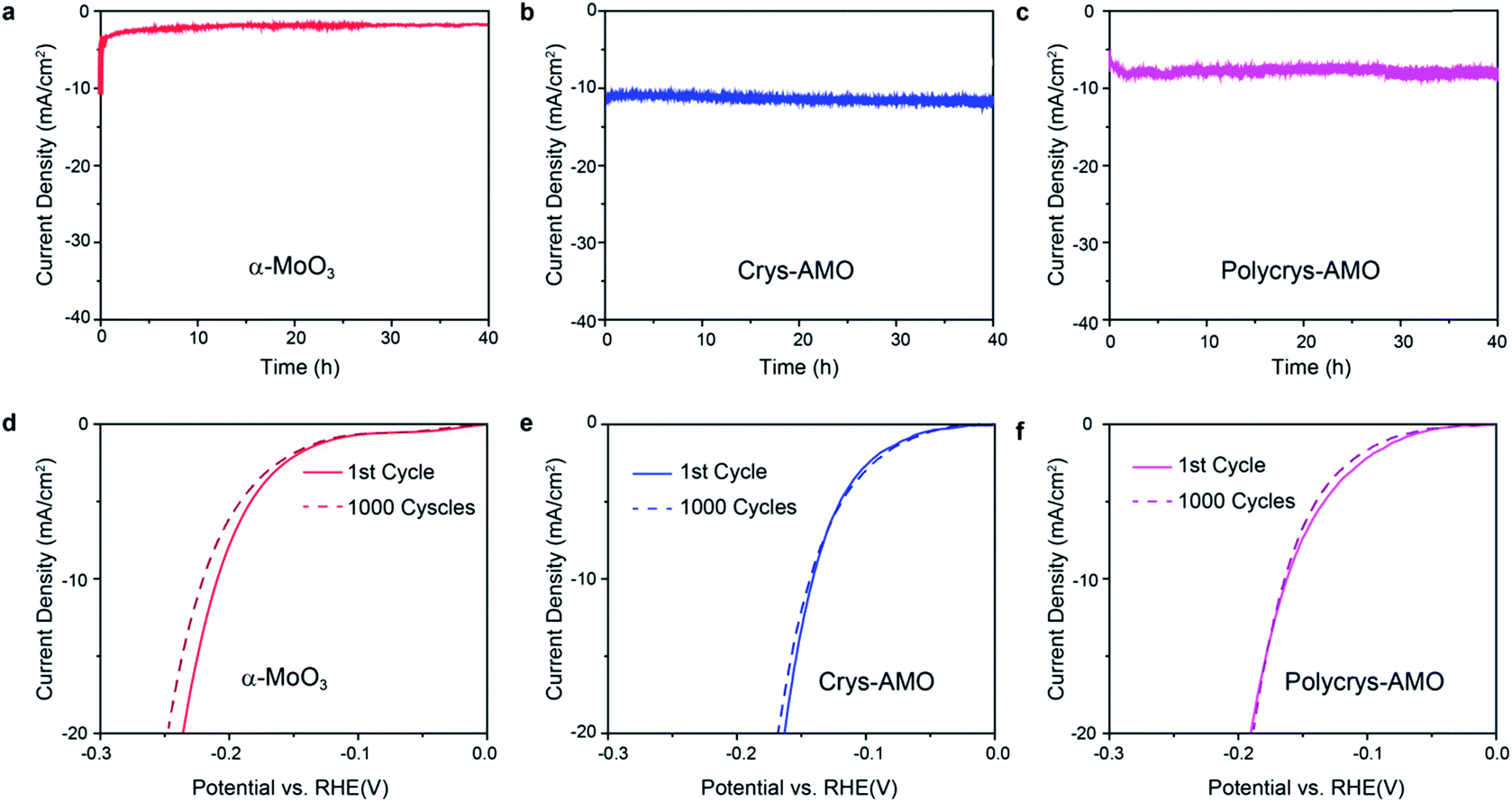 | ||
| Fig. 6 Time dependent HER current density of (a) α-MoO3, (b) Crys-AMO and (c) Polycrys-AMO at fixed overpotentials of 210, 138 and 160 mV, respectively. The polarization curve before (straight line) and after (dotted line) 1000 CV cycles of (d) α-MoO3, (e) Crys-AMO and (f) Polycrys-AMO. | ||
In comparison, excellent stabilities for both Crys-AMO and Polycrys-AMO are observed for more than 40 h with no obvious degradation (Fig. 6b and c), indicating that the NH4+ dopants and hexagonal intracrystalline pores are stable in an alkaline solution. Here, NH4+ dopants play a critical role to stabilize such a hexagonal intracrystalline porous structure (ESI Note 2). This also shows that the reduction of the stress and release of gaseous products, due to the presence of intracrystalline pores had a positive impact on the longevity of the system. There is no significant morphological change or peel-off occurring in both samples during the test (Fig. S9, ESI†). For the durabilities of the samples, there is no observable change in the HER polarization curves of both Crys-AMO and Polycrys-AMO before and after running 1000 continuous CV cycles, while significant degradation is found in α-MoO3 (Fig. 6d–f). It has been reported that the introduction of oxygen vacancies in MoO3 significantly enhances the HER performance in terms of required overpotential and reaction kinetics.3,35
However, its long term stability is weak as demonstrated previously and in this paper (Fig. 6a).3,35,36 Although phosphorus doping in MoO3−x is reported to overcome its instability in acidic solution, the overpotential and reaction kinetics are degraded and its HER stability in alkaline solution is unknown.36
In comparison, Crys-AMO is a much more suitable and commercially viable HER catalyst with an exceptional stability of at least 40 h in alkaline solution, while its overpotential and Tafel slope value are improved simultaneously (Table 1). Furthermore, the overall alkaline HER performance of Crys-AMO is superior in comparison to that of single molybdenum chalcogenide, nitride, phosphide, boride and carbide compounds without compositing with carbon- and metal-based materials.20,28,29
| Material | Overpotential@10 mA cm−2 (mV) | Tafel slope (mV dec−1) | Stability | Durability (CV cycles) | Ref. |
|---|---|---|---|---|---|
| 2D Crys-AMO | 138 | 50 | 40 h@138 mV | 1000 | This work |
| Mesoporous MoO3−x | 148 | 56 | < 1 h@170 mV | — | 35 |
| 2D MoO3−x | 142 | 58 | 2 h@142 mV | 1000 | 3 |
| 2D MoS2 | 255 | 119 | — | — | 20 |
| MoP NP | 275 | 94 | 2 h@275 mV | 2000 | 28 |
| MoP2 NP | 175 | 93 | — | 4000 | 31 |
| 2D MoN | 139 | 68 | 4 h@150 mV | 2000 | 30 |
| Mo2N NP | 350 | 100 | — | 1000 | 71 |
| Mo2C NP | 160 | 54 | 30 h@160 mV | 3000 | 29 |
| MoB NP | 220 | 59 | 2 h@200 mV | 3000 | 29 |
The HER overpotential of single 2D molybdenum nitride (139 mV) is similar to that of Crys-AMO. However, the static potential required for its stable HER performance is much higher (150 mV).30 In the case of heavily reduced single MoSx, the large number of sulphur vacancies in the crystal structure results in the significant degradation of the charge transfer kinetics (105 mV dec−1) as well as weak long term stability in alkaline environments, which are counterproductive similar to the case of single MoO3−x.
