DOI:
10.1039/C8SM02172D
(Paper)
Soft Matter, 2019,
15, 102-108
Self-complementary hydrogen-bond interactions of guanosine: a hub for constructing supra-amphiphilic polymers with controlled molecular structure and aggregate morphology†
Received
25th October 2018
, Accepted 20th November 2018
First published on 20th November 2018
Abstract
A supra-amphiphilic polymer (SAP) with controlled molecular structures is constructed, in this work, via self-complementary hydrogen bonding of guanosine groups between a hydrophilic block, poly(N-isopropylacrylamide), and a hydrophobic block, poly(ε-caprolactone). By simply changing the mixing ratio of the guanosine-capped hydrophilic and hydrophobic blocks, a series of SAPs with tailored nanostructures are constructed, which can further self-assemble into different nano-aggregates in solution, including spheres, vesicles and large vesicle micelles. The thermo-induced phase transition of the hydrophilic block induces the fusion and aggregation of the nanoparticles into irregular particles upon heating, which further transform to large compound vesicles after cooling.
Introduction
Supra-amphiphiles (SAs) refer to amphiphiles connected with non-covalent interactions and/or dynamic covalent bonds, including hydrogen bonding, coordination, host–guest interactions and π–π packing, etc.1,2 Without tedious organic synthesis, various topological structures, such as single chains, double chains, bolaforms, gemini and rotaxane types, and so on, can be constructed by tuning the linkages as well as the hydrophilic and hydrophobic segments, which can further control self-assembly/disassembly behaviors.2 Therefore, exploring new and appropriate driving forces is essential for fabricating SAs as well as providing environmental stimuli-responsive functions.3,4
Among various supramolecular interactions, hydrogen bonds, such as cyanuric acid and melamine,5 pyrimidinone and naphthyridine,6 uracyl and pyridyl moieties,7,8 nucleobases (A–U, A–T and C–G),9–12 and isoorotic acid and diaminopyridine moieties,13–15 have been frequently exploited to synthesize supramolecular polymers. Nevertheless, these intermolecular hydrogen bonds are hetero-complementary interactions, which in general involve complicated and differentiated syntheses. In comparison, using homo-/self-complementary hydrogen bonds is of more convenience and benefit from obtaining supramolecular polymers. Guanosine, which has two hydrogen-bond acceptors and two hydrogen-bond donors, can self-associate into dimers, ribbons, or macrocycles under different conditions.16 Based on self-complementary interactions, Tam's group17 reported the preparation of a supramolecular star polymer by the post-polymerization of a self-assembled guanosine-containing initiator or by the self-assembly of guanosine-capped linear polymers; Khan and co-workers18,19 presented the synthesis of supramolecular star polymers with heterogeneous compositions by the potassium cation-mediated assembly of guanosine; very recently, as reported by Carducci, Shi, Davis and their coworkers,20–22 supramolecular hydrogels were fabricated via the self-assembly of guanosine; additionally, Dash et al. exploited the self-assembly of guanosine derivatives to develop transmembrane ion channels.23–25 Despite impressive progress of the self-assembly of guanosine in supramolecular science, using such a supramolecular strategy to construct supra-amphiphilic polymers (SAPs) as well as to tune the aggregate morphology remains important but rarely reported. Herein, the assembly precursors are prepared by capping hydrophilic (poly(N-isopropylacrylamide), PNIPAM) and hydrophobic (poly(ε-caprolactone), PCL) chains, respectively, with guanosine. Owing to self-association, the guanosine-terminated building blocks, G-PNIPAM and G-PCL, can self-assemble into nanoparticles in water. Furthermore, different aggregate morphologies are obtained easily by controlling the physically mixing ratio of the two blocks. Because of the conformational transition of PNIPAM upon heating, the temperature-stimulus morphological evolution of the SAP nanoparticles is traced.
Experimental section
Materials
2′,3′-O-Isopropylidene guanosine (G, J&K, 98%) was purified twice by evaporating from dried pyridine. N-Isopropyl acrylamide (NIPAM, J&K, 99%) was purified by recrystallization from hexane. 2,2′-Azobis(isobutyronitrile) (AIBN, TCI, 98%) was purified by recrystallization from ethanol. Propargyl alcohol (Xiya (Shandong), 99%) and ε-caprolactone (ε-CL, J&K, 99%) were distilled twice under reduced pressure, respectively. Besides, all solvents taking part in the reactions were distilled to dewater, i.e. pyridine, dichloromethane (DCM), N,N-dimethylformamide (DMF) and toluene. Other unmentioned chemicals were used directly without further treatment, i.e. 4-dimethylaminepyridine (DMAP, KeLong (Chengdu), AR), Cu(I)Br (J&K, 98%), 1,1,4,7,7-pentamethyldiethylenetriamine (PMDETA, J&K, 98%), 2-bromoisobutyryl bromide (BiBB, J&K, 98%), and 2-aminoethanethiol (AET·HCl, Alfa Aesar, 98%).
Synthesis of 5′-O-isobromobutyryl-2′,3′-O-isopropylidene guanosine (G-Br)
According to literature procedures,20,24 5′-O-isobromobutyryl-2′,3′-O-isopropylidene guanosine was obtained by the reaction of 2′,3′-O-isopropylidene guanosine (G) and 2-bromoisobutyryl bromide (BiBB) employing DMAP as the catalyst under a nitrogen atmosphere at room temperature for 18 h. After quenching with 2 mL MeOH and removal of the solvent, the product was obtained from chromatography (silica gel) with 2% MeOH in CH2Cl2 as an eluent. 1H-NMR (400 MHz, DMSO-d6), δ (TMS, ppm): 10.73 (1H, NH), 7.86 (1H, H-1), 6.56 (2H, NH2), 6.04 (1H, H-1′), 5.23 (1H, H-2′), 5.18 (1H, H-3′), 4.38, 4.27 (3H, H-4′, H-5′), 3.36 (H2O), 2.50 (DMSO-d6), 1.86 (6H, H-6′), 1.51 (3H, H-2), 1.32 (3H, H-3).
Synthesis of guanosine-terminal poly(N-isopropylacrylamide) (G-PNIPAM)
G-PNIPAM was prepared via an ATRP reaction.26,27 A mixture of G-Br (0.5 mmol) and NIPAM (45 mmol) was added to the pre-dried Schlenk flask, previously deoxygenated by degassing and backfilling with argon three times. Dry DMF was then injected, and after homogenization, Cu(I)Br (0.75 mmol) was added. The mixed solution was degassed by three freeze–pump–thaw cycles, and when it maintained at room temperature, PMDETA was added. Then the reaction mixture was degassed repeatedly and stirred under an argon atmosphere in a pre-heated oil-bath at 70 °C for 24 h. The reaction solution diluted with THF was induced through a neutral Al2O3 column to remove the catalyst. The filtrate was removed under reduced pressure, and the product was recovered as a light yellow solid after precipitating in diethyl ether three times. 1H-NMR (400 MHz, DMSO-d6), δ (TMS, ppm): 0.89 (6H, –C(CH3)2), 1.04 (–NHCH(CH3)2), 1.47 (–CH–CH2– on the main chain), 1.96 (–CH–CH2– on the main chain), 3.85 (–NHCH(CH3)2), 7.21 (–NHCH(CH3)2), 7.96 (1H, H-1).
Synthesis of 5′-O-isoazidobutyryl-2′,3′-O-isopropylidene guanosine (G-N3)
A procedure for the preparation of G-N3 was as follows.24,26 1 mmol G-Br and 5 mmol NaN3 were dissolved in 40 mL of DMF, and stirred at ambient temperature overnight. The mixture was filtered and the filtrate was evaporated to dryness. The product was obtained after precipitation in 95% ethanol. After drying in a vacuum oven overnight at room temperature, it was obtained as a white powder. 1H-NMR (400 MHz, DMSO-d6), δ (TMS, ppm): 10.71 (1H, NH), 7.84 (1H, H-1), 6.54 (2H, NH2), 6.03 (1H, H-1′), 5.24 (1H, H-2′), 5.17 (1H, H-3′), 4.37 (2H, H-4′), 4.28 (1H, H-5′), 3.32 (H2O), 2.50 (DMSO-d6), 1.51 (3H, H-2), 1.37 (6H, H-6′), 1.32 (3H, H-3).
Synthesis of alkyne-terminal poly(ε-caprolactone) (PCL-alkyne)
PCL-alkyne was prepared by the ROP reaction with propargyl alcohol as an initiator.28 Novozyme 435 was added to a dried 100 mL round-bottom flask, which was previously deoxygenated by degassing and backfilling with nitrogen three times. Dry toluene, dry propargyl alcohol and dry ε-caprolactone were then injected under a nitrogen atmosphere. When homogenized, the transparent solution was stirred at 70 °C for 24 h under a N2 atmosphere. After cooling to room temperature, Novozyme 435 was removed by filtration. The filtrate was removed under reduced pressure and then dissolved in chloroform. The product was obtained after precipitation in cold diethyl ether. 1H-NMR (400 MHz, CDCl3), δ (TMS, ppm): PCL-alkyne: 1.38 (H-4), 1.65 (H-3, H-5), 2.31 (H-2), 2.47 (HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–), 3.65 (H-7), 4.06 (H-6), 4.57 (HCC–CH2–).
C–), 3.65 (H-7), 4.06 (H-6), 4.57 (HCC–CH2–).
Synthesis of guanosine-functionalized poly(ε-caprolactone) (G-PCL)
According to a procedure as described,29,30 a mixture of 0.3 mmol G-N3 and 0.1 mmol PCL-alkyne was added into the pre-dried Schlenk flask, previously deoxygenated by degassing and backfilling with argon three times. When homogenized with DMF (50 mL), catalytic amounts of Cu(I)Br and PMDETA were added. The mixture was stirred under a nitrogen atmosphere in a pre-heated oil-bath at 60 °C for 24 h. The reaction was quenched with 2 mL of MeOH, and the solvent was removed under reduced pressure. The product was obtained as a white powder after precipitation in cold diethyl ether. 1H-NMR (400 MHz, DMSO-d6), δ (TMS, ppm): G-PCL: 1.29 (H-3), 1.53 (H-2, H-4), 2.27 (H-1), 2.50 (DMSO-d6), 3.33 (H2O), 3.98 (H-5), 4.34 (H-4′), 4.54 (H-5′), 5.21 (H-3′), 5.30 (H-2′), 5.91 (H-1′).
As a control, guanosine-free poly(N-isopropylacrylamide) was also synthesized. The free-radical polymerization was conducted in DMF with AIBN and AET·HCl as the initiator and the chain transfer reagent, respectively.311H-NMR (400 MHz, D2O), δ (TMS, ppm): PNIPAM-NH2: 1.04 (–NHCH(CH3)2), 1.47 (–CH–CH2– on the main chain), 1.89 (–CH–CH2– on the main chain), 3.79 (–NHCH(CH3)2), 4.70 (D2O).
Synthesis of 5′-O-acetyl-2′,3′-O-isopropylidene guanosine (G-OAc)
G-OAc was synthesized via a procedure as reported.32,33 A mixture of 1 mmol 5′-O-isobromobutyryl-2′,3′-O-isopropylidene guanosine, 2 mmol acetic anhydride, Et3N and DMAP was dissolved in MeCN. After stirring for 24 h at r.t., the reaction was quenched with MeOH, and a white solid was obtained after filtration and washing with MeCN/H2O. 1H-NMR (400 MHz, DMSO-d6), δ (TMS, ppm): G-OAc: 1.32 (H-3), 1.52 (H-3, H-2), 1.99 (H-6′), 2.50 (DMSO), 3.35 (H2O), 4.11 (H-4′), 4.25 (H-5′), 5.14 (H-3′), 5.26 (H-2′), 6.02 (H-1′), 6.56 (NH2), 7.86 (H-1), 10.73 (N–H).
Preparation of micelles
Mixtures with different weight ratios were dissolved in a good solvent, DMSO, at room temperature. After complete dissolution, the solutions were added dropwise into deionized water under vigorous stirring. The mixtures were dialyzed against 1 L of deionized water using a dialysis bag (Mw cutoff: 3500 Da) for 24 h, during which the water was renewed every 4 h. Finally, after dialysis solutions were filtered, the samples of P1, P2 and P3 were obtained respectively by adjusting the volume of water to a total concentration of 1 mg mL−1. G-PNIPAM was directly dissolved into deionized water at 1 mg mL−1 to obtain the P0 sample.
Proton nuclear magnetic resonance (1H-NMR)
The 1H-NMR spectra were recorded on a Bruker Advance 400 MHz spectrometer (Germany). DMSO-d6 was chosen as the solvent (DMSO-d6: δ 3.33 ppm and H2O: δ 2.50 ppm) with TMS as the internal standard for G-Br, G-N3, G-PNIPAM and G-PCL at room temperature, and CDCl3 (CDCl3: δ 7.26 ppm) was used for PCL-alkyne, and D2O (D2O: δ 4.70 ppm) was used for PNIPAM-NH2.
MALDI-TOF mass spectrometry (MALDI-TOF MS)
The molecular weights of the samples were determined by MALDI-TOF mass spectrometry (MALDI-TOF MS, AXIMA Performance 2.9.4.1, Shimadzu, Japan), equipped with a 337 nm N2 laser at an acceleration voltage of 20 kV. α-Cyano-4-hydroxycinnamic acid (CHCA) was used as a matrix for molecular weight determination.
Fourier transform infrared spectroscopy (FT-IR)
Fourier transform infrared spectroscopy (FT-IR) analysis was performed on a Nicolet 6700 infrared spectrometer with KBr cells. The spectra were recorded with 64 scans at a resolution of 4 cm−1 in the region of 4000–400 cm−1.
Circular dichroism spectroscopy (CD)
To identify the conformation of guanosines, the circular dichroism (CD) spectrum of G-OAc (0.04 mg mL−1 in CHCl3) was measured using a JASCO J1500 spectrophotometer (Japan) with a quartz cuvette of 1 cm path length in the region of 200–360 nm.
Critical micelle concentration (CMC)
The critical micelle concentrations (CMCs) of the samples were determined using the fluorescent probe method with pyrene on a Varian Cary Eclipse fluorescence spectrometer (Varian Co., USA). The sample concentration varied from 1.0 × 10−4 to 0.1 mg mL−1, and the final pyrene concentration was maintained at 6 × 10−4 mol L−1. The experiments were carried out at room temperature under an emission wavelength of 372 nm. The CMC values of P1–P3 were calculated from the inflection point of polymer concentration where the relative excitation fluorescence intensity ratio (I337/I333) was changed.
Dynamic light scattering (DLS)
The hydrodynamic diameters of micelles (1 mg mL−1 in aqueous solution) were measured using a Zeta-sizer Nano ZS90 (Malvern Instruments, Ltd, UK) with a He–Ne laser (633 nm) and 90° collecting optics in the region of 25 °C–40 °C. All results were the average values of the three tests.
Transmission electron microscopy (TEM)
The micrographs of the polymer micelles were obtained using a Hitachi H600 transmission electron microscope (TEM) at an operated voltage of 75 kV. Samples (1 mg mL−1) were dropped on carbon-coated copper grids and stained with 2% (w/v) phosphotungstic acid.
Turbidity measurements
The optical transmittances of the aqueous solutions (0.5 mg mL−1) were acquired on a UV-vis spectrophotometer with a Varian Cary® 50 (Varian Co., USA) and measured at a wavelength of 500 nm using a thermostatically controlled cuvette.
Results and discussion
Prior to the construction of SAPs, the hydrophilic and hydrophobic blocks, G-PNIPAM and G-PCL, are synthesized by atom transfer radical polymerization (ATRP), ring-opening polymerization (ROP), and azide–alkyne cyclo-addition reactions (Scheme 1). Their chemical structures are characterized by 1H-NMR and FT-IR measurements. 5′-O-Isobromobutyryl-2′,3′-O-isopropylidene guanosine, of which the 1H-NMR spectrum is shown in Fig. S1 (ESI†), is selected as an initiator of ATRP to synthesize G-PNIPAM. As shown in Fig. S2 (ESI†), the proton peaks (1.47 and 1.96 ppm for CH2–CH–; 1.04 and 3.85 ppm for the isopropyl group) attributing to PNIPAM26,27,34 as well as the H-1 signal of the guanosine group are obviously observed for G-PNIPAM. Additionally, the characteristic FT-IR peaks corresponding to the amide group of PNIPAM and the C–O–C group of guanosine are determined to be at 1655 cm−1, 1545 cm−1 and 1131 cm−1 (Fig. S3, ESI†). These results confirm the successful synthesis of the hydrophilic block, PNIPAM, with the terminal guanosine group. On the other hand, once the alkyne–azide conjugation between G-N3 and PCL-alkyne occurs, the proton peaks of PCL15,29 at 1.29, 1.53, 2.27 and 3.98 ppm as well as those of ribose of guanosine at 4–6 ppm are detected in G-PCL (Fig. S4–S6, ESI†), and the FT-IR peaks of the azide group near 2119 cm−1 disappear while the new stretching peak of the tri-azole ring appears at 3126 cm−1 (Fig. S7, ESI†).35,36 These results indicate the successful conversion of the terminal group of PCL from alkyne to guanosine. Meanwhile, compared with PCL-alkyne, a shorter elution time with a unimodal peak is observed for G-PCL as shown in Fig. S8a (ESI†), which further confirms the successful click chemistry reaction. Furthermore, to obtain the accurate molecular weights of the homopolymers, the MALDI-TOF-MS measurement is performed for G-PNIPAM and G-PCL. The molecular weights of the two blocks are determined to be around 3.3 × 103 and 2.8 × 103 g mol−1 (Fig. S8b and c, ESI†). According to some previous reports, the lengths of the two blocks are suitable for further development of SAPs.37
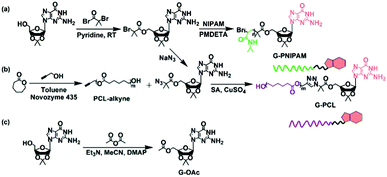 |
| | Scheme 1 Schematic illustration of the synthetic routes to (a) G-PNIPAM, (b) G-PCL, and (c) the small molecular model, G-OAc. | |
Considering the low resolution of the terminal guanosine group on polymeric blocks during the investigation on non-covalent interactions, a small model molecule, 5′-O-acetyl-2′,3′-O-isopropylidene guanosine (G-OAc) synthesized by the transesterification between 2′,3′-O-isopropylideneguanosine and acetic anhydride (Scheme 1c), is used to verify the association between the guanosine groups. A proton signal, corresponding to the COOCH3 structure, is detected at 1.99 ppm (Fig. S9, ESI†), and a FT-IR band attributing to the ester group is determined to be at 1745 cm−1 (Fig. S10, ESI†). These results confirm the successful synthesis of G-OAc. As has been reported,38,39 the C(6)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group plays a crucial role in forming inter-base hydrogen bonding, of which the IR peak can be observed below 1700 cm−1. Therefore, G-OAc is dissolved in CHCl3 and characterized by FT-IR after removing the solvent. Notably, the peak belonging to the C(6)O group of G-OAc appears at 1691 cm−1 instead of 1706 cm−1 at the original state, suggesting the occurrence of hydrogen bonds between the model molecules. Further evidence provided by CD measurement also confirms the intermolecular interaction because of the determination of two opposite CD signatures at 265 nm and 293 nm (Fig. S11, ESI†).40,41
O group plays a crucial role in forming inter-base hydrogen bonding, of which the IR peak can be observed below 1700 cm−1. Therefore, G-OAc is dissolved in CHCl3 and characterized by FT-IR after removing the solvent. Notably, the peak belonging to the C(6)O group of G-OAc appears at 1691 cm−1 instead of 1706 cm−1 at the original state, suggesting the occurrence of hydrogen bonds between the model molecules. Further evidence provided by CD measurement also confirms the intermolecular interaction because of the determination of two opposite CD signatures at 265 nm and 293 nm (Fig. S11, ESI†).40,41
As is well known, the hydrophilic–hydrophobic balance of SAPs is a key issue to morphology control.42–45 Based on the hydrogen bonding of the guanosine group, different weight ratios of G-PNIPAM/G-PCL (10.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.0, 9.0:1.0, 7.0:3.0 and 4.0:6.0 as designed) are set to prepare SAPs, which are named P0, P1, P2 and P3, respectively. To provide more scientific structural information of the nanoparticles, the actual mixing ratios are determined from the 1H-NMR spectra of the P1, P2 and P3 nanoparticles after dialysis, that is, 9.3:0.7, 7.8:2.2 and 4.4:5.6, respectively (Table S1, ESI†). As shown in Fig. 1, the critical micelle concentration (CMC) of the SAPs decreases with the increased amount of G-PCL, which can be easily explained by the fact that a thin hydrophilic shell is conducive to self-assembly.47 Consequently, the Tyndall effect becomes more obvious at a fixed concentration of 1 mg mL−1 (Fig. 2a), primarily indicating the formation of the nanoparticles.46 The hydrodynamic diameter is then investigated by dynamic light scattering (DLS) (Fig. 2b). A trimodal peak is observed at 26, 74, and 203 nm for P0, suggesting the presence of inhomogeneous nanoparticles as a result of the lack of hydrophobic blocks.47,48 In contrast, unimodal DLS peaks are detected for P1–P3, and the hydrodynamic diameter increases from 144 nm (P1) to 246 nm (P3). The increased particle size as a result of the decrease of the hydrophilic block proportion has been frequently described before.48 To further illustrate the supramolecular interaction effect of the terminal guanosine group on the self-assembly performance, a control sample, PNIPAM-NH2, is also synthesized via free-radical polymerization, whose chemical structure is confirmed by the 1H-NMR spectrum (Fig. S12a, ESI†). From the MALDI-TOF MS analysis, its molecular weight is determined to be 2.5 × 103 g mol−1. Similarly for the preparation of P1–P3 nanoparticles, PNIPAM-NH2 and G-PCL at a weight ratio of 7.0:3.0 are mixed in DMSO and subsequently dialyzed against water. However, during the dialysis process, obvious precipitation is observed (Fig. S12b, ESI†), suggesting that the supramolecular conjugation and self-assembly do not occur for the two homopolymer blocks. As a result, the guanosine-terminal is confirmed as a requisite for the amphiphilic self-assembly performance.
:0.0, 9.0:1.0, 7.0:3.0 and 4.0:6.0 as designed) are set to prepare SAPs, which are named P0, P1, P2 and P3, respectively. To provide more scientific structural information of the nanoparticles, the actual mixing ratios are determined from the 1H-NMR spectra of the P1, P2 and P3 nanoparticles after dialysis, that is, 9.3:0.7, 7.8:2.2 and 4.4:5.6, respectively (Table S1, ESI†). As shown in Fig. 1, the critical micelle concentration (CMC) of the SAPs decreases with the increased amount of G-PCL, which can be easily explained by the fact that a thin hydrophilic shell is conducive to self-assembly.47 Consequently, the Tyndall effect becomes more obvious at a fixed concentration of 1 mg mL−1 (Fig. 2a), primarily indicating the formation of the nanoparticles.46 The hydrodynamic diameter is then investigated by dynamic light scattering (DLS) (Fig. 2b). A trimodal peak is observed at 26, 74, and 203 nm for P0, suggesting the presence of inhomogeneous nanoparticles as a result of the lack of hydrophobic blocks.47,48 In contrast, unimodal DLS peaks are detected for P1–P3, and the hydrodynamic diameter increases from 144 nm (P1) to 246 nm (P3). The increased particle size as a result of the decrease of the hydrophilic block proportion has been frequently described before.48 To further illustrate the supramolecular interaction effect of the terminal guanosine group on the self-assembly performance, a control sample, PNIPAM-NH2, is also synthesized via free-radical polymerization, whose chemical structure is confirmed by the 1H-NMR spectrum (Fig. S12a, ESI†). From the MALDI-TOF MS analysis, its molecular weight is determined to be 2.5 × 103 g mol−1. Similarly for the preparation of P1–P3 nanoparticles, PNIPAM-NH2 and G-PCL at a weight ratio of 7.0:3.0 are mixed in DMSO and subsequently dialyzed against water. However, during the dialysis process, obvious precipitation is observed (Fig. S12b, ESI†), suggesting that the supramolecular conjugation and self-assembly do not occur for the two homopolymer blocks. As a result, the guanosine-terminal is confirmed as a requisite for the amphiphilic self-assembly performance.
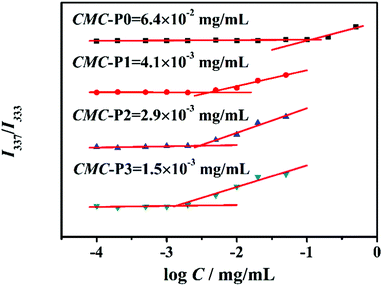 |
| | Fig. 1 Critical micelle concentrations of P0–P3. | |
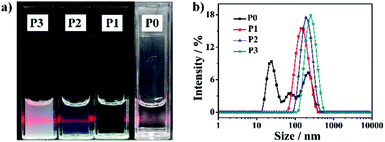 |
| | Fig. 2 (a) Digital photographs and (b) size distribution of P0–P3. | |
From the transmission electron microscopy (TEM) images shown in Fig. 3a–d, a spherical morphology is observed for P0 and P1. With a further increase in G-PCL, besides the spherical micelles, vesicles are observed for P2, and upon increasing the weight ratio of G-PNIPAM/G-PCL to 4.4:5.6, large compound vesicles are formed. Compared with the DLS results, the particle sizes determined from TEM are relatively small because of the dehydration. To understand the mechanism of the formation of different morphologies, a concept, such as hydrophilic volume fraction (f) (see the ESI† for details) described before,46,49,50 is used. Theoretically, spherical micelles should be formed when f > 50%; worm-like micelles when 40% < f < 50%; and vesicle/lamellar structure when f < 40%. Regarding the respective hydrophilic and hydrophobic roles of G-PNIPAM and G-PCL, the f values are calculated to be ∼100% (P0), 79% (P1), 65% (P2) and 35% (P3). It is easy, therefore, to understand the transition from the spherical morphology to complex ones, and the slightly different f-dependent morphological transition, as compared with previous studies,51 might be attributed to the architecture of the resultant SAPs.
 |
| | Fig. 3 TEM images of P0 (a), P1 (b), P2 (c), and P3 (d) at room temperature. Inset: Schematic representation of different nanoparticles, where green parts are the hydrophilic PNIPAM chains, purple parts are the hydrophobic PCL chains, and small white spheres represent the pores. | |
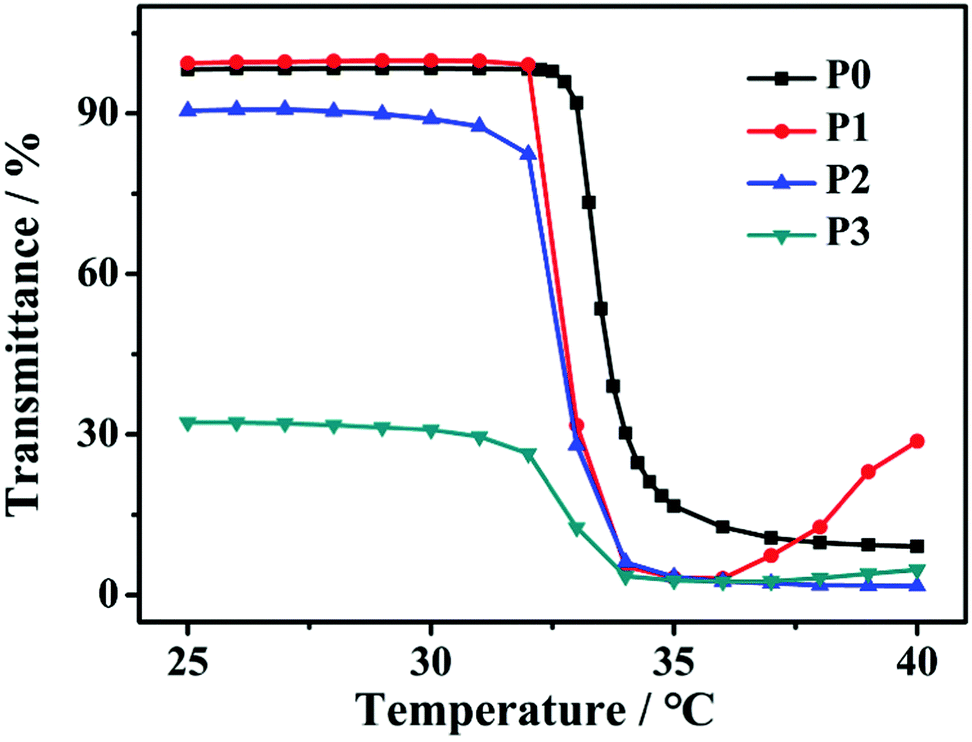
In addition, considering the thermo-induced phase transition of PNIPAM,52,53 the nanoparticle morphology is further tuned above the lower critical solution temperature (LCST). As shown in Fig. 4, the LCST of all samples is measured to be about 33 °C. Here, two representative SAPs, P1 and P3, are selected to investigate the temperature-dependent morphological transformation. Upon heating to 40 °C, the hydrodynamic diameters of P1 and P3 increase to 432 nm and 800 nm, respectively (Fig. 5a). In accordance with the DLS result, at the temperature above LCST, obvious aggregation of the nanoparticles is detected, forming irregular particles (Fig. 6a and b). Besides the presence of particle aggregation with boundaries (marked by yellow arrows), we can also see some fused micelles without clear boundaries (marked by red arrows). As a result, the increased particle size is reasonably determined. To further understand the reversibility of the morphological transformation, the colloidal solution is cooled back to 25 °C. Apparently, a drop of particular size is observed, that is, 161 nm for P1 and 268 nm for P3 (Fig. 5b), which can also be due to the reversible phase transition of PNIPAM. With respect to morphology, however, large compound vesicles are observed for both P1 and P3 after cooling (Fig. 6c and d). This phenomenon can be explained by the aggregation and fusion of individual particles at 40 °C, as well as the incomplete dissociation or cleavage of “large” clusters.52 Based on the results, a mechanism illustrating the self-assembly of G-PNIPAM and G-PCL into different nano-objects is proposed in Scheme 2. The mixing ratio of hydrophilic G-PNIPAM and hydrophobic G-PCL blocks determines the morphology of resultant nanoparticles, that is, from spheres to vesicles and further to large compound vesicles with the increased contents of G-PCL. Besides, benefiting from the thermo-induced phase transition of G-PNIPAM, obvious aggregation and fusion of nanoparticles occur for spheres and large compound vesicles after heating, resulting in the formation of irregular particles with increased sizes. Upon cooling, dissociation of larger irregular particles occurs; however, their morphology cannot change reversibly back to nano-spheres but to relatively small compound vesicles.
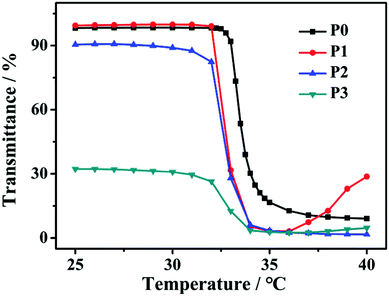 |
| | Fig. 4 Temperature-dependent optical transmittance curves of P0–P3 aqueous solutions at 500 nm with a concentration of 0.5 mg mL−1. | |
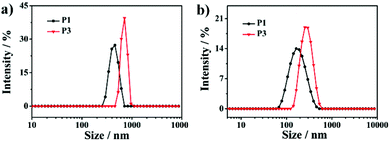 |
| | Fig. 5 Size distribution of P1 and P3 (a) at 40 °C and (b) at 25 °C after a heating–cooling cycle. | |
 |
| | Fig. 6 TEM images of (a) P1 at 40 °C, (b) P3 at 40 °C, (c) P0 at 25 °C, and (d) P3 at 25 °C after a heating–cooling cycle. | |
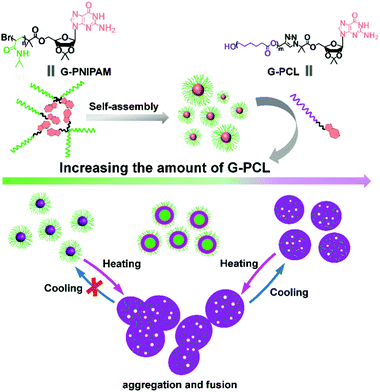 |
| | Scheme 2 A proposed mechanism for the formation of different nano-objects by the self-assembly of G-PNIPAM and G-PCL. Green parts are the hydrophilic PNIPAM chains, purple parts are the hydrophobic PCL chains, and small white spheres represent the pores. | |
Conclusions
In summary, self-complementary hydrogen bonding of guanosine is successfully employed to fabricate a series of SAPs. Owing to the adjusted amphiphilicity by changing the mixing ratio of the guanosine-capped hydrophilic and hydrophobic blocks, the SAPs can self-assemble into nano-aggregates with different morphologies, including spheres, vesicles, and large compound vesicles. Benefiting from the temperature-induced phase transition of the hydrophilic blocks, the aggregation and fusion of the nanoparticles display a temperature-dependent morphological transition, that is, from spheres or vesicles to irregular particles after heating, and then to large compound vesicles after cooling. These results indicate that the supramolecular assembly strategy is effective to build complicated molecular topology as well as adjust aggregate morphology.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the National Key R&D Program of China (2016YFB0302102), the National Natural Science Foundation of China (Grants 51773133 and 51721091), and the Program for Changjiang Scholars and Innovative Research Teams in University of China (IRT 1026).
Notes and references
- Y. T. Kang, X. Y. Tang, Z. G. Cai and X. Zhang, Adv. Funct. Mater., 2016, 26, 8920 CrossRef CAS.
- X. Zhang and C. Wang, Chem. Soc. Rev., 2011, 40, 94 RSC.
- Y. P. Wang, H. P. Xu and X. Zhang, Adv. Mater., 2009, 21, 2849 CrossRef CAS.
- K. Liu, Y. T. Kang, Z. Q. Wang and X. Zhang, Adv. Mater., 2013, 25, 5530 CrossRef CAS.
- N. Kimizuka, T. Kawasaki and T. Kunitake, J. Am. Chem. Soc., 1993, 115, 4387 CrossRef CAS.
- K. E. Feldman, M. J. Kade, T. F. A. de Greef, E. W. Meijer, E. J. Kramer and C. J. Hawker, Macromolecules, 2008, 41, 4694 CrossRef CAS.
- K. Yoosaf, A. Belbakra, N. Armaroli, A. Llanes-Pallasb and D. Bonifazi, Chem. Commun., 2009, 2830 RSC.
- A. Goujon, G. Y. Du, E. Moulin, G. Fuks, M. Maaloum, E. Buhler and N. Giuseppone, Angew. Chem., Int. Ed., 2016, 55, 703 CrossRef CAS.
- D. L. Wang, Y. Su, C. Y. Jin, B. S. Zhu, Y. Pang, L. J. Zhu, J. Y. Liu, C. L. Tu, D. Y. Yan and X. Y. Zhu, Biomacromolecules, 2011, 12, 1370 CrossRef CAS.
- D. L. Wang, X. Y. Huan, L. J. Zhu, J. Y. Liu, F. Qiu, D. Y. Yana and X. Y. Zhu, RSC Adv., 2012, 2, 11953 RSC.
- D. L. Wang, H. Y. Chen, Y. Su, F. Qiu, L. J. Zhu, X. Y. Huan, B. S. Zhu, D. Y. Yan, F. L. Guo and X. Y. Zhu, Polym. Chem., 2013, 4, 85 RSC.
- R. Iwaura, T. Iizawa, H. Minamikawa, M. Ohnishi-Kameyama and T. Shimizu, Small, 2010, 6, 1131 CrossRef CAS PubMed.
- F. Li, K. G. Yager, N. M. Dawson, J. Z. Yang, K. J. Malloy and Y. Qin, Macromolecules, 2013, 46, 9021 CrossRef CAS.
- F. Li, K. G. Yager, N. M. Dawson, Y. B. Jiang, K. J. Malloy and Y. Qin, Polym. Chem., 2015, 6, 721 RSC.
- F. Li, N. M. Dawson, Y. B. Jiang, K. J. Malloy and Y. Qin, Polymer, 2015, 76, 220 CrossRef CAS.
- J. T. Davis and G. P. Spada, Chem. Soc. Rev., 2007, 36, 296 RSC.
- A. Likhitsup, S. Yu, Y. H. Ng, C. L. Chai and E. K. W. Tam, Chem. Commun., 2009, 4070 RSC.
- I. Gadwal, S. De, M. C. Stuparu, R. J. Amir, S. G. Jang and A. Khan, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1844 CrossRef CAS.
- I. Gadwal, S. De, M. C. Stuparub and A. Khan, Polym. Chem., 2012, 3, 2615 RSC.
- F. Carducci, J. S. Yoneda, R. Itri and P. Mariani, Soft Matter, 2018, 14, 2938 RSC.
- Y. F. Li, Y. Liu, R. J. Ma, Y. L. Xu, Y. L. Zhang, B. X. Li, Y. L. An and L. Q. Shi, ACS Appl. Mater. Interfaces, 2017, 9, 13056 CrossRef CAS.
- T. N. Plank and J. T. Davis, Chem. Commun., 2016, 52, 5037 RSC.
- Y. P. Kumar, R. N. Das, O. M. Schütte, C. Steinem and J. Dash, Nat. Protoc., 2016, 11, 1039 CrossRef CAS PubMed.
- Y. P. Kumar, R. N. Das, S. Kumar, O. M. Schütte, C. Steinem and J. Dash, Chem. – Eur. J., 2014, 20, 3023 CrossRef CAS.
- R. N. Das, Y. P. Kumar, S. A. Kumar, O. M. Schütte, C. Steinem and J. Dash, Chem. – Eur. J., 2018, 24, 4002 CrossRef CAS.
- J. C. Chen, M. Z. Liu, H. H. Gong, Y. J. Huang and C. Chen, J. Phys. Chem. B, 2011, 115, 14947 CrossRef CAS.
- C. W. Tu, S. W. Kuo and F. C. Chang, Polymer, 2009, 50, 2958 CrossRef CAS.
- J. C. Chen, M. Z. Liu, H. H. Gong, G. J. Cui, S. Y. Lu, C. M. Gao, F. Huang, T. T. Chen, X. Y. Zhang and Z. Liu, Polym. Chem., 2013, 4, 1815 RSC.
- V. Darcos, S. El Habnouni, B. Nottelet, A. El Ghzaoui and J. Coudane, Polym. Chem., 2010, 1, 280 RSC.
- M. A. Tasdelen, Eur. Polym. J., 2011, 47, 937 CrossRef CAS.
- T. Giorgi, F. Grepioni, I. Manet, P. Mariani, S. Masiero, E. Mezzina, S. Pieraccini, L. Saturni, G. P. Spada and G. Gottarelli, Chem. – Eur. J., 2002, 8, 2143 CrossRef CAS.
- J. X. Zhang, L. Y. Qiu, K. J. Zhu and Y. Jin, Macromol. Rapid Commun., 2004, 25, 1563 CrossRef CAS.
- G. N. Reddy, A. Huqi, D. Iuga, S. Sakurai, A. Marsh, J. T. Davis, S. Masiero and S. P. Brown, Chem. – Eur. J., 2017, 23, 2315 CrossRef CAS.
- X. X. Ke, L. Wang, J. T. Xu, B. Y. Du, Y. F. Tu and Z. Q. Fan, Soft Matter, 2014, 10, 5201 RSC.
- Y. Q. Zhu, Y. G. Huang, W. D. Meng, H. Q. Li and F. L. Qing, Polymer, 2006, 47, 6272 CrossRef CAS.
- Y. Bai, F. Y. Xie and W. Tian, Chin. J. Polym. Sci., 2018, 36, 406 CrossRef CAS.
- Z. X. Du, J. T. Xu and Z. Q. Fan, Macromolecules, 2007, 40, 7633 CrossRef CAS.
- I. Yoshikawa, S. Yanagi, Y. Yamaji and K. Araki, Tetrahedron, 2007, 63, 7474 CrossRef CAS.
- T. Sato, M. Seko, R. Takasawa, I. Yoshikawa and K. Araki, J. Mater. Chem., 2001, 11, 3018 RSC.
- S. Masiero, R. Trotta, S. Pieraccini, S. De Tito, R. Perone, A. Randazzo and G. P. Spada, Org. Biomol. Chem., 2010, 8, 2683 RSC.
- C. Graziano, S. Masiero, S. Pieraccini, M. Lucarini and G. P. Spada, Org. Lett., 2008, 10, 1739 CrossRef CAS PubMed.
- X. F. Ji, H. Wang, Y. Li, D. Y. Xia, H. Li, G. P. Tang, J. L. Sessler and F. H. Huang, Chem. Sci., 2016, 7, 6006 RSC.
- M. M. Zhu, F. Song, W. C. Nie, X. L. Wang and Y. Z. Wang, Polymer, 2016, 93, 159 CrossRef CAS.
- F. Song, W. T. Shi, X. T. Dong, X. Han, X. L. Wang, S. C. Chen and Y. Z. Wang, Colloids Surf., B, 2014, 113, 501 CrossRef CAS PubMed.
- D. L. Tang, F. Song, C. Chen, X. L. Wang and Y. Z. Wang, Nanotechnology, 2013, 24, 145101 CrossRef.
- X. F. Cheng, Y. Jin, B. Z. Fan, R. Qi, H. P. Li and W. H. Fan, ACS Macro Lett., 2016, 5, 238 CrossRef CAS.
- A. S. Picco, B. Yameen, W. Knoll, M. R. Ceolín and O. Azzaroni, J. Colloid Interface Sci., 2016, 471, 71 CrossRef CAS PubMed.
- J. Z. Du, H. Willcock, J. P. Patterson, I. Portman and R. K. O’Reilly, Small, 2011, 7, 2070 CrossRef CAS PubMed.
- Q. Yan and Y. Zhao, Chem. Sci., 2015, 6, 4343 RSC.
- L. Chen, M. M. Xu, J. Hu and Q. Yan, Macromolecules, 2017, 50, 4276 CrossRef CAS.
- H. P. Li, Y. Jin, B. Z. Fan, S. Q. Lai, X. P. Sun and R. Qi, Macromol. Rapid Commun., 2017, 38, 1600646 CrossRef PubMed.
- F. Y. K. Wang and J. Z. Du, Chem. Commun., 2015, 51, 11198 RSC.
- N. Morimoto, Y. Sasaki, K. Mitsunushi, E. Korchagina, T. Wazawa, X. P. Qiu, S. M. Nomura, M. Suzukia and F. M. Winnik, Chem. Commun., 2014, 50, 8350 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, synthesis procedures, samples’ preparation, NMR, FTIR and CD spectra, SEC traces, etc. See DOI: 10.1039/c8sm02172d |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *,
Wu-Cheng
Nie
,
Xiu-Li
Wang
*,
Wu-Cheng
Nie
,
Xiu-Li
Wang