
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Creating gradients of amyloid fibrils from the liquid–liquid interface†
Kathleen Beth
Smith‡
 a,
Miguel Ángel
Fernandez-Rodriguez‡
b,
Lucio
Isa
b and
Raffaele
Mezzenga
*ab
a,
Miguel Ángel
Fernandez-Rodriguez‡
b,
Lucio
Isa
b and
Raffaele
Mezzenga
*ab
aDepartment of Health Sciences and Technology, Swiss Federal Institut of Technology Zürich, Schmelzbergstrasse 9, 8092 Zürich, Switzerland. E-mail: kathleen.smith@hest.ethz.ch; raffaele.mezzenga@hest.ethz.ch
bDepartment of Materials, Swiss Federal Institut of Technology Zürich, Vladimir-Prelog-Weg 1-5/10, 8093, Zürich, Switzerland. E-mail: ma.fernandez@mat.ethz.ch; lucio.isa@mat.ethz.ch
First published on 2nd October 2019
Abstract
We report a method to deposit amyloid fibrils on a substrate creating gradients in orientation and coverage on demand. For this purpose, we adapt a colloidal self-assembly method at liquid–liquid interfaces to deposit amyloid fibrils on a substrate from the water–hexane interface, while simultaneously compressing it. The amyloid fibril layers orient perpendicularly to the compression, ranging from isotropic to nematic distributions. We furthermore observe reproducible transitions from a monolayer to a bilayer and from a bilayer to multilayers with increasing surface pressures. The creation of each new layer is accompanied by a systematic drop in the structural order of the system, which is however regained upon further compression. This method shows great potential for overcoming the thin-film engineering challenges associated with the manipulation of sticky amyloid fibrils, and allows their ex situ visualisation under compression at the fluid–fluid interface, a situation relevant to understand the propagation of amyloid-related diseases, their functional role in biological systems, and their potential for technological applications.
Studying the behaviour of amyloid fibrils at fluid–fluid interfaces is of three-fold importance:1 (i) for certain amyloid-related diseases, such as Alzheimer's, atrial amyloidosis of the heart or type II diabetes mellitus, the accumulation of amyloid fibrils at biological interfaces has been linked to plaque formation;2,3 (ii) functional amyloid fibrils have been shown to be active at interfaces, e.g. Escherichia Coli excretes curli fibrils to colonize inert surfaces;4 (iii) they display great technological potential, e.g. as bio-compatible emulsion stabilizing agents,5 or for drug encapsulation.6,7 Studies have been conducted to explore the morphology of their constituents,1 their growth,8 and their interfacial rheology9,10 at fluid–fluid interfaces. The well-established Langmuir–Schaefer deposition technique is commonly used to probe the interface. It is obtained by horizontally touching an amyloid fibril-laden water–air interface once it has reached a certain surface pressure. Transferring the amyloid fibrils from the interface to solid substrates without disrupting their configuration is, however, a challenging task. Indeed, the fibrils are soft and the process of de-wetting at the water–air meniscus tends to cause aggregation due to capillary forces during drying produced at the receding meniscus. Jordens et al.1 overcame this challenge by introducing an intermediate step of washing the substrate with ethanol in order to lower surface tension during the final drying. This method provides a snapshot of the interface, but lacks a direct visualisation of the dynamics of the compression and crowding of the amyloid fibrils confined at the interface. To overcome both these challenges, the colloidal self-assembly at liquid–liquid interfaces (SALI) method shows great potential.11 In this protocol, the interface used is water–hexane, where the hexane provides a low-viscosity and high-volatility medium that reduces most of the drying effects; hexane evaporates faster than the speed at which material is deposited, so that the substrate is immediately dried as it finally crosses the hexane-air interface in a water–oil Langmuir trough. Therefore, this protocol enables the deposition of amyloid fibrils confined at the water–hexane interface on a silicon substrate, while simultaneously compressing it and maintaining their configuration at the interface (see Fig. 1). The deposited patterns thus allow the ex situ visualization of the interface as it undergoes compression by atomic force microscopy (AFM), providing useful information on the structure formation of amyloid fibril monolayers at fluid–fluid interfaces.
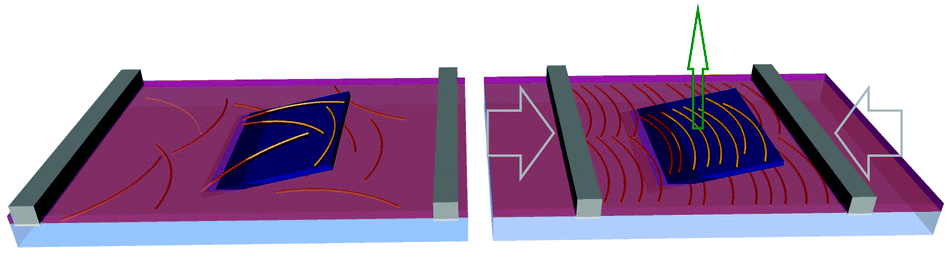 | ||
| Fig. 1 Schematic of the experimental setup where an amyloid fibril solution is deposited at the water–hexane interface. Pulling a tilted (30°) silicon substrate through the interface while simultaneously compressing it, allows transfer of a density gradient of amyloid fibrils onto the substrate. The gray arrows depict the direction of the monolayer compression and the green arrow the upward lifting of the substrate. | ||
This method shows additional potential from a technological point of view, as it can be used to deposit thin films and/or scaffolds of amyloid fibrils on a substrate. Indeed, thin films of amyloid fibrils have already been shown to display interesting cell adsorption properties.12,13 So far, these have been observed on films of a single morphology. However, it has been discussed that obtaining a gradient on a single substrate further enables the study of adhesion properties as a function of the film orientation or even the number of multilayers.14,15 To this effect, our method shows great promise. Indeed, unlike their native folded protein counterparts, for which gradients can be induced by adsorbing them to previously modified substrates,14,16,17 fully-formed amyloid fibrils are significantly harder to manipulate: because of their strong adhesion properties,29 they become unresponsive to such modified substrates. Therefore, the deposition of amyloid fibrils from a fluid interface, where they can be externally manipulated at the same time as they are deposited on a substrate, constitutes a breakthrough in their manipulation. To this end, we adapted the SALI method to deposit betalactoglobulin (BLG) amyloid fibrils from the liquid–liquid interface in a single-step process, thus highlighting the potential of this protocol to address both the continuous compression and crowding of amyloid fibrils at the interface and the creation of amyloid fibril thin-film gradients.
The SALI method has been fully described elsewhere.11,19,20 Briefly, a MilliQ water–hexane (Sigma Aldrich, 99%) interface is formed in a custom-made KSV5000 Langmuir trough. A 1 × 2 cm2 silicon wafer (Siltronix, P-Boron doped, single side polished), thoroughly cleaned by ultrasonication in toluene (Fluka Analytical, 99.7%), isopropanol (Fisher Chemical, 99.97%) and MilliQ water for 15 min each, is held at an angle of 30° and positioned below the interface, with its top edge crossing it. A freshly dialyzed 0.2 wt% solution of BLG amyloid fibrils, prepared according to a previously reported protocol,21 is then mixed with ethanol (Sigma Aldrich, 99%) at a ratio 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Ethanol is chosen as a spreading agent as it is known to have no short-term effects on BLG amyloid fibrils.1,22 Hexane is also known to have no significant effects on BLG proteins,23 thus constituting an appropriate choice for the non-polar phase. Using a Hamilton 10 μl microsyringe, the desired quantity of mixture is deposited directly at the interface and left to equilibrate for 10 minutes. The pressure at the interface is measured using a Wihelmy plate. Then, the substrate is lifted through the interface at a speed of 0.3 mm min−1, while simultaneously compressing the interface (speed of the Delrin barriers: 2.3 mm min−1). Therefore, fibrils at different pressures and area fractions at the water/hexane interface are deposited at different positions on the substrate along the axis perpendicular to the compression direction. Finally, the compression is stopped before the substrate fully crosses the interface. This creates a sudden drop in the pressure and a subsequent change in fibril concentration on the substrate, allowing us to correlate the end pressure with a specific position on the sample and thus obtain the surface pressure as a function of position along the substrate. Using this method we could correlate the AFM images to the pressure curve with an error of ±500 μm. The substrates were imaged by AFM (Bruker FastScan) in soft tapping mode (Bruker FastScan-A cantilevers, resonance frequency: 1400 kHz, spring constant: 17 N m−1). Images of 33 μm and 3072 pixels were taken every mm along the long axis of the sample. Such large scans were chosen in order to have the largest overview possible of the coverage and orientation of the amyloid fibril layers.
:1. Ethanol is chosen as a spreading agent as it is known to have no short-term effects on BLG amyloid fibrils.1,22 Hexane is also known to have no significant effects on BLG proteins,23 thus constituting an appropriate choice for the non-polar phase. Using a Hamilton 10 μl microsyringe, the desired quantity of mixture is deposited directly at the interface and left to equilibrate for 10 minutes. The pressure at the interface is measured using a Wihelmy plate. Then, the substrate is lifted through the interface at a speed of 0.3 mm min−1, while simultaneously compressing the interface (speed of the Delrin barriers: 2.3 mm min−1). Therefore, fibrils at different pressures and area fractions at the water/hexane interface are deposited at different positions on the substrate along the axis perpendicular to the compression direction. Finally, the compression is stopped before the substrate fully crosses the interface. This creates a sudden drop in the pressure and a subsequent change in fibril concentration on the substrate, allowing us to correlate the end pressure with a specific position on the sample and thus obtain the surface pressure as a function of position along the substrate. Using this method we could correlate the AFM images to the pressure curve with an error of ±500 μm. The substrates were imaged by AFM (Bruker FastScan) in soft tapping mode (Bruker FastScan-A cantilevers, resonance frequency: 1400 kHz, spring constant: 17 N m−1). Images of 33 μm and 3072 pixels were taken every mm along the long axis of the sample. Such large scans were chosen in order to have the largest overview possible of the coverage and orientation of the amyloid fibril layers.
The resulting compression curves and selected ex situ AFM scans of the resulting gradients deposited on silicon substrates can be seen in Fig. 2 (full AFM images in Fig. S1 (ESI†) and data to identify the drop in Π as reference position in Fig. S2, ESI†). We varied the initial pressure of the amyloid fibrils at the interface by depositing different quantities of the stock solution at the interface. This was necessary in order to span a broad range of surface pressures and resulting fibril morphologies. The compression curves start at a fairly large surface pressure due to the dynamic equilibrium between peptides and fibrils in the stock solution,21,22 which further contribute to the surface pressure.24 At the beginning of the compression curve, i.e. Π ∼ 15 mN m−1, the corresponding AFM image shows a sparse fibril coverage, analogous to an isotropic regime, but with the additional presence of the free peptides surrounding the fibrils, as mentioned before. Then, upon compression, the surface coverage increases and a nematic (i.e. aligned) phase arises, followed by the appearance of multilayers, with a plateau around 40 mN m−1, where a highly disordered multilayer is observed.
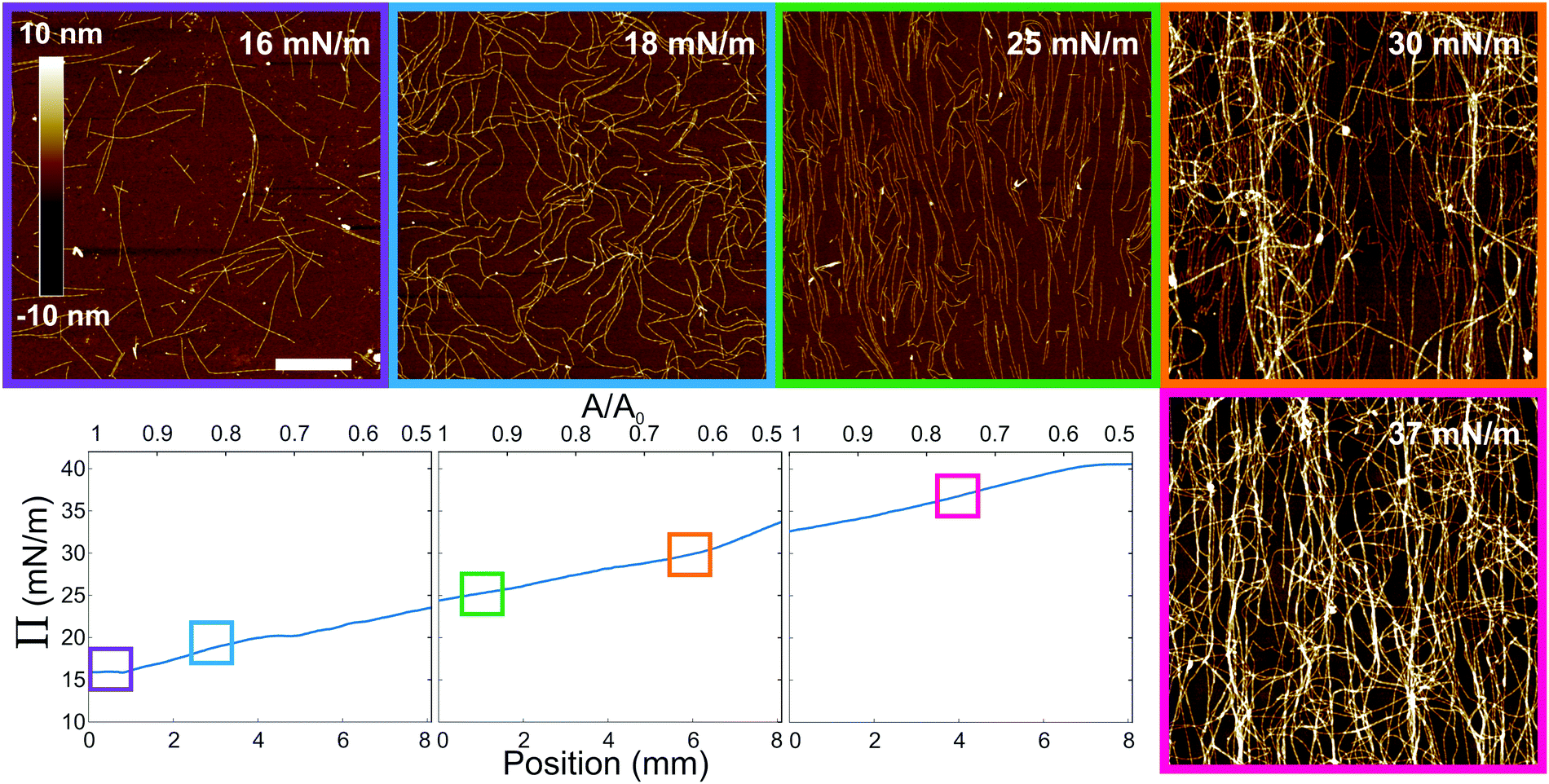 | ||
| Fig. 2 Compression curves of three experiments with increasing initial concentration of amyloid fibrils deposited at the water/hexane interface. The lower x-axis corresponds to the position on the sample after deposition, and top x-axis to Langmuir trough area, where A0 is the starting area. Corresponding cropped AFM images (full images available in Fig. S1, ESI†) are displayed from left to right (scale bar 1 μm), as indicated by the boxes on the compression curves. The compression is perpendicular to the vertical axes of the images. | ||
We further characterized the amyloid fibrils orientation and coverage as a function of surface pressure. As expected for anisotropic particles under compression, we observe a net increase in orientation in the AFM images as the nematic phase arises.26 To quantify this orientation, we used the OrientationJ software.18,27 Examples of two fully processed images can be seen in Fig. 3 (see Fig. S3 (ESI†) for full data set), where the net increase in orientation of the monolayer as the nematic appears under compression is displayed by the orientation-coded colouring of the images and their equivalent orientation distribution. We see that the fibrils vary from isotropic (multi-coloured) to nematic (mono-coloured), aligning perpendicularly to the direction of compression. This visual observation is further accentuated in the orientation distributions, for which the isotropic and nematic case show, respectively, a circular distribution and a narrow elongated distribution around 92°. Thus, the fibrils align and stretch (see Fig. S4, ESI†) as the pressure increases.
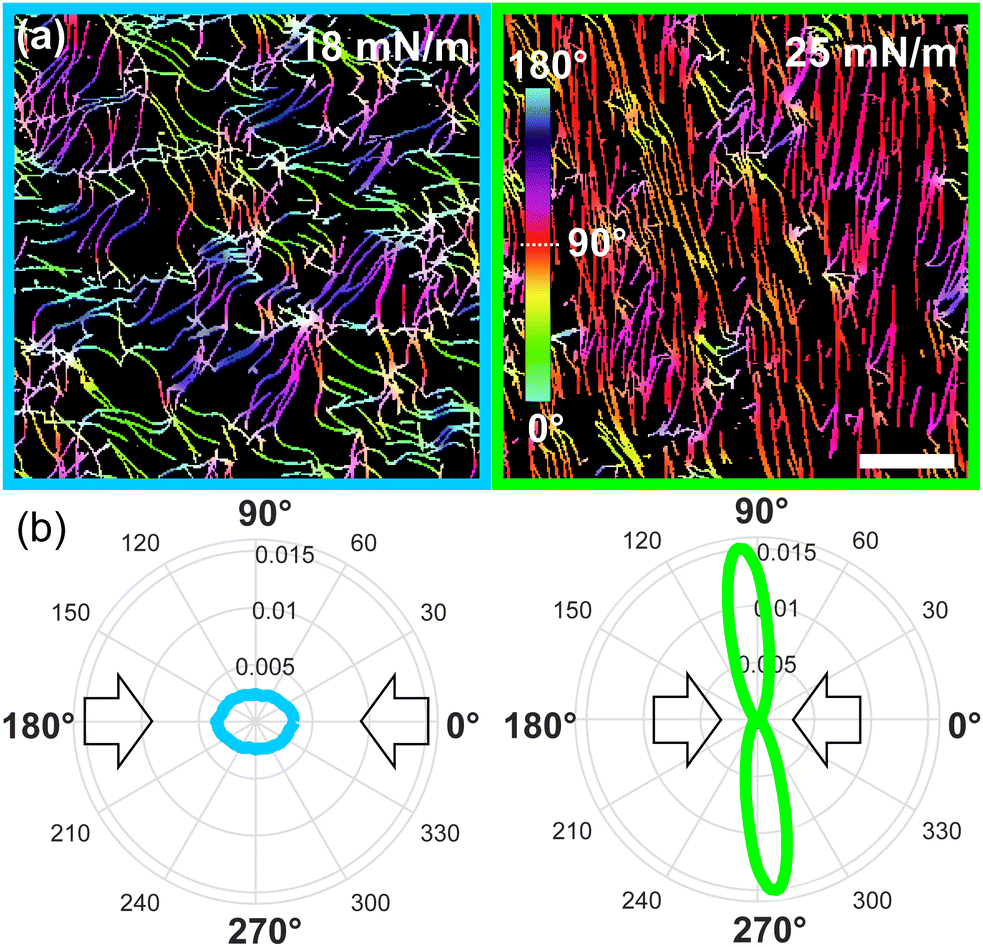 | ||
| Fig. 3 (a) Example of cropped and processed AFM images (scale bar 1 μm), showing randomly distributed (left) and aligned (right) amyloid fibrils deposited on a silicon substrate. An angle from 0 to 180° is attributed to each colour. (b) The corresponding orientation distributions are displayed below each image. The rings and corresponding labels in the radial plots correspond to the normalized frequency of the distribution of angles. | ||
Furthermore, as seen in Fig. 2, we observe an increase in the number of layers as a function of the compression. Therefore, cross-sections of every AFM image were taken, in order to classify them as monolayers, bilayers or multilayers, based on the observed height, Fig. 4(a). Moreover, we extracted a 2D order parameter S2D28 from the orientation distribution of each image. This parameter is in theory 1 when objects are aligned and 0 when randomly distributed. We note, however, that in their work Jordens et al.1 observed a length-scale-dependent behavior of this parameter, leading only to a zero value of the S2D for an infinite box-size. We therefore carried out the statistics on large-scale images, of 33 μm, to minimize this effect. Finally, compiling these parameters into one diagram, we could create a phase diagram of the different layers, the order and the compression curve, Fig. 4(b). From this, we see that Π displays a shoulder at 20 mN m−1 (see inset Fig. 4(b)), corresponding to the fibrils transitioning from an isotropic to a nematic monolayer, as shown by S2D. In the case of bilayers and multilayers, an increase in order is observed at around 28 mN m−1 and 35 mN m−1, respectively, although these transitions are not evident from the compression curves.
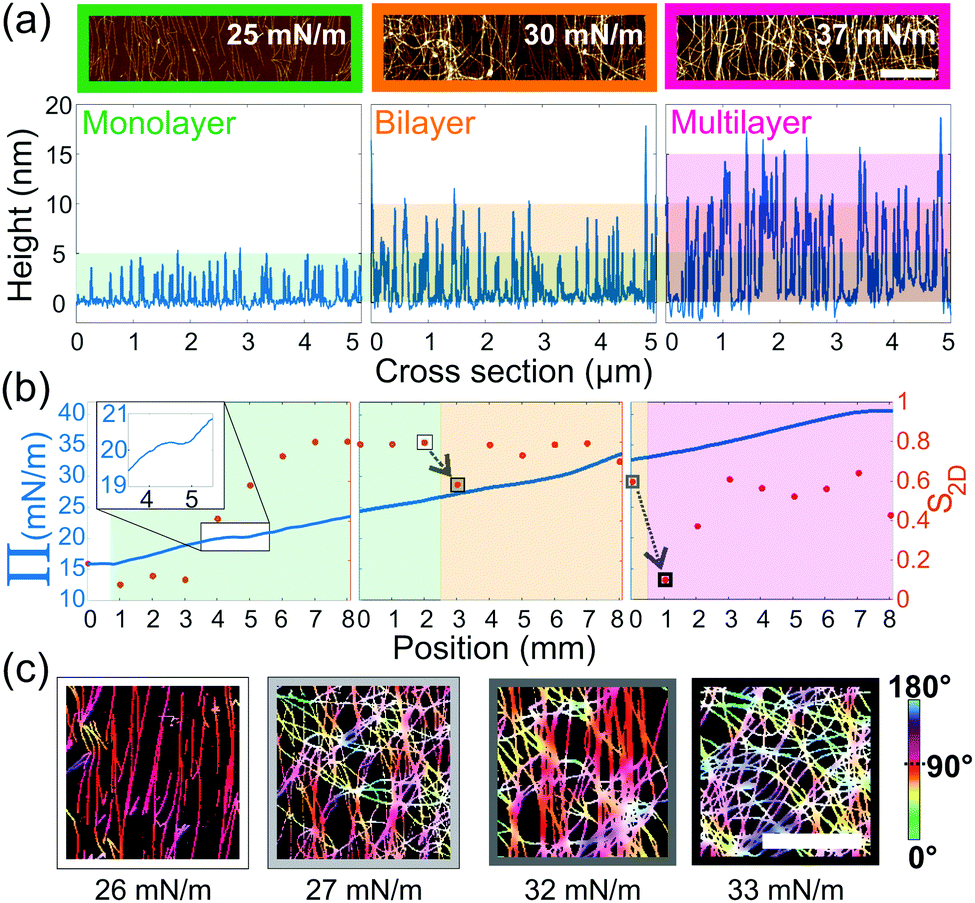 | ||
| Fig. 4 (a) Cropped AFM images (scale bar 1 μm) of a monolayer, bilayer and multilayer (left to right) and their corresponding cross sections (underneath). Each colour band corresponds to a height of 5 nm, the expected average height of the fibrils.25 (b) Phase diagram of the monolayer (green), bilayer (orange) and multilayer (magenta) overlaid on the three compression curves (left y-axis) and corresponding S2D (right y-axis (red)) as a function of sample position. The arrows highlight the sudden drop in orientation at the beginning of a new layer. Inset in first panel is a zoom of the shoulder marking the transition from isotropic to nematic. (c) Cropped and processed AFM images (scale bar 1 μm) corresponding the highlighted S2D points in (b). The color coding indicates the order of the system. | ||
In addition, we observe systematically that the creation of a new layer of fibrils, costs the system order. This happens at both the monolayer-to-bilayer and the bilayer-to-multilayer transition, as can be further seen in the corresponding AFM images, Fig. 4(c). When further continuing the compression, the order of the system is regained until a new layer is formed. The creation of a bilayer and of a multilayer happens at roughly 26.5 ± 0.5 mN m−1 and 33 ± 0.5 mN m−1, respectively. Although the pressures at which we observe these transitions were reproducible for each sample (see Fig. S5, ESI†), they are sample and sample-age-dependent. Nonetheless, the compression curves show the same transitions and demonstrate the potential of this technique to produce gradients of deposited amyloid fibrils with different degrees of ordering, from isotropic to nematic order, and surface coverage.
In conclusion, this method shows great potential for manipulating amyloid fibrils from a thin-film perspective, with potential applications for biological scaffolds, and surface functionalization and bio-activation. Indeed, this process overcomes the problem of how to manipulate or influence sticky amyloid fibrils, by controlling their distribution before deposition on a substrate. In addition, our method can also maintain a specific morphology by keeping the pressure constant, and the orientation of the fibrils can be changed by orienting the sample at an angle to the compression (Fig. S6 and S7, ESI†). This technique can further help expand our basic understanding of amyloid fibril layer formation and aggregation at interfaces, as it is relevant for amyloid-fibril-related diseases, where aggregation processes occur at interfaces. It can also contribute to elucidate their functional roles in biological systems where, for instance, their interfacial activity promotes surface colonization by bacteria excreting such fibrils. We therefore expect that our results will be of interest for a broad community of scientists interested in studying and manipulating fibrils and other large anisotropic macromolecules in controlled conditions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Dr Jozef Adamcik for his assistance in taking AFM images and insightful conversations, as well as Pascal Bertsch for scientific discussions. M. A. F. R. and L. I. acknowledge the financial support of the Swiss National Science Foundation grant no. PP00P2 172913/1.Notes and references
- S. Jordens, L. Isa, I. Usov and R. Mezzenga, Nat. Commun., 2013, 4, 1917 CrossRef PubMed.
- D. J. Selkoe, Nature, 2003, 426, 900–904 CrossRef CAS PubMed.
- R. Paparcone, S. W. Cranford and M. J. Buehler, Nanoscale, 2011, 3, 1748–1755 RSC.
- M. R. Chapman, L. S. Robinson, J. S. Pinkner, R. Roth, J. Heuser, M. Hammar, S. Normark and S. J. Hultgren, Science, 2002, 295, 851–855 CrossRef CAS PubMed.
- Z. Gao, J. Zhao, Y. Huang, X. Yao, K. Zhang, Y. Fang, K. Nishinari, G. O. Phillips, F. Jiang and H. Yang, LWT – Food Sci. Technol., 2017, 76, 1–8 CrossRef CAS.
- K. N. P. Humblet-Hua, G. Scheltens, E. van der Linden and L. M. C. Sagis, Food Hydrocolloids, 2011, 25, 569–576 CrossRef CAS.
- E. Ansarifar, F. Shahidi, M. Mohebbi, N. Ramezanian, A. Koocheki and A. Mohamadian, LWT, 2019, 115, 107884 CrossRef CAS.
- S. Campioni, G. Carret, S. Jordens, L. Nicoud, R. Mezzenga and R. Riek, J. Am. Chem. Soc., 2014, 136, 2866–2875 CrossRef CAS PubMed.
- P. A. Rühs, C. Affolter, E. J. Windhab and P. Fischer, J. Rheol., 2013, 57, 1003–1022 CrossRef.
- N.-P. K. Humblet-Hua, E. van der Linden and L. M. C. Sagis, Soft Matter, 2013, 9, 2154–2165 RSC.
- L. Isa, K. Kumar, M. Müller, J. Grolig, M. Textor and E. Reimhult, ACS Nano, 2010, 4, 5665–5670 CrossRef CAS PubMed.
- N. P. Reynolds, K. E. Styan, C. D. Easton, Y. Li, L. Waddington, C. Lara, J. S. Forsythe, R. Mezzenga, P. G. Hartley and B. W. Muir, Biomacromolecules, 2013, 14, 2305–2316 CrossRef CAS PubMed.
- N. P. Reynolds, M. Charnley, R. Mezzenga and P. G. Hartley, Biomacromolecules, 2014, 15, 599–608 CrossRef CAS PubMed.
- J. Genzer and R. R. Bhat, Langmuir, 2008, 24, 2294–2317 CrossRef CAS PubMed.
- S. Erramilli and J. Genzer, Soft Matter, 2019, 15, 4045–4067 RSC.
- G. B. Sigal, M. Mrksich and G. M. Whitesides, J. Am. Chem. Soc., 1998, 120, 3464–3473 CrossRef CAS.
- J. H. Lee and H. B. Lee, J. Biomater. Sci., Polym. Ed., 1993, 4, 467–481 CrossRef CAS PubMed.
- R. Rezakhaniha, A. Agianniotis, J. T. C. Schrauwen, A. Griffa, D. Sage, C. V. C. Bouten, F. N. van de Vosse, M. Unser and N. Stergiopulos, Biomech. Model. Mechanobiol., 2012, 11, 461–473 CrossRef CAS PubMed.
- M. Rey, M. Á. Fernández-Rodríguez, M. Steinacher, L. Scheidegger, K. Geisel, W. Richtering, T. M. Squires and L. Isa, Soft Matter, 2016, 12, 3545–3557 RSC.
- M. Á. Fernández-Rodríguez, R. Elnathan, R. Ditcovski, F. Grillo, G. M. Conley, F. Timpu, A. Rauh, K. Geisel, T. Ellenbogen, R. Grange, F. Scheffold, M. Karg, W. Richtering, N. H. Voelcker and L. Isa, Nanoscale, 2018, 10, 22189–22195 RSC.
- J. M. Jung and R. Mezzenga, Langmuir, 2010, 26, 504–514 CrossRef CAS PubMed.
- S. Jordens, J. Adamcik, I. Amar-Yuli and R. Mezzenga, Biomacromolecules, 2011, 12, 187–193 CrossRef CAS PubMed.
- J. Bergfreund, P. Bertsch, S. Kuster and P. Fischer, Langmuir, 2018, 34, 4929–4936 CrossRef CAS PubMed.
- J.-M. Jung, D. Z. Gunes and R. Mezzenga, Langmuir, 2010, 26, 15366–15375 CrossRef CAS PubMed.
- J. Adamcik, J.-M. Jung, J. Flakowski, P. De Los Rios, G. Dietler and R. Mezzenga, Nat. Nanotechnol., 2010, 5, 423–428 CrossRef CAS PubMed.
- L. Onsager, Ann. N. Y. Acad. Sci., 1949, 51, 627–659 CrossRef CAS.
- Z. Püspöki, M. Storath, D. Sage and M. Unser, Transforms and Operators for Directional Bioimage Analysis: A Survey, Advances in Anatomy, Embryology and Cell Biology, Springer International Publishing, Cham, 2016, pp. 69–93, DOI:10.1007/978-3-319-28549-8_3.
- I. Usov and R. Mezzenga, Macromolecules, 2015, 48, 1269–1280 CrossRef CAS.
- A. S. Mostaert, M. J. Higgins, T. Fukuma, F. Rindi and S. P. Jarvis, J. Biol. Phys., 2006, 32, 393–401 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sm01826c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |