
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Transient measurement and structure analysis of protein–polysaccharide multilayers at fluid interfaces
Pascal
Bertsch
 *a,
Alexandra
Thoma
a,
Jotam
Bergfreund
a,
Thomas
Geue
b and
Peter
Fischer
*a
*a,
Alexandra
Thoma
a,
Jotam
Bergfreund
a,
Thomas
Geue
b and
Peter
Fischer
*a
aInstitute of Food Nutrition and Health, ETH Zurich, 8092 Zurich, Switzerland. E-mail: pascal.bertsch@hest.ethz.ch; peter.fischer@hest.ethz.ch; Tel: +41 44 632 85 36
bLaboratory of Neutron Scattering and Imaging, Paul Scherrer Institut, 5232 Villigen PSI, Switzerland
First published on 9th July 2019
Abstract
The formation of electrostatic protein–polysaccharide multilayers has attracted attention for the design of fluid interfaces with enhanced stability and functionality. However, current techniques are often limited to measuring final multilayer properties. We present an interfacial shear rheology setup with simultaneous subphase exchange, allowing the transient measurement of biopolymer multilayers by their viscoelasticity. The successive and simultaneous adsorption of β-lactoglobulin (β-lg) and low-methoxyl pectin were investigated at the n-dodecane/water interface at pH 4. The successive injection of pectin increased the viscoelasticity of an adsorbed β-lg layer by electrostatic complexation. On the other hand, simultaneous adsorption impeded adsorption kinetics and interfacial layer strength due to complexation in the bulk phase prior to adsorption. Neutron reflectometry at the air–water interface confirmed the formation of an initial β-lg layer and electrostatic complexation of a secondary pectin layer, which desorbed upon pH-induced charge inversion. The layer formed by simultaneous adsorption mainly consisted of β-lg. We conclude that protein–polysaccharide complexes show limited surface activity and result in a lower effective protein concentration available for adsorption.
1 Introduction
The stabilization of fluid interfaces by proteins is a common approach in colloid and interface science. Proteins adsorb at the air/water (A/W) or oil/water (O/W) interface, induce a decrease in interface tension, and form a viscoelastic interfacial network.1 The successive adsorption of a polysaccharide has emerged as a promising approach to improve the performance of protein stabilized interfaces, such as increased resistance against heat and ionic strength,2,3 applicability in a broad pH-range,3,4 increased interfacial moduli,5 or stability under gastric conditions.6,7Here, we use β-lactoglobulin (β-lg), a globular protein fractionated from whey.8 The adsorption of globular proteins at fluid interfaces comprises three consecutive subprocesses: (i) diffusion limited transport to the interface,9 (ii) conformational changes and adsorption limited by the proteins' thermodynamic stability,1,10 and (iii) interfacial rearrangement.11 The functional structure of β-lg regarding interactions with lipids is a hydrophobic β-strand barrel called calyx.12 Because of the affinity of the calyx towards hydrophobic substances, the adsorption of β-lg strongly depends on the hydrophobicity of the employed subphase, as shown in molecular dynamics simulations13 and experimentally.14 β-lg has a pH-dependent charge with an isoelectric point ≈5, where it forms the most compact interfacial layers due to minimum intermolecular repulsion.15
Most polysaccharides are not surface active and do not adsorb at fluid interfaces. An established approach is the adsorption of a primary protein layer followed by electrostatic complexation of a secondary polysaccharide layer, as employed here. Alternatively, proteins and polysaccharides may be covalently conjugated.16–18 In case of electrostatic complexation, the formation of a stable protein layer prior to polysaccharide addition is crucial, as complexation in the bulk impedes adsorption kinetics and results in weaker interfacial layers compared to pure protein layers.5,19 On the other hand, the successive adsorption of a polysaccharide at an equilibrated protein layer increases interfacial layer strength, as demonstrated by Ganzevles et al.5 by an increase in interfacial shear moduli. However, interfacial rheology measurements are usually restricted to measuring final layer properties, stressing the need for transient interfacial rheology measurements during multilayer formation.
Here, we employ an interfacial shear rheology setup with simultaneous subphase exchange. This allowed the formation of pure β-lg layers followed by injection of low-methoxyl pectin while measuring interfacial shear rheology. The interfacial viscoelasticity during simultaneous and successive injection of β-lg and low-methoxyl pectin was investigated and complemented with dynamic interface tension, interfacial dilatation, and neutron reflectivity measurements.
2 Materials and methods
2.1 Materials
β-lg was obtained from the Food and Bioprocess Engineering group at the Technical University of Munich, Germany.8 Low-methoxyl pectin (32% degree of esterification (DE)) was kindly provided by CP Kelco, Germany. Citric acid monohydrate was purchased from Fisher Scientific, Switzerland. Disodium hydrogen phosphate, n-dodecane, and NaOH were purchased from Sigma Aldrich, Switzerland. HCl (37% fuming) was purchased from Merck, Switzerland. Milli-Q water was obtained from a Merck Millipore system, Germany.Experiments were performed at pH 4 in 1/10 McIlvaine buffer mixed from 10 mM citric acid and 20 mM disodium hydrogen phosphate. The protein or pectin was added to the buffer an the pH adjusted using 1 M HCl and NaOH, respectively. The zeta-potential of β-lg and pectin as a function of pH was determined by a Zetasizer nano ZS (Malvern, U.K.).
2.2 Methods
3 Results and discussion
3.1 Electrostatic complexation of β-lg and pectin
As proteins and most polysaccharides have a pH-dependent charge, the pH is crucial in order to form electrostatic protein–polysaccharide complexes. Fig. 1 depicts the zeta-potential, the magnitude of electrostatic potential, for β-lg and pectin as a function of pH. β-lg is charged negatively at neutral pH and shows a charge inversion at the isoelectric point ≈4.9. The charge of pectin on the other hand derives from its carboxyl groups, and can thus only be charged negatively. The pKa of the carboxyl groups is ≈3,25 resulting in a decreasing net charge at low pH. To form electrostatic interfacial multilayers, all further experiments were performed at pH 4 where the zeta-potential of β-lg and pectin is +14 and −34, respectively. A low-methoxyl pectin (32% DE) was chosen to have a higher number of potentially charged carboxyl groups and increase the protein–pectin binding affinity. The electrostatic complexation of β-lg and pectin as a function of pH and pectin DE was addressed in detail by Girard et al.26 An association constant of 1.4 × 103 M−1 was reported at pH 4 for a comparable pectin with 28% DE. The formation of electrostatic β-lg–pectin complexes is reversible.27 Such electrostatic complexes may be exploited to manipulate the charge in protein stabilized emulsions, yielding emulsion droplets with negative surface charge in a broad pH-range.2,4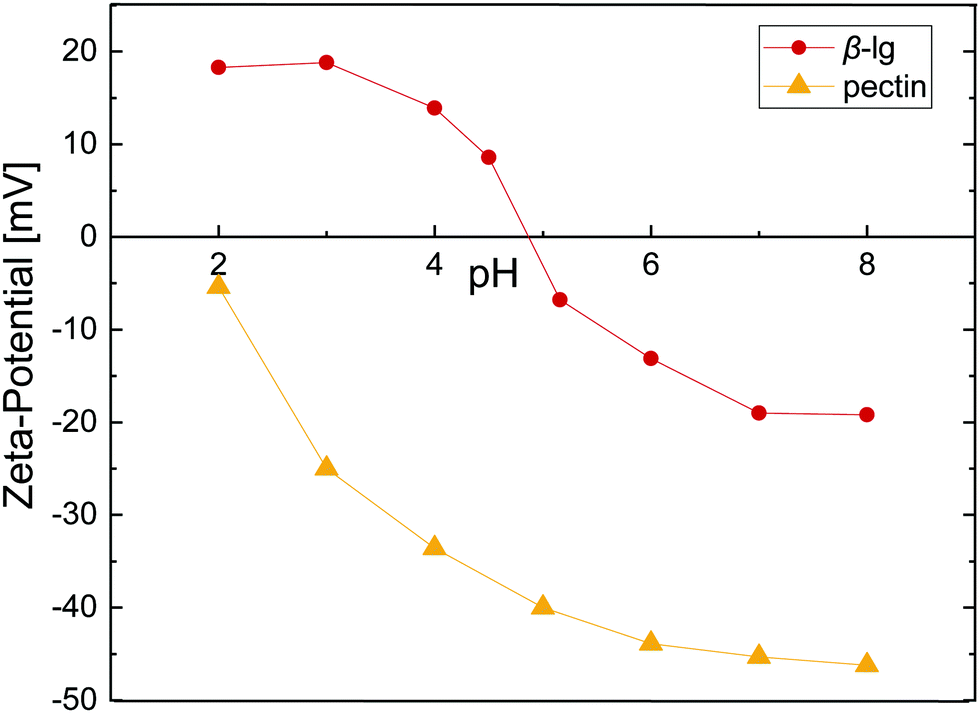 | ||
| Fig. 1 Zeta-potential as a function of pH for 1 wt% β-lg and 0.1 wt% pectin in 1/10 McIlvaine buffer. Lines are to guide the eye. | ||
3.2 Transient interfacial rheology measurement during multilayer formation
The adsorption of proteins is a common approach to stabilize fluid interfaces. As indicated above, the successive adsorption of a polysaccharide can increase the stability and functionality of adsorbed protein layers. Dynamic interface tension measurements are commonly employed to assess the formation of interfacial layers, as shown in Fig. 2A. β-lg adsorbs at the n-dodecane/water interface and decreases its interface tension to 20 mN m−1. The successive adsorption of pectin had a minor impact on surface tension as it is not surface active itself. On the other hand, in case of simultaneous adsorption β-lg–pectin complexes are formed in the bulk prior to adsorption, resulting in slower adsorption kinetics and a higher final surface tension compared to pure β-lg. Although these measurements are useful for the determination of adsorption kinetics and changes in interface tension, they fail to detect the adsorption of non-surface active polysaccharides, and neglect the potential increase in viscoelasticity upon polysaccharide adsorption.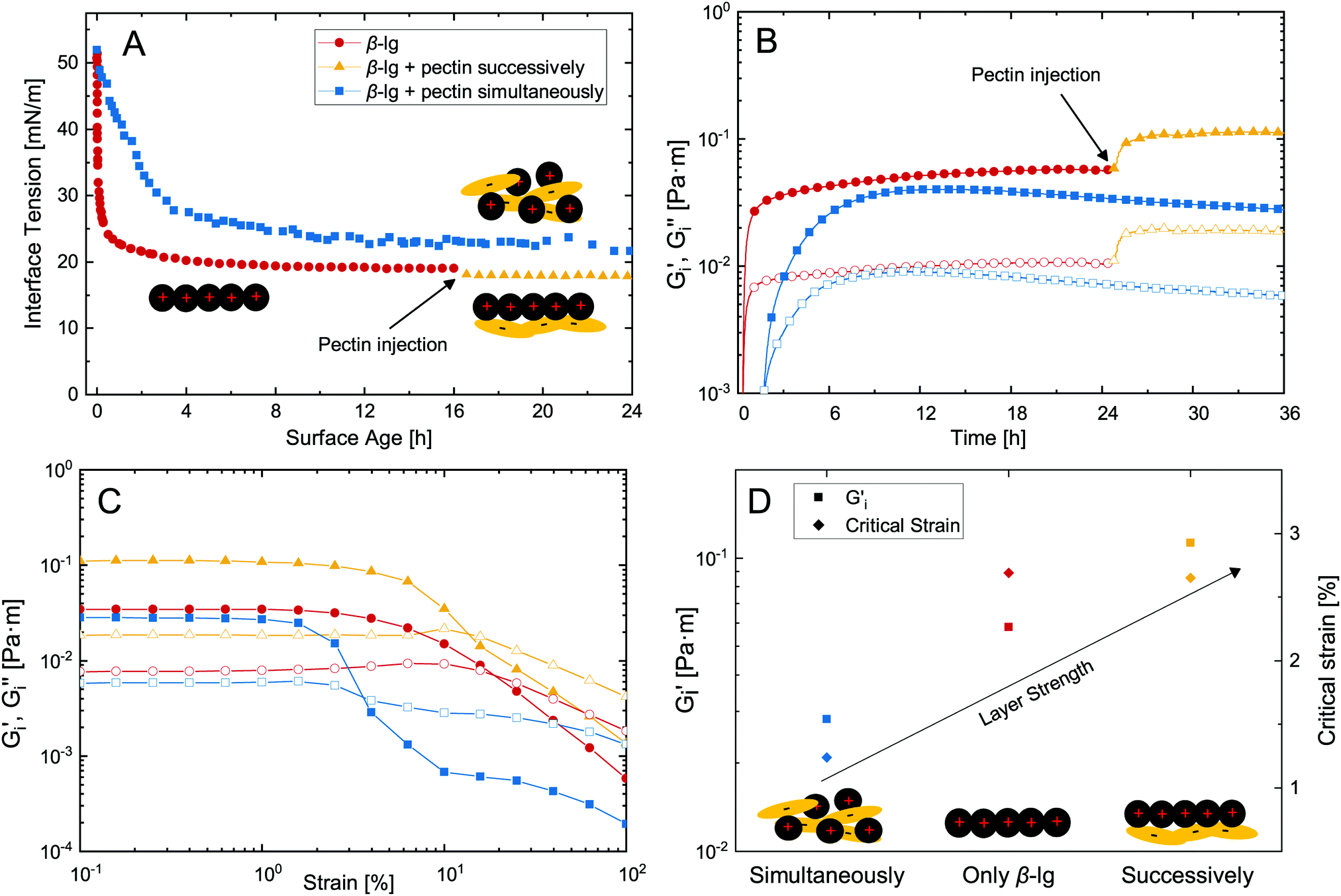 | ||
| Fig. 2 (A) Dynamic interface tension measurements of an n-dodecane droplet formed in 1/10 McIlvaine buffer at pH 4 with 0.001 wt% β-lg and 0.01 wt% pectin added before drop formation or after 16 h. (B) Interfacial shear rheology measurements at the n-dodecane/water interface with simultaneous subphase exchange showing interfacial storage Gi′ (full) and loss modulus Gi′′ (empty) as a function of time during injection of β-lg with successive pectin injection or a β-lg/pectin mixture. (C) Strain sweep experiments and (D) equilibrium Gi′ and critical strain of the formed biopolymer layers. Experiments were performed at 1 rad s−1 angular frequency, 0.5% strain, and 20 °C in 1/10 McIlvaine buffer at pH 4. Lines are to guide the eye. | ||
We employed an interfacial shear rheology setup with simultaneous subphase exchange,21 allowing the successive injection of β-lg and pectin while measuring interfacial shear rheology. This method was recently demonstrated to allow transient measurements of dynamic interfacial moduli Gi′ and Gi′′ during β-lg adsorption.14Fig. 2B depicts the interfacial layer formation of pure β-lg with successive pectin addition compared to their simultaneous injection. The injection of 50 mL 0.1 wt% β-lg solution (0.5 mL min−1) was initiated after 5 min and a measurable viscoelastic network was formed after 20 min. Note that a peristaltic pump was attached to provide constant mixing, resulting in a significantly shorter lag-phase compared to our previous investigation of diffusion limited β-lg adsorption.14 After 24 hours, 20 mL 0.1 wt% pectin were injected to the equilibrated β-lg layer, resulting in a twofold increase in interfacial moduli. Thus, the electrostatic complexation of pectin at an adsorbed β-lg layer increases its interfacial viscoelasticity. This is in contrast to simultaneous injection of β-lg and pectin. The electrostatic complexation in the bulk prior to adsorption results in a prolonged lag-phase, slower adsorption, and lower Gi′ and Gi′′ compared to the pure β-lg layer. The adverse effect of protein–polysaccharide complexation on adsorption kinetics is in agreement with dynamic interface tension measurements and was reported before.5,19 It is attributed to the slower diffusion, lower affinity for adsorption, or even precipitation of the β-lg–pectin complexes. In our case with constant mixing diffusion probably played a minor role, underlining the importance of the latter two. The adsorption limiting effect of polysaccharides is enhanced at increasing polysaccharide charge density,28 and may be counteracted by increasing the β-lg/pectin ratio.5
Fig. 2C depicts strain sweep experiments of the formed biopolymer layers. The mixed β-lg–pectin layer formed after simultaneous adsorption showed the lowest critical strain with yield at 1%. Pure β-lg and the successively adsorbed β-lg–pectin layer showed similar critical strains at ≈2.7%. Thus, the additional adsorption of pectin increases the dynamic interfacial moduli without lowering the critical strain, which is often observed for layers at increasing interfacial moduli.7 An overview of equilibrated Gi′ and critical strain of the different biopolymer layers is provided in Fig. 2D. Compared to pure β-lg, Gi′ was increased twofold by the successive adsorption of pectin. Comparable results were obtained for the successive adsorption of other proteins and polysaccharides under associative conditions.16,29,30 The interfacial elasticity of successively adsorbed β-lg and pectin was five times higher compared to simultaneous adsorption, in good agreement with findings of Ganzevles et al.5 who found up to sixfold stronger interfacial layers. We used a β-lg/pectin mixing ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Ganzevles et al.5 found that at β-lg/pectin ratios higher than five the adverse effects of pectin on adsorption kinetics and interfacial viscoelasticity diminish. For successive adsorption on the other hand, the mixing ratio does not affect layer properties. In contrast to our results, the complexation of longer protein fibrils and pectin in the bulk may even increase the interfacial viscoelasticity compared to pure protein fibril layers.31 The presence of polysaccharides may also increase the strength of protein layers at neutral pH when both are charged negatively. This is attributed to a limited thermodynamic compatibility which favors protein aggregation at the interface.32–34 On the other hand, interfacial protein layers are weakened if polysaccharides are surface active themselves33 or covalently conjugated to adsorbed proteins.35 Interfacial rheological measurements with simultaneous subphase exchange presented here facilitate the determination of such complex synergistic and antagonistic biopolymer interactions, as it allows the transient assessment of interfacial moduli and is not limited to measuring final layer properties.
:1. Ganzevles et al.5 found that at β-lg/pectin ratios higher than five the adverse effects of pectin on adsorption kinetics and interfacial viscoelasticity diminish. For successive adsorption on the other hand, the mixing ratio does not affect layer properties. In contrast to our results, the complexation of longer protein fibrils and pectin in the bulk may even increase the interfacial viscoelasticity compared to pure protein fibril layers.31 The presence of polysaccharides may also increase the strength of protein layers at neutral pH when both are charged negatively. This is attributed to a limited thermodynamic compatibility which favors protein aggregation at the interface.32–34 On the other hand, interfacial protein layers are weakened if polysaccharides are surface active themselves33 or covalently conjugated to adsorbed proteins.35 Interfacial rheological measurements with simultaneous subphase exchange presented here facilitate the determination of such complex synergistic and antagonistic biopolymer interactions, as it allows the transient assessment of interfacial moduli and is not limited to measuring final layer properties.
The successive adsorption of a protein and a polysaccharide is exploited in so-called layer-by-layer emulsions. Besides increased interfacial moduli, the secondary polysaccharide layer increases steric repulsion which may enhance emulsion stability.36 However, adsorbed polysaccharides may also destabilize emulsions by bridging flocculation or depletion.4,17,34 Besides emulsion stability, layer-by-layer adsorption can be employed to create functional emulsions for specific needs. The adsorption of a secondary polysaccharide layer increases heat resistance,2 allows the modulation of emulsion zeta-potential,3,4 and can prevent the adsorption of enzymes for gastric stable emulsions.6,7
3.3 Multilayer dilatational rheology and structure at the A/W interface
Besides O/W interfaces and emulsions, protein–polysaccharide complexes may be employed to improve the stability of foams and respective A/W interfaces.37 In this section, we characterize β-lg–pectin multilayers at the A/W interface by dilatational rheology and elucidate their structure using neutron reflectivity.Fig. 3A depicts dilatational amplitude sweeps of pure β-lg layers compared to multilayers formed by successive and simultaneous pectin injection. All layer showed mostly linear behavior with only a slight decrease in dilatational storage modulus E′ at increasing deformation. The loss modulus E′′ was lower at all deformation, but was not depicted in Fig. 3A for clarity. The successive electrostatic complexation of pectin resulted in an increase in E′ compared to the pure β-lg layer. In contrast, the complexation in the bulk prior to adsorption resulted in lower E′. Thus, the same effects of simultaneous and successive pectin complexation are observed in dilatational rheology at the A/W interface as in shear rheology at the O/W interface discussed above. The interfacial elasticity can not be further enhanced by increasing the concentration of successively added pectin, as demonstrated in Fig. 3B. It is known from interfacial shear rheology that the β-lg/pectin ratio does not alter the interfacial properties if pectin is added successively.5
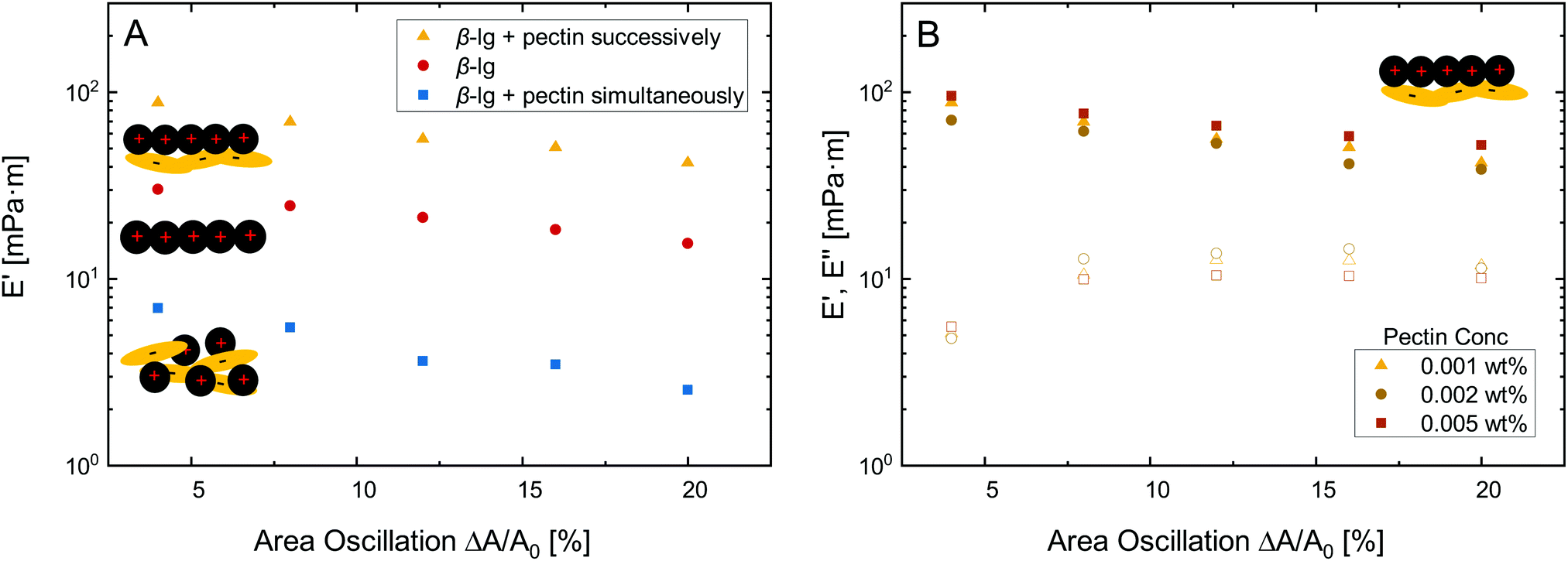 | ||
| Fig. 3 Dynamic interfacial dilatational storage E′ (full) and loss modulus E′′ (empty) as a function of area deformation for (A) 0.001 wt% β-lg and 0.001 wt% pectin added simultaneously and successively and (B) increasing pectin concentration added successively. Experiments were performed at the A/W interface with 1/10 McIlvaine buffer at pH 4 and 22 °C. Oscillation frequency was 10 mHz. | ||
The adsorption of biopolymer layers at the A/W interface further facilitates to probe their structure using neutron reflectivity. Fig. 4 shows the fitted neutron reflectivity, reflectivity relative to the reference RRel, and scattering length density (SLD) profiles of the respective layers at the air/D2O interface.
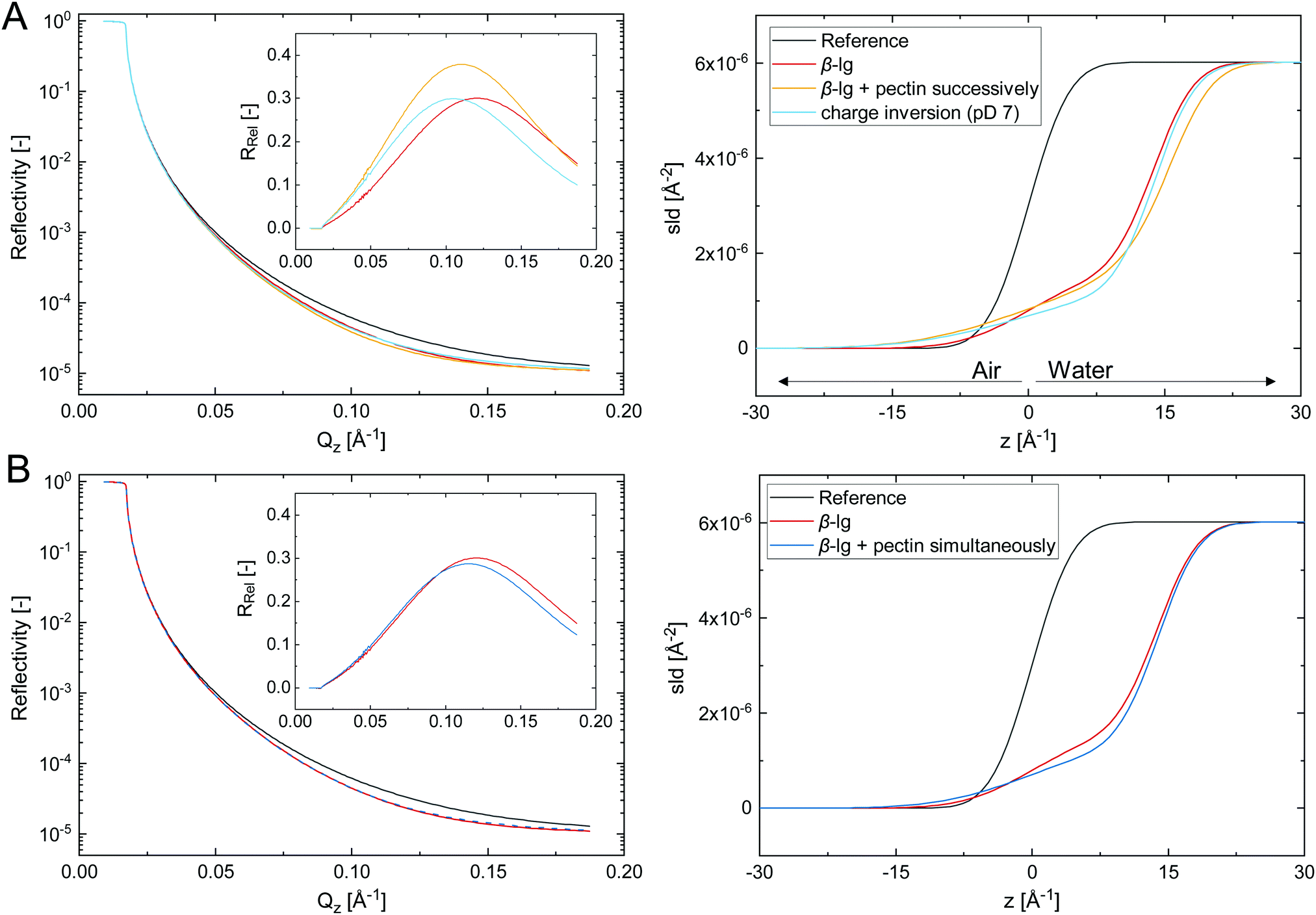 | ||
| Fig. 4 Fitted neutron reflectivity, reflectivity relative to the reference RRel, and scattering length density SLD of biopolymer layers adsorbed at the air/D2O interface. Experiments were performed in 1/10 McIlvaine buffer at pD 4 and 20 °C, which was measured as reference. (A) A pure β-lg layer formed at 0.001 wt%, successive addition of 0.002 wt% pectin, followed by β-lg charge inversion by increasing the pD to 7. (B) Comparison of a pure protein layer formed at 0.001 wt% β-lg and simultaneously adsorbed β-lg and pectin at 0.005 wt% each. Reflectivity curves were fitted with the Parratt algorithm and had a χ2 < 7.5 × 10−3. | ||
The biopolymer layers depicted in Fig. 4A were formed in three consecutive steps. (i) A pure β-lg layer was formed at 0.001 wt%, which was best described by a monolayer with a thickness of 14 Å. This corresponds to a loosely packed monolayer, considering the diameter of native β-lg in solution of approximately 40 Å. However, protein unfolding and spreading at the interface may result in a more compact β-lg structure at the interface.10,14 Previous studies reported β-lg monolayers in the range of 20–40 Å.7,38,39 The thinner layer formed here can be attributed to a tenfold lower β-lg concentration, which was chosen to avoid excess β-lg in the bulk and prevent complexation with pectin. (ii) 0.002 wt% pectin was successively injected to the adsorbed β-lg layer. The obtained interfacial layer was best described by a two-layer model consisting of the primary β-lg layer followed by the consecutively added pectin. The β-lg layer was compressed to 11 Å and had a higher surface roughness compared to the initial layer, as denoted by a shift in the SLD profile towards the air phase. Hence, the initial β-lg layer is compressed and pushed towards the hydrophobic subphase by the consecutive adsorption of pectin. The adsorbed pectin layer was 5 Å in thickness. The thickness of the pectin backbone is 3 Å,40 indicating a compact pectin layer with limited extension into the water phase. The dense pectin layer is probably the reason why higher pectin concentrations did not increase interfacial dilatational moduli (Fig. 3B). The structure of successive β-lg–pectin layers may be different in emulsions, as pectins can adsorb at more than one droplet, particularly at high molecular weight. This may induce bridging flocculation, but could also provide additional bulk stabilization. In a third step (iii), the pD was increased to 7 to investigate if the electrostatic complexation of pectin is reversible upon charge inversion of β-lg. The obtained layer was indeed best described by a β-lg monolayer with a thickness of 14 Å, same as the initial β-lg layer. Thus, pectin desorbed due to electrostatic repulsion. The remaining β-lg layer was rougher compared to the initial layer. It can not be conclusively stated if this derives from irreversible structural changes due to pectin adsorption or traces of remaining pectin. Adsorbed protein layers are known to undergo structural changes at varying pH15 or in presence of like-charged polysaccharides.32–34 Thus, interfacial electrostatic β-lg–pectin complexes are reversible, as previously demonstrated in bulk experiments.27 This pH-triggered adsorption and desorption of pectin at protein-stabilized interfaces could allow the production of pH-sensitive colloidal materials. For example, it has been demonstrated that secondary polysaccharide layers can prevent the digestion of protein-stabilized emulsions under gastric conditions.6,7 The pH-triggered charge inversion in the duodenum could induce the desorption of this protective layer for controlled delivery applications.
In Fig. 4B the neutron reflectivity of a β-lg layer formed at 0.001 wt% is compared to a layer of simultaneously adsorbed β-lg and pectin at each 0.005 wt%. The β-lg layer was a 14 Å monolayer as discussed in detail above. Interestingly, the layer formed during simultaneous adsorption of 0.005 wt% β-lg and pectin was also best described by a monolayer 14 Å in thickness, and fit goodness increased when the SLD was changed towards the one of β-lg. This suggests that the interfacial layer formed during simultaneous adsorption consists mostly of β-lg. The layer roughness was slightly increased similar to the layer after charge inversion at pD 7 discussed above, potentially due to the presence of a minor pectin fraction. Overall, the layer was similar to the one formed with pure β-lg at fivefold lower concentrations. We conclude that β-lg–pectin complexes formed in the bulk show a low tendency towards adsorption, and the complexation reduces the effective β-lg concentration available for adsorption. Thus, the impeded adsorption kinetics and interfacial viscoelasticity discussed above could derive from a decreased effective protein concentration. This theory is supported by previous findings who demonstrated that the adverse effects of pectin on adsorption and interfacial layer strength can be overcome by increasing the β-lg concentration.5 However, interfacial layers formed at higher β-lg/pectin ratios also showed a different structure.39 A dense β-lg layer followed by a loosely packed secondary layer of β-lg–pectin complexes was observed at tenfold higher β-lg concentrations and β-lg/pectin ratios of two to six. This is in contrast to the β-lg dominated monolayer observed here for a mixing ratio of one.
4 Conclusions
The formation of protein–polysaccharide multilayers at fluid interfaces is an interesting approach to increase the stability and widen the field of applications of protein-stabilized interfaces. However, the transient measurement of protein–polysaccharide multilayer formation is often difficult. We introduced an interfacial shear rheology setup with simultaneous subphase exchange, allowing the transient measurement of biopolymer multilayers by their viscoelasticity. Thereby, we could demonstrate that the successive adsorption of pectin to an established β-lg layer increases its interfacial layer strength, whereas simultaneous adsorption impedes adsorption kinetics and viscoelasticity compared to pure β-lg layers.Structure analysis by neutron reflectometry confirmed the formation of an initial β-lg layer followed by electrostatic complexation of a secondary pectin layer. The complexation of pectin was reversible upon pH-induced charge inversion, posing interesting applications for pH-sensitive colloidal materials. For the simultaneous adsorption of β-lg and pectin we observed a thin monolayer which consists mostly of β-lg. We concluded that electrostatic complexes decrease the effective protein concentration available for adsorption and show limited surface activity themselves.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Neutron reflectivity experiments were performed at the Swiss Spallation Neutron Source SINQ, Paul Scherrer Institute, Villigen PSI, Switzerland. This project was funded by the Swiss National Science Foundation, Project No. 200021-137941 and 200021-175994.References
- V. Mitropoulos, A. Mütze and P. Fischer, Adv. Colloid Interface Sci., 2014, 206, 195–206 CrossRef CAS.
- T. Harnsilawat, R. Pongsawatmanit and D. J. McClements, J. Agric. Food Chem., 2006, 54, 5540–5547 CrossRef CAS.
- D. Guzey and D. J. McClements, J. Agric. Food Chem., 2007, 55, 475–485 CrossRef CAS.
- Y. H. Cho and D. J. McClements, Langmuir, 2009, 25, 6649–6657 CrossRef CAS.
- R. A. Ganzevles, K. Zinoviadou, T. van Vliet, M. A. Stuart and H. H. De Jongh, Langmuir, 2006, 22, 10089–10096 CrossRef CAS.
- N. Scheuble, T. Geue, E. J. Windhab and P. Fischer, Biomacromolecules, 2014, 15, 3139–3145 Search PubMed.
- N. Scheuble, M. Lussi, T. Geue, F. Carrière and P. Fischer, Biomacromolecules, 2016, 17, 3328–3337 CrossRef CAS.
- J. Toro-Sierra, A. Tolkach and U. Kulozik, Food Bioprocess Technol., 2013, 6, 1032–1043 CrossRef CAS.
- F. MacRitchie and A. E. Alexander, J. Colloid Interface Sci., 1963, 18, 453–457 CrossRef.
- L. Day, J. Zhai, M. Xu, N. C. Jones, S. V. Hoffmann and T. J. Wooster, Food Hydrocolloids, 2014, 34, 78–87 CrossRef CAS.
- E. M. Freer, K. S. Yim, G. G. Fuller and C. J. Radke, J. Phys. Chem. B, 2004, 108, 3835–3844 CrossRef CAS.
- S. Brownlow, J. H. Morais Cabral, R. Cooper, D. R. Flower, S. J. Yewdall, I. Polikarpov, A. C. North and L. Sawyer, Structure, 1997, 5, 481–495 CrossRef CAS.
- D. Zare, J. R. Allison and K. M. McGrath, Biomacromolecules, 2016, 17, 1572–1581 CrossRef CAS.
- J. Bergfreund, P. Bertsch, S. Kuster and P. Fischer, Langmuir, 2018, 34, 4929–4936 CrossRef CAS.
- K. Engelhardt, M. Lexis, G. Gochev, C. Konnerth, R. Miller, N. Willenbacher, W. Peukert and B. Braunschweig, Langmuir, 2013, 29, 11646–11655 CrossRef CAS.
- E. Dickinson, Soft Matter, 2008, 4, 932–942 RSC.
- E. Dickinson, Food Hydrocolloids, 2011, 25, 1966–1983 CrossRef CAS.
- P. Fischer, Eur. Phys. J.-Spec. Top., 2013, 222, 73–81 CrossRef CAS.
- R. A. Ganzevles, M. A. Cohen Stuart, T. van Vliet and H. H. de Jongh, Food Hydrocolloids, 2006, 20, 872–878 CrossRef CAS.
- P. Erni, P. Fischer, E. J. Windhab, V. Kusnezov, H. Stettin and J. Läuger, Rev. Sci. Instrum., 2003, 74, 4916–4924 CrossRef CAS.
- P. A. Rühs, N. Scheuble, E. J. Windhab, R. Mezzenga and P. Fischer, Langmuir, 2012, 28, 12536–12543 CrossRef.
- D. Clemens, P. Gross, P. Keller, N. Schlumpf and M. Könnecke, Phys. B, 2000, 276–278, 140–141 CrossRef CAS.
- M. Gupta, T. Gutberlet, J. Stahn, P. Keller and D. Clemens, Pramana, 2004, 63, 57–63 CrossRef.
- L. G. Parratt, Phys. Rev., 1954, 95, 359–369 CrossRef.
- R. Kohn and P. Kovác, Chem. Zvesti, 1978, 32, 478–485 CAS.
- M. Girard, S. L. Turgeon and S. F. Gauthier, J. Agric. Food Chem., 2003, 51, 6043–6049 CrossRef CAS.
- B. L. Sperber, M. A. Cohen Stuart, H. A. Schols, A. G. Voragen and W. Norde, Biomacromolecules, 2009, 10, 3246–3252 CrossRef CAS.
- R. A. Ganzevles, H. Kosters, T. van Vliet, M. A. Stuart and H. H. De Jongh, J. Phys. Chem. B, 2007, 111, 12969–12976 CrossRef CAS.
- L. Piazza, N. Dürr-Auster, J. Gigli, E. J. Windhab and P. Fischer, Food Hydrocolloids, 2009, 23, 2125–2131 CrossRef CAS.
- J. N. Miquelim, S. C. Lannes and R. Mezzenga, Food Hydrocolloids, 2010, 24, 398–405 CrossRef CAS.
- N. P. K. Humblet-Hua, E. van Der Linden and L. M. Sagis, Soft Matter, 2013, 9, 2154–2165 RSC.
- R. Baeza, C. Carrera Sanchez, A. M. Pilosof and J. M. Rodríguez Patino, Food Hydrocolloids, 2004, 18, 959–966 CrossRef CAS.
- R. Baeza, A. M. R. Pilosof, C. C. Sanchez and J. M. Rodríguez Patino, AIChE J., 2006, 52, 2627–2638 CrossRef CAS.
- L. S. Jourdain, C. Schmitt, M. E. Leser, B. S. Murray and E. Dickinson, Langmuir, 2009, 25, 10026–10037 CrossRef CAS.
- T. J. Wooster and M. A. Augustin, Food Hydrocolloids, 2007, 21, 1072–1080 CrossRef CAS.
- A. Benichou, A. Aserin and N. Garti, J. Dispersion Sci. Technol., 2002, 23, 93–123 CrossRef CAS.
- E. Dickinson and E. Izgi, Colloids Surf., A, 1996, 113, 191–201 CrossRef CAS.
- P. J. Atkinson, E. Dickinson, D. S. Horne and R. M. Richardson, J. Chem. Soc., Faraday Trans., 1995, 91, 2847–2854 RSC.
- R. A. Ganzevles, R. Fokkink, T. van Vliet, M. A. Cohen Stuart and H. H. de Jongh, J. Colloid Interface Sci., 2008, 317, 137–147 CrossRef CAS PubMed.
- T. Imaizumi, M. Szymańska-Chargot, P. M. Pieczywek, M. Chylińska, A. Kozioł, D. Ganczarenko, F. Tanaka, T. Uchino and A. Zdunek, LWT–Food Sci. Technol., 2017, 84, 658–667 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |