
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStyrene maleic acid copolymer induces pores in biomembranes
Marcella
Orwick Rydmark
a,
Mikkel Killingmoe
Christensen
b,
Elif Senem
Köksal
b,
Ilayda
Kantarci
b,
Kiryl
Kustanovich
c,
Ventsislav
Yantchev
c,
Aldo
Jesorka
 c and
Irep
Gözen
*bcd
c and
Irep
Gözen
*bcd
aDepartment of Biosciences, University of Oslo, 0316 Oslo, Norway
bCentre for Molecular Medicine Norway, Faculty of Medicine, University of Oslo, 0318 Oslo, Norway. E-mail: irep@uio.no
cDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, 412 96 Göteborg, Sweden
dFaculty of Mathematics and Natural Sciences, University of Oslo, 0315 Oslo, Norway
First published on 20th September 2019
Abstract
We investigated the interactions between styrene–maleic acid (SMA) copolymers and phospholipid bilayers, using confocal microscopy and surface acoustic wave resonance (SAR) sensing. For the first time we experimentally observed and followed pore formation by SMA copolymers in intact supported bilayers and unilamellar vesicles, showing that fluorescein, a water-soluble organic compound with a mean diameter of 6.9 Å, can traverse the membrane. Our findings are in agreement with recent theoretical predictions, which suggested that SMA copolymers may insert along the plane of the bilayer to form stable toroidal pores.
Tonge et al. showed in 2001 that styrene maleic acid (SMA) copolymers consisting of hydrophobic styrene and hydrophilic maleic acid monomer units have the ability to solubilize lipids without the necessity for detergents, and in 2006 patented formulations that efficiently transformed lipid bilayers into stable nano-sized bilayer disks at neutral pH.1,2 Subsequently, SMA copolymers were utilized for detergent-free extraction of proteins directly from native cell bilayers into stable bilayer disks, commonly referred to as ‘native nanodiscs’.3,4 Since then, interest in SMA copolymers has increased, and they have been successfully employed in a variety of functional, biophysical, and structural studies with membrane proteins in their native, or native-like lipidic environment.5–8 The elucidation of the nature of the interaction of the SMA copolymer with the bilayers benefits membrane protein isolation and investigation. The SMA copolymer-assisted, detergent-free isolation of membrane proteins preserves native interactions of membrane proteins with lipid species and other proteins, and provides insights into protein structure and function. In this capacity, SMA copolymers are particularly versatile compounds, which have already made a significant contribution to membrane protein research.
To date, most studies examining the physicochemical aspects of lipid–SMA copolymer interactions have focused on the fully formed bilayer discs, where information related to the ordering of lipid acyl chains, the orientation of the styrene group with respect to the bilayer normal, and the approximate width of polymer that encircles the lipid bilayer was obtained.9,10 Several works have also reported on how lipid physiochemical properties (lipid acyl chain length, dynamics, and headgroup type) affect disk formation.11 Still, the formation mechanism of the discs is not known, though possible sequences of steps have been predicted under consideration of initial SMA copolymer–lipid headgroup interactions, intercalation into, and saturation of the membrane, as well as solubilization and formation of discs.12 Very recently, molecular dynamics simulations predicted that SMA copolymers may induce water-filled pores in membranes prior to their solubilization into lipid particles.13 These pores are nanometer-sized and should allow passage of small water-soluble molecules.
We devised a series of experiments to verify the predictions of the theoretical study, and investigated lipid–SMA copolymer interactions on solid-supported planar lipid bilayer patches, and surface-adhered giant unilamellar liposomes. To be able to characterize the interactions at the mesoscale, we applied SMA–copolymer to the supported bilayers and the vesicles in a controlled manner, using an open-space microfluidic device, and observed the resulting transformations under a confocal microscope. We further characterized the interaction of SMA with the supported bilayers by means of a surface acoustic wave resonance (SAR) sensor device, which responds to mass deposition/film formation and accompanied viscosity changes on a surface. These observations pointed to alterations in supported membrane morphology including swelling and pore formation upon exposure to the polymer. In experiments involving surface-adhered giant vesicles with fluorescein encapsulated in the interior volume we directly observed loss of internal contents upon exposure to SMA copolymer solution, while the volume and shape of the spherical vesicle were maintained, indicating that poration precedes membrane solubilization.
Materials and methods
SUV formation and polymer preparation
All lipids were purchased from Avanti Polar Lipids (Alabama, USA), and in all cases, preparations were formed freshly to avoid vesicle fusion and aggregation. Table 1 summarizes the different lipid species used in the experiments with the corresponding polymer concentrations tested on each species. Briefly, lipids dissolved in chloroform (10 mg ml−1) were dispensed into a round-bottomed flask, and chloroform was removed under reduced pressure (∼20 kPa) using a rotary evaporator to produce a dry lipid film. The film was rehydrated in buffer (PBS buffer pH 7.4 containing 5 mM Trisma Base, 30 mM K3PO4, 30 mM KH2PO4, 3 mM MgSO4·7H2O, 0.5 mM Na2EDTA) resulting in a 1 mg ml−1 suspension (dehydration/rehydration method).14,15 50 μl of the desired vesicle stock solutions were diluted (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) with TRIS buffer (10 mM Trisma base, 125 mM NaCl, 1 mM Na2EDTA, pH 7.4), and sonicated for 10 min in an ultrasonic cleaner USC-TH (35 kHz, +30 °C, VWR) and extruded through a 0.1 μm pore size filter 11 times using the Avanti mini extruder. The pre-hydrolyzed form of styrene–maleic anhydride, (Lipodisq® Styrene:Maleic Anhydride Copolymer 3:1, pre-hydrolyzed) with the styrene and maleic acid monomers present in a 3:1 ratio16 was purchased from Sigma and prepared in the same buffers containing the lipid suspensions to achieve the desired final concentration. Other specific details of the polymer are as follows: Mw (weight average): ∼9500 Da; Mn (number average): ∼3050 Da; the polydispersity index is accordingly ∼3.11.
:10) with TRIS buffer (10 mM Trisma base, 125 mM NaCl, 1 mM Na2EDTA, pH 7.4), and sonicated for 10 min in an ultrasonic cleaner USC-TH (35 kHz, +30 °C, VWR) and extruded through a 0.1 μm pore size filter 11 times using the Avanti mini extruder. The pre-hydrolyzed form of styrene–maleic anhydride, (Lipodisq® Styrene:Maleic Anhydride Copolymer 3:1, pre-hydrolyzed) with the styrene and maleic acid monomers present in a 3:1 ratio16 was purchased from Sigma and prepared in the same buffers containing the lipid suspensions to achieve the desired final concentration. Other specific details of the polymer are as follows: Mw (weight average): ∼9500 Da; Mn (number average): ∼3050 Da; the polydispersity index is accordingly ∼3.11.
