
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSuperlattice assembly by interpolymer complexation†
Nathan
Horst
 a,
Srikanth
Nayak
b,
Wenjie
Wang
c,
Surya
Mallapragada
ab,
David
Vaknin
d and
Alex
Travesset
*ad
a,
Srikanth
Nayak
b,
Wenjie
Wang
c,
Surya
Mallapragada
ab,
David
Vaknin
d and
Alex
Travesset
*ad
aAmes Laboratory, and Iowa State University Department of Materials Science and Engineering, Ames, Iowa 50011, USA. E-mail: trvsst@ameslab.gov
bAmes Laboratory, and Iowa State University Department of Chemical and Biological Engineering, Ames, Iowa 50011, USA
cDivision of Materials Science and Engineering Ames Laboratory, USDOE, Ames, Iowa 50011, USA
dAmes Laboratory, and Iowa State University Department of Physics and Astronomy, Ames, Iowa 50011, USA
First published on 5th November 2019
Abstract
We present a coarse grained model for a system where nanocrystals are functionalized with a polymer that is a hydrogen bond acceptor, such as polyethylene glycol (PEG), and are dispersed in a solution with a polymer whose monomers consist of a hydrogen bond donor, such as polyacrylic acid (PAA) at low pH (interpolymer complexation). We determine the minimum concentration of the polymer donor to induce aggregation and the structure and dynamics of the induced (fcc) superlattice. Our results are compared to previous and new experiments.
1 Introduction
Nanocrystals (NCs) are usually functionalized with organic or inorganic molecules, the ligands, which, by a number of different strategies control the NC assembly into structures with different degrees of ordering and physical properties. In this way, sophisticated NC arrangements displaying long-range order, superlattices (SLs), have been reported.1 Most commonly, ligands are short molecules such as hydrocarbon surfactants, but many examples also exist where they are polymers such as polystyrene, polyethylene glycol (PEG) or poly(N-isopropylacrylamide).2–6 DNA is also a common ligand,7,8 although the binding moiety that actually drives self-assembly, the linker, is typically a short oligomer.Interpolymer Complexation (IPC), where two different polymers interact through noncovalent interactions, such as hydrogen bonds,9 has been suggested as a possible strategy to direct SL assembly.10,11 Only very recently,12 however, have the first SLs been reported, consisting of PEG-ylated gold NCs and polyacrylic acid (PAA) in solution at sufficiently low pH such that the latter is electrically neutral. Hereon, we will define a generic interpolymer complex as consisting of generic hydrogen bond acceptor (PA) and donor (PD) polymers. Our model will be such that the PA (=PEG in the experimental system) is grafted to the NC core, as shown in Fig. 1.
 | ||
| Fig. 1 Illustrated example of interpolymer complexation (IPC), where hydrogen bonds drive the complexation of PEG and PAA chains. | ||
In addition to reporting the first successful SLs by IPC, the results in ref. 12 concluded that although crystallization of NCs is initiated by hydrogen bonds, the final equilibrium state is almost solvent free, suggesting that van der Waals forces play a significant role in overall stability. Thus, although both IPC and DNA-mediated assembly are driven by hydrogen bonding, the resultant structures are completely different; while DNA SLs consist of 90% water,7 little to no water is present in the final structure induced by IPC, which more closely resembles SLs obtained from solvent evaporation.1 Hence, the potential advantage of IPC as an assembly strategy is that it shares the (a) versatility of the programmability often associated with DNA, (b) cost-effectiveness and bio-compatibility of polymers and (c) high stability typical of hydrocarbon systems, where SLs retain their structure after being removed from solution and completely dried.
The goal of this study is to develop a rigorous coarse-grained (CG) model that complements experimental observations of IPC-mediated assemblies, such as Fig. 2, answers structural questions not accessible in experiments and predicts new phenomena associated with IPC-mediated self-assembly. It is our expectation that it will become the standard model for the prediction of other general assembly methods involving hydrogen bonding. As it relates to the experiments presented here and from ref. 12, the following questions will be addressed: (1) Is there a critical concentration of PD necessary to bridge NCs and drive assembly? (2) What is the structure and composition of IPCs in the assemblies? and (3) how dynamics of assembly occur; how critical is the exchange of PD bound to the assemblies with the PD in solution in order to reach equilibrium?
 | ||
| Fig. 2 Results from Small Angle X-ray Scattering (SAXS) showing the structure factors for IPC-mediated NC assemblies consisting of AuNP-PEG5k with varying lengths of PAA. Propionic acid (monomer equivalent of PAA), PAA2k, PAA5k show structure factors corresponding to FCC structure. For longer PAA100k the assembly shows short-range correlations with poor crystallinity. The structure factors have been shifted vertically for clarity. | ||
2 Model
We consider a suspension of spherical NCs grafted with PA chains in a θ-solution containing PD chains. The solvent (water) is considered implicitly. Each CG PD and PA bead includes an internal site that mediates donor–acceptor attraction through a Lennard-Jones (LJ) potential, thus modeling hydrogen bond hybridizations by a directional short-ranged interaction, as illustrated in Fig. 3. This model is a natural generalization of a previous DNA model,13 but the representation of the “internal hybridizer” allows for full rotational freedom of each CG bead, such that the polymer chains behave as bona fide CG polymer systems when the hybridization interaction is switched off. | ||
| Fig. 3 Model setup, where complementary sites for hydrogen bonding are internal to the polymer backbone, and the hybridization interaction is mediated by a non-bonded potential between internal hybridizers, εH. | ||
Drawing from our previous experience modeling hydrogen bonding,13 it is clear that a balance must be found between the strength of a hybridization and its lifetime, which allows our system to sample the energy landscape without quenching, while making efficient use of computational resources. To explore this interaction, we first consider a solution of PA and PD chains (without NCs) where we vary the strength of the hybridization interaction, εH as shown in Fig. 4a.
 | ||
| Fig. 4 The effect of varying hybridization strength, (εH) on the (a) hybridization interaction potential, (b) potential of mean force (PMF) between two NCs. Orange lines highlight behavior at εH = 15, the value used in the remainder of our study. The dotted black line indicates the range of the hybridizer monomer potential. | ||
We define a hybridization event as an interaction between complementary hybridizers within 1.5σH, where hybridizers have a diameter σH = 0.5, and all other CG beads in the system have a diameter σAB = 1.0. As expected, there is an increase in the number of hybridization events as εH increases, but additionally, we discover εH has clear correlation with system stoichiometry, such that an excess of one CG monomer species leads to increased hybridization of the other CG monomer, see Fig. S2 in the ESI.†
In fact, we find that for sufficiently high PD concentrations, the average number of hybridizations per CG PA bead exceeds one, with some typical situations discussed in detail in the supporting information (Fig. S3, ESI†). However, we note that each CG bead in our model represents many monomeric units of the polymer, so we interpret these situations as reflecting that many hybridizations have been established among two CG beads.
For the remainder of this work, we consider NCs of radius Rc = 2σAB grafted with NA = 35 PA chains consisting of nA = 10 monomers that serve as acceptors in the hydrogen bond interaction, see Fig. 3. PD chains consist of nD = 5 monomers (h-bond donors), and in light of the results discussed above, where hybridization is dependent on system stoichiometry, all ligand lengths are kept fixed in this study to address the primary goal of characterizing hybridization quality. The relevant parameters varied in this work are εH (hydrogen bonding strength) and PD concentration, expressed as
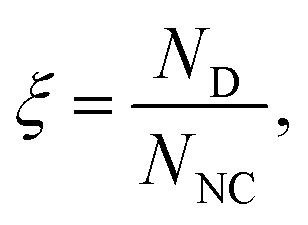 | (1) |
3 Results
3.1 Potential of mean force calculations
We determine the value of (the coarse-grained parameter) hydrogen bonding energy εH, by establishing under what conditions hybridizations result in an attractive interaction between NCs. As shown in Fig. 4b, the PMF is repulsive for εH ≤ 11, indicating that hybridization attraction is not yet strong enough to induce aggregation. Considering all the results compiled in Fig. 4, we need to choose a value of εH where the PMF energy well is sufficiently deep that the NC pair is stable against thermal fluctuations, the number of hybridization events is high enough to induce aggregation, and the directional, short-ranged nature of hybridization interactions is preserved. To this end, all simulations are hereon performed with εH = 15, as marked by orange lines in Fig. 4. We emphasize that this coarse-grained energy represents many simultaneous single hydrogen bonds at the molecular level.
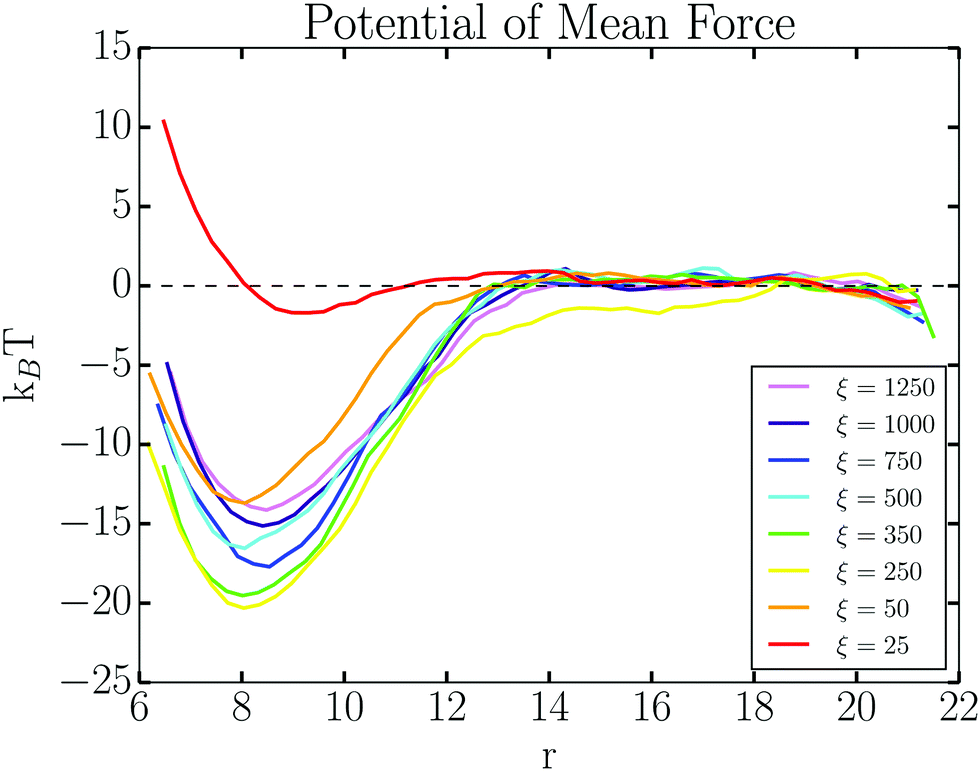 | ||
| Fig. 5 PMF calculations for a pair of NCs with εH = 15 show a non-monotonic dependence on ξ. | ||
 | ||
| Fig. 6 (a) Percentage of CG PA beads involved in hybridization events increases with ξ but (inset) total hybridizations remain constant as NCs approach one another. (b) Cartoon representation shows intra-particle hybridizations transitioning to bridging events (BE) as NCs approach. (c) Internal energy for the entire system decreases with NC separation (r) due to more efficient packing of complementary hybridizers. (d) The number of bridging events (BE) is proportional to the depth of the PMF in Fig. 5. | ||
We query the polymer species present in the corona during a pair interaction, by observing all occupants of a prescribed cylinder with radius σ = 1 along the pair axis (Fig. 7a), and reporting the ratio of the species found within (Fig. 7b). This analysis clearly shows the homogeneity of the PD distribution, as this quantity does not differ between isolated NCs at large r and NCs at the PMF minimum. This effect is also independent of ξ.
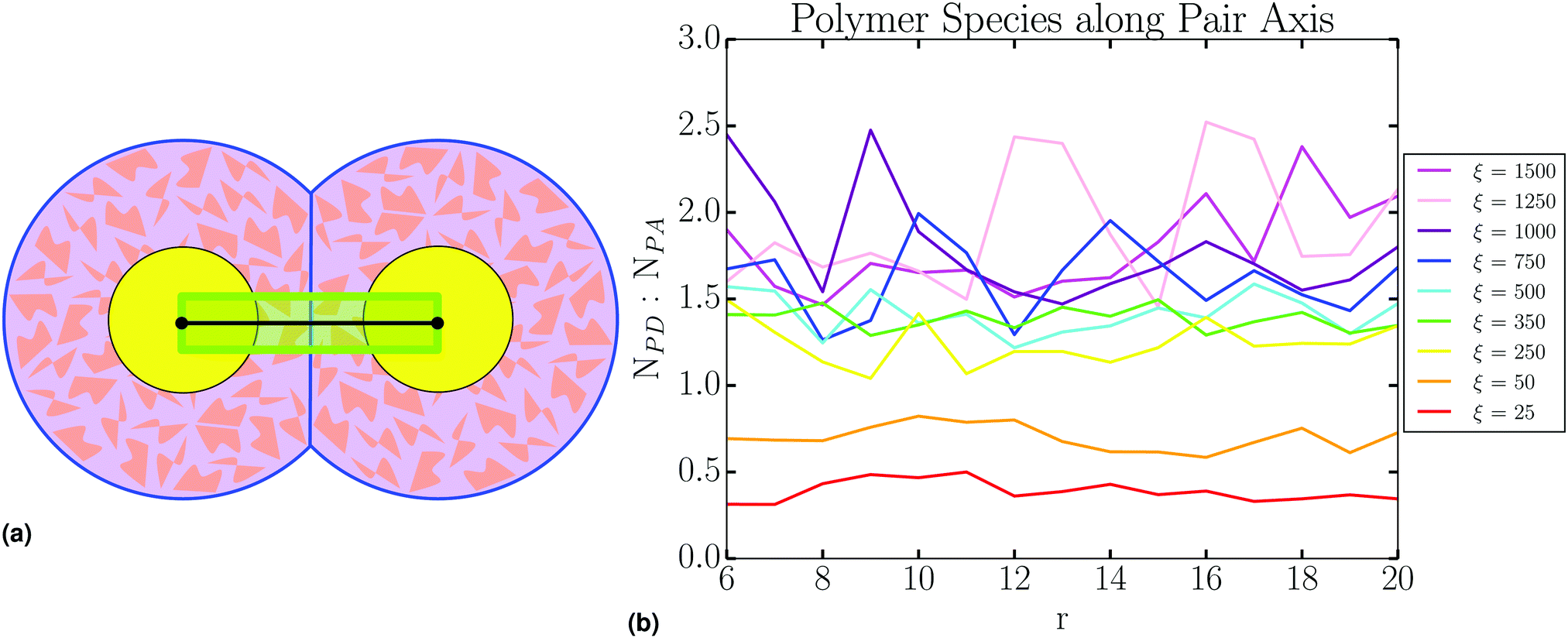 | ||
| Fig. 7 (a) Corona analysis is performed by observing polymer species within a cylinder, shown in lime, along the pair interaction axis. (b) Analysis shows that relative species density along the pair axis remains constant, from large r “isolated particles” to the PMF minimum. | ||
Hybridization lifetimes τH play a very important role; if they exceed the simulation time, the system is quenched into an out of equilibrium state. In Fig. 8a we show hybridization lifetime as a function of ξ at the minimum of the PMF. Short lifetimes at low concentrations imply a drastic reduction in entropy, while the reduction in lifetime at larger concentrations reflects the competition among different PDs to bind to PA. In Fig. 8b we show that even in an isolated NC, hybridizations are highly dynamic and are being exchanged between different regions of the corona, depicted in Fig. 8c. We ran our simulations more than 100 times the mean hybridization lifetime to ensure proper equilibration.
 | ||
| Fig. 8 (a) Mean hybridization lifetime for a pair of complementary hybridizers at the PMF minimum as a function of ξ (in timestep units). The dashed vertical line indicates the value of ξ at which there are the same number of PA and PD beads. (b) Percentage of PD beads exchanged between adjacent regions of the corona of a isolated NC for varying ξ. (c) Graphic detailing the regions of the corona, where inner and outer shells are of equal width, splitting the region from the hydrodynamic radius to the core surface. | ||
An instance where a single PD chain hybridizes to PA chains which are grafted to at least two distinct NCs is defined as a bridging event (BE). These type of hybridizations are very important as they drive attraction between NCs. In Fig. 6d we show the number of BE as a function of NC separation for varying ξ. As expected, the number of BE increases with ξ. Note that, according to Fig. 6a the number of hybridizations per PA is constant, thus, Fig. 6d implies that a number of hybridized PD beads transition from intra-NC hybridizations to BE as the NCs become closer, as shown in Fig. 6b.
3.2 FCC superlattice
The free energy of a fcc SL is obtained by following the same methods as previous work,16 where it is computed as the reversible work of compressing the lattice at a given nearest-neighbor separation aNN (or lattice constant a), see Fig. 10a. The free energy as a function of ξ is shown in Fig. 9a. A minimum clearly developes at sufficiently high PD concentration, indicating a thermodynamically stable fcc SL. In ref. 12 it was shown that this minimum is consistent with the predictions of the OPM17 and OTM18,19 models, where the dimensionless equilibrium separation (rmin by τ = rmin/(2Rc) is given as| τOPM = (1 + 3λν)1/3 | (2) |
 | ||
| Fig. 9 (a) Fcc lattice free energy shows a non-monotonic dependence on ξ. (inset) Osmotic repulsion at large ξ and large aNN is evident for otherwise isolated NCs. (b) OPM prediction agrees well with the experimental IPC results presented above, experimental results in ref. 12, and our simulated lattice systems at varied ξ. | ||
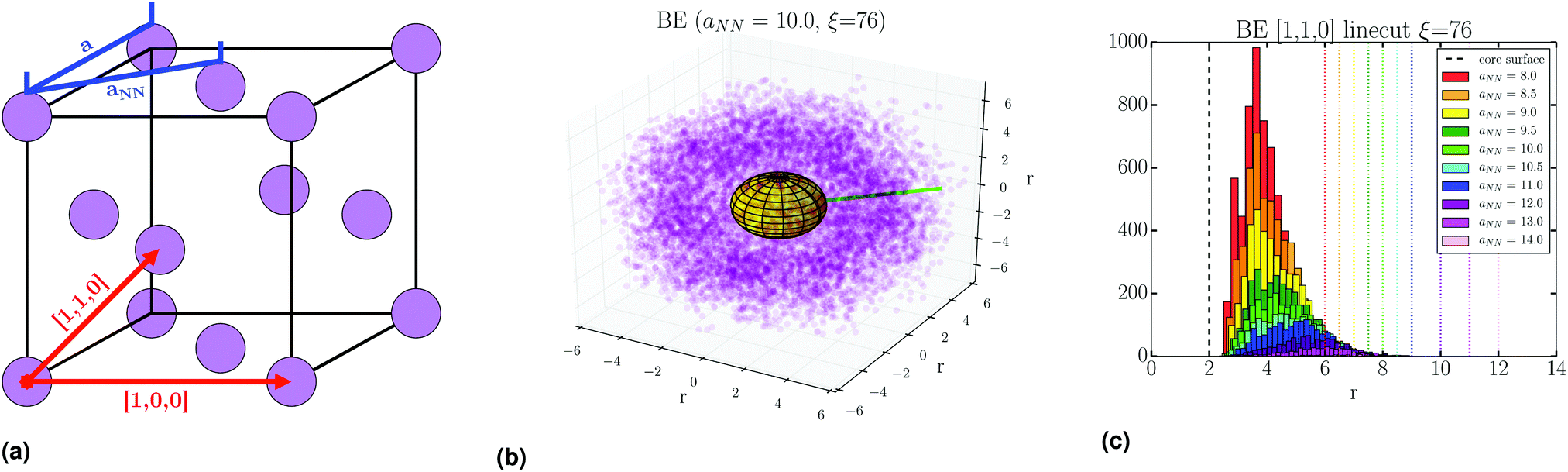 | ||
| Fig. 10 (a) FCC unit cell parameters, including relevant symmetry directions. (b) 3D rendering of mapped tether points involved in BE, at the PMF minimum. (c) Distribution of bridging event tether points along NN [1,1,0] line cut through the mapped data. | ||
The depth of the free energy eventually decreases as a function of ξ, a behaviour that we also observed in the analysis of the PMF. To understand this decrease, it is paramount to first note the difference in the variable ξ for the lattice and pair cases. During a pair interaction there can exist a large excess of PD even after full saturation of the corona. Indeed, to calculate the PMF, ξ must be sufficiently high that absorption into the corona is likely to occur along the interaction axis, as NCs have no local coordination in other directions. Therefore, direct comparisons with the periodic lattice are not meaningful, as local coordination is high and such PD excess cannot exist. However, non-monotonic dependence of the interaction energy on the variable ξ can be attributed to excess PD absorption in both cases, despite the difference in absolute value of ξ. In fact, the effect of PD excess is on full display in the lattice, where repulsion arises from the osmotic pressure exerted by non-hybridized PD, as shown in Fig. 11 and 12, where for small values of ξ, PD is completely absorbed into the corona, but with increasing ξ fills the voids absent of PA and exerts a significant osmotic pressure, also noticeable in the long distance tail of the free energy, Fig. 9a.
 | ||
| Fig. 11 Snapshots of systems for aNN = 18 with varying ξ. PD (orange) is absorbed into the NC corona until fully saturated, then occupies remaining interstitial space. | ||
 | ||
| Fig. 12 Snapshots of systems for aNN = 10 (around free energy minima). PD (orange) is well-dispersed in the SL. | ||
In our previous work,16 we calculated many body effects in the lattice by subtracting from the free energy the collection of calculated pair PMFs. Unfortunately, this cannot be done here as different values of ξ in periodic lattices and pair systems correspond to number densities that are not easily related. Instead, we find that the PMF serves as a case study for an isolated interaction, clearly displaying the relationship between linker-mediated hybridizations and interaction energy, as well as the non-monotonic dependence of linker-mediated interactions on the quantity ξ.
3.3 Lattice hybridizations and BE
Stability of the SL is dependent on the number of BE. As detailed in Fig. S4 (ESI†), we collect all tether points involved in BE by using lattice vectors to translate information from all NCs to a single lattice site. This gives a distribution of BE as shown in Fig. 10b. Then, the location of these BE is analyzed through line cuts along specific directions, an example shown in Fig. 10c, where there is a clear increase in the number of BE as aNN is lowered. From these line cuts, we garner spatial information about BE in the SL. Namely, Fig. 13 shows that tethers involved in BE are located centrally between NCs along the NN [1,1,0] direction, and shifted off-center along the 2NN [1,0,0] axis. This indicates that many CG monomers along the PD chain mediate the NN interaction, while 2NN BE consist of configurations which involve the ends of the PD chains, closer to the configuration we illustrate in Fig. 1.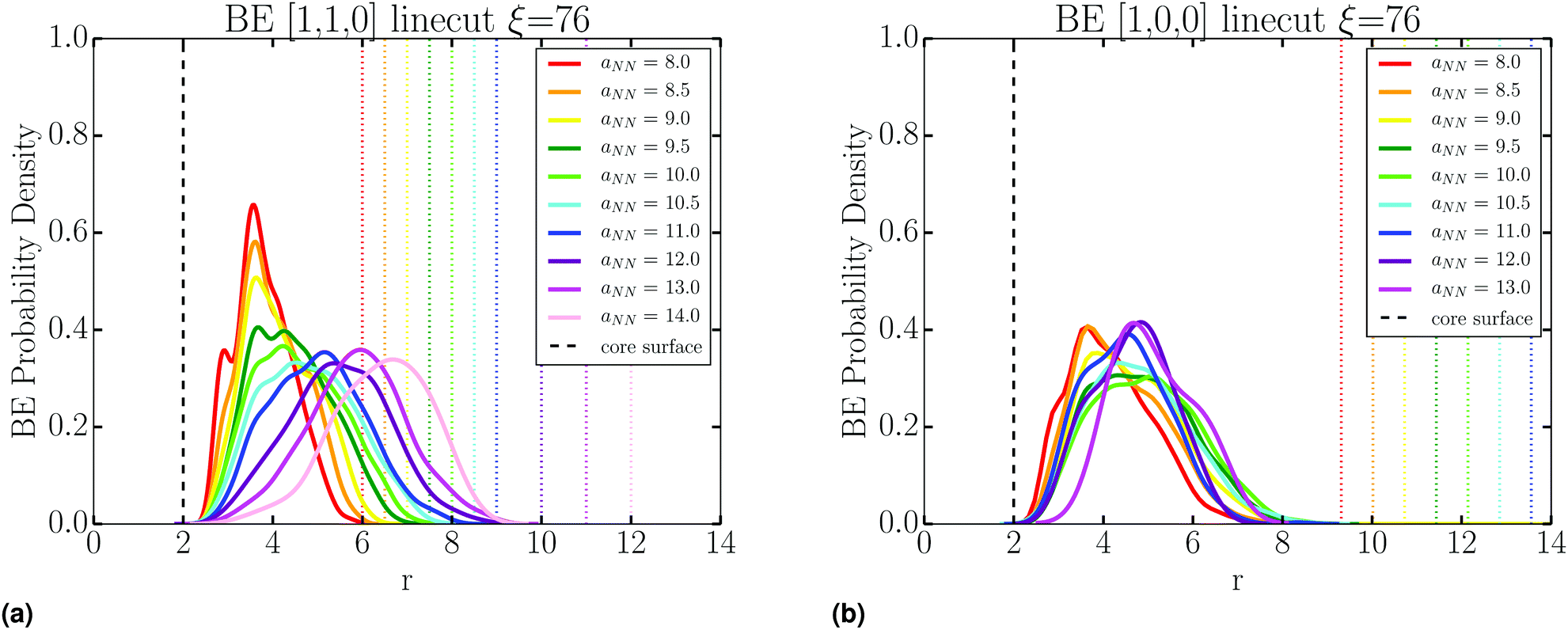 | ||
| Fig. 13 These figures show the probability density of finding a hybridization tether point involved in a bridging event, along NN (a) and 2NN (b) symmetry lines. Dotted lines indicate the location of the core surface at the corresponding value of aNN. | ||
It should be noted that a single PD chain may bridge more than 2 NCs. Therefore, we define the “order” of the bridging event by O = Nteth − 1, where Nteth is the number of NCs tethered to a unique PD chain (Fig. 14b). Here, O = 1 corresponds to PD connecting just two NCs (simple bridge). Unsurprisingly, due to geometric constraints, high ordered bridging events form at small aNN, and are absent at large aNN, as shown in Fig. 14a. High ordered bridging events also seem to come at an entropic cost, as O = 1 bridges make up a higher percentage of the total bridges at large ξ, see Fig. 14c. The configurational characteristics elucidated by our analysis persist regardless of PD concentration, as evident by Fig. S5–S15 in the ESI,† clearly indicating general configurational behavior in the systems studied.
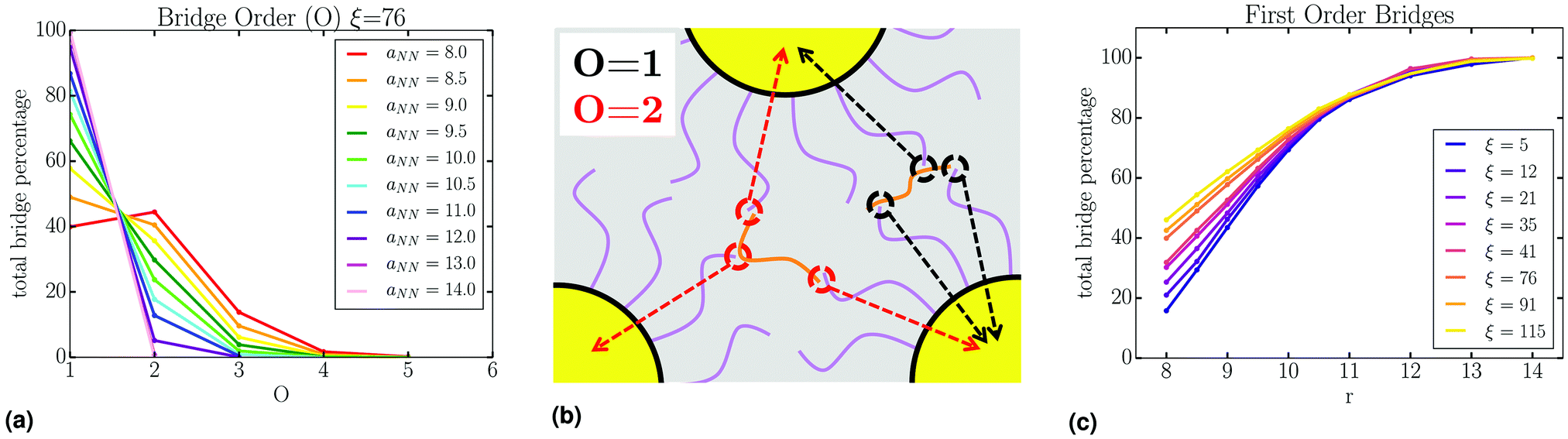 | ||
| Fig. 14 (a) Order analysis of bridging events shows the normalized order of bridges for varying aNN at the minimum of the lattice free energy. (b) Cartoon representation of bridge order, with hybridizations colored by order. (c) Percentage of O = 1 bridging events, where there is a preference for high order bridges at low ξ. | ||
4 Conclusions
We have presented a model that describes assembly by interpolymer complexation (IPC), between grafted polymer acceptors (PA) and linker ligand donors (PD), see Fig. 1. The main results of the paper are:1. There is a PD concentration in the lattice where NC attractions are optimal (ξ ≈ 76); small and large values of ξ prevent attraction through absorption and osmotic pressure, respectively, see Fig. 5 and 9.
2. Interpolymer complexation is driven by characteristic bridging events consisting of PD linkers that bridge two or more NCs in a largely homogeneous distribution and the number of these events is directly related with the strength of NC interaction, see Fig. 6d, 10, 13 and 14.
3. PD is very mobile during IPC, leading to dynamic configurations that can efficiently break and re-establish hydrogen bonds, see Fig. 8.
As shown in Fig. 2, the position of the Bragg peaks is approximately independent of PAA length for molecular weights between 2k and 5k, at least. This implies that the lattice constant is also independent of PAA length, a result that is consistent with our theoretical calculations, as shown in Fig. 9b. In addition, we find very good agreement with the OPM and OTM models,17,20 both from theory and experiment, which reaffirms that IPC, although apparently similar to DNA driven assembly, results in an equilibrium structure closer to solvent evaporation, characterized by the high packing efficiency of the ligands.
We note that agreement of experimental and simulation results with OPM/OTM models, see Fig. 9b, is approximate. There is a noticeable deviation towards larger lattice constants for longer PAA (in experiments) or for higher PD concentrations (simulation). This is indicative of the presence of significant amounts of PAA/PD that make the lattice expand. Experiments for much longer lengths such as 100k, see Fig. 2, show a much larger lattice constant (peak at lower values of q), consistent with this observation, even though the structure itself is more disordered, as evidenced from the width of the Bragg peaks. Varying the length of PD would facilitate more direct comparison of experiment and simulation, but require a more extensive study that is outside the scope of this work.
We have also conducted experiments with the shortest possible PAA chains, i.e. propionic acid, which consists of a single monomer. Rather surprisingly, assembly of a high quality FCC superlattice is observed. The lattice constant, however, is clearly very large, larger than even the 100k PAA chain, see Fig. 2, thus pointing to a different form of assembly, as propionic acid cannot form bridging events, as described in this study. We may speculate that a different type of bridging events, assisted by water molecules may drive assembly. This will require a model where solvent is considered explicitly. In summary, we have provided a predictive coarse-grained model and shown that interpolymer complexation is a very robust strategy for engineering superlattices, with properties intermediate between DNA assembly and those obtained by solvent evaporation.
5 Simulation details
All simulations used HOOMD-Blue21 and modeled the NC cores as rigid bodies.22 Simulations were conducted through the HOODLT23 package, with a Langevin integrator and time step dt = 0.01 in a dimensionless unit system.Non-bonded interactions were treated through Lennard-Jones interactions where most pairs interacted through Sigma repulsions with a cut-off of  . The hybridization interaction was the only extended-range potential in our system, with rcut = 10σH to ensure timely NC aggregation. Intra-chain bonds were treated as harmonic oscillators, with r0 = 0.84.
. The hybridization interaction was the only extended-range potential in our system, with rcut = 10σH to ensure timely NC aggregation. Intra-chain bonds were treated as harmonic oscillators, with r0 = 0.84.
Pair PMF simulations were carried out in similar fashion to our prior work,14 where final data collection was carried out over 106–107 timesteps, depending on system size. Lattice PMF simulations were carried out by the following method: a fully coordinated 2 × 2 × 2 fcc lattice of NCs, held together by harmonic bonds, was initialized at aNN large enough so that no NCs interact through non-bonded potentials. PD chains were then added to the interstitial space of the lattice, at the appropriate concentration. This lattice was gradually reduced in size, maintaining periodicity, and stopping for system equilibration at a range of aNN values for which configurations were saved. These configurations were used to run further simulations, where equilibrium data for that configuration was taken. Using this compression routine gave us confidence that we had avoided metastable minima, and allowed for free diffusion of PD through the lattice at large lattice constant.
6 Experimental methods
Citrate capped gold nanoparticles with 10 nm nominal diameter were purchased from Ted Pella. Poly(ethylene glycol)methyl ether thiol (PEG-SH) with molecular weight, Mn ∼ 5000 was purchased from CreativePEGWorks Inc. Propionic acid, and poly(acrylic acid) with Mn ∼ 2000, 5000, and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 were purchased from Sigma Aldrich. Polyethylene glycol functionalized gold nanoparticles (AuNP-PEG) were obtained by ligand exchange procedure. Briefly, aqueous solution of PEG-SH was added to citrate-capped gold nanoparticle suspension at 1:25000 molar ratio of nanoparticles to polymer. The mixture is kept under constant roto-shaking for two days after which, the AuNP-PEG are separated from unbound PEG-SH by centrifugation. Samples (nanoparticle assemblies) were prepared by mixing AuNP-PEG with appropriate amount of PAA solutions and then the pH was lowered by adding HCl. Final concentration of HCl in the samples was 1 mM. Concentration of PAA is maintained such that all the samples have same concentration of the monomer of 55.6 mM. So, the assemblies were prepared with 2 mM of PAA2k, or 0.8 mM of PAA5k, or 0.04 mM of PAA100k, or 55.6 mM of propionic acid. The mixture was left to stand for two days. Agglomerates/precipitates formed in the vial were then transferred to quartz capillaries for SAXS measurements.
000 were purchased from Sigma Aldrich. Polyethylene glycol functionalized gold nanoparticles (AuNP-PEG) were obtained by ligand exchange procedure. Briefly, aqueous solution of PEG-SH was added to citrate-capped gold nanoparticle suspension at 1:25000 molar ratio of nanoparticles to polymer. The mixture is kept under constant roto-shaking for two days after which, the AuNP-PEG are separated from unbound PEG-SH by centrifugation. Samples (nanoparticle assemblies) were prepared by mixing AuNP-PEG with appropriate amount of PAA solutions and then the pH was lowered by adding HCl. Final concentration of HCl in the samples was 1 mM. Concentration of PAA is maintained such that all the samples have same concentration of the monomer of 55.6 mM. So, the assemblies were prepared with 2 mM of PAA2k, or 0.8 mM of PAA5k, or 0.04 mM of PAA100k, or 55.6 mM of propionic acid. The mixture was left to stand for two days. Agglomerates/precipitates formed in the vial were then transferred to quartz capillaries for SAXS measurements.
Small angle X-ray scattering measurements were carried out at sector 12-ID B of Advanced Photon Source, Argonne National Laboratory. Samples were contained in 2 mm OD quartz capillaries and the measurements were done at room temperature. Measurements were made in transmission mode with photon energy, E = 13.3 keV using 2D Pilatus2m detector. Scattering angle was calibrated with silver behenate powder standard. Standard data reduction techniques were used to obtain the structure factor curves for the samples.12
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research was supported by the U. S. Department of Energy (U. S. DOE), Office of Basic Energy Sciences, Division of Materials Sciences and Engineering. Ames Laboratory is operated for the U. S. DOE by Iowa State University under Contract DE-AC02-07CH11358. This research used resources of the Center for Functional Nanomaterials, which is a U. S. DOE Office of Science Facility, and the Scientific Data and Computing Center, a component of the Computational Science Initiative, at Brookhaven National Laboratory under Contract No. DE-SC0012704. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U. S. DOE Office of Science by Argonne National Laboratory, was supported by the U. S. DOE under Contract No. DE-AC02-06CH11357.Notes and references
- M. A. Boles, M. Engel and D. V. Talapin, Chem. Rev., 2016, 116, 11220–11289 CrossRef CAS PubMed.
- X. Ye, C. Zhu, P. Ercius, S. N. Raja, B. He, M. R. Jones, M. R. Hauwiller, Y. Liu, T. Xu and A. P. Alivisatos, Nat. Commun., 2015, 6, 10052 CrossRef PubMed.
- H. Zhang, W. Wang, S. Mallapragada, A. Travesset and D. Vaknin, Nanoscale, 2017, 9, 164–171 RSC.
- H. Zhang, W. Wang, S. Mallapragada, A. Travesset and D. Vaknin, J. Phys. Chem. C, 2017, 121, 15424–15429 CrossRef CAS.
- G. Zhang, F. H. Stillinger and S. Torquato, Phys. Rev. E, 2017, 96, 1–8 Search PubMed.
- W. Wang, J. J. Lawrence, W. Bu, H. Zhang and D. Vaknin, Langmuir, 2018, 34, 8374–8378 CrossRef CAS PubMed.
- D. Nykypanchuk, M. M. Maye, D. van der Lelie and O. Gang, Nature, 2008, 451, 549–552 CrossRef CAS PubMed.
- S. Y. Park, A. K. R. Lytton-Jean, B. Lee, S. Weigand, G. C. Schatz and C. A. Mirkin, Nature, 2008, 451, 553–556 CrossRef CAS PubMed.
- V. A. Kabanov and I. M. Papisov, Polymer Sci., 1979, 21, 261–307 Search PubMed.
- G. S. Irmukhametova, B. J. Fraser, J. L. Keddie, G. A. Mun and V. V. Khutoryanskiy, Langmuir, 2012, 28, 299–306 CrossRef CAS PubMed.
- Y. Torii, N. Sugimura, H. Mitomo, K. Niikura and K. Ijiro, Langmuir, 2017, 33, 5537–5544 CrossRef CAS PubMed.
- S. Nayak, N. Horst, H. Zhang, W. Wang, S. Mallapragada, A. Travesset and D. Vaknin, J. Phys. Chem. C, 2019, 123, 836–840 CrossRef CAS.
- C. Knorowski, S. Burleigh and A. Travesset, Phys. Rev. Lett., 2011, 106, 215501 CrossRef CAS PubMed.
- C. Waltmann, N. Horst and A. Travesset, ACS Nano, 2017, 11, 11273–11282 CrossRef CAS PubMed.
- S. Kumar, J. M. Rosenberg, D. Bouzida, R. H. Swendsen and P. A. Kollman, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef CAS.
- X. Zha and A. Travesset, J. Phys. Chem. C, 2018, 122, 23153–23164 CrossRef CAS.
- U. Landman and W. D. Luedtke, Faraday Discuss., 2004, 125, 1–22 RSC.
- A. Travesset, Soft Matter, 2017, 13, 147–157 RSC.
- A. Travesset, Phys. Rev. Lett., 2017, 119, 1–5 CrossRef PubMed.
- A. Travesset, ACS Nano, 2017, 11, 5375–5382 CrossRef CAS PubMed.
- J. A. Anderson, C. D. Lorenz and A. Travesset, J. Comput. Phys., 2008, 227, 5342–5359 CrossRef.
- T. D. Nguyen, C. L. Phillips, J. A. Anderson and S. C. Glotzer, Comput. Phys. Commun., 2011, 182, 2307–2313 CrossRef CAS.
- A. Travesset, J. Chem. Phys., 2014, 141, 164501 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sm01659g |
| This journal is © The Royal Society of Chemistry 2019 |