Conclusions
In summary, we demonstrated that the formation of highly-ordered intracrystalline pores of 2D molybdenum oxide enables its efficient and extremely stable HER activity in an alkaline medium. The key to realizing this unique strategy was to induce crystal phase transition through NH4+ doping at an optimum level. At low-level doping of NH4+ (Crys-AMO), α-MoO3 with initial belt-shaped morphology was exfoliated into few-layered 2D nanoflakes. Simultaneously, its orthorhombic crystal coordination was transformed into a hexagonal phase, leading to the formation of highly ordered intracrystalline pores through the entire 2D lateral plane which facilitates the diffusion of water molecules and the release of HER gaseous products. These pores also reduced the stress on the electrodes, facilitating long term stability. More importantly, HER active sites were identified inside these intracrystalline pores with a significantly lowered hydrogen adsorption energy compared to that of α-MoO3, and were easily accessible and therefore improved the HER reaction kinetics. The resulting HER overpotential and Tafel slope of 2D Crys-AMO were 138 mV and 50 mV dec−1, respectively, and were both stable for at least 40 h in the alkaline medium. The increased concentration of NH4+ doping resulted in the production of mono-layered polycrystalline metallic nanoflakes (Polycrys-AMO), which therefore increased the surface active area and improved the charge conductivity. However, the corresponding HER performance was slightly degraded compared to that of 2D Crys-AMO due to the reduction in the number of accessible pores and relocation of the HER catalytic sites. Compared to other reported single molybdenum compound based alkaline HER catalysts, our 2D Crys-AMO demonstrated a significantly enhanced overall HER activity, confirming that the doping-driven formation of highly-ordered intracrystalline pores on 2D materials could be a feasible design strategy for high-performance non-metal catalysts for alkaline hydrogen evolution.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the facilities and scientific and technical assistance of the RMIT MicroNano Research Facility (MNRF), the RMIT Microscopy and Microanalysis Facility (RMMF) and Frank Antolasic, Applied Chemistry. The authors would also like to acknowledge the support from the Australian Research Council (DE160100715). N. W. would also like to thank the support from The Finance Science and technology project of Hainan province (No. ZDYF2018004) and The National Natural Science Foundations of China (No. 51775152, 61761016). N. V. M. and Y. Y would like to acknowledge the support from the Center of Excellence in Future Low Energy Electronics (CE170100039). N. V. M. and Y. Y also gratefully acknowledge the computational support from the Monash Campus Cluster, NCI National Facility at the Australian National University and Pawsey Supercomputing Facility. The authors would like to thank Anthony S. R. Chesman, CSIRO Manufacturing, Ian Wark Laboratories, Clayton, Victoria, Australia for performing the PESA experiments.Notes and references
- D. Voiry, H. Yamaguchi, J. Li, R. Silva, D. C. Alves, T. Fujita, M. Chen, T. Asefa, V. B. Shenoy and G. Eda, Nat. Mater., 2013, 12, 850 CrossRef CAS PubMed.