Lipid film formation and polymer exposure
A schematic of the experimental set-up for forming supported bilayer patches is shown in Fig. 1. A laser scanning confocal microscope (Leica TCS SP8, Leica Microsystems GmbH, Wetzlar, Germany) was used. The supported lipid bilayers were formed in situ by transforming SUVs on a glass substrate (WillCo Wells B.V. Amsterdam, NL), using an open-space microfluidic multichannel pipette17 (Fluicell AB, Sweden). For deposition of the lipids, the microfluidic pipette was positioned using a 3-axis water hydraulic micromanipulator (Narishige, Japan) 10–20 μm above the surface and the recirculation of SUVs (0.1 mg ml−1) was initiated (Fig. 1a). This leads to the adhesion of SUVs onto the solid surface, rupturing and eventual merging of the individual ruptured lipid patches into a circular homogeneous planar bilayer17 (Fig. 1a). Approximately 2 minutes after forming the lipid patch, this time SMA copolymer was applied to the newly formed bilayer via the pipette which in some instances lead to pore formation (Fig. 1b). All experiments were performed at constant room temperature of 18 °C.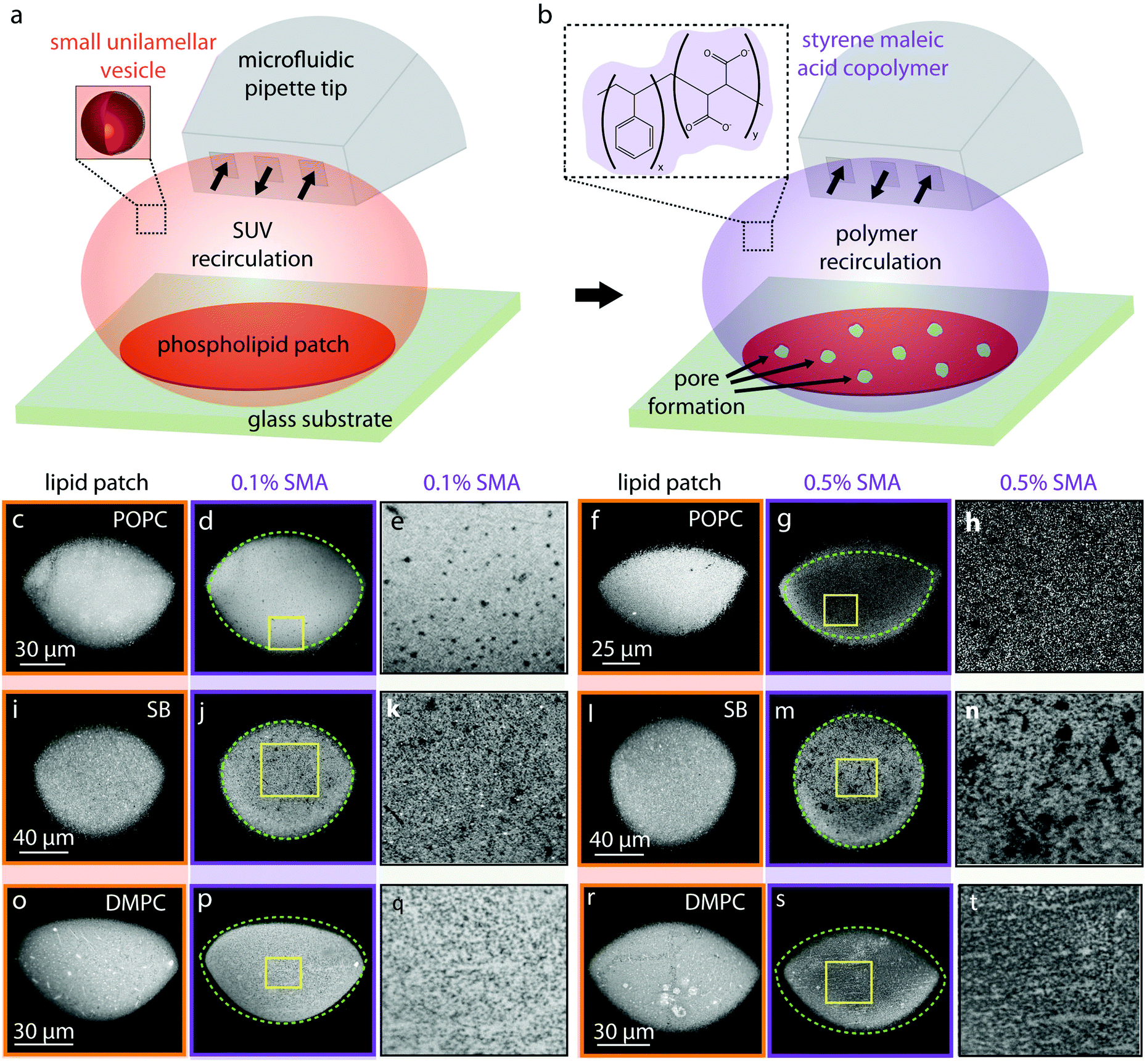 | ||
| Fig. 1 Formation of molecular lipid films and their exposure to SMA copolymer. (a and b) Schematic drawing summarizing the experimental set-up. (a) Lipid particles were recirculated above a glass substrate using a microfluidic pipette which results in formation of a circular lipid bilayer patch. (b) Upon formation of the lipid film, the SMA copolymer solution was applied which in some instances lead to pore formation. The styrene to maleic acid monomer ratio x:y is 3:1. (c–t) Confocal microscopy images of circular lipid bilayer patches from top view. (c and f; i and l; o and s) POPC, soybean, DMPC bilayers, respectively; formed on the surface before polymer exposure. (d, j and p) Lipid patches after exposure to 0.1% SMA copolymer. (e, k and q) magnified versions of yellow frames in (d, j and p). (g, m and s) Lipid patches after exposure to 0.5% SMA copolymer. (h, n and t) magnified versions of yellow frames in (g, m and s). The green dashed-lines representing the contour of the lipid films before polymer exposure have been superimposed over their polymer-exposed versions. | ||
Fluorescence recovery after photobleaching (FRAP)
FRAP experiments were performed on lipid films using the FRAP module in the Leica confocal software, using a 40× 1.25 oil objective and the white light laser at 488 nm, with 100% intensity. A circular region of interest (ROI) of (d = 10 μm) on each lipid patch was defined. The ROIs were sequentially exposed to steps of pre-bleaching (12 s) and bleaching (40 s). The recovery was monitored for 257 s post-bleaching. All experiments were performed at constant room temperature of 18 °C.Surface acoustic resonance measurements
A microfluidics-integrated surface acoustic wave (SAW) sensor in surface acoustic resonance (SAR) configuration, as described in Kustanovich et al.18,19 was used to follow membrane and polymer surface deposition. Fabrication, performance data and measurement environment are specified in the same work.19 Briefly, the microelectromechanical (MEMS) device utilizes surface acoustic wave resonance (SAR), which is conceptually similar to the quartz crystal microbalance (QCM) with respect to resonance frequency downshifts of the resonator upon mass deposition on its surface. The SAW resonator/transducer acts as a combined transmitter and read-out element. The mass load-sensing elements of the SAR sensor are the reflective gratings of the device, and changes in wave characteristics (phase velocity and reflectivity) largely determine the SAW resonance frequency shifts. This allows the simultaneous study of two complementary sensing parameters: the shift of resonance frequency and the shift of the conductance magnitude at resonance, revealing details about structural changes in the sensing layer. The viscoelastic properties of the layer strongly depend on the layer morphology. More specifically, mass deposition results in resonant frequency downshifts, while interactions that alter the viscoelastic properties of the sensing layer can be characterized by changes in the magnitude of the conductance peak, and vice versa. The SAR sensor used here operates at a frequency of ∼186 MHz, with noise levels below 0.5 ppm for resonance frequencies, and below 6 ppm for the conductance peak. These numbers determine the limit of frequency detection (3σ < 1.5 ppm) and the limit of conductance magnitude detection (3σ < 18 ppm). All experiments were performed using a temperature-regulated stage set to 21 °C.The sensor surface is composed of SiO2, which supports the transformation of a lipid suspension into a supported POPC bilayer film similarly to the glass (borosilicate) surfaces used in the confocal microscopy experiments. Although the polymer interaction strengths and kinetics may differ between the two surfaces, the mobility of the lipids largely determines the behavior of the membrane on a solid support. Lipids on glass and SiO2 form bilayers of similar mobility.
