
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIon-selective binding as a new trigger for micellization of block copolyelectrolytes with two anionic blocks†
Nico
Carl
 *ab,
Sylvain
Prévost
a,
Ralf
Schweins
a and
Klaus
Huber
b
*ab,
Sylvain
Prévost
a,
Ralf
Schweins
a and
Klaus
Huber
b
aInstitut Laue-Langevin, 71 Avenue des Martyrs, 38042 Grenoble, France. E-mail: carlno@ill.fr; Tel: +33 (0) 476207021
bUniversity of Paderborn, Warburger Str. 100, 33098 Paderborn, Germany
First published on 27th September 2019
Abstract
This work presents well-defined and switchable micelles of block copolymers consisting of the two anionic polyelectrolytes sodium polyacrylate (NaPA) and sodium polystyrene sulfonate (NaPSS). Micellization occurs due to the specific binding of Ca2+ to acrylate groups, which results in neutralization of the corresponding block and thereby formation of the hydrophobic core of the micelles. In contrast, the PSS block remains charged and forms the stabilizing shell. Micellization is triggered by variations of the Ca2+ concentration or the temperature and is a fully reversible and repeatable process. Small-angle neutron scattering (SANS) could unambiguously reveal the structure of the micelles, using a partially deuterated polymer and the contrast variation technique. Considering the variety of metal cations and their broad spectrum of interactions with polyelectrolytes, this new class of like-charged block copolymers opens the door to a broad range of switchable and responsive polyelectrolyte-based systems.
1 Introduction
Block copolymers establish a family of versatile macromolecules, capable of combining diverse properties and response patterns in one polymer chain.1,2 They can mediate among different phases, acting as surfactants in aqueous media or compatibilizing two different non-miscible polymers.2–4 Most of the works published so far on block copolymers deal with the combination of either two neutral polymers5–8 or one neutral and one charged block.9–13 The present work establishes an entirely new strategy, where characteristic features of two different anionic polyelectrolytes shall be introduced into block copolymeric systems. The motivation for pursuing such a strategy is based on a rich variety of interactions between polyelectrolytes and metal cations in aqueous solutions. Such interactions may be purely electrostatic in nature, leading to counterion condensation and charge screening or they may be chemically specific. Such specific interactions corresponding to complex binding of the cations to the anionic residues neutralize the polyelectrolyte chains partly or entirely and drastically change their solution behavior.14 We employ this specific interaction to form micelles.Previous work addressed the specific interaction of Ca2+ with sodium polyacrylate (NaPA),15–21 where the cations form complex bonds with the negatively charged COO− residues of the PA chain. Upon increase of the Ca2+ concentration the polyelectrolytes get increasingly neutralized by Ca2+ until a critical concentration [Ca2+]c is reached, beyond which a precipitation threshold according to
| [Ca2+]c = m + r0[PE] | (1) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22 due to the release of two sodium cations and several water molecules upon binding. Consequently, temperature variations can be used as a second mode to control the phase behavior in addition to a variation of the Ca2+ concentration.23 An increase in temperature is expected to promote further binding of Ca2+ to anionic PA chains and thus to induce precipitation of a CaPA phase.
22 due to the release of two sodium cations and several water molecules upon binding. Consequently, temperature variations can be used as a second mode to control the phase behavior in addition to a variation of the Ca2+ concentration.23 An increase in temperature is expected to promote further binding of Ca2+ to anionic PA chains and thus to induce precipitation of a CaPA phase.
In contrast, sodium polystyrenesulfonate (NaPSS) and Ca2+ do not show specific interactions. No matter how large the concentration of Ca2+ gets PSS remains soluble in water. Light scattering and isothermal titration calorimetry demonstrated that PSS chains do not aggregate in the presence of Ca2+ and no binding takes place.24,25
Blockcopolymers of PSS and PA combine this diverging response pattern toward Ca2+ cations in one polymer. Whereas the PA block is expected to become decorated with Ca2+ cations, thus changing its solution behavior in water drastically, the PSS block only interacts non-specifically with Ca2+, thus staying soluble. Only one publication is known to the authors, which reports on the synthesis of a PA-b-PSS block copolymer and applies it to form multi-layers by a layer-by-layer assembly.26
In the present work, PA-b-PSS block copolymers are synthesized in order to investigate how the combination of the two different response patterns towards Ca2+ affect the solution behavior of such block copolyelectrolytes. The investigation is motivated by the chance to unravel a new toolbox for the design of switchable materials to be controlled via the selection of block lengths, type of cation or temperature.
2 Results and discussion
2.1 Polymer characterization
Four different block copolyelectrolytes of PSS and PA are investigated in this work. Two polymers have a deuterated PA block, one with a long PA and a short PSS block (d3-PA1190PSS70) and one with a symmetric block ratio (d3-PA360PSS400). Deuteration facilitates a meaningful small angle neutron scattering experiment (SANS) based on contrast variation, which shall provide clear insight in the block copolyelectrolyte morphology induced by Ca2+ cations. One further polymer serves as the fully hydrogenated reference for the asymmetrical case (h3-PA1190PSS70) and the fourth polymer has a long PSS and a short PA block (h3-PA100PSS750). The subscript denotes the degree of polymerization. Synthesis of the block copolymers proceeded via RAFT polymerization (Section 1 of the ESI†). In all cases, the shorter block served as macro RAFT agent, which was successively exposed to the RAFT polymerization of the second monomer. Characterization of the resulting block copolyelectrolytes was carried out with NMR and static and dynamic light scattering (SLS and DLS). NMR revealed the ratio between PA and PSS blocks, fully in agreement with the anticipated values. Although compatible with the formation of block copolymers, NMR cannot entirely exclude coexistence of the blocks as separate coils. SLS and DLS further support the formation of the proper block copolyelectrolytes as the values for the respective weight averaged molar mass values Mw are in agreement with the values calculated from NMR and DLS in no case showed a second mode. Characterisation of polyelectrolytes with size exclusion chromatography (SEC), particularly in cases of more complex morphologies such as block copolyelectrolytes, is extremely challenging.27 We tried to develop a SEC-protocol in aqueous 0.1 mol L−1 Na2HPO4 and failed to obtain meaningful results. As shall be outlined later on, SANS at variable scattering contrast, which is the major tool of the present work in unravelling the morphology of the resulting aggregates, provides an independent proof of the successful connection of the two blocks. The characterization of all four block copolyelectrolytes are summarized in Table 1. Detailed data are given in Section 2 of the ESI.†| Sample | PA:PSS/mol% |
M w/kg mol−1 | R h/nm |
|---|---|---|---|
| h3-PA1190PSS70 | 94:6 |
160 ± 3 | 13.8 ± 0.6 |
| d3-PA1190PSS70 | 94:6 |
181 ± 3 | 13.6 ± 0.5 |
| d3-PA360PSS400 | 47:53 |
127 ± 1 | 10.4 ± 0.2 |
| h3-PA100PSS750 | 12:88 |
220 ± 2 | 14.0 ± 0.2 |
2.2 Phase behavior in the presence of Ca2+
The set of block copolyelectrolytes reveals a remarkable response pattern in aqueous solutions of CaCl2. At low Ca2+ concentrations, all samples show single chain behaviour. With increasing CaCl2 concentration, the polymer with the longest PA block (PA1190PSS70) reaches a threshold beyond which stable aggregates are formed (Fig. 1A). This is in contrast to pure PA, which exhibits a precipitation threshold instead. The line of aggregation shows the same trend as the respective precipitation threshold in case of pure PA.15 Upon further increase of the Ca2+ concentration, a second threshold line appears depicting precipitation of the aggregates.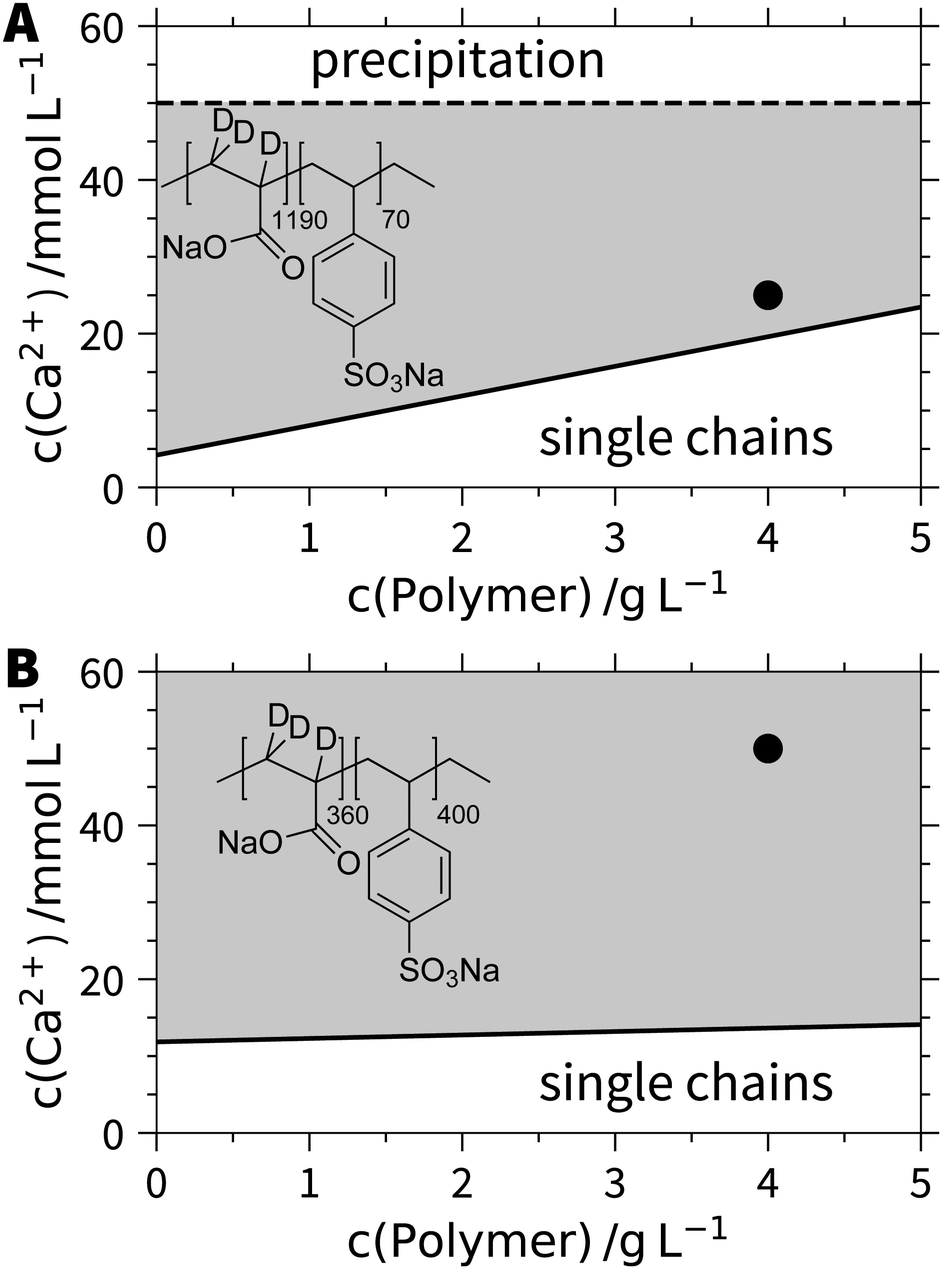 | ||
| Fig. 1 Phase diagrams of (A) d3-PA1190PSS70/h3-PA1190PSS70 and (B) d3-PA360PSS400 in the presence of Ca2+. The black points indicate the points where SANS experiments with contrast variation were performed. | ||
Noteworthy, the exchange of deuterated by hydrogenated PA (d3-PA1190PSS70versus h3-PA1190PSS70) does not affect this solution behavior. By decreasing the PSS/PA block ratio to the symmetric case (d3-PA360PSS400), the regime of stable single coils is even extended and the aggregation threshold gets almost horizontal. No second phase boundary indicating precipitation is detected in this case. Strikingly, the polymer with the long PSS and short PA block (h3-PA100PSS750) remains stable in the state of single coils throughout the entire probed phase diagram. This complex pattern can be nicely reconciled with the response pattern of the two pure polyelectrolytes in solution. As PSS does not show any specific interactions within aqueous solution remaining soluble at any Ca2+ concentration, this feature also dominates as long as the PSS block is long enough. With increasing length of the PA block, specific interactions among PA and Ca2+ come into play, which induce a precipitation in case of pure PA and which lead to aggregates in case of block copolyelectrolytes, obviously stabilized by the PSS blocks. At short enough PSS blocks, the aggregation threshold starts to resemble the precipitation line of pure PA. Thus, the PSS blocks confine the typical precipitation threshold of pure PA (eqn (1)) now to domains of stable aggregates keeping them in solution. However, the short PSS blocks cannot prevent precipitation of the polymers at very high Ca2+ content establishing a second phase boundary.
These findings suggest that the aggregates adopt a micellar shape where the Ca2+/PA complexes caused by specific interactions form the less soluble core with this core being stabilized by the dangling and still soluble anionic PSS blocks. This morphology justifies the use of the term micelles from now and the structure of those micelles will be intensively characterized by means of SANS at variable contrasts. However, before addressing this important question, an exciting feature, which can be inferred from our hypothesis shall be outlined. If the specific binding of Ca2+ to the PA block is responsible for the formation of stable aggregates, formation of micelles and subsequent re-dissolution may occur in reversible cycles upon repeated addition of Ca2+ and subsequent removal by addition of the disodium salt of ethylenediaminetetraacetic acid (EDTA).
In order to demonstrate this anticipated reversibility of micelle formation induced by Ca2+, we added Na2EDTA to a solution of h3-PA1190PSS70 micelles. Fig. 2A shows the molecular weight Mw from SLS for multiple subsequent additions of Ca2+ and EDTA. The first point of Mw corresponds to the polymer solution in 0.1 mol L−1 NaCl in the absence of Ca2+. Each Ca2+ aliquot addition induces an increase of the molecular weight by more than two orders of magnitude. Upon subsequent addition of EDTA, Mw drops to close the initial value where Ca2+ was absent. Hence, the micelle formation can be quantitatively reversed by the addition of a complexing agent such as EDTA, which removes Ca2+ from the equilibrium and subsequent addition of Ca2+ re-triggers micellization. The hydrodynamic radius Rh shows a similar trend and is shown in the ESI† (Fig. S14).
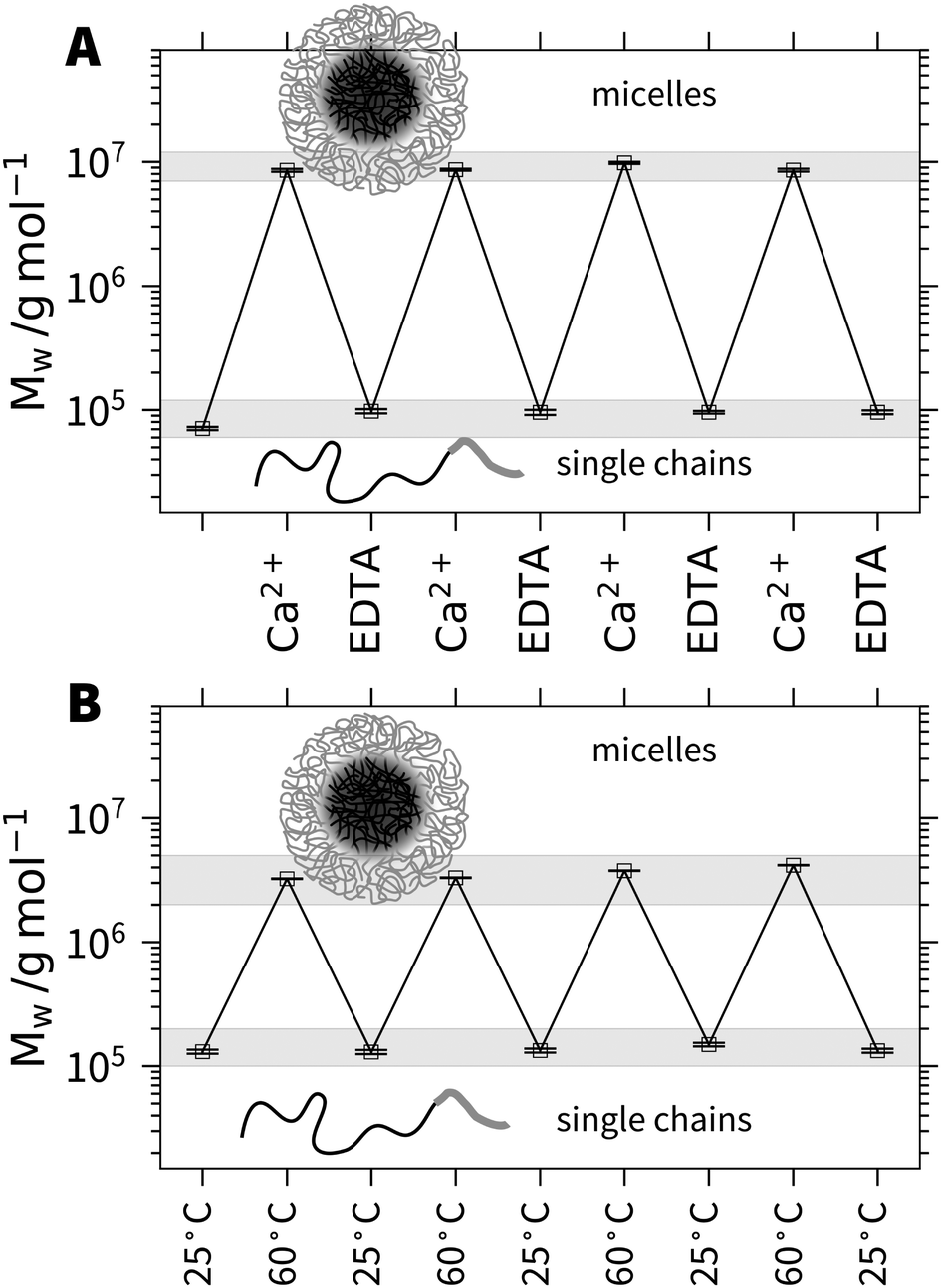 | ||
| Fig. 2 (A) Molecular weight Mw as a function of subsequent injections of CaCl2 and Na2EDTA. The first point refers to a solution of 1 g L−1 h3-PA1190PSS70 in 0.1 mol L−1 NaCl. (B) Molecular weight Mw as a function of temperature cycles between 25 °C and 60 °C for h3-PA1190PSS70 at a polymer concentration of 1 g L−1 and a Ca2+ concentration of 6.1 mmol. | ||
The responsivity of the PA block towards Ca2+ cations offers another way to trigger the process of micellization. As the specific binding of Ca2+ to COO− is driven by entropy due to the release of several water molecules and two sodium cations,22 temperature can be used to influence the binding and therefore the solubility of the PA block. An increase of temperature at a given level of decoration by Ca2+ is expected to increase the amount of bound Ca2+ and thus promote micellization.23 In order to demonstrate this, we prepared a sample of h3-PA1190PSS70 close to the threshold line separating single chains from micelles. As demonstrated in Fig. 2B, we can repeatedly trigger the micelle formation and reverse it by increasing and lowering the temperature in cycles. The temperature effect arises from the entropy driven binding of Ca2+ to PA.22,23 Both experiments revealed the reversible but specific binding of a metal cation selectively to only one of the two anionic blocks as a new system to trigger micellization at ambient conditions. This high versatility of the presented block copolyelectrolyte system should open the route to functional materials for sensing or schizophrenic micelles or for cargo transportation and release.
2.3 Micelle structure probed by SANS
Extended SANS and small-angle X-ray scattering (SAXS) experiments have been carried out on all four polymer samples in order to reveal the morphology of the micelles hold together by Ca2+ cations. The present work shall focus on the detailed contrast variation experiments carried out with the samples d3-PA360PSS400 and d3-PA1190PSS70. Two striking features can already be extracted from these contrast variation experiments without making any assumptions on a particular model of the aggregates. The first feature refers to the integrity of the block copolyelectrolytes and the second addresses the question whether the PA blocks establish the core or the corona of the micelles.All parameters necessary to describe the scattering contrasts of the PSS blocks and of the PA blocks in terms of scattering length densities at variable H2O/D2O ratios are available in the Section 4 of the ESI.† By using the chemical composition and the scattering length densities, the match point of the forward scattering can be predicted for both block copolyelectrolytes (Fig. 3A). Since the chemical composition and therefore the match point is independent on whether block copolymers are present as single chains or as micelles, we can use this information and the experimental forward scattering to verify the success of the block copolymer synthesis. As is outlined in Fig. 3A, the experimentally found match point is identical within experimental uncertainty with the predicted one. This gets noticeable in the most straightforward manner with sample d3-PA360PSS400 because the match point in this case lies in the middle of the probed contrast range spanned (corresponding to the entire range of H2O/D2O ratios). This rules out the existence of individual PA and PSS polymers as they would not show well-defined scattering patterns together with the predicted match point. Although the match point lies out of the range available by variation of the H2O/D2O ratio for d3-PA1190PSS70, extrapolation of the data again reveals a point in full agreement with the value predicted by the corresponding block copolyelectrolyte chain. Thus, all samples under consideration are block copolyelectrolytes as characterized by NMR, SLS and DLS.
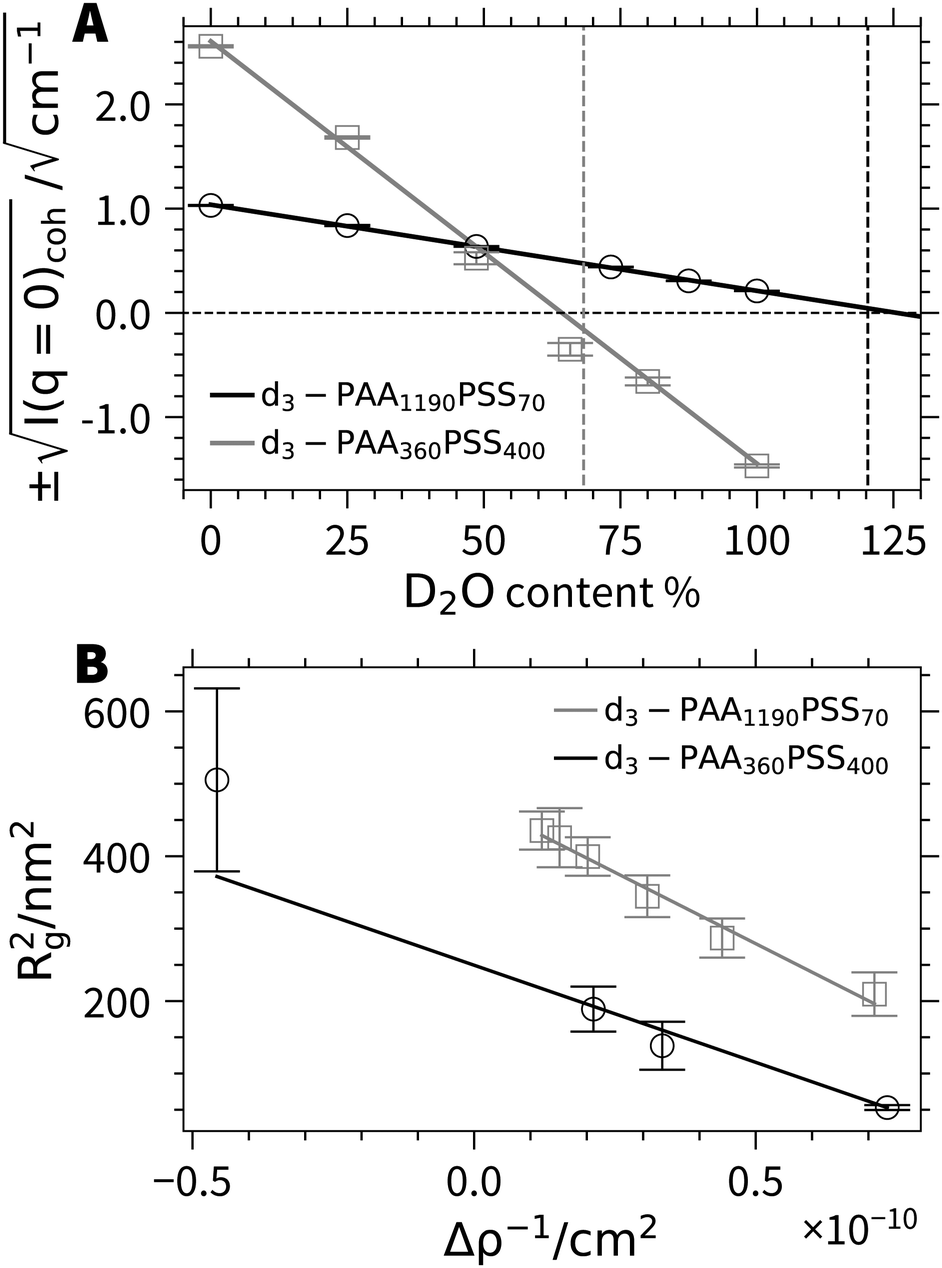 | ||
| Fig. 3 (A) Coherent forward scattering from SANS as a function of D2O content for d3-PA360PSS400 and d3-PA1190PSS70. The vertical dashed lines indicate the theoretical match points calculated from the chemical composition. (B) Squared radius of gyration as a function of inverse scattering contrast for d3-PA360PSS400 and d3-PA1190PSS70.28 | ||
As a next step, the chemical nature of the core and corona shall unambiguously be resolved by model independent data from SANS at variable contrast. All SANS (Fig. 4) and SAXS (Fig. S15 and S16, ESI†) scattering curves exhibit characteristic oscillations indicating particles with a regular shape like a spherical symmetry.30 In the limit of high q, power laws may indicate fractal-like features. An exponent of −4, which is observed close to 48.6% D2O, the match point for PSS chains, signals a sharp boundary of a core surface,31 in line with a PA core. On the other hand, an exponent close to −2, which indicates scattering dominated by polymer coils,32 is found in pure D2O. As the scattering of deuterated PA is very weak in pure D2O this exponent can be assigned to the PSS chains closing the corona with their dangling ends. Even stronger hints for the distribution of the two blocks among core and corona is received from the low q-limit. The radius of gyration extracted from the initial slopes of the scattering curves from particles with components or compartments differing in their scattering contrast is an apparent value. The variation of this radius of gyration Rg with varying scattering contrast can be represented as a plot of Rg2versus the inverse averaged excess scattering length density of the total particle with respect to the solvent. Stuhrmann28 demonstrated that this correlation depends on the morphology of the scattering particles. Trends for core–shell structures give a negative slope if the core has a higher local scattering length density than the shell or corona and a positive slope if the shell (or corona) has a higher density. As is shown in Fig. 3B, the trends observed with the two present polymers is negative. This is a clear-cut evidence for the fact that PA forms the core.
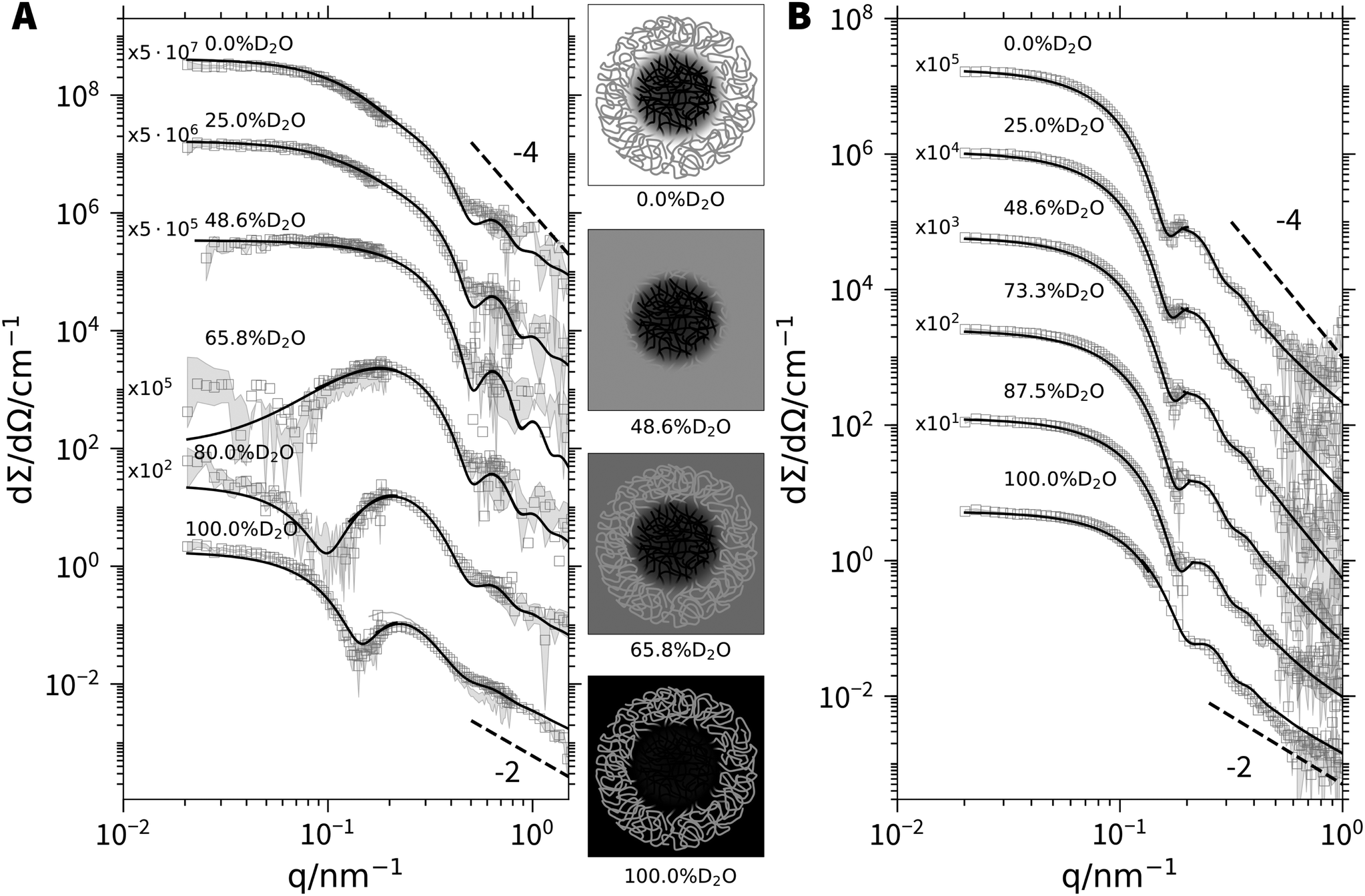 | ||
| Fig. 4 SANS profiles of (A) d3-PA360PSS400 (cpolymer = 4, cCa2+ = 50 mM) and (B) d3-PA1190PSS70 micelles (cpolymer = 4, cCa2+ = 25 mM) at various H2O/D2O ratios. The solid lines represent fits to the form factor of a polydisperse block copolymer micelle.29 An overview of the fit results can be found in Table S3 of the ESI.† The sketches in the middle illustrate the scattering contrast of the d3-PA360PSS400 micelles for selected H2O/D2O ratios. | ||
Having provided compelling evidence for the Ca2+-induced formation of micellar like aggregates of PA-b-PSS-block copolyelectrolytes with PA blocks in the core, the detailed SANS and SAXS data at variable scattering contrast shall finally be interpreted with a suitable micellar model being composed of block copolymers. Such a model has been provided by Pedersen et al.,29 where one block species is forming the core and the other one the corona of a micellar like aggregate. The model parameters include an aggregation number Nagg based on a log-normal distribution, the radius of the core Rcore, the molar volume of the Ca2+ cations and a variable content of water molecules in the core. Using the proper scattering contrast from Table S2 (ESI†), a global fit with common fitting parameters of the SANS and SAXS curves has been performed. The procedure has been carried out for both polymer samples with deuterated PA blocks. A detailed outline of the fit procedure together with a complete list of fit parameters is given in Section 5 of the ESI.†
In short, polymer d3-PA360PSS400 reveals an aggregation number of Nagg = 19.6, independent of the composition of the solvent, resulting in a core radius Rcore = 8.8 nm. Strikingly, aggregation numbers for the polymer with the longer PA block d3-PA1190PSS70 shows a much larger aggregation number of Nagg = 168 and along with it, a larger core of the micelles Rcore = 26 nm compared to the micelles of the polymer with symmetric block lengths. Noteworthy, Nagg and Rcore of the polymer with the much longer (and thus dominating) PA block show a weak dependence on the H2O/D2O ratio of the solvent, decreasing from 168 (H2O) to 126 (D2O) and from 26 nm (H2O) to 24 nm (D2O) respectively as the D2O content increases from 0 to 100%. This weak dependence is most likely an isotope effect on the solvent quality. Such effects are common in polymer science and are for instance observed for the theta temperature of polymers in ideal solvents, which also slightly changes if either the solvent or the polymer is deuterated.33,34 Fits to all SANS curves are included in Fig. 4A and B. Fits where the core is formed by the PSS blocks instead of the PA blocks have also been carried out. However, this model is not able to describe the data (see Section 5 of the ESI† for details) thus further supporting core formation by Ca2+/PA complexes and not by PSS.
Finally, one observation revealed by the fits deserves special attention. The scattering curves could only be successfully reproduced with the present core–corona model29 once a considerable amount of water molecules is incorporated into the core. The values received for both polymers are 12 water molecules per acrylate monomer, amounting to some 85% by volume of the core. This nicely shows that the mechanism of the PA/Ca2+ core formation is a liquid–liquid phase separation as already found for the PA homopolymer and Ca2+.35,36
3 Conclusion
To summarize, we succeeded in presenting block copolymers based on two anionic polyelectrolyte blocks as a new class of double hydrophilic block copolymers with an unusual solution behavior in aqueous solution containing Ca2+ cations. Whereas pure PA precipitates at large enough concentrations of Ca2+ due to specific interactions of the COO− residues with Ca2+,15–21 pure PSS only shows counter ion condensation and electrostatic screening.24,25 A combination of these two features in one block copolyelectrolyte leads to micelle formation instead of PA precipitation, with the PA blocks forming the core. Micelle formation is triggered by alternately adding Ca2+ cations and Na2EDTA to a polymer solution. Even more striking is the feature that the entropic nature of the Ca2+ binding to the NaPA-blocks can be used to switch micellization. Application of small temperature changes reversibly form (ΔT > 0) and dissolve (ΔT < 0) micelles. The micelles are stable and soluble due to the PSS blocks. Micelle formation is entirely reversible, indicating a new route for up-taking and releasing cargo. Increasing the ratio of the block length of PA/PSS increases the aggregation number and weakens the solubilizing power of the PSS corona. Micellization can be tuned by varying the amount of Ca2+ cations and by varying the temperature.22,23 Given the large variety of metal cations, capable of interacting specifically with one or both blocks in the same or different way14,15,24,37,38 and taking further into account the impact of environmental conditions like pH, ionic strength and temperature, an extremely rich pattern of responses can be expected from PA-b-PSS block copolymers. This prospective makes PA-b-PSS block copolymers a highly promising new material. Schizophrenic micelles,39 new contrast agents by utilizing Gd3+ or Eu3+ or new strategies for ion separation are just a few of the possible applications of like-charged block copolyelectrolytes.4 Experimental details
Polymer synthesis, characterization, details on the SANS and SAXS experiments, the used form factor model and scattering length densities can be found in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
In memoriam Isabelle Grillo. The authors thank the Institut Laue-Langevin (https://doi.ill.fr/10.5291/ILL-DATA.9-11-1888) and the European Synchrotron Radiation Facility for the provision of beam time, the Partnership for Soft Condensed Matter (PSCM) for supply of the light scattering, Dr Hans Egold and Karin Stolte of the University of Paderborn for NMR measurements, Isabelle Morfin on D2AM for local contacting and Narayanan Theyencheri and the ID02 team for the supply of in-house time at ID02. Nico Carl acknowledges funding for a PhD scholarship from Institut Laue-Langevin. The open access fee was covered by FILL2030, a European Union project within the European Commission's Horizon 2020 Research and Innovation programme under grant agreement No. 731096.Notes and references
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969 RSC.
- L. Zhang, R. J. Barlow and A. Eisenberg, Macromolecules, 1995, 28, 6055–6066 CrossRef CAS.
- K. Khougaz, I. Astafieva and A. Eisenberg, Macromolecules, 1995, 28, 7135–7147 CrossRef CAS.
- S. Förster, A. K. Khandpur, J. Zhao, F. S. Bates, I. W. Hamley, A. J. Ryan and W. Bras, Macromolecules, 1994, 27, 6922–6935 CrossRef.
- A. Knoll, A. Horvat, K. S. Lyakhova, G. Krausch, G. J. Sevink, A. V. Zvelindovsky and R. Magerle, Phys. Rev. Lett., 2002, 89, 355011 CrossRef PubMed.
- T. P. Lodge, B. Pudil and K. J. Hanley, Macromolecules, 2002, 35, 4707–4717 CrossRef CAS.
- K. J. Hanley, T. P. Lodge and C. I. Huang, Macromolecules, 2000, 33, 5918–5931 CrossRef CAS.
- M. Almgren, W. Brown and S. Hvidt, Colloid Polym. Sci., 1995, 273, 2–15 CrossRef CAS.
- S. Förster, N. Hermsdorf, C. Böttcher and P. Lindner, Macromolecules, 2002, 35, 4096–4105 CrossRef.
- S. Förster, V. Abetz and A. H. E. Müller, Polyelectrolytes with Defined Molecular Architecture II, 2004, vol. 166, pp. 173–210 Search PubMed.
- H. Endo, D. Schwahn and H. Cölfen, J. Chem. Phys., 2004, 120, 9410–9423 CrossRef CAS PubMed.
- J. Bolze, D. Pontoni, M. Ballauff, T. Narayanan and H. Cölfen, J. Colloid Interface Sci., 2004, 277, 84–94 CrossRef CAS PubMed.
- K. Yokota, M. Morvan, J. F. Berret and J. Oberdisse, Europhys. Lett., 2005, 69, 284–290 CrossRef CAS.
- N. Volk, D. Vollmer, M. Schmidt, W. Oppermann and K. Huber, Advances in Polymer Science, 2004, vol. 166, pp. 29–65 Search PubMed.
- R. Schweins, G. Goerigk and K. Huber, Eur. Phys. J. E: Soft Matter Biol. Phys., 2006, 21, 99–110 CrossRef CAS PubMed.
- R. Schweins, P. Lindner and K. Huber, Macromolecules, 2003, 36, 9564–9573 CrossRef CAS.
- R. Schweins and K. Huber, Eur. Phys. J. E: Soft Matter Biol. Phys., 2001, 5, 117–126 CrossRef CAS.
- K. Huber, J. Phys. Chem., 1993, 97, 9825–9830 CrossRef CAS.
- A. Ikegami and N. Imai, J. Polym. Sci., 1962, 56, 133–152 CrossRef CAS.
- P. J. Flory and J. E. Osterheld, J. Phys. Chem., 1954, 58, 653–661 CrossRef CAS.
- I. Sabbagh and M. Delsanti, Eur. Phys. J. E: Soft Matter Biol. Phys., 2000, 1, 75–86 CrossRef CAS.
- C. G. Sinn, R. Dimova and M. Antonietti, Macromolecules, 2004, 37, 3444–3450 CrossRef CAS.
- S. Lages, R. Schweins and K. Huber, J. Phys. Chem. B, 2007, 111, 10431–10437 CrossRef CAS PubMed.
- M. Hansch, B. Hämisch, R. Schweins, S. Prévost and K. Huber, J. Chem. Phys., 2018, 148, 014901 CrossRef PubMed.
- M. Hansch, H. P. Kaub, S. Deck, N. Carl and K. Huber, J. Chem. Phys., 2018, 148, 114906 CrossRef PubMed.
- H. P. Yap, X. Hao, E. Tjipto, C. Gudipati, J. F. Quinn, T. P. Davis, C. Barner-Kowollik, M. H. Stenzel and F. Caruso, Langmuir, 2008, 24, 8981–8990 CrossRef CAS PubMed.
- R. J. Bruessau, Makromol. Chem., Macromol. Symp., 1992, 61, 190–218 CrossRef.
- H. B. Stuhrmann, J. Appl. Crystallogr., 1974, 7, 173–178 CrossRef CAS.
- C. Svaneborg and J. S. Pedersen, Macromolecules, 2002, 35, 1028–1037 CrossRef CAS.
- J. S. Pedersen and M. C. Gerstenberg, Macromolecules, 1996, 29, 1363–1365 CrossRef CAS.
- L. Rayleigh, Proc. R. Soc. London, Ser. A, 1911, 84, 25–38 CrossRef.
- J. S. Pedersen, Adv. Colloid Interface Sci., 1997, 70, 171–210 CrossRef CAS.
- M. K. Braun, M. Wolf, O. Matsarskaia, S. Da Vela, F. Roosen-Runge, M. Sztucki, R. Roth, F. Zhang and F. Schreiber, J. Phys. Chem. B, 2017, 121, 1731–1739 CrossRef CAS PubMed.
- C. Strazielle and H. Benoit, Macromolecules, 1975, 8, 203–205 CrossRef CAS.
- L. Dai, E. P. Douglas and L. B. Gower, J. Non-Cryst. Solids, 2008, 354, 1845–1854 CrossRef CAS.
- L. B. Gower and D. J. Odom, J. Cryst. Growth, 2000, 210, 719–734 CrossRef CAS.
- V. Prabhu, M. Muthukumar, G. Wignall and Y. Melnichenko, Polymer, 2001, 42, 8935–8946 CrossRef CAS.
- Y. Ikeda, M. Beer, M. Schmidt and K. Huber, Macromolecules, 1998, 31, 728–733 CrossRef CAS.
- C. Papadakis, P. Müller-Buschbaum and A. Laschewsky, Langmuir, 2019, 35, 9660–9676 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Polymer synthesis, characterization, details on the SANS and SAXS experiments, the used form factor model and scattering length densities. See DOI: 10.1039/c9sm01138b |
| This journal is © The Royal Society of Chemistry 2019 |