- N. Mahmood, Y. Yao, J. W. Zhang, L. Pan, X. Zhang and J. J. Zou, Adv. Sci., 2017, 5, 1700464 CrossRef PubMed.
- R. Datta, F. Haque, M. Mohiuddin, B. Carey, N. Syed, A. Zavabeti, B. Zhang, H. Khan, K. Berean and J. Ou, J. Mater. Chem. A, 2017, 5, 24223–24231 RSC.
- B. Wang, Z. Wang, X. Wang, B. Zheng, W. Zhang and Y. Chen, J. Mater. Chem. A, 2018, 6, 12701–12707 RSC.
- B. Yu, F. Qi, B. Zheng, J. Zhou and Y. Chen, Electrochim. Acta, 2018, 277, 161–167 CrossRef CAS.
- J. A. Turner, Science, 2004, 305, 972–974 CrossRef CAS PubMed.
- L. Chen, X. Dong, Y. Wang and Y. Xia, Nat. Commun., 2016, 7, 11741 CrossRef CAS PubMed.
- W. Hou, B. Yu, F. Qi, X. Wang, B. Zheng, W. Zhang, Y. Li and Y. Chen, Electrochim. Acta, 2017, 255, 248–255 CrossRef CAS.
- J. Lin, J. He, F. Qi, B. Zheng, X. Wang, B. Yu, K. Zhou, W. Zhang, Y. Li and Y. Chen, Electrochim. Acta, 2017, 247, 258–264 CrossRef CAS.
- H. Wang, X. Wang, D. Yang, B. Zheng and Y. Chen, J. Power Sources, 2018, 400, 232–241 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K.-C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- J. Wang, F. Xu, H. Jin, Y. Chen and Y. Wang, Adv. Mater., 2017, 29, 1605838 CrossRef PubMed.
- H. Yue, B. Yu, F. Qi, J. Zhou, X. Wang, B. Zheng, W. Zhang, Y. Li and Y. Chen, Electrochim. Acta, 2017, 253, 200–207 CrossRef CAS.
- B. Yu, Y. Hu, F. Qi, X. Wang, B. Zheng, K. Liu, W. Zhang, Y. Li and Y. Chen, Electrochim. Acta, 2017, 242, 25–30 CrossRef CAS.
- B. Yu, F. Qi, B. Zheng, W. Hou, W. Zhang, Y. Li and Y. Chen, J. Mater. Chem. A, 2018, 6, 1655–1662 RSC.
- J. D. Benck, Z. Chen, L. Y. Kuritzky, A. J. Forman and T. F. Jaramillo, ACS Catal., 2012, 2, 1916–1923 CrossRef CAS.
- B. Yu, F. Qi, Y. Chen, X. Wang, B. Zheng, W. Zhang, Y. Li and L.-C. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 30703–30710 CrossRef CAS PubMed.
- W. Hou, B. Zheng, F. Qi, B. Yu and Y. Chen, Electrochim. Acta, 2018, 269, 155–162 CrossRef CAS.
- W. Hou, B. Zheng, F. Qi, J. He, W. Zhang and Y. Chen, Electrochim. Acta, 2018, 283, 1146–1153 CrossRef CAS.
- M. Anjum, H. Jeong, M. Lee, H. Shin and J. Lee, Adv. Mater., 2018, 30, 1707105 CrossRef PubMed.
- M. A. Lukowski, A. S. Daniel, F. Meng, A. Forticaux, L. Li and S. Jin, J. Am. Chem. Soc., 2013, 135, 10274–10277 CrossRef CAS PubMed.
- J. Xie, H. Zhang, S. Li, R. Wang, X. Sun, M. Zhou, J. Zhou, X. W. D. Lou and Y. Xie, Adv. Mater., 2013, 25, 5807–5813 CrossRef CAS PubMed.
- B. Yu, F. Qi, X. Wang, B. Zheng, W. Hou, Y. Hu, J. Lin, W. Zhang, Y. Li and Y. Chen, Electrochim. Acta, 2017, 247, 468–474 CrossRef CAS.
- X. Wang, B. Zheng, B. Yu, B. Wang, W. Hou, W. Zhang and Y. Chen, J. Mater. Chem. A, 2018, 6, 7842–7850 RSC.
- J. D. Cain, E. D. Hanson and V. P. Dravid, J. Appl. Phys., 2018, 123, 204304 CrossRef.
- X. Wang, J. He, B. Zheng, W. Zhang and Y. Chen, Electrochim. Acta, 2018, 283, 1660–1667 CrossRef CAS.
- Z. Xing, Q. Liu, A. M. Asiri and X. Sun, Adv. Mater., 2014, 26, 5702–5707 CrossRef CAS PubMed.
- Z. Wu, J. Wang, J. Zhu, J. Guo, W. Xiao, C. Xuan, W. Lei and D. Wang, Electrochim. Acta., 2017, 232, 254–261 CrossRef CAS.
- H. Vrubel and X. Hu, Angew. Chem., Int. Ed., 2012, 124, 12875–12878 CrossRef.
- J. Xiong, W. Cai, W. Shi, X. Zhang, J. Li, Z. Yang, L. Feng and H. Cheng, J. Mater. Chem. A, 2017, 5, 24193–24198 RSC.
- T. Wu, M. Pi, D. Zhang and S. Chen, J. Power Sources, 2016, 328, 551–557 CrossRef CAS.
- B. Zheng, Z. Wang, Y. Chen, W. Zhang and X. Li, 2D Mater., 2018, 5, 045011 CrossRef.
- X. Sha, L. Chen, A. C. Cooper, G. P. Pez and H. Cheng, J. Phys. Chem. C, 2009, 113, 11399–11407 CrossRef CAS.
- H.-S. Kim, J. B. Cook, H. Lin, J. S. Ko, S. H. Tolbert, V. Ozolins and B. Dunn, Nat. Mater., 2017, 16, 454–460 CrossRef CAS PubMed.
- Z. Luo, R. Miao, T. D. Huan, I. M. Mosa, A. S. Poyraz, W. Zhong, J. E. Cloud, D. A. Kriz, S. Thanneeru and J. He, Adv. Energy Mater., 2016, 6, 1600528 CrossRef.
- L. Li, T. Zhang, J. Yan, X. Cai and S. F. Liu, Small, 2017, 13, 1700441 CrossRef PubMed.
- Y. Xu, L. Xie, Y. Zhang and X. Cao, Electron. Mater. Lett., 2013, 9, 693–696 CrossRef CAS.
- X. Xiao, H. Song, S. Lin, Y. Zhou, X. Zhan, Z. Hu, Q. Zhang, J. Sun, B. Yang and T. Li, Nat. Commun., 2016, 7, 11296 CrossRef CAS PubMed.
- L. Zheng, Y. Xu, D. Jin and Y. Xie, Chem. Mater., 2009, 21, 5681–5690 CrossRef CAS.
- N. Caiger, S. Crouch-Baker, P. Dickens and G. James, J. Solid State Chem., 1987, 67, 369–373 CrossRef CAS.
- A. Chithambararaj, N. Rajeswari Yogamalar and A. C. Bose, Cryst. Growth Des., 2016, 16, 1984–1995 CrossRef CAS.
- V. Kumar, X. Wang and P. S. Lee, Nanoscale, 2015, 7, 11777–11786 RSC.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- B. Mendoza-Sánchez, T. Brousse, C. Ramirez-Castro, V. Nicolosi and P. S. Grant, Electrochim. Acta, 2013, 91, 253–260 CrossRef.
- M. M. Alsaif, K. Latham, M. R. Field, D. D. Yao, N. V. Medehkar, G. A. Beane, R. B. Kaner, S. P. Russo, J. Z. Ou and K. Kalantar-zadeh, Adv. Mater., 2014, 26, 3931–3937 CrossRef CAS PubMed.
- B. Y. Zhang, A. Zavabeti, A. F. Chrimes, F. Haque, L. A. O'Dell, H. Khan, N. Syed, R. Datta, Y. Wang and A. S. Chesman, Adv. Funct. Mater., 2018, 28, 1706006 CrossRef.
- X. W. Lou and H. C. Zeng, J. Am. Chem. Soc., 2003, 125, 2697–2704 CrossRef CAS PubMed.
- A. Chithambararaj and A. C. Bose, Beilstein J. Nanotechnol., 2011, 2, 585–592 CrossRef CAS PubMed.
- J. Z. Ou, J. L. Campbell, D. Yao, W. Wlodarski and K. Kalantar-Zadeh, J. Phys. Chem. C, 2011, 115, 10757–10763 CrossRef CAS.
- N. Sotani, K. Eda, M. Sadamatu and S. Takagi, Bull. Chem. Soc. Jpn., 1989, 62, 903–907 CrossRef CAS.
- V. Atuchin, T. Gavrilova, V. Kostrovsky, L. Pokrovsky and I. Troitskaia, Inorg. Chem., 2008, 44, 622–627 CAS.
- K. Eda, J. Solid State Chem., 1992, 98, 350–357 CrossRef CAS.
- Y. Leung, P. Wong, M. Zhou, K. Mitchell and K. Smith, Appl. Surf. Sci., 1998, 136, 178–188 CrossRef CAS.
- G.-T. Kim, T.-K. Park, H. Chung, Y.-T. Kim, M.-H. Kwon and J.-G. Choi, Appl. Surf. Sci., 1999, 152, 35–43 CrossRef CAS.
- D. Ruan, R. Lin, K. Jiang, X. Yu, Y. Zhu, Y. Fu, Z. Wang, H. Yan and W. Mai, ACS Appl. Mater. Interfaces, 2017, 9, 29699 CrossRef CAS PubMed.
- T. A. Patterson, J. C. Carver, D. E. Leyden and D. M. Hercules, J. Phys. Chem. A, 1976, 80, 1700–1708 CrossRef CAS.
- R. Nadimicherla, W. Chen and X. Guo, Mater. Res. Bull., 2015, 66, 140–146 CrossRef CAS.
- V. Kumar, X. Wang and P. S. Lee, CrystEngComm, 2013, 15, 7663–7669 RSC.
- A. S. Etman, H. N. Abdelhamid, Y. Yuan, L. Wang, X. Zou and J. Sun, ACS Omega, 2018, 3, 2193–2201 CrossRef CAS.
- J. Swiatowska-Mrowiecka, S. de Diesbach, V. Maurice, S. Zanna, L. Klein, E. Briand, I. Vickridge and P. Marcus, J. Mater. Chem. C, 2008, 112, 11050–11058 CAS.
- H. Zhang, Y. Wang, E. R. Fachini and C. R. Cabrera, ECS Solid State Lett., 1999, 2, 437–439 CrossRef CAS.
- T. Barbara, G. Gammie, J. W. Lyding and J. Jonas, J. Solid State Chem., 1988, 75, 183–187 CrossRef CAS.
- B. Hu, L. Mai, W. Chen and F. Yang, ACS Nano, 2009, 3, 478–482 CrossRef CAS PubMed.
- F. Haque, T. Daeneke, K. Kalantar-zadeh and J. Z. Ou, Nano-Micro Lett., 2018, 10, 23 CrossRef PubMed.
- M. T. Greiner, L. Chai, M. G. Helander, W. M. Tang and Z. H. Lu, Adv. Funct. Mater., 2012, 22, 4557–4568 CrossRef CAS.
- M. Gong, W. Zhou, M.-C. Tsai, J. Zhou, M. Guan, M.-C. Lin, B. Zhang, Y. Hu, D.-Y. Wang and J. Yang, Nat. Commun., 2014, 5, 4695 CrossRef CAS PubMed.
- Z. Pu, I. S. Amiinu, M. Wang, Y. Yang and S. Mu, Nanoscale, 2016, 8, 8500–8504 RSC.
- J. Tian, Q. Liu, N. Cheng, A. M. Asiri and X. Sun, Angew. Chem., Int. Ed., 2014, 53, 9577–9581 CrossRef CAS PubMed.
- C. W. Li, J. Ciston and M. W. Kanan, Nature, 2014, 508, 504 CrossRef CAS PubMed.
- L. Ma, L. R. L. Ting, V. Molinari, C. Giordano and B. S. Yeo, J. Mater. Chem. A, 2015, 3, 8361–8368 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional details on PESA, XPS analysis, UV-vis spectra, SEM images, equivalent circuits and tables. See DOI: 10.1039/c8ta08330d |
| This journal is © The Royal Society of Chemistry 2019 |