Fluorescein encapsulating giant unilamelar vesicle (GUV) formation on solid supported bilayer
Fluorescein-encapsulating GUVs on a solid support were formed as it was described by Köksal et al.20 MLVs containing 50% soybean polar lipid extract, 49% E. coli polar lipid extract and 1% rhodamine-PE were prepared with the dehydration and rehydration method described above. Lipid reservoirs were placed on 84 nm SiO2 deposited glass substrates, fabricated with E-beam and thermal PVD using EvoVac (Ångstrom Engineering), in HEPES buffer containing 10 mM HEPES, 100 mM NaCl and 4 mM CaCl2, 100 μM fluorescein disodium salt, Sigma Aldrich), pH 7.8. After the lipid reservoirs self-spread as a double bilayer and rupture,21 GUVs emerge spontaneously from the surface-adhered nanotubular networks.20 During growth of the vesicles within several hours, fluorescein is encapsulated, which was confirmed by confocal microscopy. Thereafter, the excess dye is removed from the ambient buffer by exchanging it with fluorescein-free HEPES buffer. The open-volume microfluidic device/pipette described above positioned by a 3-axis water hydraulic micromanipulator was used to expose the surface adhered GUVs on a limited area to SMA copolymer solutions of 0.1%, 0.5% and 1% w/v in 10 mM HEPES, 100 mM NaCl pH 7.8. A 20× (NA: 0.75) air objective of the confocal laser scanning microscope was used for acquisition of fluorescence images. The utilized excitation/emission wavelengths for fluorophore imaging were 560/583 nm for rhodamine, and 488/515 nm for fluorescein. Median filtering was applied to fluorescence micrographs by means of the NIH Image-J Software.Transmission electron microscopy (TEM) experiments
Samples were prepared for transmission electron microscopy as described in Asadi et al.22 Briefly, 0.5 mg ml−1 SUVs made from soybean polar lipids, DMPC or POPC, in the presence and absence of SMA copolymer, were adsorbed on 200 mesh formvar coated copper grids for five minutes followed by washing on 6 drops with milliQ H2O. Grids were then incubated for five minutes on ice on drops with methylcellulose-uranylacetate mixture (800 μl 2% methylcellulose and 200 μl 4% uranyl acetate), looped out and dried. Images were recorded at 120 kV on a JEOL 1400plus Transmission Electron Microscope.Results and discussion
Effect of SMA copolymer on molecular lipid films
We investigated the effects of the SMA copolymer by directly dispensing it onto fluorescently labeled molecular lipid films formed on solid supports by direct writing with an open-space microfluidic device, and observing the changes in membrane structure and morphology with a confocal microscope (Fig. 1). This experiment provided visual evidence that the membrane is locally compromised through the formation of defects, the size of which is related to the concentration of the polymer solution used.Briefly, fluorescently labeled small unilamellar vesicles (SUVs) were circulated above a glass substrate using an open space microfluidic device (microfluidic pipette23), which results in adhesion of the SUVs to the surface of the exposed substrate area, followed by rupturing of the SUVs and formation of a planar lipid bilayer patch17,24,25 (Fig. 1a).
Three different lipid species were tested: 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC) (Fig. 1c–h), soybean lipids (Fig. 1i–n), and 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) (Fig. 1o–u). These lipids were chosen based upon their known ability to form fluid, planar bilayers on glass substrates (POPC),17 their importance in biological, biophysical and structural studies (soybean lipids),21,26,27 and their extensive use in SMA-related physicochemical studies, most notably DMPC.9,10,12 Note that POPC and DMPC lipids are zwitterionic. At room temperature, POPC is above its gel phase transition temperature28 and fluid, DMPC is below it and in its gel phase.29 Soybean polar lipid extract is a mixture of various lipids present in soybean lipids e.g. phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylinositol (PI) and phosphatidic acid (PA). Soybean phospholipids reveal no transitions in the range between 3 to 80 °C.30 Membranes composed of soybean lipids possess an overall negative charge, due to the 25 wt% contents of the negatively charged lipids PA and PI.
The average fluorescence intensities of each lipid patch presented in Fig. 1, before and after the application of SMA copolymers are presented in Table 2. For each lipid patch before and after polymer exposure, the mean intensity in a circular region of interest positioned at the center of the membrane has been determined. The 2D lipid membrane is incompressible; the fluorescence intensity of the membrane is therefore proportional to the lipid molecules/lipid material affected.
| Average patch intensity before polymer exposure | Average patch intensity after 0.1% SMA exposure | Average patch intensity before polymer exposure | Average patch intensity after 0.5% SMA exposure | |
|---|---|---|---|---|
| a Each cell shows the mean intensity of the selected region of interest (ROI, top), and the affected area (bottom). The intensity is denoted in levels of an 8bit image (0–255 levels) in the associated selected ROI. The second row contains the area of each circular ROI in μm2. | ||||
| POPC | 122a (AROI: 2895) | 114 (AROI: 2757) | 107 (AROI: 2221) | 38 (AROI: 2333) |
| SB | 100 (AROI: 3410) | 90 (AROI: 3644) | 108 (AROI: 3680) | 76 (AROI: 3842) |
| DMPC | 140 (AROI: 1738) | 130 (AROI: 1615) | 158 (AROI: 1709) | 101 (AROI: 1682) |
The fluidity of the lipid patches, an indicator for the establishment of a continuous biomimetic lipid membrane, was initially determined by means of fluorescence recovery after photobleaching (FRAP) measurements (Fig. 2). The FRAP results for the membranes created from POPC lipid vesicles show quick recovery and confirm fluidity, whereas the lipid patches composed of soybean and DMPC lipids do not fully recover.
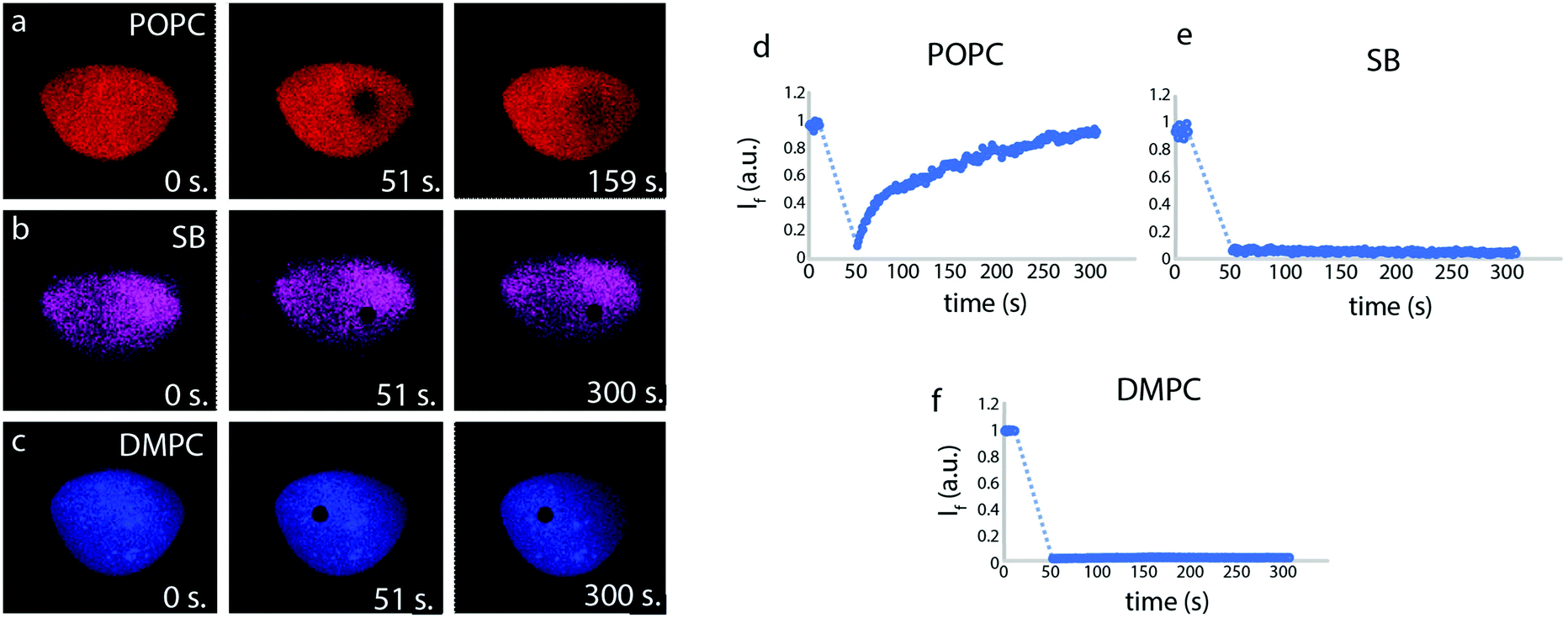 | ||
| Fig. 2 (a–c) Snapshots from the fluorescence recovery after photobleaching (FRAP) experiments performed on solid supported membranes composed of POPC, soybean, and DMPC lipids. (d–f) Plots showing FRAP data of the membranes shown in (a–c): POPC, SB and DMPC lipids, respectively. POPC membranes recover their overall fluorescence over time, whereas SB and DMPC remain completely photo-bleached. | ||
Fig. 1b shows the second part of the experiment schematically, where by switching from lipid particle to SMA copolymer exposure, pores are introduced into the surface-supported membrane. We observed small pores on POPC bilayers after the addition of the 0.1% polymer (Fig. 1d and e). After exposure to 0.5% polymer solution (Fig. 1g and h), the average fluorescence intensity of the membrane drops significantly (65%, Table 2) accompanied by an area expansion of the lipid patch (dashed line in Fig. 1g). The lipid patch in Fig. 1f grows in size (Fig. 1g), suggesting the SMA copolymer strongly acts upon the membrane, and displaces lipids from the initial patch on the solid support.
Fig. 1i and l shows lipid patches created from soybean polar lipid particles. Soybean polar lipids are a complex lipid mixture composed primarily of PC, PE, PI and PA, possessing an overall net negative charge. While it is possible to deposit the lipid particles onto the glass substrate, the circular patches have a grainy appearance (Fig. 1i and l), indicating that formation of a homogeneous lipid bilayer does not fully occur. FRAP confirms this, as no recovery of the photo-bleached regions is observed (Fig. 2). This means that the SUVs remain semi-fused on the glass substrate, but not all of them rupture and form a planar bilayer in the time frame of the experiment. After exposure to the 0.1% (Fig. 1j and k) and 0.5% SMA copolymer (Fig. 1m and n), large uncovered surface areas between 0.5–3.9 μm in size become visible in the lipid films resulting in a 30% decrease in mean fluorescence intensity of the initial lipid patch (Table 2). Since the lipid patch is not continuous and consists of small vesicles adhering to the surface, the SMA copolymer must be reversing the adhesion of these vesicles to the substrate. This fraction of the lipid patch dissolves, leading to larger uncovered regions on the surface (Fig. 1k and n). Since the patch is not fluid as a whole, there are no distinct edge effects as observed with POPC, and the patch size does not increase over time.
In a third series (Fig. 1o–t), we performed the same procedure with DMPC lipid vesicles. The formed patches resulted in bilayers that are also not continuous, as the photo-bleached regions do not recover (Fig. 2). With 0.1% SMA copolymer solution (Fig. 1p and q), the formation of small defects is observed. The poration, i.e., removal of lipid material from the patch, becomes more apparent with 0.5% SMA copolymer solution. The DMPC lipid film shown in Fig. 1o and r visibly decreases in size upon polymer exposure (Fig. 1p and s). Simultaneously, the mean fluorescence intensity of the patch is decreased by 36% (Table 2).
Next, a microfluidics-integrated surface acoustic wave resonance (SAR) sensor, effectively a microscale equivalent of a quartz crystal microscale (QCM)18,19 was used to analyze surface deposition and surface-based transformations of POPC lipids and SMA copolymer (Fig. 3). The SAR sensor measures changes in surface acoustic wave resonance frequency (mass deposition) and conductance (resistance to transfer of acoustic energy) that can be related to lipid film formation and depletion, as well as changes in viscoelastic properties of the deposited material, respectively.
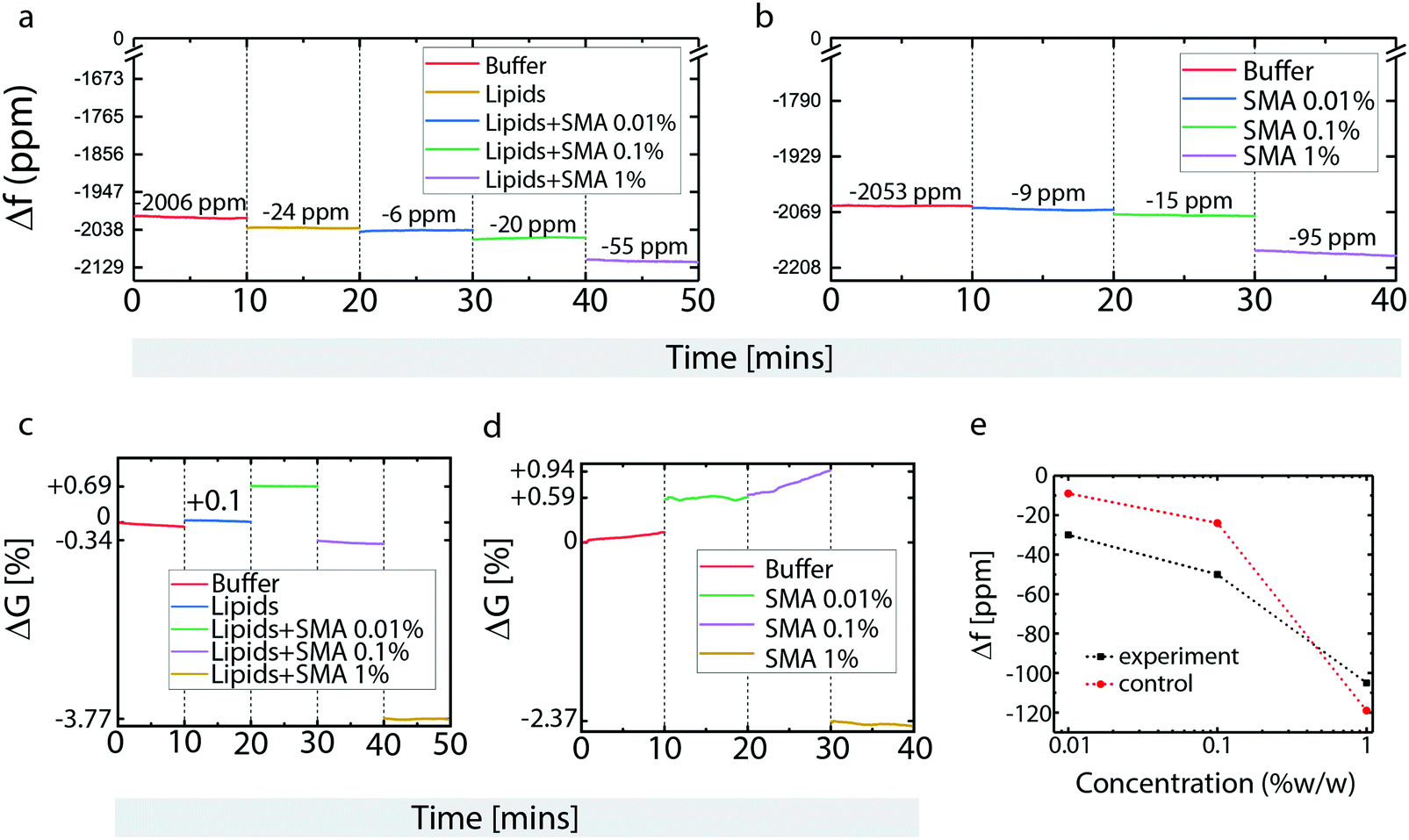 | ||
| Fig. 3 SAR sensor data for POPC lipid and SMA copolymer deposition onto an SiO2 support. (a and b) Time-resolved frequency response (Δf, in parts per million) of the SAR sensor upon sequential loading with lipids and SMA copolymer (a), and SMA copolymer only (b). The colors encode the signals for the individual deposits. (c and d) Time-resolved conductance (ΔG, in percentage) response of the SAR sensor upon sequential loading with lipids and SMA copolymer (c), and SMA copolymer only as control (d). The signals are normalized to the conductance of the lipid coated and bare sensor, respectively, before the exposure to SMA copolymer solution. (e) Calibration curve for the lipid membrane/SMA copolymer interaction (black line) and the reference on the unmodified sensor surface (red line). | ||
Buffer loading resulted in an initial frequency decrease with respect to the unloaded dry device (Fig. 3a). Lipid deposition causes further frequency reduction, due to mass increase from bilayer formation (Fig. 3a). Three different concentrations of SMA copolymer were successively applied next: 0.01%, 0.1% and 1% (Fig. 3a). Compared to the sensor, our microscopy results are less detailed, e.g., the size of the pores forming after 0.1% polymer exposure are quite small, and not directly visible due to the diffraction limit; 0.5% lead to significantly larger pores, and 1% complete disassociation of the membrane. In contrast, the sensor can detect mass changes as small as at 0.01%. In order to obtain a wider view on the concentration range of effective interaction, a concentration below 0.1% was chosen for one of the sensor measurements. The magnitude of frequency downshifts after each SMA copolymer exposure indicates that the polymer deposits with each application, and that the amount of immobilized SMA copolymer on the membrane depends on the SMA copolymer concentration in the supplied solution. This can mean the surface is slowly saturated, or that the polymer builds up on an already saturated surface.
The bare, lipid-free sensor surface was next exposed to the same SMA copolymer concentration series (Fig. 3b). Here, a similar concentration dependent build-up of polymer was observed between 0.01 and 0.1% SMA copolymer concentration, while at 1% w/v SMA copolymer, the frequency down-shift on the bare surface exceeds that on the lipid-coated surface shown in Fig. 3a. This suggests that the lipid bilayer blocks SMA copolymer association with the SiO2 surface, or that the mass lost as a result of membrane solubilization exceeds the mass gained from polymer deposition in the surface areas exposed in the pore regions. The crossover point between the two graphs in Fig. 3e points to a concentration between 0.4 and 0.5% w/v, in which this coverage limitation sets in. To distinguish whether this is the point at which surface saturation occurs or whether it is related to a transformation of surface properties, analysis of the simultaneously recorded conductance data (Fig. 3c and d) must also be taken into account.
Upon lipid deposition, the conductance of the sensor increases, i.e., surface acoustic wave (SAW) energy confinement within the device is enhanced (Fig. 3c). This indicates a change in viscoelastic properties that reduces the losses of SAW energy through the surface into the liquid. This typically occurs upon mass addition under increasing coverage of the surface, or structural changes such as increased rigidity, or healing of surface defects. Deposition of 0.01% SMA copolymer results in a further increase of the conductance, suggesting that the SMA copolymer initially increases membrane rigidity and/or reduces surface defects (Fig. 3c).
Upon deposition of 0.1% w/v SMA copolymer solution, the conductance sharply drops, suggesting structural changes in the lipid bilayer, which is in agreement of the findings on supported lipid films, where lipid solubilization and disruption occurs at this concentration. (Fig. 3c). Addition of 1% w/v SMA copolymer solution results in the conductance dropping even further (Fig. 3c).
Experiments on the bare sensor surfaces show different behavior in one key aspect (Fig. 3d). Between 0.01% and 0.1% w/v SMA copolymer concentrations, the conductance values are unstable and increase steadily over time (Fig. 3d), which is not observed in the presence of a lipid film (Fig. 3d). This indicates that the SMA copolymer becomes more rigid, possibly due to a structural/conformational re-arrangement, or association/entanglement of polymer chains. At 1% w/v the conductance drops to a degree comparable to what was observed on the lipid membrane (Fig. 3d). The similarity of these two readings suggests that the polymer–surface interactions are similar in both cases, with the polymer–surface interaction dominating even in the presence of the (already strongly disrupted) lipid membrane. The increased mass, coupled to conductance reduction, suggests that the polymer film causes viscous loading to the acoustic waves, possibly through swelling and formation of a hydrogel layer. The polymer/water network coupled to the SiO2 coating is sensed as a mass increase within the limits of the SAW penetration depth, while actually the SAW losses through the less rigid hydrogel matrix increase, and are recorded as a conductance decrease.
The sensor measurements confirm the findings from the patch deposition/SMA exposure experiments shown in Fig. 1. In the experiment on POPC film shown in Fig. 1f and g, lipid material is removed from the film, but an area expansion of the lipid bilayer occurs at the same time. This indicates that SMA copolymer is displacing, but not removing lipid material adhered to the surface, which leads to expansion of the patch border outwards. It appears that polymer is adhering to the surface in the patch areas that have been freed from lipid material, i.e., polymer successfully competes with lipids for surface area coverage. Sensor and confocal data are in full agreement. The rough calibration curve depicted in Fig. 3e shows a mass increase on both the lipid film and the bare surface. The relative mass increase on the bare surface exceeds that on the lipid-coated surface substantially at higher polymer concentrations, indicating that the polymer interaction with the surface in this concentration range is dominant in the presence of a surface support. The study on supported membranes also shows that polymer interaction with the membrane are “one-sided”, meaning that open space on the opposite side of the membrane is not necessary for the opening of membrane pores, indirectly supporting the theoretical findings that the polymer largely lines the rim of the generated pores.
Effect of SMA copolymer on free-standing GUV membranes
To confirm that pore formation occurs in free-standing lipid bilayers, and is not primarily driven by the increased gain of free surface energy associated with the displacement of lipids from surface regions by polymer, adhered GUVs composed of soybean polar lipid and E. coli polar lipids (50/50% w/w) encapsulating a fluorescein solution were produced and exposed to SMA copolymer (Fig. 4). In contrast to the solid-supported lipid film experiments, here only a limited fraction of the surface area is adhered to the solid substrate (Fig. 4a). Note that this area is, unlike in a supported membrane, obscured by the vesicle body and thus not accessible to the liquid environment. This allows for observing the lipid membranes in a quasi-suspended manner, where the position of the GUVs is maintained on the support, and polymer effects can be observed in real-time over extended time spans. The GUVs used in the experiments contain fluorescein (mean diameter 6.9 Å) in their internal volume. The encapsulation of fluorescein occurs during the formation of the vesicles in fluorescein-enriched external solution, which is exchanged for dye-free buffer after maturation of the vesicles. Due to the difference in fluorescence emission between the rhodamine-labeled GUVs and encapsulated fluorescein, it is possible to observe both the lipid vesicles and the internalized fluorescein simultaneously. We applied the SMA copolymer to the vesicles via the microfluidic pipette (Fig. 4b) used in the previous experiments on supported membranes. This mode of application in a hydrodynamically confined volume ensures that the polymer is present in the volume around selected vesicles only during exposure, and consistently at the specified concentration. We chose three regions on the solid support populated with GUVs and applied three different concentrations of the SMA copolymer to the GUVs in each region: 0.1% (Fig. 4c–h), 0.5% (Fig. 4i–n) and 1% (Fig. 4o–t). At 0.1% SMA copolymer concentration, no significant changes were observed with respect to fluorescence signal collected from the internalized fluorescein (Fig. 4d–g). The vesicles appeared intact, and the dye remained encapsulated (Fig. 4f and g). The fluorescence intensities along the arrows depicted in Fig. 4e and j, corresponding to the intensities before (yellow color) and after (blue color) exposure to the 0.1% polymer solution, are shown in Fig. 4h, indicating no effective alterations of the membrane. Increasing the concentration to 0.5% did not lead to the disruption of the GUVs, but caused rapid release of internalized fluorescein (Fig. 4j–m). The emission intensities along the arrows depicted in Fig. 4k and m, corresponding to the intensities before (yellow color) and after (blue color) the 0.5% polymer exposure, are shown in Fig. 4n, indicating the presence of water-filled pores, large enough to allow for the passage of fluorescein molecules (≥7 Å). 1% w/v SMA copolymer fully solubilized the GUVs (Fig. 4o–t). Note that the vesicular membranes in Fig. 4m fluoresce in the fluorescein channel. This is due to the slight overlap between the absorption spectra of fluorescein and rhodamine B. The weak absorption of membrane-associated Rho B at 488 nm also causes a minute amount of fluorescence, leading to the weak cross-over signal, i.e., a green ring. During exposure to 1% SMA copolymer we observed drastic shape deformations (Fig. 4t). The peculiar shape of the vesicles, visible in this figure, points towards a single giant pore forming in the membrane during exposure, which will eventually lead to osmotic breakdown of the vesicle, and collapse. Note that the vesicles in this system are in a low tension regime, due to the connection to a large lipid nanotube network on the surface (fluid lipid reservoir). That entails that unstabilized small pores would rapidly contract and close, according to the line tension in the pore edge.31 The collapse of the vesicles indicates that the polymer-lipid interaction has a stabilizing effect on the pore edge; it compensates the line tension and thus keeps the pore open. It can be assumed that the polymer removes lipid material continuously from the edge of the pore, increasing its size, until the vesicle finally folds onto the surface (Fig. 4r and s). Alternatively, smaller pores could be formed in large numbers and continuously merge, reducing the overall pore edge tension. During this process, lipid material could be simultaneously separated from the vesicle membrane (Fig. 6). The observation is in principle in agreement with the prediction of Xue et al.,13 and also supported by the finding that adhered DMPC membrane patches dissolve noticeably from the edges (Fig. 1o, p and r, s). We suggest that the polymer adheres to the membrane, increases its rigidity (which is strongly supported by the increase in sensor conductance at low SMA concentrations), then penetrates the membrane to form defects, where it lines the pore perimeter and dissolves lipid material from the edge, causing pore enlargement to an extent dependent on polymer concentration. The formation of a single large pore is consistent with earlier reports on flat giant unilamellar vesicles in an experimental system which employs Ca2+ to stabilize the pore by pinning.32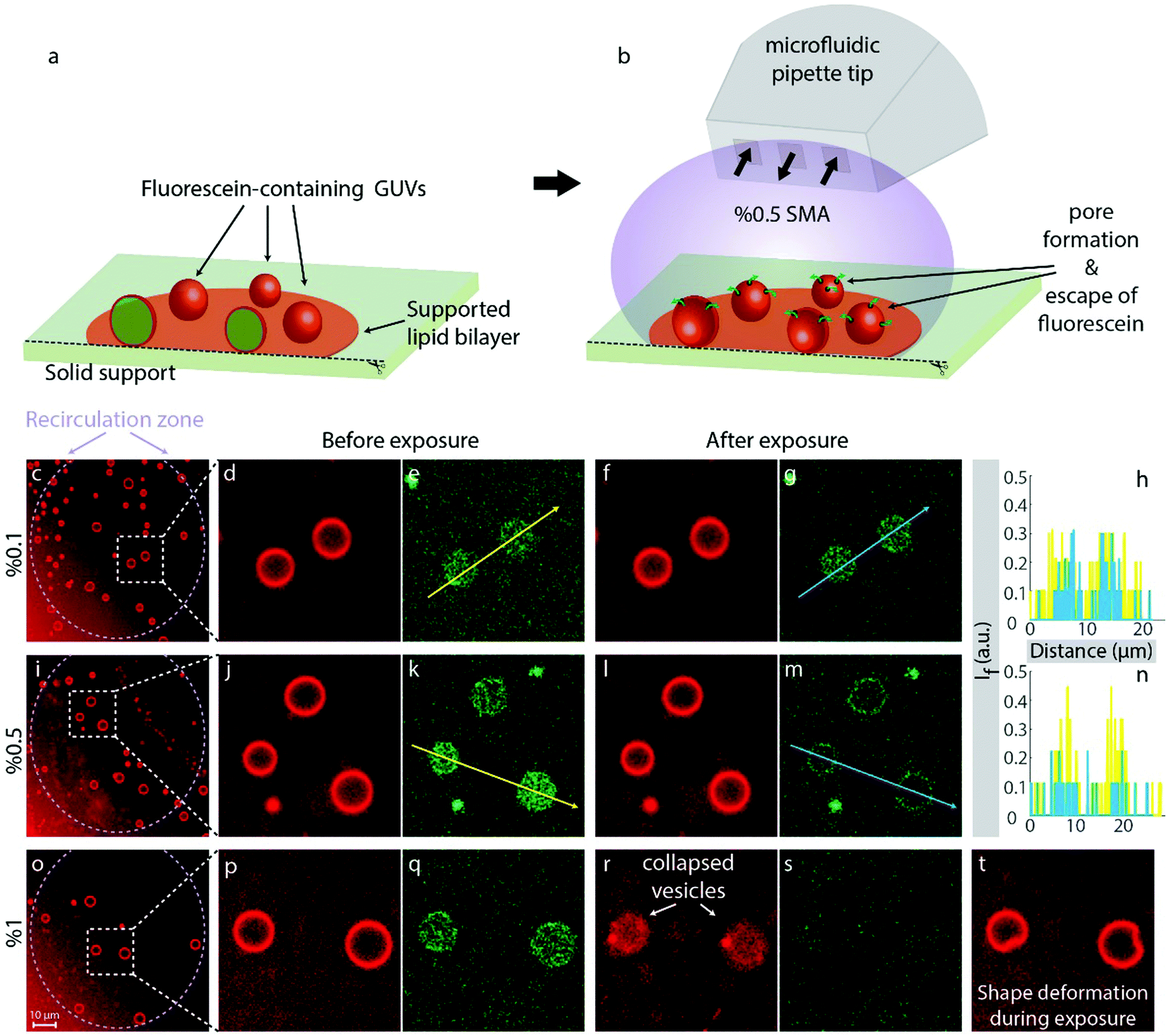 | ||
| Fig. 4 Poration of fluorescein-encapsulating surface-adhered GUVs with SMA copolymer. (a and b) Experimental setup. GUVs are formed on the solid support, and a microfluidic pipette tip is used to add the SMA copolymer. (c–t) Confocal micrographs of the experiment summarized in (a and b). (c, d, i, j, o and p) Three different regions on the lipid patch where the SMA copolymer was applied at varying concentrations: (c–h) 0.1%, (i–n) 0.5% and (o–t) 1% w/v. The plots in (h and n) show the fluorescent intensity of encapsulated fluorescein (arbitrary unit) before (yellow) and after (blue) SMA exposure. (t) shows the drastic shape deformations observed during 1% polymer exposure depicted in (o–s). | ||
Supported membranes vs. spherical membranes (vesicles)
0.1% SMA copolymer creates visible membrane defects in supported membranes, but no noticeable pore formation was observed on the same concentration when applied to spherical containers. 0.5% SMA copolymer porates the vesicle membrane efficiently, as is evident from the loss of internalized contents. Planar supported membranes show a major loss of lipid material upon exposure.According to the SAR sensor measurements, SMA copolymer associates with the membrane, but equally strongly to the bare, lipid-free surface. Between 0.01 and 0.1% SMA copolymer, the conductance measurements, which report on membrane rigidity and general transparency to acoustic energy, indicate a strong change in membrane properties that is associated with a mass increase at the surface. In conjunction with confocal microscopy on supported fluid membranes, the sensor findings suggest that there is a mass increase even though lipid is removed, resulting from interaction of the polymer with the surface. Lipid is removed from the surface (decrease in fluorescence intensity), and simultaneously displaced (increase of patch outer diameter). Our findings on supported membranes clearly indicate that the polymer-induced pore formation does not require the availability of both hydrophilic faces of the membrane, but benefits from the presence of a rigid bottom layer. The presence of the surface appears to have a supporting influence on the impact of the polymer on the membrane. This finding might also be relevant to the cytoskeleton supporting the cell membranes under the influence of the polymer. Studies of SMA interaction on cells with weakened cytoskeleton, e.g. by zeiosis-inducing reagents, could reveal more information on such connection.
Effect of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :1 SMA copolymer on small unilamellar vesicle (SUV) suspensions
:1 SMA copolymer on small unilamellar vesicle (SUV) suspensions
To further characterize polymer-induced morphological changes, negative-stain electron microscopy was performed on a small unilamellar vesicles (SUVs) suspension (Fig. 5). Since the final lipid:polymer ratios in the confocal microscopy experiments are unknown, but the exact concentration of lipid and polymer need to be established in order to only partially solubilize the vesicle membrane, we first recorded turbidity curves (Fig. 5a–c). Turbidity assays are a routine way to determine detergent-solubilization curves for liposomes, and have also previously been used monitor the solubilization of lipids by SMA copolymers.33 In all samples a slight increase in turbidity occurs, which is less obvious for POPC and DMPC (Fig. 5a and c) than for soybean lipids (Fig. 5b), followed by a decrease to baseline levels. In detergent-solubilization curves of liposomes, small changes in turbidity, similar to those seen here, are linked to disrupted bilayer vesicles, rather than more complex worm-like structures that scatter more light.34 The small absorbance increases that are observed along the overall decrease (marked with asterisks) are likely to be due to some unspecified structural intermediates that may form along the solubilization pathway.
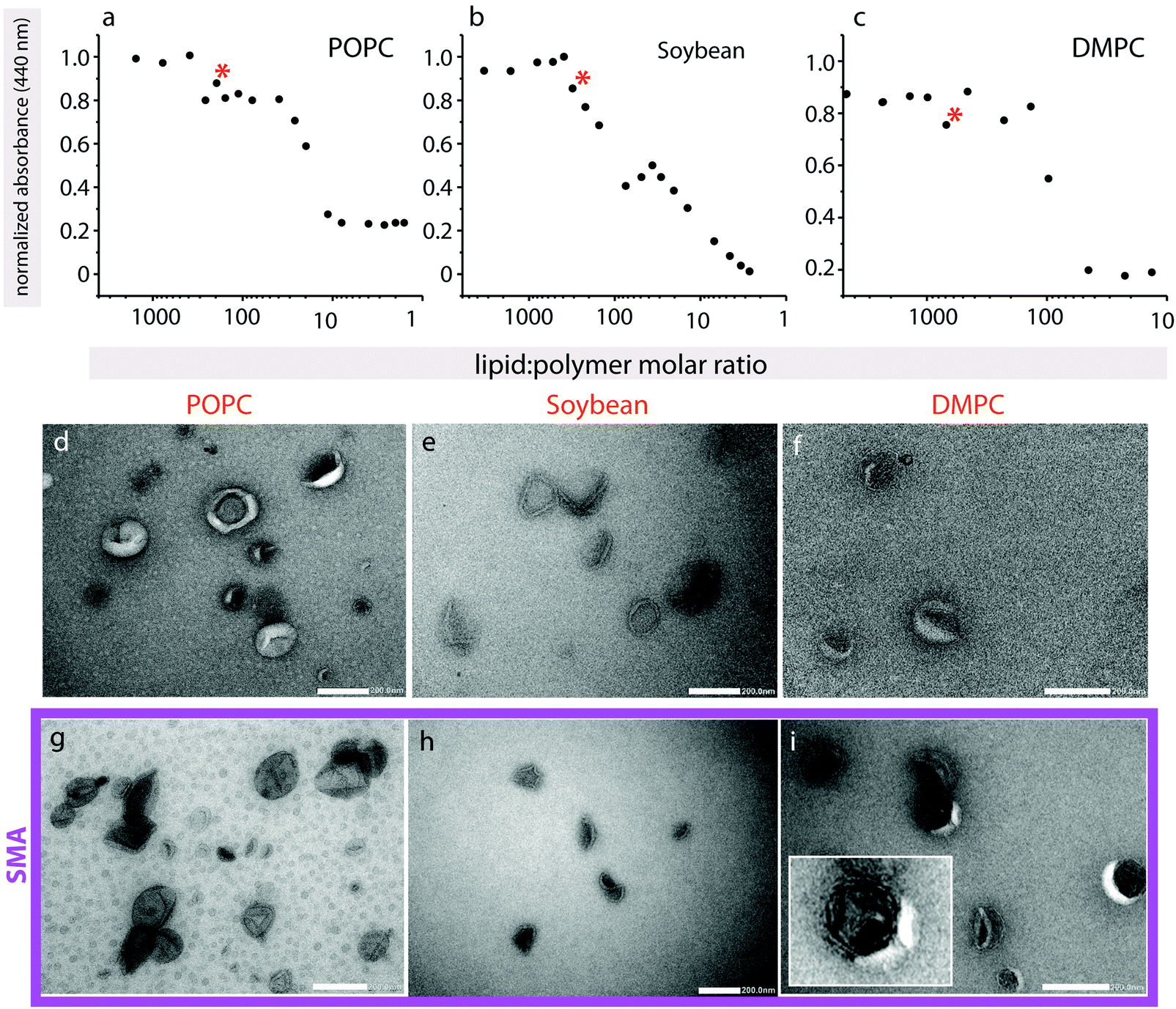 | ||
| Fig. 5 Lipid solubilization. (a–c) Turbidity curves in the presence of SMA copolymer for POPC, Soyben and DMPC lipids. The polymer:lipid ratio that was chosen for EM images is marked with a red asterisk in each plot. (d–i) TEM images showing the vesicles produced from different lipids in the absence (d–f) and presence (g–i) of 3:1 SMA copolymer. Differences in background color are largely attributed to uneven staining, where background in POPC in the presence of SMA copolymer appears to have less stain present than for POPC only. The inset in panel (i) is a magnified image of a multilamellar vesicle. Scale bars: 200 nm. | ||
For TEM, we chose lipid:polymer ratios where we expect partial polymer-induced vesicle disruption of the lipid vesicles, but not their full solubilization. Soybean SUVs in the absence of SMA copolymer appear well-formed (Fig. 5e), with a visible bilayer, and are approximately 70–120 nm in diameter. SMA copolymer addition results in a collapsed structure, where the vesicles display an inward concavity (Fig. 5h). They appear generally still intact, and the bilayer is present. These observations are in agreement with the turbidity assay that also indicates a simple disruption of the bilayer rather than the formation of a more complex lipid–polymer phase. These observations suggest that SMA-induced disruption of the bilayer leads to escape of the liquid within the samples upon dehydration and application of the vacuum, leading to their collapse. This is in excellent agreement with fluorescein encapsulation assays on quasi-suspended GUVs, where at intermediate polymer concentrations the fluorescein is also able to escape the intact vesicle. POPC liposomes without polymer are approximately 70–150 nm in diameter, and are more donut-shaped in appearance (Fig. 5d). The liposomes appear to burst in the presence of polymer (Fig. 5g), and the average SUV size appears somewhat smaller (40–110 nm). DMPC SUVs in the absence of polymer were heterogeneous in size, and many were already collapsed, possibly due to the inclusion of sonication in preparing them, making the observation of SUV morphological changes upon SMA copolymer addition more difficult (Fig. 5f). However, we did observe a significant increase in vesicle multilamellarity (see Fig. 5i inset) that was not observed in the other lipid types (Fig. 5i).
One possible assumption for the pathway to the formation of pores in membranes under the influence of SMA copolymer can be formulated. This involves polymer chains lining the edge of the pores formed (as suggested by Xue et al.), compensating for the edge line tension. It is possible that as the number of pores increase, they may merge to form isolated lipid islands that would break out to form SMA-supported lipid bilayer discs (Fig. 6). SAR sensor data on the supported membranes indeed show that a mass loss occurs at higher polymer concentrations in excess of the mass gain explainable by adhesion of polymer in open pore areas. External data from biomembranes which form lipid discs including membrane proteins,5–8 support the island formation pathway, while the confocal microscopy data also provide strong support for the edge-dissolution pathway.
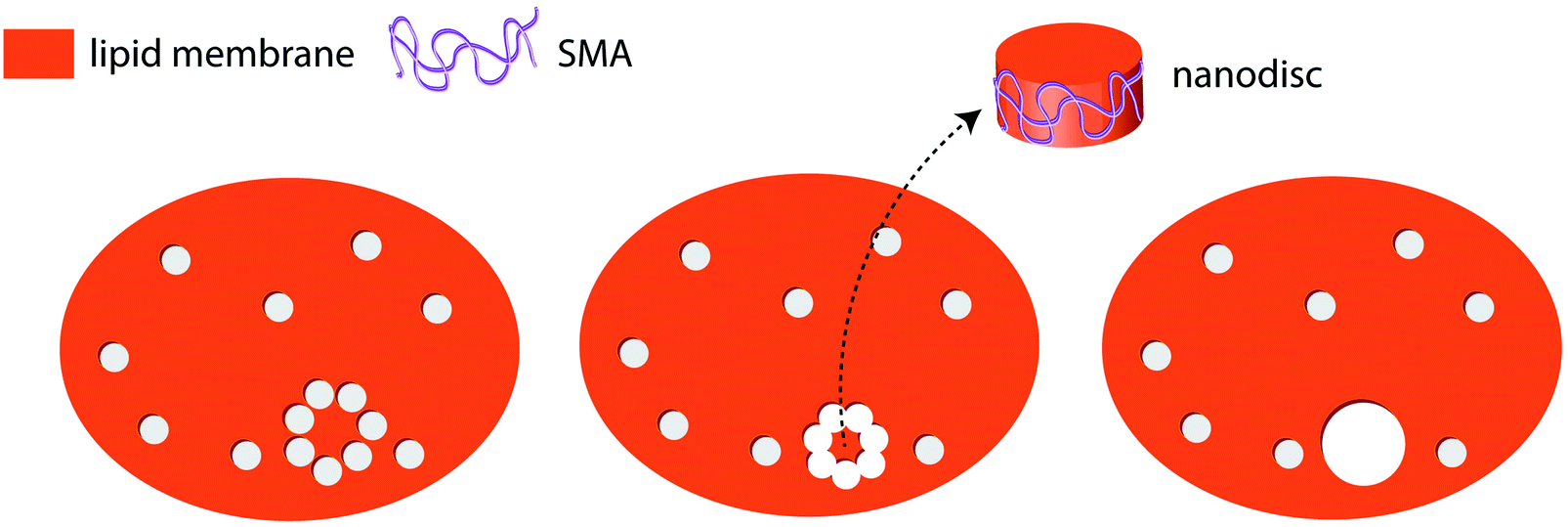 | ||
| Fig. 6 A possible mechanism for SMA–nanodisc formation from lipid bilayers (top view). Small pores induced by the SMA copolymer (left panel) eventually merge forming a lipid island (middle panel). The lipid island moves out of the lipid bilayer as a SMA–lipid disc complex (middle and right panel). | ||
Conclusion
We found experimental evidence that SMA copolymer forms pores in lipid bilayers, both on solid-supported membranes and in free-standing bilayers of unilamellar vesicles. The work supports the theoretical prediction of SMA copolymer-induced pores, published by Xue et al. in 2018.13 We do not have, however, sufficient data to claim with certainty that the mechanism proposed by the authors is the exact one found in the experimental system. The results altogether confirm that SMA copolymers cause water-filled pores in lipid bilayers, well complementing the recent theoretical predictions.Conflicts of interest
The authors declare the following competing financial interest(s): AJ is co-inventor of the microfluidic multichannel pipette, and minority share holder of Fluicell AB, the company that markets the microfluidic pipette. No payments or financial gain were a reason for, or a direct consequence of, the research contained within the manuscript.Acknowledgements
The authors thank Norbert Roos and Antje Hofgaard (Electron Microscopy Laboratory, Dept. of Biosciences, University of Oslo) for technical assistance. This work was made possible through financial support obtained from the Research Council of Norway (Forskningsrådet) Project Grant 274433, UiO: Life Sciences Convergence Environment, the Swedish ResearchCouncil (Vetenskapsrådet) Project Grant 2015-04561 as well as the startup funding provided by the Centre for Molecular Medicine Norway &Faculty of Mathematics and Natural Sciences at the University of Oslo. A.J. acknowledges H2020 ITN “Chemical Reaction Networks – CReaNET” – Ref. 812868 and the Swedish Foundation for Strategic Research (SSF) (GMT14-0077). M.O.R. gratefully acknowledges funding from the Research Council of Norway Project Grant 240909.References
- S. R. Tonge and B. J. Tighe, Adv. Drug Delivery Rev., 2001, 53, 109–122 CrossRef CAS PubMed.
- S. R. Tonge, Compositions comprising a lipid and copolymer of styrene and maleic acid, UK Pat., WO/2006/129127, 2006 Search PubMed.
- T. J. Knowles, R. Finka, C. Smith, Y. P. Lin, T. Dafforn and M. Overduin, J. Am. Chem. Soc., 2009, 131, 7484–7485 CrossRef CAS PubMed.
- M. Orwick-Rydmark, J. E. Lovett, A. Graziadei, L. Lindholm, M. R. Hicks and A. Watts, Nano Lett., 2012, 12, 4687–4692 CrossRef CAS PubMed.
- Z. Stroud, S. C. L. Hall and T. R. Dafforn, Methods, 2018, 147, 106–117 CrossRef CAS PubMed.
- J. M. Dörr, S. Scheidelaar, M. C. Koorengevel, J. J. Dominguez, M. Schäfer, C. A. van Walree and J. A. Killian, Eur. Biophys. J., 2016, 45, 3–21 CrossRef PubMed.
- S. C. Lee, T. J. Knowles, V. L. G. Postis, M. Jamshad, R. A. Parslow, Y. P. Lin, A. Goldman, P. Sridhar, M. Overduin, S. P. Muench and T. R. Dafforn, Nat. Protoc., 2016, 11, 1149–1162 CrossRef CAS PubMed.
- C. Sun, S. Benlekbir, P. Venkatakrishnan, Y. Wang, S. Hong, J. Hosler, E. Tajkhorshid, J. L. Rubinstein and R. B. Gennis, Nature, 2018, 557, 123–126 CrossRef CAS PubMed.
- M. Jamshad, V. Grimard, I. Idini, T. J. Knowles, M. R. Dowle, N. Schofield, P. Sridhar, Y. Lin, R. Finka, M. Wheatley, O. R. T. Thomas, R. E. Palmer, M. Overduin, C. Govaerts, J. M. Ruysschaert, K. J. Edler and T. R. Dafforn, Nano Res., 2015, 8, 774–789 CrossRef CAS PubMed.
- M. C. Orwick, P. J. Judge, J. Procek, L. Lindholm, A. Graziadei, A. Engel, G. Gröbner and A. Watts, Angew. Chem., Int. Ed., 2012, 51, 4653–4657 CrossRef CAS PubMed.
- J. J. Domínguez Pardo, C. A. van Walree, M. R. Egmond, M. C. Koorengevel and J. A. Killian, Chem. Phys. Lipids, 2019, 220, 1–5 CrossRef PubMed.
- S. Scheidelaar, M. C. Koorengevel, C. A. van Walree, J. J. Dominguez, J. M. Dörr and J. A. Killian, Biophys. J., 2016, 111, 1974–1986 CrossRef CAS PubMed.
- M. Xue, L. Cheng, I. Faustino, W. Guo and S. J. Marrink, Biophys. J., 2018, 115, 494–502 CrossRef CAS PubMed.
- E. S. Köksal, P. F. Belletati, G. Reint, R. Olsson, K. D. Leitl, I. Kantarci and I. Gözen, J. Visualized Exp., 2019, 2019, e58923 Search PubMed.
- M. Karlsson, K. Nolkrantz, M. J. Davidson, A. Stromberg, F. Ryttsen, B. Akerman and O. Orwar, Anal. Chem., 2000, 72, 5857–5862 CrossRef CAS PubMed.
- A. D. Goddard, P. M. Dijkman, R. J. Adamson, R. I. Dos Reis and A. Watts, Methods Enzymol., 2015, 556, 405–424 CAS.
- A. Ainla, I. Gözen, B. Hakonen and A. Jesorka, Sci. Rep., 2013, 3, 2743 CrossRef PubMed.
- K. Kustanovich, V. Yantchev, V. Kirejev, G. D. M. Jeffries, T. Lobovkina and A. Jesorka, J. Micromech. Microeng., 2017, 27, 114002 CrossRef.
- K. Kustanovich, V. Yantchev, A. Olivefors, B. Ali Doosti, T. Lobovkina and A. Jesorka, J. Micromech. Microeng., 2019, 29, 024001 CrossRef CAS.
- E. S. Koksal, S. Liese, I. Kantarci, R. Olsson, A. Carlson and I. Gozen, ACS Nano, 2019, 13, 6867–6878 CrossRef CAS PubMed.
- I. Gözen, P. Dommersnes, I. Czolkos, A. Jesorka, T. Lobovkina and O. Orwar, Nat. Mater., 2010, 9, 908–912 CrossRef PubMed.
- J. Asadi, S. Ferguson, H. Raja, C. Hacker, P. Marius, R. Ward, C. Pliotas, J. Naismith and J. Lucocq, Micron, 2017, 99, 40–48 CrossRef CAS PubMed.
- A. Ainla, G. D. M. Jeffries, R. Brune, O. Orwar and A. Jesorka, Lab Chip, 2012, 12, 1255–1261 RSC.
- E. Reimhult, F. Höök and B. Kasemo, Langmuir, 2003, 19, 1681–1691 CrossRef CAS.
- P. S. Cremer and S. G. Boxer, J. Phys. Chem. B, 1999, 103, 2554–2559 CrossRef CAS.
- X. Zhuang, A. Ou and J. B. Klauda, J. Chem. Phys., 2017, 146, 215103 CrossRef PubMed.
- R. G. Efremov, C. Gatsogiannis and S. Raunser, Methods Enzymol., 2017, 594, 1–30 CAS.
- S. Leekumjorn and A. K. Sum, J. Phys. Chem. B, 2007, 111, 6026–6033 CrossRef CAS PubMed.
- W. Chen, F. Duša, J. Witos, S. K. Ruokonen and S. K. Wiedmer, Sci. Rep., 2018, 8, 14815 CrossRef PubMed.
- S. D. Oneill and A. C. Leopold, Plant Physiol., 1982, 70, 1405–1409 CrossRef CAS PubMed.
- E. Karatekin, O. Sandre, H. Guitouni, N. Borghi, P. H. Puech and F. Brochard-Wyart, Biophys. J., 2003, 84, 1734–1749 CrossRef CAS PubMed.
- I. Gözen, B. Ortmen, I. Põldsalu, P. Dommersnes, O. Orwar and A. Jesorka, Soft Matter, 2013, 9, 2787–2792 RSC.
- C. Vargas, R. C. Arenas, E. Frotscher and S. Keller, Nanoscale, 2015, 7, 20685–20696 RSC.
- M. C. A. Stuart and E. J. Boekema, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 2681–2689 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |