DOI:
10.1039/C9SM00795D
(Paper)
Soft Matter, 2019,
15, 5234-5242
Modeling the assembly of oppositely charged lock- and key-colloids†
Received
18th April 2019
, Accepted 29th May 2019
First published on 1st June 2019
Abstract
The interaction of oppositely charged lock- and key-colloids is investigated using computer simulations. We show that indented spheres, i.e., lock-particles, can be specifically assembled with spherical key-particles using solely electrostatic interactions in addition to a hard overlap potential. An analytic description of the entropic and energetic contributions is derived and supported by simulations and explicit energy calculations, respectively. The analytic expression of the electrostatic contribution is further employed to build up a schematic model allowing for efficient large-scale Monte Carlo simulations. The influence of the charge/ionic strength, the degree of indentation, and the size/number ratio is discussed by analyzing the specific and unspecific associations from the simulations. Herein, both particle design and mixing conditions lead to the formation of stable specific clusters analogous to colloidal molecules whose valence is defined by the number of lock-particles associated with a key-particle. In addition, the approach is extended to the encapsulation of an excess of small key-particles in largely indented lock-particles. These two examples exemplify that highly specific pairwise interactions can be implemented by using solely oppositely charged particles with complementary geometries, which opens the road for a rational design of complex hierarchical self-assemblies of complementary building blocks.
Introduction
Self-assembly is a key construction principle that Nature uses so successfully to create often highly elaborate structures.1 Proteins and peptides have, for instance, the intrinsic ability to spontaneously self-assemble into complex structures such as fibrils, shells or nanotubes, as observed in the formation of cytoskeletal filaments or viral assemblies.2–6 While spherical colloids have been established as models to investigate various aspects of phase transitions and self-assembly processes,7–10 colloidal particles with centrosymmetric interaction potentials are often unable to capture the intrinsic complexity of diverse biological materials. This represents a major limitation in any attempts to quantitatively understand their self-assembly and motivates the use of new synthetic colloids such as anisotropic colloids, patchy particles, or well-defined colloidal clusters11–17 as novel model systems to study more complex associative processes and develop bioinspired applications and materials.
The lock and key assembly principle, introduced to describe the specific interactions of enzymes with substrates,18–21 illustrates the exquisite ability of biological entities to fabricate and regulate their molecular machinery.4 Herein, the molecular recognition mechanism stemming from shape complementary and pairwise interactions constitutes as well one of the foundations of modern supramolecular chemistry within the formation of specific host–guest and donor–acceptor complexes.22 Besides, even at the molecular level, assemblies are often dynamic and the constituents may adapt to their local environment so as to maximize their interactions. This marks the evolution of the lock and key theory into the induced fit theory,20,21 which can be seen in analogy to the more recent development of supramolecular chemistry towards adaptive chemistry.22,23
A synthetic colloidal analog of a lock, or an acceptor, follows recent developments of indented spheres or bowl-shaped particles. These anisotropic colloids have been investigated in detail due to their rich phase diagram and with respect to their specific self-assembly with complementary spherical key-particles.24 The prevailing strategies to trigger short range specific interactions driving the assembly of complementary shaped particles have so far relied on depletion interactions. The first experimental realizations could be rationalized by excluded volume theory, which has been extensively supported by computer simulations.25–28 However, other interactions for colloidal lock and key self-assembly have rarely been explored so far, with the exception of some works on electric field induced lock- and key-interactions,29 cell imprint interactions,30 and point-charge models.31 Recently, it has been demonstrated that thermoresponsive microgel-based lock- and key-particles may interact to form defined colloidal molecules simply by considering them as oppositely charged building blocks.32 Other studies also point to electrostatics being an important factor where assembly between lock- and key-particles is concerned.33 By taking advantage of the fact that both the geometrical parameters and interactions could be tuned with temperature, the valence of the clusters can be controlled from a tetravalent “methane”- to a divalent “carbon dioxide”-like configuration. Oppositely charged microgels can, hence, be regarded as a colloidal analog of adaptive chemistry.
Those experimental findings have motivated this work to further model such complex lock and key assembly. Given the complexity of the experimental systems, we only focus on oppositely charged “hard” lock- and key-particles where we assume a uniformly distributed charge throughout the surface of the colloidal molecule, which in many respects makes this model a more realistic alternative to a simple point charge model. We present an analytic expression to describe the energetic contribution to the interaction potential and further build up a schematic model allowing for large scale Monte Carlo simulations of the assembly. The specificity of the association illustrated by the formation of stable specific colloidal molecules is then discussed in relation to the ionic strength, size and number ratio between the lock- and the key-particles, and the degree of lock-indentation.
Results and discussion
Lock- and key-particle definition
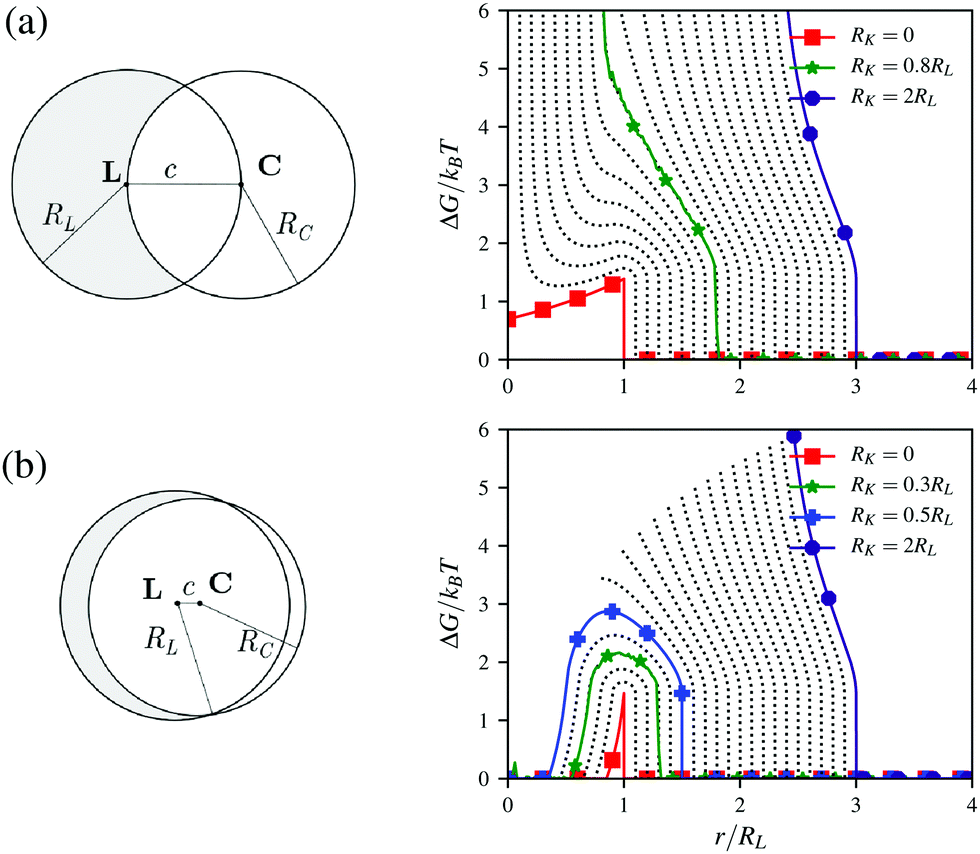
Let us consider a lock-particle defined as a spherical particle with a radius RL indented by a spherical cavity with a radius RC as schematically depicted in Fig. 1 (left) for two different indentations. Herein, the two sphere centers, L and C, are separated by the distance c. The complementary spherical key-particle is defined using its center position K and its radius RK. Finally, we note that for c > RC + RL a lock-particle has no indentation and is equivalent to a sphere. In the following we will, from an analytic perspective, explore the entropic and energetic (electrostatic) contributions that will later be included in the simulations.
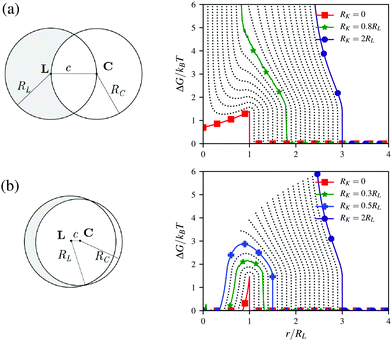 |
| | Fig. 1 Schematic illustration of a lock-particle and the corresponding free energy due to excluded (rotational) volume effects. The lock-particle is defined by (a) c = RC = RL and (b) c = 0.1RL and RC = 0.95RL. The lines represent different RK-values from RK = 0 (red line) to RK = 2RL (indigo line) in steps of 0.1RL. The green line (star) indicates the results from simulations using a hard lock-particle and a hard key-particle. The blue line (plus), RK = 0.5RL (≈0.5375RL), is the (rough) limit for no cost encapsulation. | |
Entropic contribution
The free energy ΔG between a hard lock- and a hard key-particle was previously32 shown to be| | 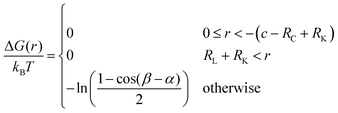 | (1) |
where r = |r| = |K − L|, α = ∠PLK and β = ∠PLC, where P defines the contact point between the particles, kB is the Boltzmann constant, and T is the temperature. Here the angular dependence has been averaged, and thus the free energy describes the rotational entropic cost at a specific distance r around L. In Fig. 1 (right), ΔG(r) is presented for two distinct indentations and different RK-values using eqn (1). The free energy increases with an increase of RK and a local minimum can be observed in ΔG(r) for small RK. That is, for some r there is an attractive entropic force pushing the particles closer together. Worth noting is that a lock-particle can encapsulate a key-particle with no (rotational) entropic cost when c + RK < RC as shown in Fig. 1b (right) for RK < 0.5375RL. These analytic results are in agreement with simulations using lock- and key-particles with hard body conditions,32 which is illustrated for two cases in Fig. 1.
Electrostatic contribution
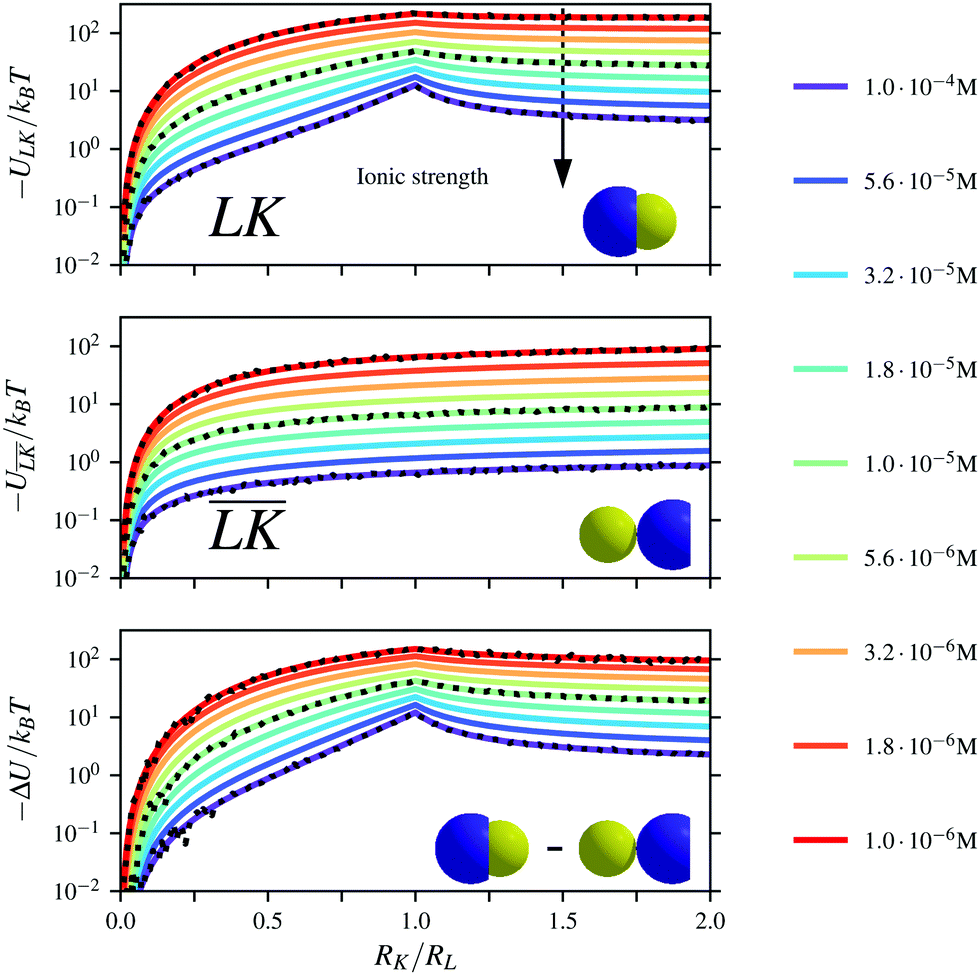
The Yukawa interaction-energy between uniformly surface-charged lock- and key-particles was analytically derived when the key-particle is perfectly in front or behind the lock-particle cavity, i.e., for |(C − L)·(K − L)|/(c·r) = 1. An approximate formula for the same setup has been derived elsewhere30 yet we present an exact expression in eqn (S1) in the ESI.† The energies in the specific LK configuration, ULK, and the unspecific 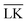 configuration,
configuration, 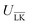 , were calculated for different ionic strengths, I, and size ratios, RK/RL, in Fig. 2. Note how the Debye-length
, were calculated for different ionic strengths, I, and size ratios, RK/RL, in Fig. 2. Note how the Debye-length 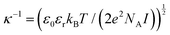 is related to the ionic strength I, where ε0 is the vacuum permittivity, εr = 72 is the relative permittivity of the dispersing medium, T = 293.15 K, e is the elementary charge, and NA is the Avogadro constant. The calculations were performed considering RL = RC = c = 500 nm, oppositely charged lock- and key-particles with a constant absolute surface charge-density |σ| = 3.183 × 10−4e nm−2. By defining the effective charge34–36 of a lock- or key-particle as
is related to the ionic strength I, where ε0 is the vacuum permittivity, εr = 72 is the relative permittivity of the dispersing medium, T = 293.15 K, e is the elementary charge, and NA is the Avogadro constant. The calculations were performed considering RL = RC = c = 500 nm, oppositely charged lock- and key-particles with a constant absolute surface charge-density |σ| = 3.183 × 10−4e nm−2. By defining the effective charge34–36 of a lock- or key-particle as| | 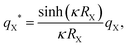 | (2) |
where X = {L, K} indexes the particle type, a simplified expression for ULK for such a setup can be retrieved as| | 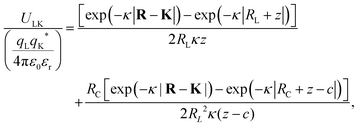 | (3) |
assuming L as the origin. Here R is a point on the rim of the lock-particle defined in the ESI,† and z = |K|. The analytic expression was found to be in agreement with explicit charge calculations performed at three different ionic strengths. Herein, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 equally charged point charges were randomly distributed at the surface of the lock-particle, which yielded the same surface charge-density |σ| as above, and the total electrostatic energy was retrieved by summing up all the point-charge interactions with the effective charge of the key-particle qK*. We note a maximum when the key-particle is in front of the lock-particle at RK = RL. So does the difference between the
000 equally charged point charges were randomly distributed at the surface of the lock-particle, which yielded the same surface charge-density |σ| as above, and the total electrostatic energy was retrieved by summing up all the point-charge interactions with the effective charge of the key-particle qK*. We note a maximum when the key-particle is in front of the lock-particle at RK = RL. So does the difference between the  and LK configurations, ΔU. Hence, RK should be equal to RL in order to maximize the interaction-energy difference between specific LK and unspecific
and LK configurations, ΔU. Hence, RK should be equal to RL in order to maximize the interaction-energy difference between specific LK and unspecific 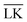 interactions. Such a configuration is quite costly from an entropic point-of-view, see Fig. 1(a) (right), and it is not straightforward to predict the optimal particle design for self-assembly. Simulations are thus needed to model the assembly process, which then also includes many-body effects. However, whereas ΔG is solely defined by the geometries of the constituents, it is important to note that ULK,
interactions. Such a configuration is quite costly from an entropic point-of-view, see Fig. 1(a) (right), and it is not straightforward to predict the optimal particle design for self-assembly. Simulations are thus needed to model the assembly process, which then also includes many-body effects. However, whereas ΔG is solely defined by the geometries of the constituents, it is important to note that ULK, 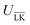 , and ΔU are all strongly dependent on I and |σ|, cf. ΔU ∝ |σ|2, and can thus be tuned over many units of kBT. A specific lock and key self-assembly is therefore expected, at least in the limit of low I and high |σ|.
, and ΔU are all strongly dependent on I and |σ|, cf. ΔU ∝ |σ|2, and can thus be tuned over many units of kBT. A specific lock and key self-assembly is therefore expected, at least in the limit of low I and high |σ|.
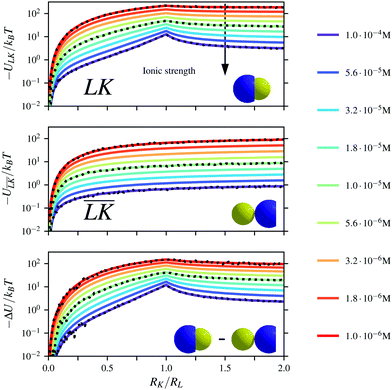 |
| | Fig. 2 Interaction-energy between a lock-particle and a key-particle at contact as a function of the size ratio RK/RL calculated at different ionic strengths (solid lines). Energy calculations for the specific LK-configuration are shown in the top panel and for the unspecific 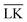 -configuration in the middle panel, and the difference between the two energies is displayed in the bottom panel. The gray dotted lines refer to explicit charge calculations using a lock-particle decorated with 10000 randomly distributed point-charges for ionic strengths of 10−4 M, 10−5 M and 10−6 M. -configuration in the middle panel, and the difference between the two energies is displayed in the bottom panel. The gray dotted lines refer to explicit charge calculations using a lock-particle decorated with 10000 randomly distributed point-charges for ionic strengths of 10−4 M, 10−5 M and 10−6 M. | |
In order to perform simulations of lock- and key-particles, one needs an angular-dependent potential accounting for the lock-particle anisotropy. This potential is constructed based on the exact expressions derived in the previous section. By denoting the exact potential in front of the lock-particle as VL(dLK(r,c)) and the analogue expression behind it as 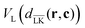 , where c = C − L, the angular-dependent potential VL(r,c) is approximated as
, where c = C − L, the angular-dependent potential VL(r,c) is approximated as
| | 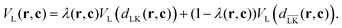 | (4) |
Note that when
VL is a function of a scalar distance, it is described by eqn (S1), ESI,
† whereas when
VL is a function of a vector distance (and
c), it is described by
eqn (4). Here
λ(
r,
c) ∈ [0,1] is a function depending on the region where the potential is evaluated in, see
Fig. 3 (top-right), and
dLK(
r,
c) and
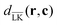
are distances to the surface of the lock-particle. A more detailed description of the potential can be found in the ESI.
† In order to assess the validity of the approximate potential, we compared it to the potential of a lock-particle where the surface is decorated with 10
000 randomly positioned point-charges, see the schematic in
Fig. 3 (top-left). The potentials stemming from these two respective models are displayed in the bottom panels of
Fig. 3. Note that along the symmetry axis (
x = 0),
eqn (4) is exact. The same calculations were repeated for different ionic strengths and are provided in the ESI
† (Fig. S3–S5). With the exception of some regions tangent to the cavity rim, the approximate potential,
VL(
r,
c), was found to provide an accurate description of the lock-particle electrostatics with a reduced number of elements as compared to the explicit charge potential, thus allowing for large-scale simulations.
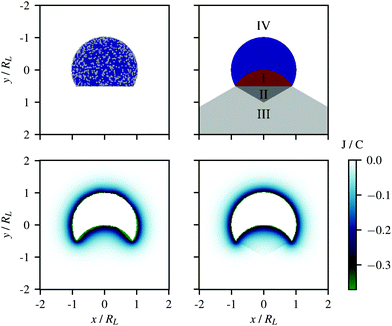 |
| | Fig. 3 Electrostatic potential from (lower-left) 10000 randomly distributed point-charges on the surface of a lock-particle and (lower-right) the approximate potential using eqn (4). The ionic strength for both figures was 10−5 M. The upper figures illustrate the respective models. | |
For the purpose of such simulations, we use the effective charge34–36 of a lock- or key-particle, see eqn (2), to define their interaction energy as
| | | U(LK)(r,c) = VL(r,c)qK*, | (5) |
while between key-particles it is
| | 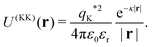 | (6) |
Finally, the interaction energy between two lock-particles was approximated using
| | 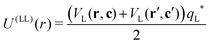 | (7) |
where the prime indicates the second lock-particle. Note that for this approximation the interaction-energy is slightly overestimated when lock-particles face each other at close distances. From this we gather that the model is at its worst for dense systems, and more appropriate for dilute ones, as it is the case in this work but also in other studies.
37
Methods
We performed Monte Carlo simulations using the Faunus software,38 where we included particle translation–rotation moves and standard cluster moves centered around key-particles.39 The probability of a single particle move was equal to the probability of a cluster move. The aim was to obtain simulations with typical acceptance in the range of 30–40%. This was achieved by using a maximum rotational displacement parameter of 0.5 radians for all moves, whereas the maximum translational displacement parameters varied between 0.01RL and 0.5RL for single particle moves and 0.5RL for cluster moves. Simulations were performed using hard lock- and key-particles and the electrostatic interaction-energy presented in eqn (5)–(7), and periodic boundary conditions. The hard overlap potential between lock-particles was modeled by checking for overlap between the surface-arcs of the particles.40 This algorithm only checks for overlapping surfaces and thus lock-particles could in principle be enclosed within one another without any detectable overlap for polydisperse systems. In our simulations, however, the lock-particles were equally sized in each system. Encasement could, however, take place between lock- and key-particles (using the previous algorithm), since the key-particles sometimes were small enough to entirely fit within a lock-particle. To prevent this, we used the volume overlap code implemented in Faunus38 between lock- and key-particles. The total number of particles in the system was N = 1320 and the number ratio between lock- and key-particles NL/NK varied from 0.5 to 10. The simulations were sampled in N × 105 steps starting from energetically equilibrated configurations (over N × 105 steps). The temperature, relative permittivity of the dispersing medium, and ionic strength were the same as mentioned earlier, and the particle number density was ρ = 0.1 μm−3. The implemented model does not account for features arising from counterion-release,41–44 and thus we chose the charge-densities σL = −σK = |σ| so as to minimize its repulsive effect. Any other |σL| ≠ |σK| would have the free energy minima shifted, which effectively corresponds to an inflated particle. A basic length-unit in the simulations was RL = 500 nm, which was kept constant throughout the simulations, in which other lengths such as RC, c, RK, and κ−1 were scaled so as to get generic units. If not mentioned otherwise, then RC = RL. A bond is defined as specific (LK) when the distance between a lock- and a key-particle is r ≤ RK + RL; that is, the key-particle is in the cavity of the lock-particle. Conversely, a bond is defined as unspecific ( ) when RK + RL < r < 1.1(RK + RL). Different values than 1.1 ranging from 1.05 to 1.20 were tested and found to provide tantamount results within the simulated systems. The simulations in Fig. 1 used NL = NK = 1, no cluster moves, N × 109 simulations steps, and only the hard overlap potential.
) when RK + RL < r < 1.1(RK + RL). Different values than 1.1 ranging from 1.05 to 1.20 were tested and found to provide tantamount results within the simulated systems. The simulations in Fig. 1 used NL = NK = 1, no cluster moves, N × 109 simulations steps, and only the hard overlap potential.
Simulations
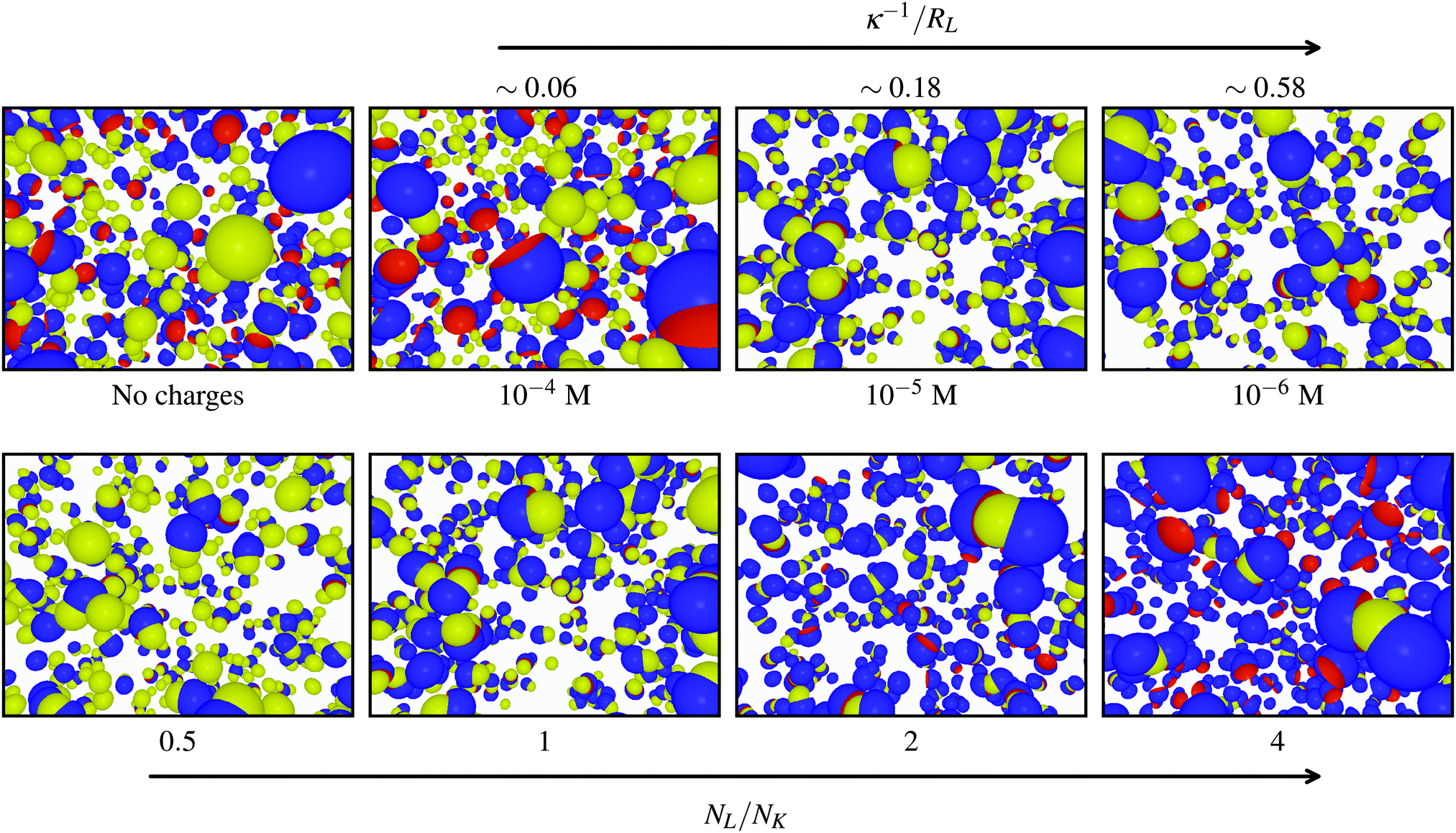
Typical simulation snapshots generated with NL/NK = 1 at different ionic strengths including the uncharged conditions (∼infinite screening) are shown in Fig. 4 (top). The size of the key-particles was set to RK = 0.8RL, which is slightly smaller than the lock-particles and their cavities. Without effective electrostatics (far left), no assembly was observed (as expected for such hard lock- and key-particles). However, by decreasing I < 10−4 M, i.e., by increasing the amplitude and range of the interactions, particles start to build up defined clusters. Fig. 4 (bottom) displays simulation snapshots captured at constant I = 10−5 M for different NL/NK-values. Starting from the left, for NL/NK ≤ 1, there are primarily ‘dipolar’ clusters consisting of a key-particle specifically associated in the cavity of one lock-particle. By increasing the number ratio (going right), higher order clusters start to form, primarily ‘CO2’ molecules, i.e., two lock-particles specifically bonded to a single key-particle. Based on these initial results, we conclude that specific clusters can be formed and tuned using the number ratio and ionic screenings.
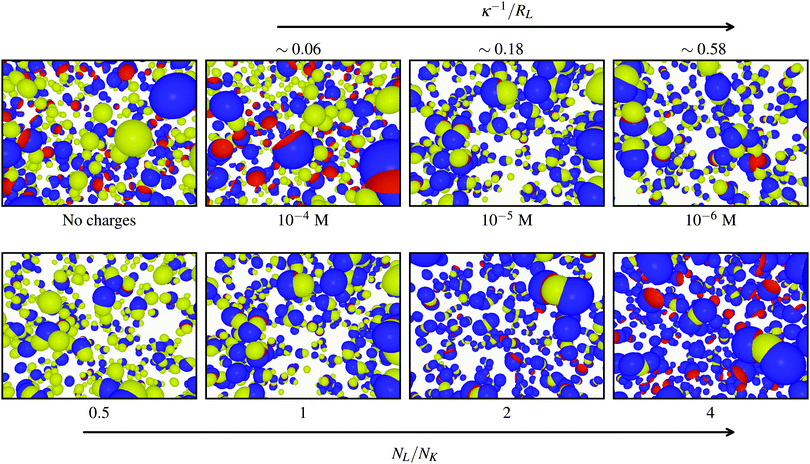 |
| | Fig. 4 (top) Configurations from simulations using NL/NK = 1 for different ionic screenings and (far left) no charges (∼infinite screening). (bottom) Configurations from simulations using I = 10−5 M for different ratios NL/NK. The lock-particles are described by RC = c = RL and the key-particles by RK = 0.8RL. | |
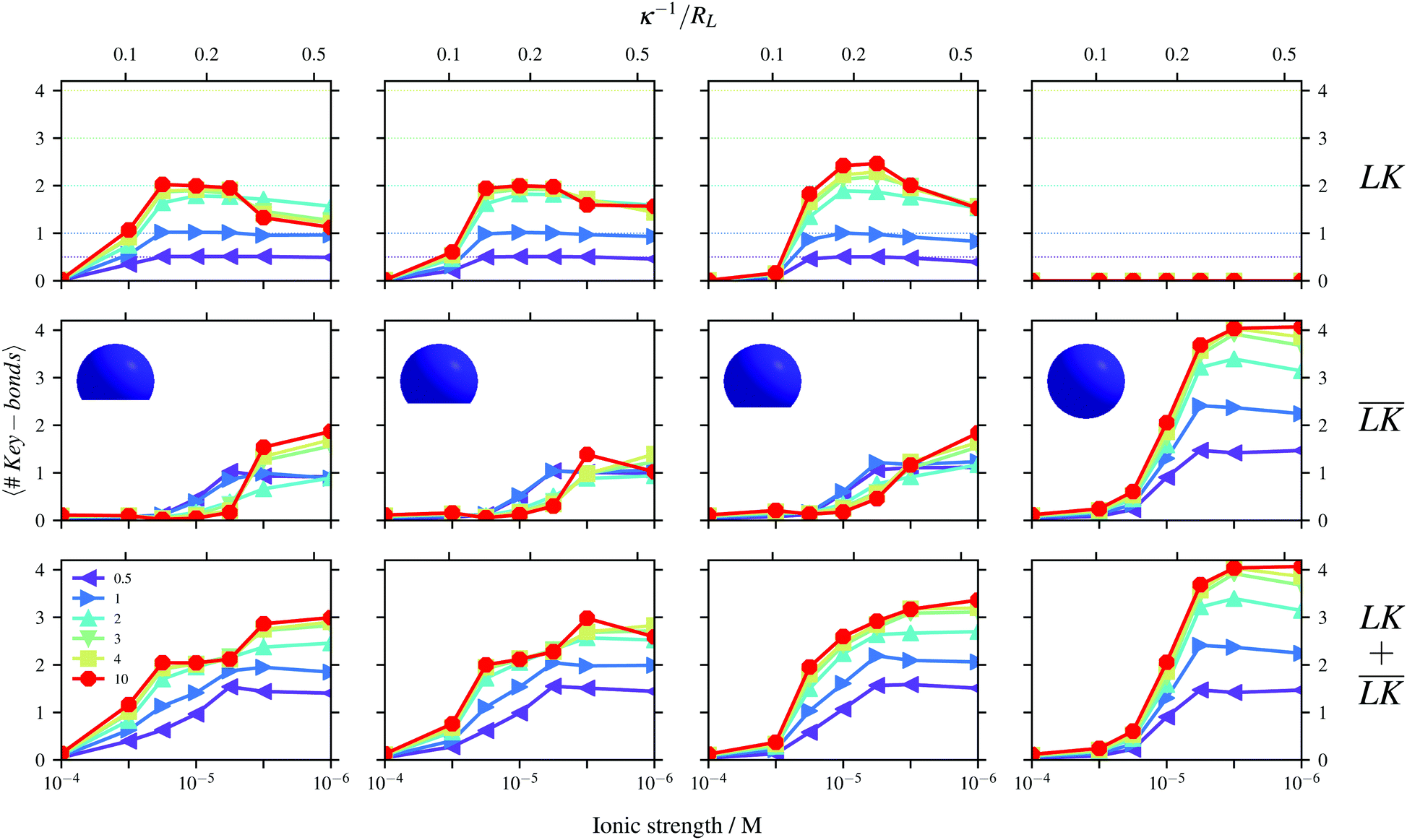
The simulation results shown in Fig. 4 are quantified in Fig. 5 considering the average numbers of specific, unspecific, and total bonds per key-particle as functions of ionic strength/Debye-length, for different indentations c. The dotted lines in all figures represent the maximas of specific bonds (assuming that each lock-particle is specifically bound to a single key-particle). Larger values thus refer to higher order clusters consisting of several key-particles. Though the total number of bonds seems to increase (fairly) monotonically with a decrease of I, the specific number of bonds does show a maximum around I ≈ 10−5 M, independent of the cavity center c. After this maximum, unspecific bonds are increasingly favored and the key-particles are effectively shifted from the cavities of the lock-particles toward their backs/sides. Conversely, upon decreasing I the key-particles shift from unspecific to specific sites. This binding-feature has been shown to occur in experimental setups.45 From geometrical arguments, we note that small c decreases the number of possible specific bonds per key-particle as compared to large c. However, for larger indentations, the area of the cavity is decreased and thus the interactions become energetically less specific. By going from left to right in Fig. 5, the maximum number of possible specific bonds increases. The top left graph shows a maximum around 2, which is in line with geometrical considerations, for which a key-particle can at most be specifically assembled with 2 lock-particles. By increasing c to 1.4RL, this number gets larger with a maximum for NL/NK = 10 at ≈2.33. The simulation results show that basically 2/3 of the clusters are linear “CO2” molecules and 1/3 have a trigonal planar geometry like SO32−. For NL/NK = 2, however, most of the clusters present a “CO2”-configuration. The “valency” of the key-particle centered clusters can therefore be tuned by adjusting the lock-indentation, the number ratio, and/or ionic strength. As has been noted before for spherical particles,10 we confirm also for lock- and key-particle assembly of various degrees of indentation (by observing configurations) that the geometrical formation of the decorated particles most often diverges from the optimal positions retrieved from using κ = 0 as is in the Thomson problem.46,47
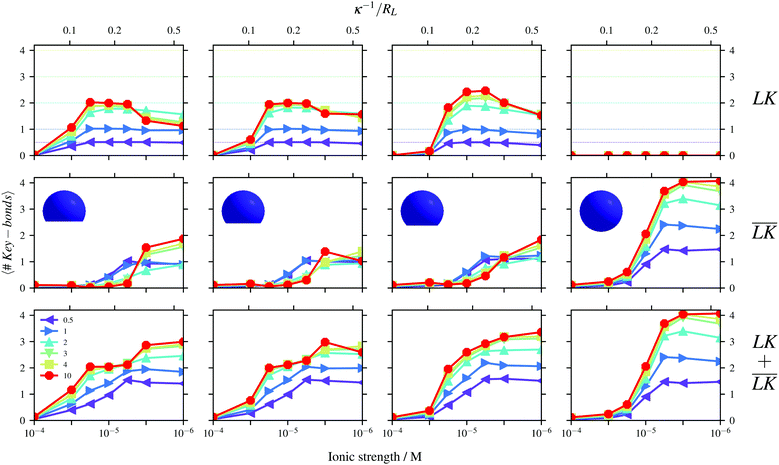 |
| | Fig. 5 Numbers of specific (top), unspecific (middle) and total (bottom) bonds per key-particle. The radius of the key-particle was RK = 0.8RL. The figures show results using: c = 1.0RL (far left), c = 1.2RL (left), c = 1.4RL (right), and c = 2RL (far right), for different ratios NL/NK according to the legend. | |
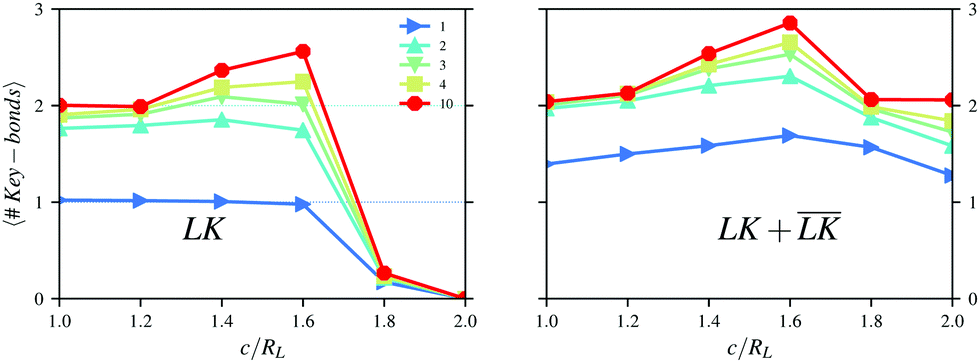
In Fig. 6, the number of bonds per key-particle is plotted as a function of the center of the cavity c. For NL/NK = 1 and c ≤ 1.6RL, almost all the lock-particles are specifically bound to a key-particle. The total number of bonds, though, is larger than unity, indicating the formation of larger order structures, such as dipolar chains as shown in a simulation snapshot in Fig. S6 (left) (ESI†). Such dipolar chains are not observed using NL/NK = 2 in Fig. S6 (right) (ESI†), where rather different higher order structures appear. This points to the importance of the stoichiometry for the generation of defined hierarchical self-assemblies. The total number of bonds has a maximum at c = 1.6RL (the same as the maxima for the specific bonds) after which the contacts become mostly unspecific. These results indicate that the presence of an indentation may promote the assembly process and increase even the total number of bonds when compared to spherical particles. We rationalize this observation by noting that the entropic cost decreases seemingly more than does the attractive electrostatic interactions as the cavity is shifted from c = RL towards c = 1.6RL. After this point the slight energetic difference between a specific and an unspecific bond is too small to overcome even moderate entropic costs. Thus, for c ≈ 1.6RL, the balance between the two contributions provides the maximum yield of specific and total bonds. In the ESI,† we present more simulation results analogous to Fig. 4–6 but using RK = RL and RK = 1.2RL.
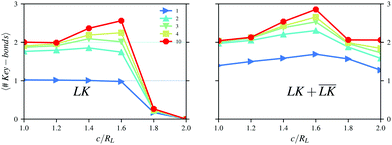 |
| | Fig. 6 Numbers of specific (left) and total (right) bonds per key-particle as functions of the degree of indentation for different number ratios NL/NK. The simulations were performed with RK = 0.8RL at I = 10−5 M. | |
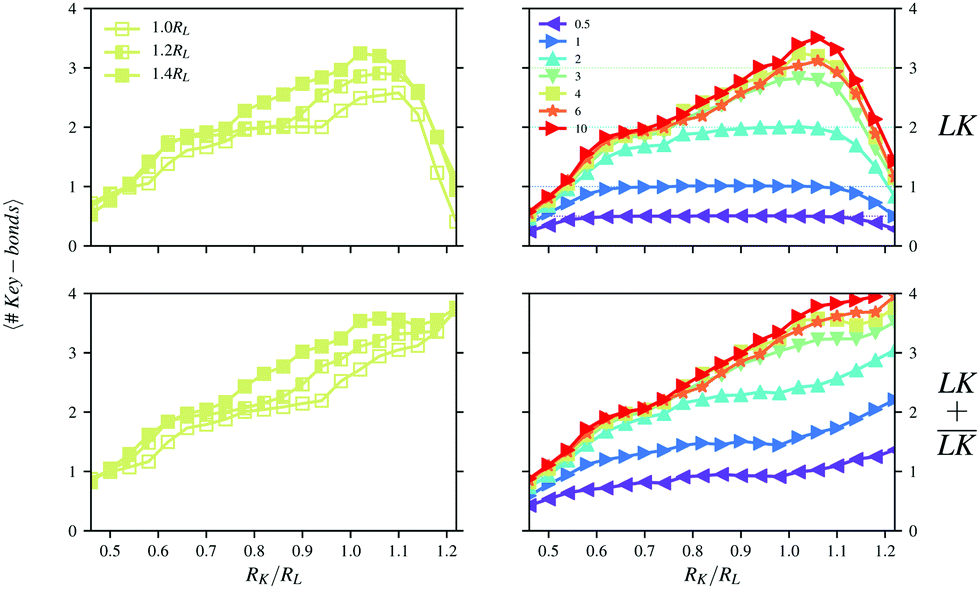
In Fig. 7, the number of bonds per key-particle is shown as a function of the radius of the key-particle. The total number of bonds increases with an increase of RK. The surface-area of the key-particle increases quadratically with RK, which geometrically allows for more lock-particles to associate. Also, since the surface charge-density is constant, the total charge of the particle as well increases quadratically with RK. Yet, going from small to large RK, there is an initial increase of the number of specific bonds, which quickly decays toward zero after its peak value at RK ≈ 1.0 − 1.1RL. This peak is in fact rather a quite broad plateau when NL/NK ≤ 1, whereas for larger NL/NK-values, a single maximum is observed. Note that for NL/NK ≥ 1, a local plateau is as well visible for the transition around 2 specific bonds per key-particle, which is geometrically strongly constrained by the degree of indentation. Thus, depending on the number ratio, the impact of the size of the key-particles on the cluster conformation may be more or less pronounced.
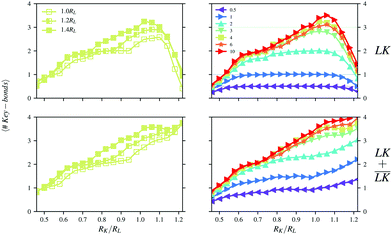 |
| | Fig. 7 Numbers of specific (top) and total (bottom) bonds per key-particle as functions of the size ratio RK/RL at I = 10−5 M. Simulations were performed using (left) NL/NK = 4 for different degrees of indentation c, and (right) using c = 1.4RL and different number ratios NL/NK. | |
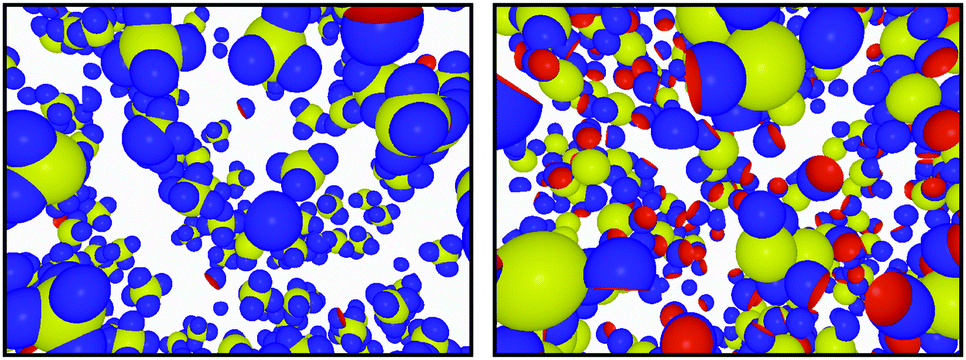
In Fig. 8 we present configurations from simulations where the radius of the cavity perfectly matches the radius of the key-particle, i.e. RC = RK (left), and when it does not, i.e. RC < RK (right). The total number of bonds per key-particle is roughly the same, ∼4; however, for a geometrically perfect match between the cavity and the key-particle, the specific number of bonds is ∼4, while the same number using RC < RK is ∼0. The RC/RK ratio defines the geometrical complementarity of the two components, and is thus of great importance for the assembly specificity, keeping in mind that the energetic contribution is maximum for matching curvatures, i.e. RC/RK = 1. Tuning this parameter therefore allows switching from specific to unspecific bonds. Similar results were obtained for RK = 2RL and NL/NK = 10 as shown in Fig. S12 in the ESI.†
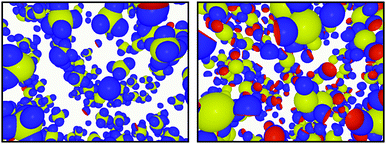 |
| | Fig. 8 Configurations from simulations using NL/NK = 4, ionic strength 10−5 M, and RK = 1.4RL. The left configuration is sampled by using c = RC = RK (i.e. perfect geometric match between the cavity and key-particle) and the right by using c = RC = RL. | |
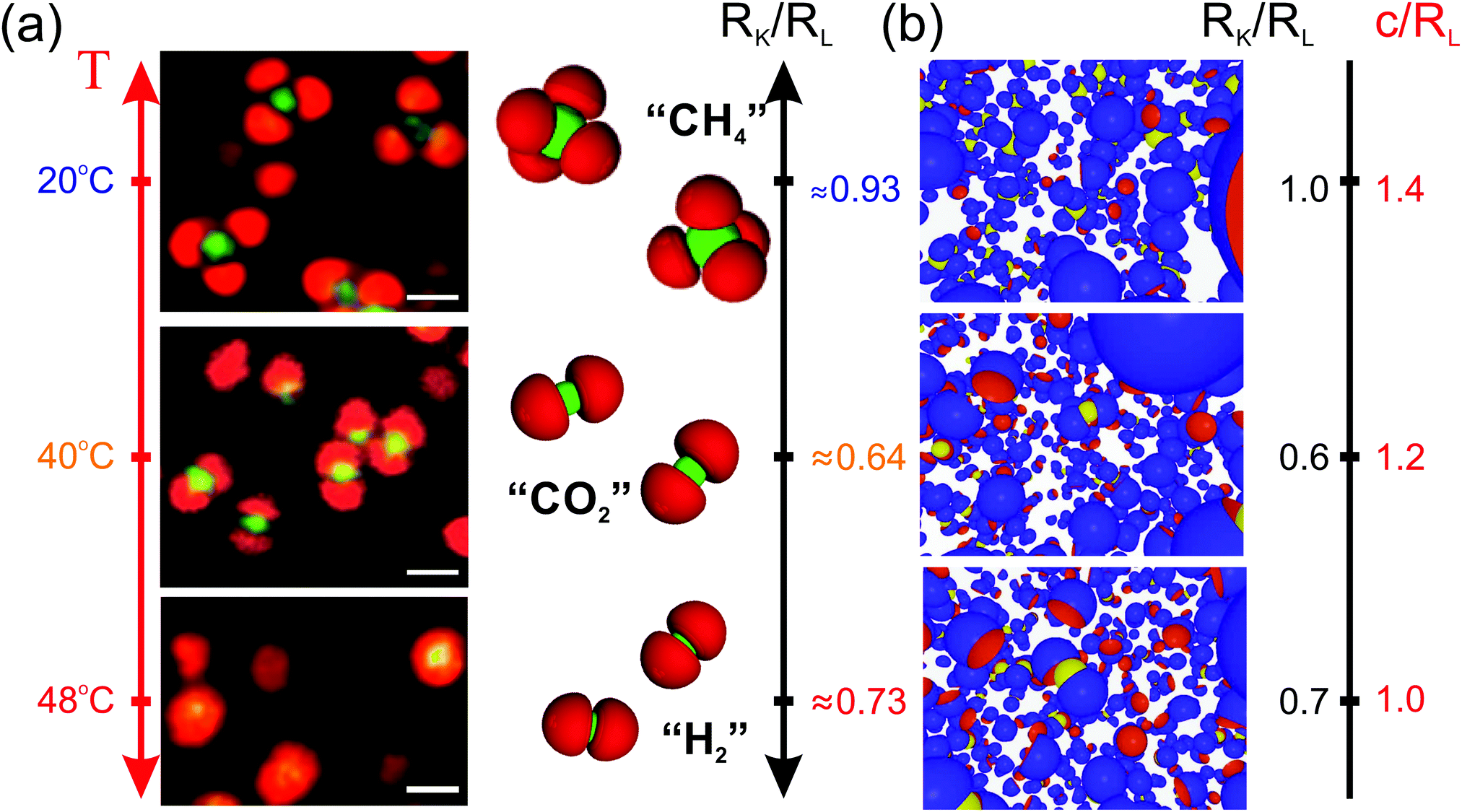
Having established the influence of diverse parameters on the specific lock- and key-particle self-assembly, simulations can now be compared to recent experimental results. Herein, oppositely charged microgel-based particles were associated into specific colloidal molecules as shown in Fig. 9a. Due to their respective thermosensitivity, the size and shape of the particles could be adjusted with temperature as well as the valence and conformation of the colloidal molecules (Fig. 9a). We refer to a previous study for further information on the experimental systems.32 Interestingly, the model presented in this work was able to capture most of the assemblies by fixing the surface charge-density and implementing a size ratio and degree of indention roughly corresponding to different deswellings, see Fig. 9b. In both experiments and simulations, trimeric, “SO32−”-like and tetrameric “CH4”-like colloidal molecules are observed in the swollen regime corresponding to RL ≈ RK and to small lock-indentations with a transition to “CO2”-like molecules for lower size ratios and larger indentations. Although the model captures most of the features, the experimental realizations differ in some aspects. The microgel based systems are soft and deformable. Consequently, the entropic cost is expected to be much lower than that in the simulations. In analogy to the induced fit theory, the microgels may as well adapt their configuration to maximize their contact area. Therefore, we speculate that the interactions for soft particles may become even more specific. In addition, such systems experience van der Waals and hydrophobic interactions at higher temperatures in their collapsed configurations, which thus promotes specific interactions even more. As an example, the key-particle is fully enclosed within two lock-particles, forming an “H2”-configuration in the experiments performed at 48 °C, whereas simulation results exhibit a much looser “CO2”-configuration.
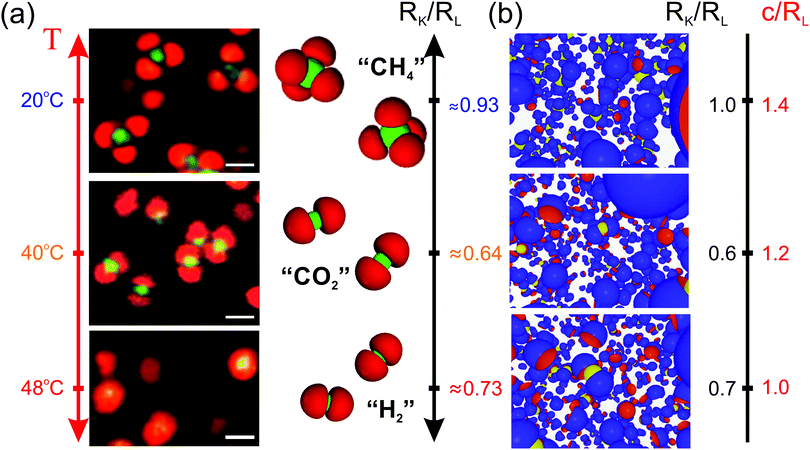 |
| | Fig. 9 (a) 2D CLSM micrographs of a dispersion using NL/V ≈ 0.068 μm−3 and NL/NK ≈ 6. To the right are schematic representations illustrating the specific self-assembly in colloidal molecules with a valency of: ∼4 (CH4; top), ∼2 (CO2; middle), and a collapsed state with ∼2 (H2; bottom).32 Scale bars in (a): 1 μm. (b) Results from simulations using NL/V ≈ 0.086 μm−3 and NL/NK ≈ 6 obtained for different size ratios and degrees of indentation. | |
To conclude this study, we have tested the possibility of encapsulating small key-particles into highly indented lock-particles schematically depicted in Fig. 1b. We use RK = 0.3RL, which is small enough for the key-particle to be enclosed with no entropic cost as discussed earlier. Two simulation snapshots obtained for two different surface charge-densities at I = 10−5 M with NK/NL = 4 are shown in Fig. 10. In the ESI,† we present similar results for NK/NL = 1 (Fig. S11, ESI†). At |σ| = 3.183 × 10−4e nm−2, most of the key-particles were found to be freely dispersed. By charging up both types of particles by a factor of 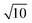 , the entropic barrier for encapsulation was overcome, which leads to an almost complete encapsulation of all key-particles by “Pac-Man”-like lock-particles.
, the entropic barrier for encapsulation was overcome, which leads to an almost complete encapsulation of all key-particles by “Pac-Man”-like lock-particles.
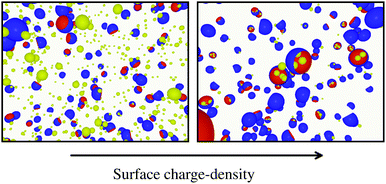 |
| | Fig. 10 Configurations from simulations using NK/NL = 4, I = 10−5 M, c = 0.1RL, RC = 0.95RL, and RK = 0.3RL. The left configuration is sampled by using the absolute surface charge-density |σ| as described earlier, whereas the right configuration used 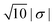 . . | |
Finally, we comment on the effect of counterion-release, which can either promote the association42 or add an additional repulsion in the case of particles presenting different absolute surface charge-densities and perfectly matching curvatures.43 To avoid the latter, we used equally magnified and oppositely polarized charge-densities. Yet, an additional attraction stemming from the counterion-release is to be expected, contributing mostly at large distances and for high charge-densities.44 Since at small distances this effect becomes negligible, such a mechanism is not the origin of the cluster stability.
Conclusion
We have derived an exact analytic expression for the electrostatic potential perfectly in front of and behind a lock-particle, and from this formula approximated the angular-dependent potential. Based on this potential, we have performed simulations of oppositely charged complementary shaped lock- and key-particles. The influence of the charge/ionic strength, the degree of indentation, and the size/number ratio was systematically investigated. We found that a combination of electrostatic interactions with a hard overlap potential is sufficient to specifically assemble and stabilize the two components and form colloidal molecules of different valences, as has been shown in experiments elsewhere. The energetic electrostatic contribution is maximized when the radius of the key-particle matches the size of the cavity; however, this also entails a high entropic cost. Nonetheless, we found that specific interactions are greatly promoted for particles presenting matching curvatures.
Our study provides an alternative to colloidal lock and key self-assembly, which usually relies on short range depletion (excluded volume) interactions. By solely altering the geometry of the particles, electrostatics can induce specific ensembles into well-defined clusters, a feature which further may be amplified or modified by the direct effects of charge-scaling and choice of screening-length. Therefore the model describes a versatile and multifaceted mechanism in understanding and directing self-assembly that could be easily applied to other pair-wise selective interactions and geometries. In addition to the model used for the simulation, the underlying analytic expressions derived in this work indeed allow extensions of the presented approach to a large variety of systems, among others multi-indented lock-particles or particles with an in-built surface roughness.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge financial support from the Knut and Alice Wallenberg Foundation (project grant KAW 2014.0052) and the Swedish Research Council (621-2014-4387).
References
- Y. Zhao, Z. Xie, H. Gu, C. Zhu and Z. Gu, Chem. Soc. Rev., 2012, 41, 3297–3317 RSC.
- A. J. Ridley and A. Hall, Cell, 1992, 70, 389–399 CrossRef CAS.
-
C. I. Brändén and J. Tooze, Introduction to protein structure, Taylor & Francis, 1999 Search PubMed.
-
B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular Biology of the cell, Garland, New York, 5th edn, 2008 Search PubMed.
- S. G. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed.
- A. K. Das, R. Collins and R. V. Ulijn, Small, 2008, 4, 279 CrossRef CAS PubMed.
- P. N. Pusey and W. van Megen, Nature, 1986, 320, 340–342 CrossRef CAS.
- V. J. Anderson and H. N. Lekkerkerker, Nature, 2002, 416, 811 CrossRef CAS PubMed.
- M. E. Leunissen, C. G. Christova, A.-P. Hynninen, C. P. Royall, A. I. Campbell, A. Imhof, M. Dijkstra, R. Van Roij and A. V. Blaaderen, Nature, 2005, 437, 235–240 CrossRef CAS PubMed.
- A. F. Demirörs, J. C. Stiefelhagen, T. Vissers, F. Smallenburg, M. Dijkstra, A. Imhof and A. van Blaaderen, Phys. Rev. X, 2015, 5, 021012 Search PubMed.
- S. C. Glotzer and M. J. Solomon, Nat. Mater., 2007, 6, 557–562 CrossRef PubMed.
- F. Huo, A. K. R. Lytton-Jean and C. A. Mirkin, Adv. Mater., 2006, 18, 2304–2306 CrossRef CAS.
- Y. Wang, Y. Wang, D. R. Breed, V. N. Manoharan, L. Feng, A. D. Hollingsworth, M. Weck and D. J. Pine, Nature, 2012, 491, 51–55 CrossRef CAS PubMed.
- L. Hong, A. Cacciuto, E. Luijten and S. Granick, Nano Lett., 2006, 6, 2510–2514 CrossRef CAS PubMed.
- Z. Zhang and S. C. Glotzer, Nano Lett., 2004, 4, 1407–1413 CrossRef CAS PubMed.
- A. Giacometti, F. Lado, J. Largo, G. Pastore and F. Sciortino, J. Chem. Phys., 2010, 132, 174110 CrossRef PubMed.
- X. Bouju, E. Duguet, F. Gauffre, C. R. Henry, M. L. Kahn, P. Mélinon and S. Ravaine, Adv. Mater., 2018, 30, 1706558 CrossRef PubMed.
- E. Fischer, Eur. J. Inorg. Chem., 1894, 2985–2993 CAS.
- L. Pauling, Chem. Eng. News, 1946, 24, 1375–1377 CrossRef CAS.
- D. E. Koshland, Angew. Chem., Int. Ed. Engl., 1995, 33, 2375–2378 CrossRef.
-
A. R. Fersht, Structure and mechanism in protein science, Macmillan, 1999 Search PubMed.
- J. M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160 RSC.
- J.-M. Lehn, Angew. Chem., Int. Ed., 2015, 54, 3276–3289 CrossRef CAS PubMed.
- S. Sacanna, W. Irvine, P. M. Chaikin and D. J. Pine, Nature, 2010, 464, 575–578 CrossRef CAS PubMed.
- G. Odriozola, F. Jiménez-Ángeles and M. Lozada-Cassou, J. Chem. Phys., 2008, 129, 111101 CrossRef CAS PubMed.
- D. J. Ashton, R. L. Jack and N. B. Wilding, Soft Matter, 2013, 9, 9661–9666 RSC.
- D. Ortiz, K. L. Kohlstedt, T. D. Nguyen and S. C. Glotzer, Soft Matter, 2014, 10, 3541–3552 RSC.
- D. J. Ashton, R. L. Jack and N. B. Wilding, Phys. Rev. Lett., 2015, 114, 237801 CrossRef PubMed.
- A. Van Blaaderen, M. Dijkstra, R. van Roij, A. Imhof, M. Kamp, B. Kwaadgras, T. Vissers and B. Liu, Eur. Phys. J.: Spec. Top., 2013, 222, 2895–2909 Search PubMed.
- J. Borovička, S. D. Stoyanov and V. N. Paunov, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2015, 92, 032730 CrossRef PubMed.
- G. Odriozola and M. Lozada-Cassou, Phys. Rev. Lett., 2013, 110, 105701 CrossRef PubMed.
- A. M. Mihut, B. Stenqvist, M. Lund, P. Schurtenberger and J. J. Crassous, Sci. Adv., 2017, 3, e1700321 CrossRef PubMed.
- J. Borovička, S. D. Stoyanov and V. N. Paunov, Nanoscale, 2013, 5, 8560–8568 RSC.
- B. Beresford-Smith, D. Y. Chan and D. J. Mitchell, J. Colloid Interface Sci., 1985, 105, 216–234 CrossRef CAS.
- R. Kjellander and D. J. Mitchell, J. Chem. Phys., 1994, 101, 603–626 CrossRef CAS.
- M. Trulsson, J. Forsman, T. Åkesson and B. Jönsson, Langmuir, 2009, 25, 6106–6112 CrossRef CAS PubMed.
- J.-M. Meijer and J. J. Crassous, Small, 2018, 1802049 CrossRef PubMed.
- B. Stenqvist, A. Thuresson, A. Kurut, R. Vacha and M. Lund, Mol. Simul., 2013, 39, 1233–1239 CrossRef CAS.
- D. Wu, D. Chandler and B. Smit, J. Phys. Chem., 1992, 96, 4077–4083 CrossRef CAS.
- M. Marechal and M. Dijkstra, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2010, 82, 031405 CrossRef PubMed.
- V. A. Parsegian and D. Gingell, Biophys. J., 1972, 12, 1192–1204 CrossRef CAS PubMed.
- M. T. Record Jr, T. M. Lohman and P. D. Haseth, J. Mol. Biol., 1976, 107, 145–158 CrossRef CAS.
- J. Staahlberg, B. Jönsson and C. Horvath, Anal. Chem., 1991, 63, 1867–1874 CrossRef CAS.
- A. Meier-Koll and C. Fleck, and H. Von Grünberg, J. Phys.: Condens. Matter, 2004, 16, 6041 CrossRef CAS.
- L. Colón-Meléndez, D. J. Beltran-Villegas, G. Van Anders, J. Liu, M. Spellings, S. Sacanna, D. J. Pine, S. C. Glotzer, R. G. Larson and M. J. Solomon, J. Chem. Phys., 2015, 142, 174909 CrossRef PubMed.
- J. J. Thomson, Philos. Mag., 1904, 7, 237–265 CAS.
- L. Föppl, J. Reine Angew. Math., 1912, 141, 251–302 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9sm00795d |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Martin
Trulsson
*a,
Martin
Trulsson
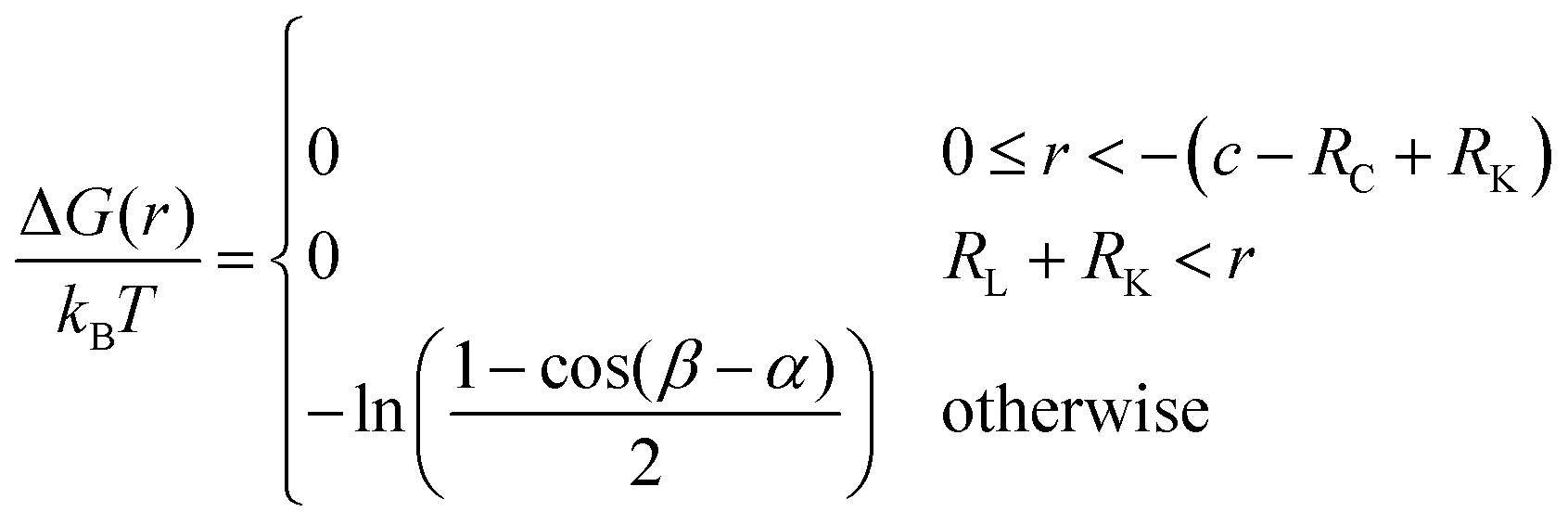
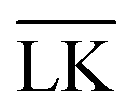 configuration,
configuration, 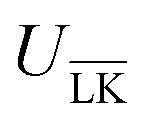 , were calculated for different ionic strengths, I, and size ratios, RK/RL, in Fig. 2. Note how the Debye-length
, were calculated for different ionic strengths, I, and size ratios, RK/RL, in Fig. 2. Note how the Debye-length 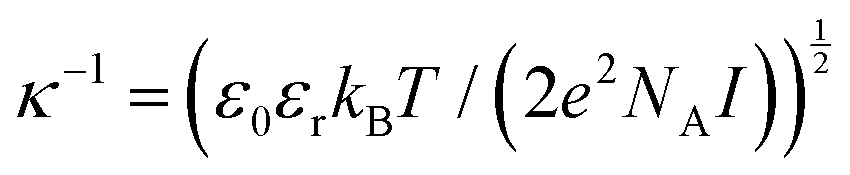 is related to the ionic strength I, where ε0 is the vacuum permittivity, εr = 72 is the relative permittivity of the dispersing medium, T = 293.15 K, e is the elementary charge, and NA is the Avogadro constant. The calculations were performed considering RL = RC = c = 500 nm, oppositely charged lock- and key-particles with a constant absolute surface charge-density |σ| = 3.183 × 10−4e nm−2. By defining the effective charge34–36 of a lock- or key-particle as
is related to the ionic strength I, where ε0 is the vacuum permittivity, εr = 72 is the relative permittivity of the dispersing medium, T = 293.15 K, e is the elementary charge, and NA is the Avogadro constant. The calculations were performed considering RL = RC = c = 500 nm, oppositely charged lock- and key-particles with a constant absolute surface charge-density |σ| = 3.183 × 10−4e nm−2. By defining the effective charge34–36 of a lock- or key-particle as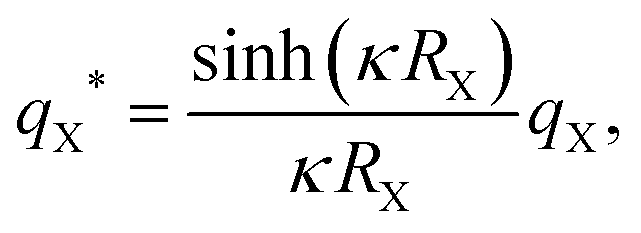
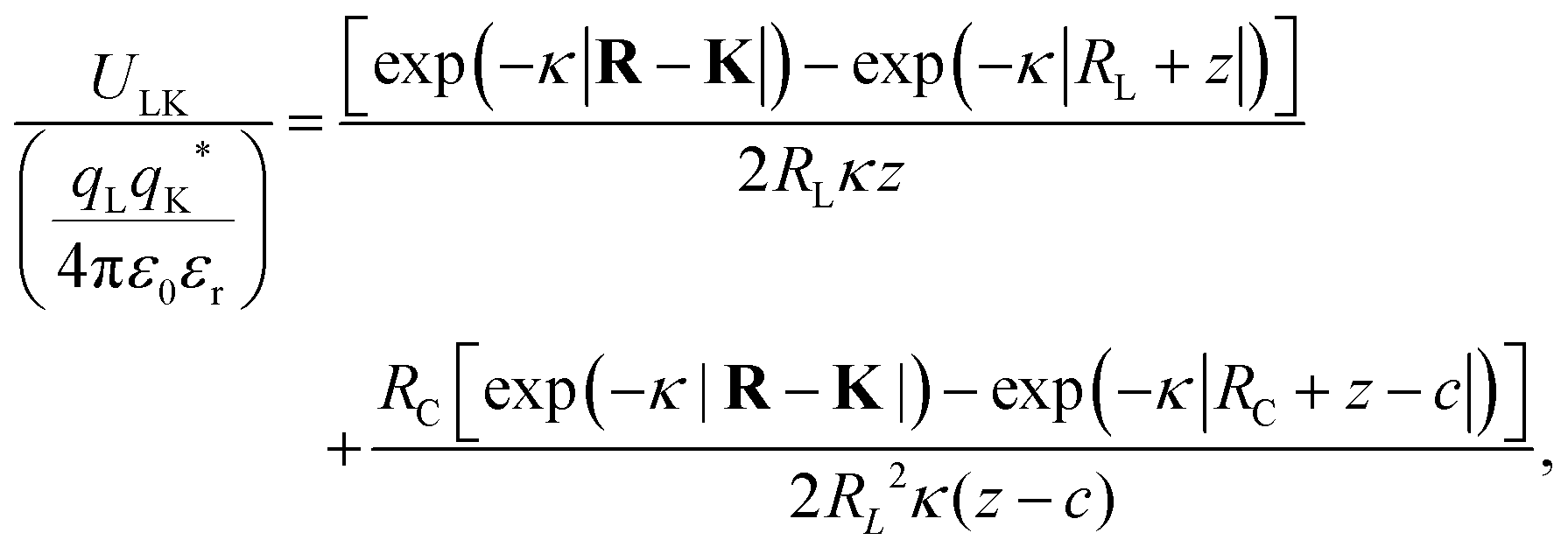
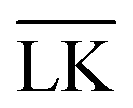 and LK configurations, ΔU. Hence, RK should be equal to RL in order to maximize the interaction-energy difference between specific LK and unspecific
and LK configurations, ΔU. Hence, RK should be equal to RL in order to maximize the interaction-energy difference between specific LK and unspecific 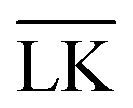 interactions. Such a configuration is quite costly from an entropic point-of-view, see Fig. 1(a) (right), and it is not straightforward to predict the optimal particle design for self-assembly. Simulations are thus needed to model the assembly process, which then also includes many-body effects. However, whereas ΔG is solely defined by the geometries of the constituents, it is important to note that ULK,
interactions. Such a configuration is quite costly from an entropic point-of-view, see Fig. 1(a) (right), and it is not straightforward to predict the optimal particle design for self-assembly. Simulations are thus needed to model the assembly process, which then also includes many-body effects. However, whereas ΔG is solely defined by the geometries of the constituents, it is important to note that ULK, 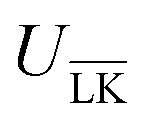 , and ΔU are all strongly dependent on I and |σ|, cf. ΔU ∝ |σ|2, and can thus be tuned over many units of kBT. A specific lock and key self-assembly is therefore expected, at least in the limit of low I and high |σ|.
, and ΔU are all strongly dependent on I and |σ|, cf. ΔU ∝ |σ|2, and can thus be tuned over many units of kBT. A specific lock and key self-assembly is therefore expected, at least in the limit of low I and high |σ|.
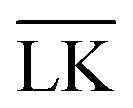 -configuration in the middle panel, and the difference between the two energies is displayed in the bottom panel. The gray dotted lines refer to explicit charge calculations using a lock-particle decorated with 10
-configuration in the middle panel, and the difference between the two energies is displayed in the bottom panel. The gray dotted lines refer to explicit charge calculations using a lock-particle decorated with 10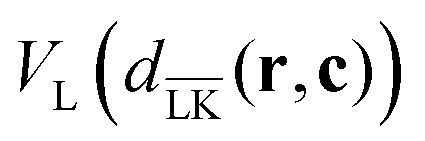 , where c = C − L, the angular-dependent potential VL(r,c) is approximated as
, where c = C − L, the angular-dependent potential VL(r,c) is approximated as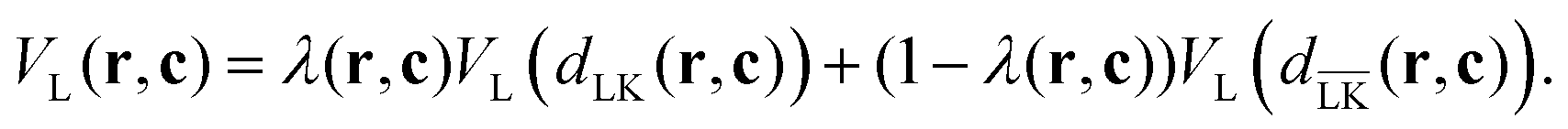
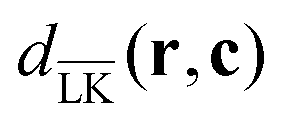 are distances to the surface of the lock-particle. A more detailed description of the potential can be found in the ESI.† In order to assess the validity of the approximate potential, we compared it to the potential of a lock-particle where the surface is decorated with 10
are distances to the surface of the lock-particle. A more detailed description of the potential can be found in the ESI.† In order to assess the validity of the approximate potential, we compared it to the potential of a lock-particle where the surface is decorated with 10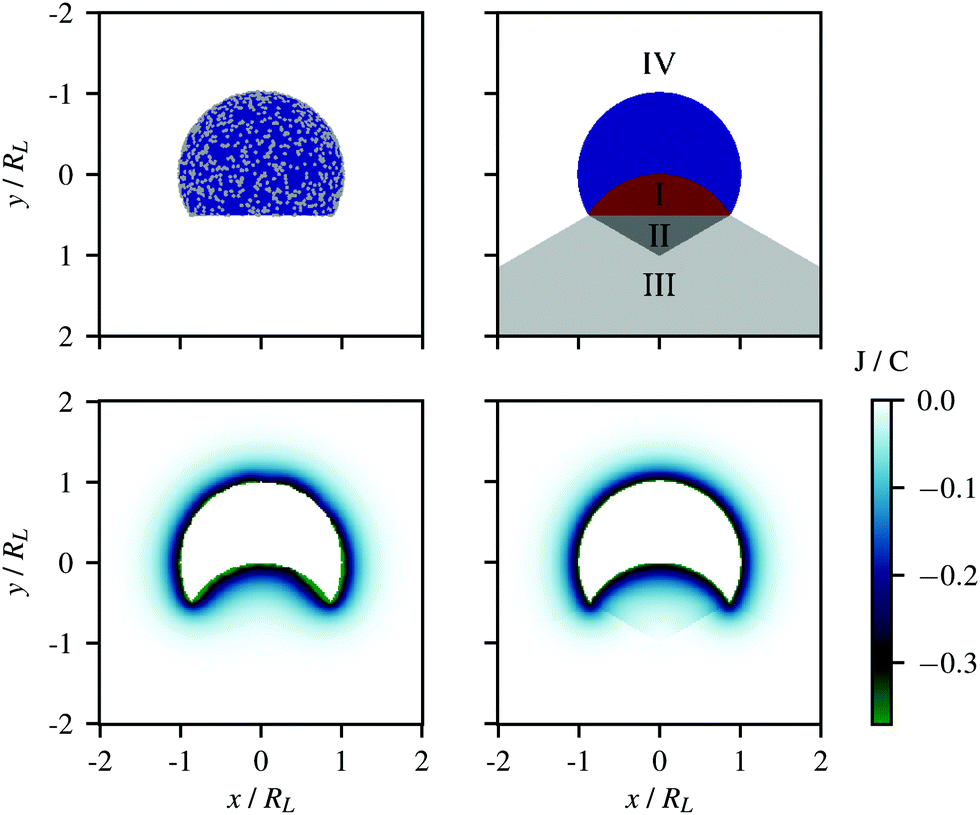
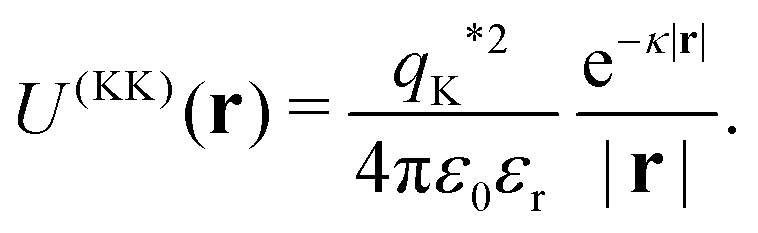
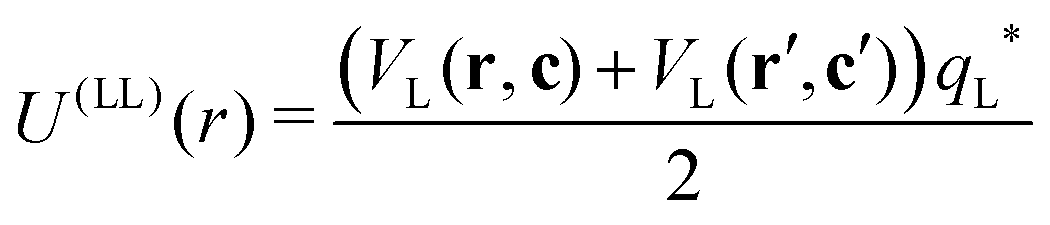
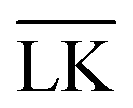 ) when RK + RL < r < 1.1(RK + RL). Different values than 1.1 ranging from 1.05 to 1.20 were tested and found to provide tantamount results within the simulated systems. The simulations in Fig. 1 used NL = NK = 1, no cluster moves, N × 109 simulations steps, and only the hard overlap potential.
) when RK + RL < r < 1.1(RK + RL). Different values than 1.1 ranging from 1.05 to 1.20 were tested and found to provide tantamount results within the simulated systems. The simulations in Fig. 1 used NL = NK = 1, no cluster moves, N × 109 simulations steps, and only the hard overlap potential.
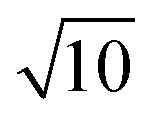 , the entropic barrier for encapsulation was overcome, which leads to an almost complete encapsulation of all key-particles by “Pac-Man”-like lock-particles.
, the entropic barrier for encapsulation was overcome, which leads to an almost complete encapsulation of all key-particles by “Pac-Man”-like lock-particles.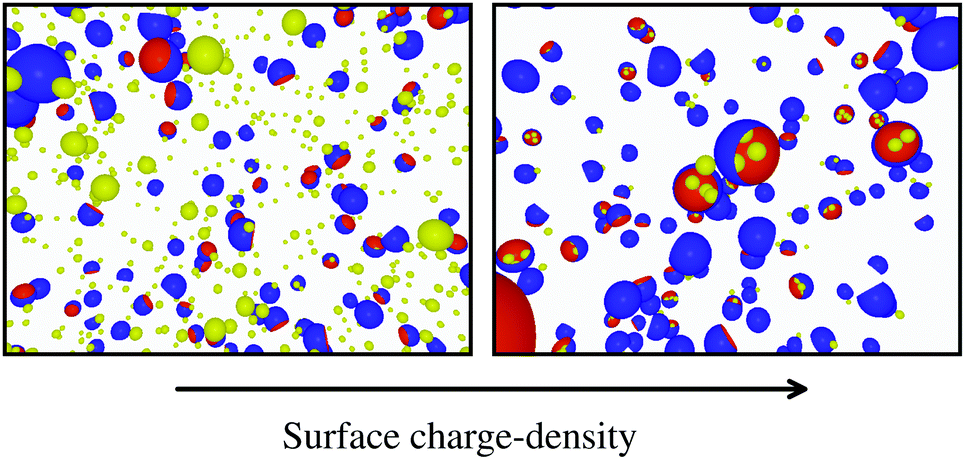
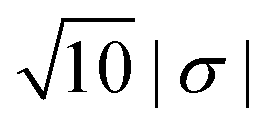 .
.