
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceColloid supported lipid bilayers for self-assembly†
Melissa
Rinaldin‡
 ab,
Ruben W.
Verweij‡
a,
Indrani
Chakraborty
c and
Daniela J.
Kraft
*a
ab,
Ruben W.
Verweij‡
a,
Indrani
Chakraborty
c and
Daniela J.
Kraft
*a
aHuygens-Kamerlingh Onnes Lab, Universiteit Leiden, P.O. Box 9504, 2300 RA Leiden, The Netherlands. E-mail: kraft@physics.leidenuniv.nl
bInstituut-Lorentz, Universiteit Leiden, P.O. Box 9506, 2300 RA Leiden, The Netherlands
cSchool of Chemistry, Raymond and Beverly Sackler Faculty of Exact Sciences, Tel Aviv University, Tel Aviv 69978, Israel
First published on 19th December 2018
Abstract
The use of colloid supported lipid bilayers (CSLBs) has recently been extended to create colloidal joints, that enable the assembly of structures with internal degrees of flexibility, and to study lipid membranes on curved and closed geometries. These novel applications of CSLBs rely on previously unappreciated properties: the simultaneous fluidity of the bilayer, lateral mobility of inserted (linker) molecules and colloidal stability. Here we characterize every step in the manufacturing of CSLBs in view of these requirements using confocal microscopy and fluorescence recovery after photobleaching (FRAP). Specifically, we have studied the influence of different particle properties (roughness, surface charge, chemical composition, polymer coating) on the quality and mobility of the supported bilayer. We find that the insertion of lipopolymers in the bilayer can affect its homogeneity and fluidity. We improve the colloidal stability by inserting lipopolymers or double-stranded inert DNA into the bilayer. We include surface-mobile DNA linkers and use FRAP to characterize their lateral mobility both in their freely diffusive and bonded state. Finally, we demonstrate the self-assembly of flexibly linked structures from the CSLBs modified with surface-mobile DNA linkers. Our work offers a collection of experimental tools for working with CSLBs in applications ranging from controlled bottom-up self-assembly to model membrane studies.
 Daniela J. Kraft | Daniela Kraft is an Associate Professor in Soft Matter Physics at the Huygens-Kamerlingh Onnes Laboratory at Leiden University, The Netherlands. She obtained her PhD cum laude from the University of Utrecht, The Netherlands, under supervision of Willem Kegel. Supported by a Rubicon grant, she then joined the Center for Soft Matter Research at New York University, USA, as a postdoctoral researcher. In 2013, she moved to Leiden, where she established her own group. Her research focuses on self-assembly in biological and soft matter systems, ranging from anisotropic colloidal particles to lipid membranes, emulsions, and viruses. Dr Kraft has been awarded a VENI fellowship from the Netherlands Organisation for Scientific Research, an ERC starting grant and the paper of the year award 2017 from Biophysical Journal. |
Introduction
Colloid supported lipid bilayers (CSLBs) are used in a diverse range of research areas and applications,1 from drug delivery,2–4 bio-sensing,5,6 membrane manipulation7 and cell biology8–10 to fundamental studies on lipid phase separation11,12 and self-assembly.13–16 The presence of a lipid bilayer around nano- or micrometer-sized solid particles or droplets provides biomimetic properties and a platform for further functionalization. One intriguing recent example used DNA-based linkers to functionalize the lipid bilayer, thereby enabling self-assembly of the underlying colloidal particles or droplets into flexible structures.13–18 Within such a structure, the colloidal elements can move over each other's surface while remaining strongly and specifically bonded. This new type of bonding enables fundamental studies on structures with internal degrees of flexibility, such as the self-assembly of novel crystal phases and their phase transitions.19–22 Furthermore, these complex colloids have great potential for smart drug delivery techniques,2 photonic band-gap materials23,24 and wet computing.25CSLBs are particularly suitable and versatile building blocks for the assembly of floppy structures, because they combine the best qualities of free standing bilayers (vesicles) and colloids. Vesicles, upon application of linkers,26–29 can connect into flexible structures, but are unstable to small disturbances, heterogenous in size and easily deformable. Colloidal particles are available in diverse materials and with a variety of stable shapes, and can be assembled after functionalization with surface-bound DNA linkers.30–32 However, the obtained structures are often rigid due to the immobility of the linking groups on the particles' surface and are non-equilibrium structures due to a “hit-and-stick” aggregation process.33 Emulsions coated with lipid monolayers and DNA-linkers that are mobile on the droplet interface possess both interaction specificity and bond flexibility.17,18,34 Therefore, they assemble into flexible structures in a controlled fashion, but their shape is limited to spheres and they deform upon binding. Conversely, CSLBs consist of colloidal particles which provide a stable support for the lipid bilayer that is tunable in shape, size and material. The range of shapes for colloidal particles comprises, among others, spheres, cubes, rods, and (a)symmetric dumbbell particles, and their sizes range from hundreds of nanometers to several micrometers. They can be produced reliably with a narrow size distribution and are commercially available. Additionally, CSLBs feature a lipid bilayer on the surface of the colloids which creates a liquid film for molecules, such as DNA linkers, to freely move in. This allows for linking particles specifically, and yet flexibly, making the assembly of floppy structures possible.13–16,34
To obtain flexible instead of rigid structures, it is vital that the linker molecules which are inserted into the lipid bilayer are free to move over the surface of the CSLBs. Their lateral mobility relies on the fluidity and homogeneity of the bilayer, which in turn depend on the linker concentration16 and lipid composition. The lipids need to be in the fluid state under experimental conditions, and this may be impeded by bilayer-surface interactions. Similarly, the success of experiments studying the phase separation of lipid bilayers on anisotropic colloidal supports relies on the fluidity and homogeneity of the bilayer.11,12 Finally, controlling the self-assembly pathway through complementary DNA linkers implies that all other nonspecific interactions need to be suppressed. In other words, CSLBs need to have sufficient colloidal stability. To the best of our knowledge, these requirements of membrane homogeneity and fluidity plus colloidal stability have not been studied simultaneously. However, they are of key importance for using CSLBs in self-assembly and model membrane studies, while possibly having wider implications for all other applications.
Here, we carefully characterize every stage in the preparation of CSLBs specifically related to these three properties. First, we study the effect of the material properties of the particles and the use of polymers on the membrane fluidity and homogeneity. Then, we investigate the influence of lipopolymers and inert double-stranded DNA on the colloidal stability of the CSLBs. Subsequently, we include DNA-based linkers connected to hydrophobic anchors and characterize their diffusion in the bilayer. Finally, we show that when using the optimal experimental parameters determined by this study, CSLBs self-assemble into flexibly linked structures that are freely-jointed.
Experimental section
Reagents
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), itaconic acid (≥99%), 3-aminopropyltriethoxysilane (99%) and acetic acid (99.8%) were purchased from Sigma-Aldrich. Magnesium chloride (MgCl2, for analysis) was purchased from Merck. All solutions were prepared with Milli-Q water (Milli-Q Gradient A10).
:1 molar ratio in HEPES buffer type 3. The solution was then heated in an oven to 90 °C for 30 min, after which the oven was turned off and the solution was allowed to cool down slowly overnight in the closed oven. Strand 1 and 2 are hybridized to form “10 nm Inert DNA”, 3 and 4 are hybridized to form “20 nm Inert DNA”, 4 and 5 form “20 nm Linker A”, 4 and 6 make “20 nm Linker A′”, 7 and 8 form “30 nm Linker A” and, finally, 7 and 9 are hybridized to form “30 nm Linker A′”. The linkers “A” have a single-stranded sticky end (indicated by cursive text in Table S1 of the ESI†) that is complementary to the single-stranded end of linkers “A′”.
000), itaconic acid (≥99%), 3-aminopropyltriethoxysilane (99%) and acetic acid (99.8%) were purchased from Sigma-Aldrich. Magnesium chloride (MgCl2, for analysis) was purchased from Merck. All solutions were prepared with Milli-Q water (Milli-Q Gradient A10).
:1 molar ratio in HEPES buffer type 3. The solution was then heated in an oven to 90 °C for 30 min, after which the oven was turned off and the solution was allowed to cool down slowly overnight in the closed oven. Strand 1 and 2 are hybridized to form “10 nm Inert DNA”, 3 and 4 are hybridized to form “20 nm Inert DNA”, 4 and 5 form “20 nm Linker A”, 4 and 6 make “20 nm Linker A′”, 7 and 8 form “30 nm Linker A” and, finally, 7 and 9 are hybridized to form “30 nm Linker A′”. The linkers “A” have a single-stranded sticky end (indicated by cursive text in Table S1 of the ESI†) that is complementary to the single-stranded end of linkers “A′”.
Preparation of CSLBs
Typically, CSLBs were made by spontaneous spreading and fusion of small unilamellar vesicles (SUVs) on the particle surface. An SUV dispersion prepared via either extrusion or sonication was mixed with the particles, allowing a bilayer to spread on the surface for at least one hour. Subsequently the CSLBs were washed to remove excess SUVs. We observed no substantial differences in the obtained CSLBs between the two methods presented here.This method was used for all experiments regarding the influence of particle material, surface roughness and the effect of polymer insertion on the spreading and mobility of the lipid bilayer.
17 μL of 0.5 g L−1 SUVs in HEPES buffer 2 or 3 were mixed with 35 μL 0.5% w/v of particles in Milli-Q water, leading to a surface ratio of SUVs:particles of 8:1. The dispersion was gently rotated for 1 h. The particles were centrifuged at 43 rcf for 2 min and the supernatant was replaced with HEPES buffer type 2 or 3 to remove any remaining free SUVs from the dispersion. Alternatively, the particles were allowed to sediment by gravity for 30 min instead of centrifuging and the supernatant was replaced.
This method was used for all experiments regarding the colloidal stability of CSLBs, the mobility of inserted DNA and the mobility of self-assembled CSLB clusters. For all experiments where DNA was inserted into the membrane of the CSLBs, buffer type 3 was used to prevent calcium-mediated DNA absorption to the lipid bilayer.42
Coating CSLBs with DNA for self-assembly
After the particles were coated with a lipid bilayer using method 2, various amounts of inert and/or linker DNA were added and the dispersion was gently rotated for 1 h. To remove any remaining free DNA strands in solution, the particles were washed by centrifugation for 2 min at 43 rcf, or alternatively, by sedimentation by gravity for 30 min, and the supernatant was replaced three times by HEPES buffer type 3.We define the amount of dsDNA that we add as a surface density σDNA,
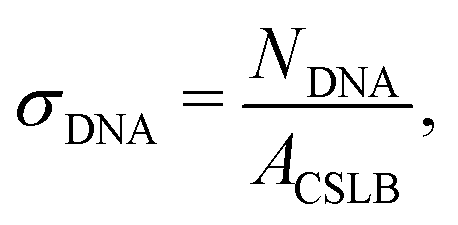 | (1) |
Particle clusters were formed by mixing two particle types coated with complementary DNA linkers in a 1:1 number ratio in a round metal sample holder on a polyacrylamide coated cover slip (see ref. 43 for details). The polyacrylamide coating keeps the particles from sticking to the glass surface, allowing them to cluster via diffusion limited aggregation.
Sample characterization
The samples were imaged with an inverted confocal microscope (Nikon Ti-E) equipped with a Nikon A1R confocal scanhead with galvano and resonant scanning mirrors. A 100× oil immersion objective (NA = 1.49) was used. A 561 nm laser was employed to excite the Lissamine rhodamine dye, a 488 nm laser was used to excite the TopFluor-Cholesterol dye. The excitation light passed through a quarter wave plate to prevent polarization of the dyes. 500–550 nm and 565–625 nm filters were used to separate the emitted light from the TopFluor and the rhodamine dyes, respectively.The charge of the particles in MilliQ water and HEPES buffer type 1 was determined via zeta potential measurements using a Malvern Zetasizer Nano ZS.
Fluorescence recovery after photobleaching (FRAP)
We used fluorescence recovery after photobleaching (FRAP) to check the mobility of the lipids in a CSLB. A circular area of the fluorescent sample was bleached, the recovery signal was collected and normalized as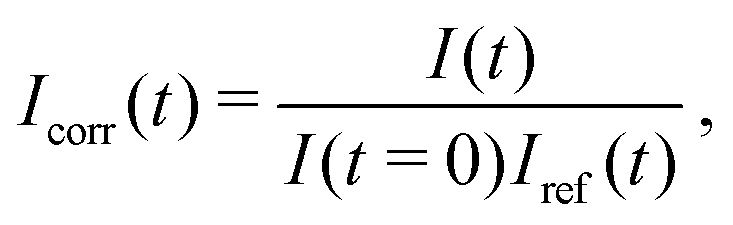 | (2) |
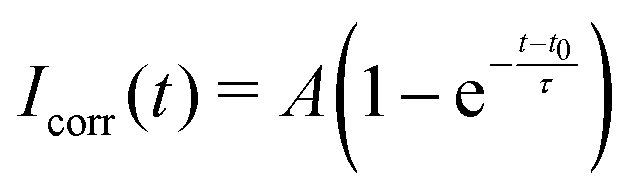 | (3) |
While there is a simple relation linking τ to D for circular bleaching areas on planar surfaces,44 we are not aware of a similar expression for a spherical surface that is partly bleached from the side, as is the case in our experiments. Therefore, we quantify the lateral mobility in terms of the recovery time τ only.
All FRAP experiments on silica particles were performed using 7 μm particles, unless stated otherwise. To measure the lateral mobility of DNA linkers using FRAP, no fluorescently labeled lipids were used and instead, we used a high linker DNA concentration (σDNA = 3 × 105 μm−2) that provided a sufficiently bright fluorescent signal.
Particle stability analysis
To estimate the colloidal stability of particles, we rotated the particles (0.4% w/w) in a test tube for at least 1 h, thereby allowing them to aggregate. We then immobilized some of the clusters on a glass substrate, allowing us to take a “snapshot” of the cluster distribution at that time. Particles were located in bright-field microscopy images of these sedimented, semi-dilute (volume fraction ϕ ≈ 0.001) samples. The cluster sizes were determined by using the bandpass, locate and cluster functions from TrackPy.45 Erroneously tracked positions were corrected manually. The separation distance below which particles are considered to be part of a cluster was chosen to be 1.1D, where D is the particle diameter. This can lead to a small overestimation of the number of clusters when particles are close together but have not irreversibly aggregated. We defined the fraction of single particles fsingle as the number of detected clusters with a size of 1 (i.e. single particles) divided by the total number of individual particles. The error on this fraction was estimated as the standard deviation of the average cluster size divided by the square root of the total number of particles. For each measurement, we analyzed between 150 to 4000 individual particles.Trimer flexibility analysis
We have analyzed three linearly-connected CSLBs that were functionalized with inert dsDNA and linker dsDNA. To quantify the mobility of the self-assembled trimers, we tracked the position of the three individual particles in bright-field movies as a function of time and calculated the opening angle θ between them. For tracking and calculating θ, we used a custom algorithm that is depicted in Fig. S2 of the ESI.†First, the user selects the particles of interest from the first frame (see Fig. S2A, ESI†). This increases the computational efficiency of tracking because it reduces the number of tracked features and allows for cropping of all frames. We then iterate over all frames to identify the positions of the selected particles. Each current frame is inverted, so that all particles have a ring of high intensity around them (see Fig. S2B, ESI†). The frame is converted to polar coordinates with the current provisional particle position at the origin (see Fig. S2C, ESI†), where the provisional position is the one that the user selected for the first frame and the previous tracked position for all subsequent frames. For each row (each polar angle), the position of maximum intensity is found (see Fig. S2D, ESI†) and these coordinates are then converted back to the original Cartesian coordinate system of the frame (see Fig. S2E, ESI†). A circle is fitted to these coordinates using a least squares method (see Fig. S2F, ESI†). After all three particles are found in this frame, the opening angle between them is determined using simple trigonometry (see Fig. S2G, ESI†). From the opening angles of all the frames, we calculated the mean squared displacement of the angle (MSAD or “joint flexibility” J).16
We analyzed the free energy of trimers as function of opening angle using two methods: (1) by converting the histogram to the free energy using Boltzmann weighing and (2) using a maximum likelihood estimation method of angular displacements.43,46 We confirmed that both methods agreed and show only the result of the maximum likelihood method, because it allows us to estimate the error in our measurement. We now describe these methods in detail.
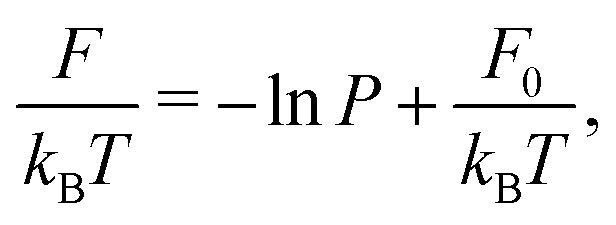 | (4) |
To summarize, we followed the method outlined in Chapter 3.4.2 of ref. 46: we find the maximum likelihood estimate of the local force field F(θ) by using a model for the transition probability P:
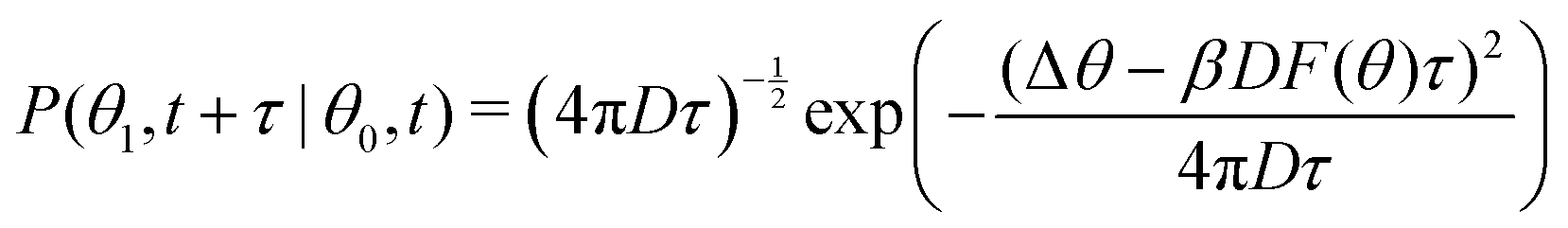 | (5) |
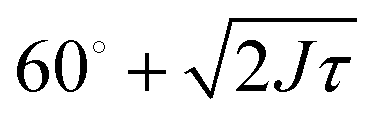 (where J is the joint flexibility) leading to a slight overestimation of the free energy for those angles.
(where J is the joint flexibility) leading to a slight overestimation of the free energy for those angles.
Results and discussion
We will now characterize every step in the formation of CSLBs. In the first section of the results we study the homogeneity and mobility of the lipid bilayer on colloidal particles made from different materials, and therefore various surface functionalities and degrees of roughness. Furthermore, we investigate the effect of PEGylated lipids on the homogeneity and mobility of the lipid bilayer and their use as steric stabilisers to prevent nonspecific aggregation.Having found conditions that yield colloidal particles with a homogeneous and mobile bilayer, we subsequently introduce double-stranded DNA connected to a hydrophobic anchor into the bilayer, as shown in Fig. 1(2). We employ DNA constructs both with and without single-stranded sticky ends, to investigate their use in DNA-mediated binding and their effect on colloidal stability, respectively. Finally, we demonstrate that CSLBs can be used for self-assembly by employing DNA linkers with complementary single-stranded sequences. We use FRAP to measure the lateral mobility of DNA linkers on the particle surface inside and outside the bond area. In this way, we show that they are mobile if the bilayer is fluid and that, in this case, the particles can freely roll over each other's surfaces when bonded.
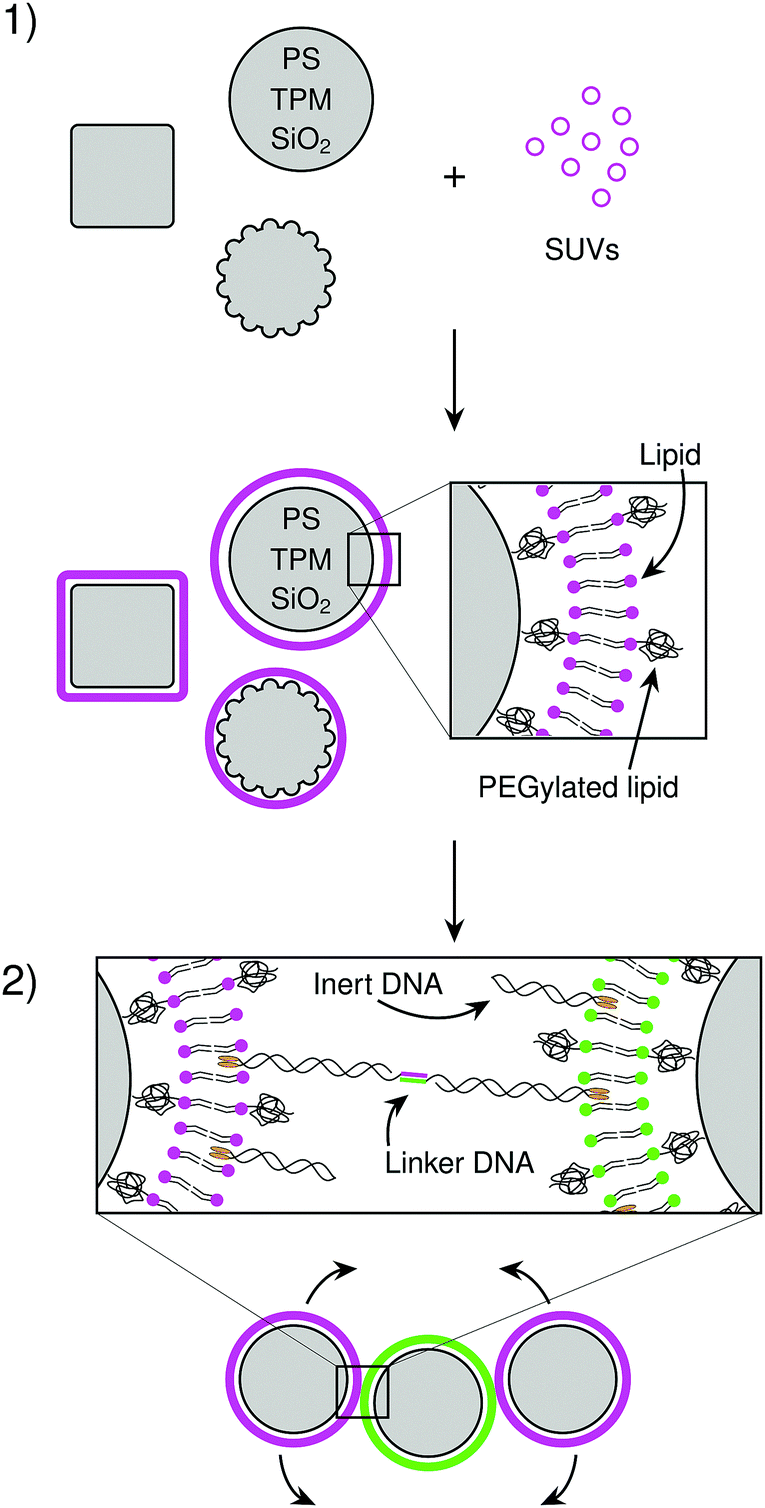 | ||
| Fig. 1 Overview of the experimental system. (step 1) Micrometer sized colloidal particles are coated with a lipid bilayer by adding small unilamellar vesicles (SUVs) that rupture and spread on the particle surface. We varied the composition of the lipids, as well as the material and shape of the particles. (step 2) DNA linkers with hydrophobic anchors can be added to make particles that are functionalized with DNA with complementary sticky ends. When the lipid bilayer is fluid, the linkers can diffuse over the particle surface and therefore also the linked particles can slide over each other. | ||
Lipid bilayer coating of colloidal particles
To use CSLBs in self-assembly studies or as model membrane systems, it is critical that a homogeneous and fluid bilayer forms on the colloidal particles. This implies successful assembly of both leaflets of the bilayer and lateral mobility of the lipids, and hence proteins, linkers, and larger lipid domains, in the membrane. The formation of lipid bilayers on solid supports can be achieved by deposition of SUVs under physiological conditions,49 as shown in Fig. 1(1). A combination of electrostatic and van der Waals forces between the SUVs and the surface of the support lead to spreading of the liposomes, which, because of hydrophobic/hydrophilic interactions, fuse together on the substrate.50–53 Between the surface of the support and the bilayer a thin layer of water remains, allowing the lipids to laterally diffuse in the absence of other motion-restricting forces. Previous studies on planar supported lipid bilayers (SLBs) reported that there are many factors which can prevent homogeneity and mobility of the bilayer.54 These factors are related to the surface that is coated (its surface charge, chemical composition and roughness), the aqueous medium (pH and ionic strength), the SUVs (composition, charge, size, hydrophobic/hydrophilic balance, transition temperature) and the temperature at which the lipid coating happens.50,55 Here we will study how some of these factors, that are inherent to the use of solid particles, influence the formation of supported lipid bilayers on colloidal substrates.After coating, we visually inspect the lipid-coated particles using confocal microscopy and consider bilayers to be homogenous if more than 50% of the particles do not show strong variations of the fluorescence intensity signal on individual colloids, which we associate to defects of the bilayer. We characterize the bilayer fluidity by measuring the mobility of the fluorescently-labeled lipids on the colloid surface using FRAP. After bleaching, we observe the recovery of the fluorescence intensity due to the diffusion of the dyed lipids in and out of the bleached area. We consider the lipids and thus the bilayer to be mobile if the intensity signal recovers homogeneously in the bleached area, otherwise we consider them to be (partially) pinned to the surface.
Our first observation was that only particles that possess a silica surface exhibited homogeneous and mobile bilayers (Table 1). For spherical silica particles, the fluorescence intensity was homogeneous (see Fig. S1 in the ESI†), which indicates that all particles are successfully coated with a homogeneous bilayer. We did not succeed in coating colloids made from polystyrene or hematite with a homogeneous bilayer. However, once such substrates were first coated with a silica shell, the bilayer was found to be both homogeneous and mobile. Unexpectedly, particles made from an organosilica compound (TPM) whose surfaces are similar to silica38 only showed homogeneous, but not mobile bilayers. Since silica, TPM and polystyrene colloids were all negatively charged (see Table 1), we conclude that the chemical composition of the substrate and not only the surface charge plays a fundamental role in the homogeneity and fluidity of the bilayer. These results agree with previous experiments on planar SLBs, in which silica-based surfaces were found to be one of the most reliable supports for homogeneous and mobile lipid bilayers.50
| Type | Material | Zeta potential [mV] | Bilayer homogeneity | Bilayer mobility | |
|---|---|---|---|---|---|
| Water | Buffer | ||||
| 1 | Silica spheres (Stöber method, microparticles GmbH) | −56 ± 6 | −23 ± 2 | Yes | Yes |
| 2 | Hematite cubic particles35 | +39 ± 5 | +12 ± 1 | No | No |
| 3 | Silica-coated hematite cubic particles36 | −32 ± 6 | −6 ± 2 | Yes | Yes |
| 4 | Polystyrene spheres (Sigma Aldrich) | −38 ± 2 | −34 ± 3 | No | No |
| 5 | Polystyrene spheres with carboxyl groups40 | −43 ± 1 | −43 ± 1 | No | No |
| 6 | Silica-coated polystyrene-TPM (an)isotropic particles11,37 | −33 ± 1 | −21 ± 2 | Yes | Yes |
| 7 | TPM spheres38 | −42 ± 1 | −20 ± 1 | Yes | No |
| 8 | TPM spheres with carboxyl groups39 | −46 ± 1 | −23 ± 2 | No | No |
| 9 | TPM spheres with amino groups39 | −12 ± 4 | −17 ± 1 | No | No |
Since colloidal particles are often functionalized with different groups on the surface, we furthermore have studied the bilayer on particles equipped with surface groups commonly used in colloidal science (Table 1). While TPM particles with an unmodified silica-like surface showed homogeneous bilayers, we found that functionalization with negatively charged carboxyl or positively charged amino groups prevented spreading and fusion of the lipid vesicles. Likewise, functionalization of polystyrene spheres with carboxyl groups did not enhance the homogeneity of the lipid bilayer. A previous study on planar SLBs reported that the spreading of SUVs depends on the combination of the molecular ions in the buffer and the type and density of surface charge.57 While amino-functionalized surfaces are hence expected to disrupt the spreading of SUVs in the presence of the negatively charged HEPES molecules of the buffer, the observation of inhomogenous bilayers on carboxyl-functionalized surfaces can likely be allocated to an insufficiently dense surface coverage. We conclude that similar to planar SLBs, the homogeneity and fluidity of the bilayer of CSLBs depends on a complex interplay of the chemical and physical properties of the lipids and the particle's surface.
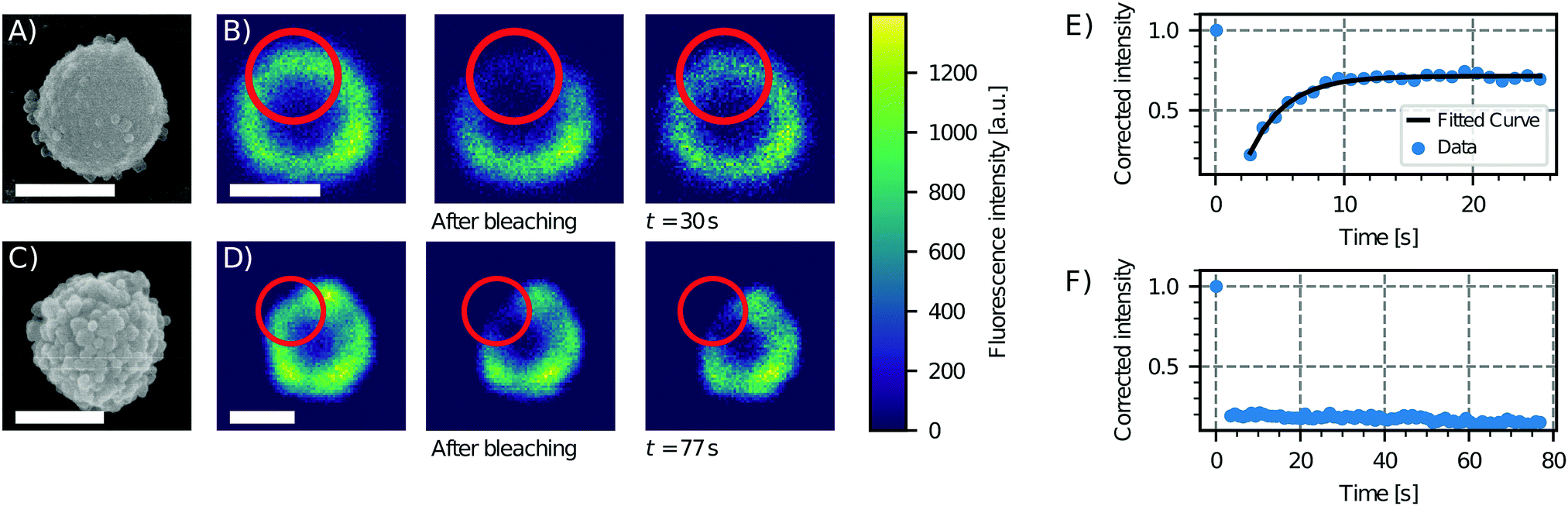 | ||
| Fig. 2 Effect of surface roughness on lipid bilayer formation. Scanning electron microscopy (SEM) images of particles of type 6 (see Table 1), showing low (A) and high (C) degrees of roughness. Sequence of a FRAP experiment before bleaching, just after bleaching and after 30 s and 77 s for the (B) less rough and (D) rougher particle, respectively. On the right, the fluorescence intensity as a function of time and an exponential fit of the data for the (E) less rough and (F) rougher particle are shown. The fluorescence recovery of the bilayer on the smooth particle shows that the bilayer is fluid, in contrast to the rough particle which does not show a recovery of the fluorescence. Scale bars are 1 μm. | ||
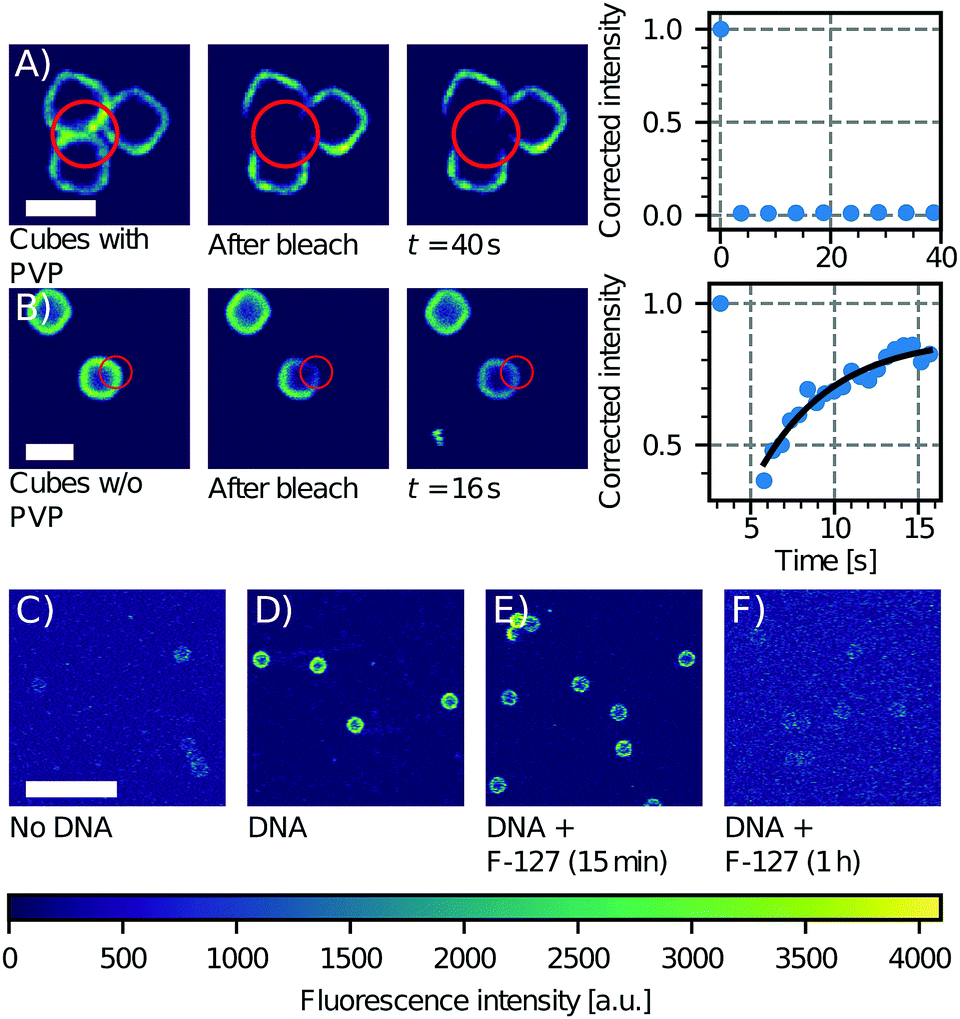 | ||
| Fig. 3 Effect of PVP on CSLB formation. FRAP experiment on a group of three cubes with (A) and without (B) PVP (particle type 3 without the hematite core, Table 1). Only the sample without PVP shows recovery of the signal in the bleached area. Scale bars are 2 μm. Influence of F-127 on linker inclusion. (C) Control image of the fluorescence of the undyed CSLBs (particle type 1, Table 1). (D) Fluorescence intensity of CSLBs coated with 20 nm linker dsDNA. (E) The same sample as in (D), but imaged 15 min after dispersion in 5% F-127 solution. The fluorescence on the particles was found to be significantly less homogeneous than in (D). (F) The same sample as in (D), but imaged 1 h after dispersion in 5% F-127 solution. The fluorescence intensity is comparable to the uncoated control in (C) so we conclude that all dsDNA has been removed from the bilayer by F-127. Scale bar is 10 μm. | ||
Moreover, the presence of polymers may not only affect the bilayer's properties, but also the incorporation of functional groups such as DNA linkers into it. We tested this by preparing CSLBs with fluorescently labeled DNA linkers connected to double cholesterol anchors and transferring an aliquot of this dispersion to a HEPES solution containing 5% w/w of Pluronic F-127, a polymer that is commonly used for the stabilization of colloidal particles. While the fluorescent signal of the CSLBs with and without F-127 were initially equal, already 15 minutes after mixing we observed less dsDNA fluorescence on the CSLBs with F-127 compared to particles without it. After 1 h, the fluorescence intensity of the CSLBs with F-127 was comparable to that of control particles not coated with linker dsDNA (Fig. 3C–F). We therefore conclude that F-127 removed the cholesterol-anchored linker DNA from the bilayer, in line with recent experiments on emulsion droplets coated with mobile DNA linkers.61
Inspired by this, we study how a specific lipopolymer tether affects the spreading and the fluidity of the bilayer in CSLBs. We used the lipopolymer DOPE-PEG, a phospholipid with a covalently bound PEG molecule. We employed PEGylated lipids with three different molecular weights: 2000, 3000 and 5000 g mol−1 in varying concentrations. It is important to note that PEGylated lipids were introduced in the system during the SUV formation by mixing them with the other lipids. This means that once the bilayer is formed, they are present in both leaflets.
We report in Fig. 4A and B the effect of varying concentration and molecular weight of the lipopolymers on the spreading and the mobility of the bilayer. In the absence of PEGylated lipids, the bilayer on the CSLBs was observed to be fluid. At increased concentrations of DOPE-PEG, the bilayer became inhomogeneous, which indicates insufficient spreading and fusion of the SUVs. This effect appeared at lower molar fractions for lipopolymers with higher molecular weights of the DOPE-PEG. For completeness, we note that a small fraction of particles in samples that are labeled as inhomogeneously coated do exhibit a homogeneous, but nevertheless immobile, bilayer. We believe that the reason for the observed inhomogeneity is two-fold. On the one hand, higher concentrations of lipopolymers lead to an increased steric stabilization, that prevents fusion of the SUVs and hinders the van der Waals interactions between the SUVs and the substrate that aid spreading. On the other hand, PEGylated lipids in the brush state increase the bending rigidity of the SUV membrane, thereby preventing rupture and spreading on the surface.67
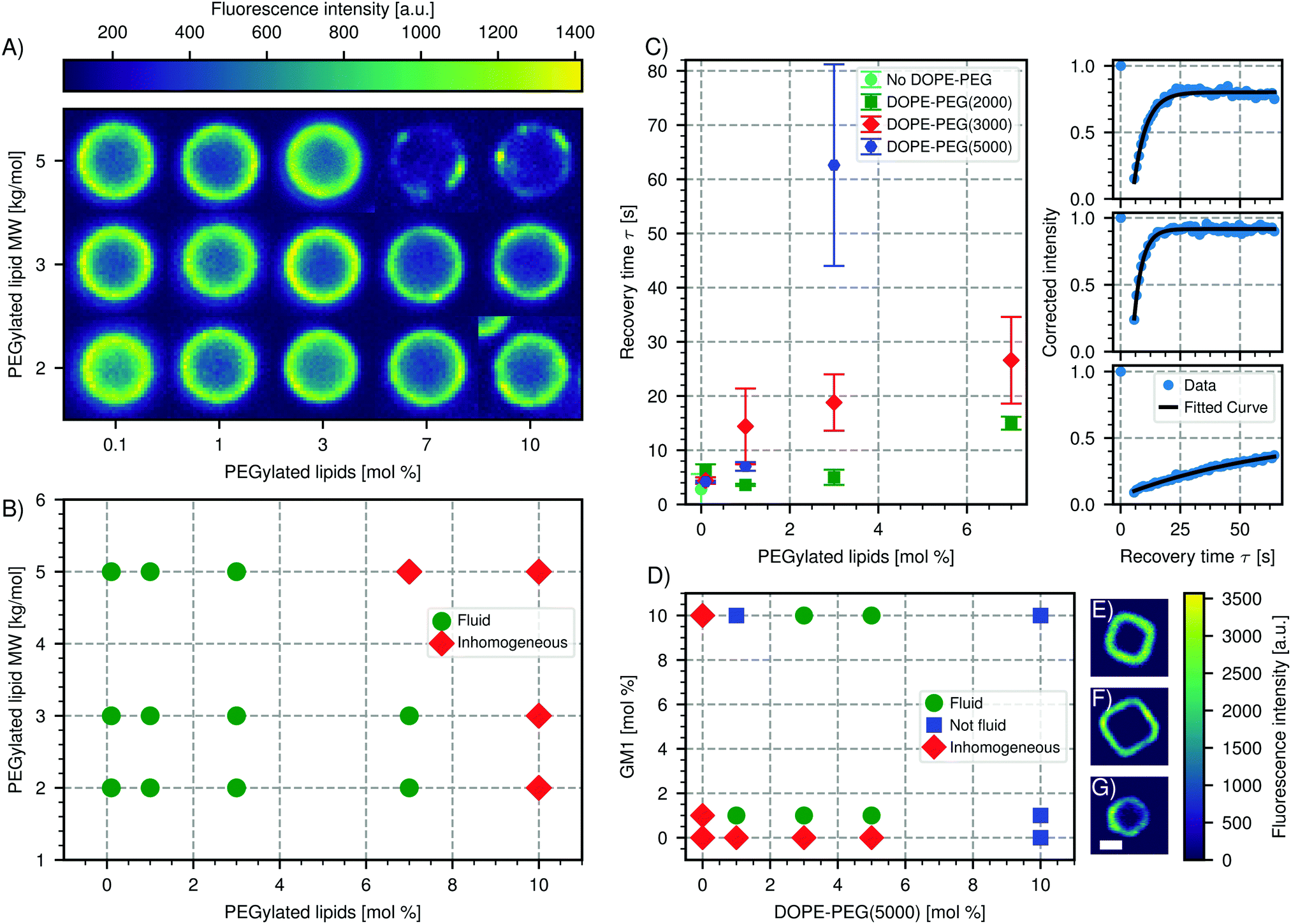 | ||
| Fig. 4 Effect of PEGylated lipids on CSLB formation and fluidity. (A) Color-map of the intensity of the bilayer of spherical CSLBs (particle type 1, Table 1) ordered by increasing molecular weight and concentration of PEGylated lipids. The images are taken in the equatorial plane. (B) Phase diagram of the state of the bilayer for varying molecular weight and concentration of PEGylated lipids. (C) Left: Plot of the recovery time after FRAP depending on molar concentration and size of the PEGylated lipids. Right: Intensity recovery curves as a function of time from FRAP experiments of, from top to bottom, CSLBs without PEGylated lipids, with 1 mol% DOPE-PEG(2000) and 3 mol% DOPE-PEG(5000). (D) Phase diagram of spreading and fluidity of the bilayer on cubic silica shells (particle type 3 without the hematite core, Table 1) depending on GM1 and PEGylated lipids. Colormap of a fluorescent image of a cubic bilayer made with (E) 1 mol% GM1 and 3 mol% DOPE-PEG(5000), (F) 1 mol% GM1 and 10 mol% DOPE-PEG(5000), (G) no GM1 or DOPE-PEG(5000). Scale bar 1.5 μm. | ||
For fluid membranes, we quantified the mobility of the lipids by calculating the recovery time from FRAP experiments, which is the time it takes a bleached area to recover its fluorescence intensity. We find that the diffusion of the lipids is faster for PEGylated lipids with a lower molecular weight and increases with decreasing amount of the lipopolymers, see Fig. 4C. This latter result agrees with a study performed on planar supported lipid bilayers.62 In the presence of lipopolymers, we find the shortest recovery time (3.2 ± 0.02 s), e.g. highest diffusivity, for 1 mol% of DOPE-PEG(2000). The decrease of the diffusion coefficient with the amount of lipopolymer indicates that the PEGylated lipids are pinned to the surface and in this way hinder the mobility of the other lipids.
We emphasize that the mobility of supported lipid bilayers in presence of polymers is dependent on many factors and one may not extend our results to other types of polymers, lipid bilayers or physiological environments.66 The complex interplay between polymers and the chemical properties of the colloidal surface can lead to surprising results. For example, and in contrast to what we reported above, we found that an homogenous bilayer on cubic silica particles could only be obtained by using both PEGylated lipids (DOPE-PEG(5000)) and a negatively charged lipid (GM1). Interestingly, at high concentrations of PEGylated lipids the bilayer is very homogeneous but not mobile (Fig. 4D). This is in contrast to silica spheres coated with the same concentrations of lipopolymers and only zwitterionic lipids, which do not possess a homogeneous bilayer, see Fig. 4A. We indicated this state in which the bilayer is homogeneous, but not fluid, with blue squares in Fig. 4D. A possible origin of this unusual behavior could be the different porosity, surface chemistry and charge of the silica cubes56 compared to the silica spheres (Table 1).
Stabilizing CSLBs against nonspecific aggregation
To build specific colloidal structures from the bottom up, careful control over the interactions between the particles is required. On the one hand, specific attractive interactions may be employed to control which particles interact. This specific binding can be achieved by using dsDNA linkers with complementary sticky ends.13,16,68 On the other hand, the particles need to be prevented from binding to each other nonspecifically: that is, not via dsDNA linker interactions but via other attractive forces that act indiscriminately between all particles, such as van der Waals forces. In other words, it is crucial to be able to control the colloidal stability of the CSLBs.69In our experiments, stabilization by repulsive electrostatic interactions is not a feasible route because surface charges are screened by the counterions in the HEPES buffer that is needed to allow the complementary DNA sticky ends to bind.68 The ionic strength of the buffer must be higher than 50 mM so that clusters are formed via DNA-mediated attractions.70 At these salt concentrations, even the bare silica particles (particle type 1, Table 1) are no longer stabilized by their negatively charged surface groups. Indeed, we found that both the bare silica particles and the silica particles coated with a lipid bilayer aggregated in all buffers, as was previously observed.71 The fraction of single particles determined from light microscopy images was fsingle = 0.67 ± 0.10 for uncoated silica particles after one hour of mixing in the buffer, while they were previously stable in deionised water. We therefore explored different ways to sterically stabilize the particles using higher concentrations of PEGylated lipids, SDS and inert dsDNA strands.
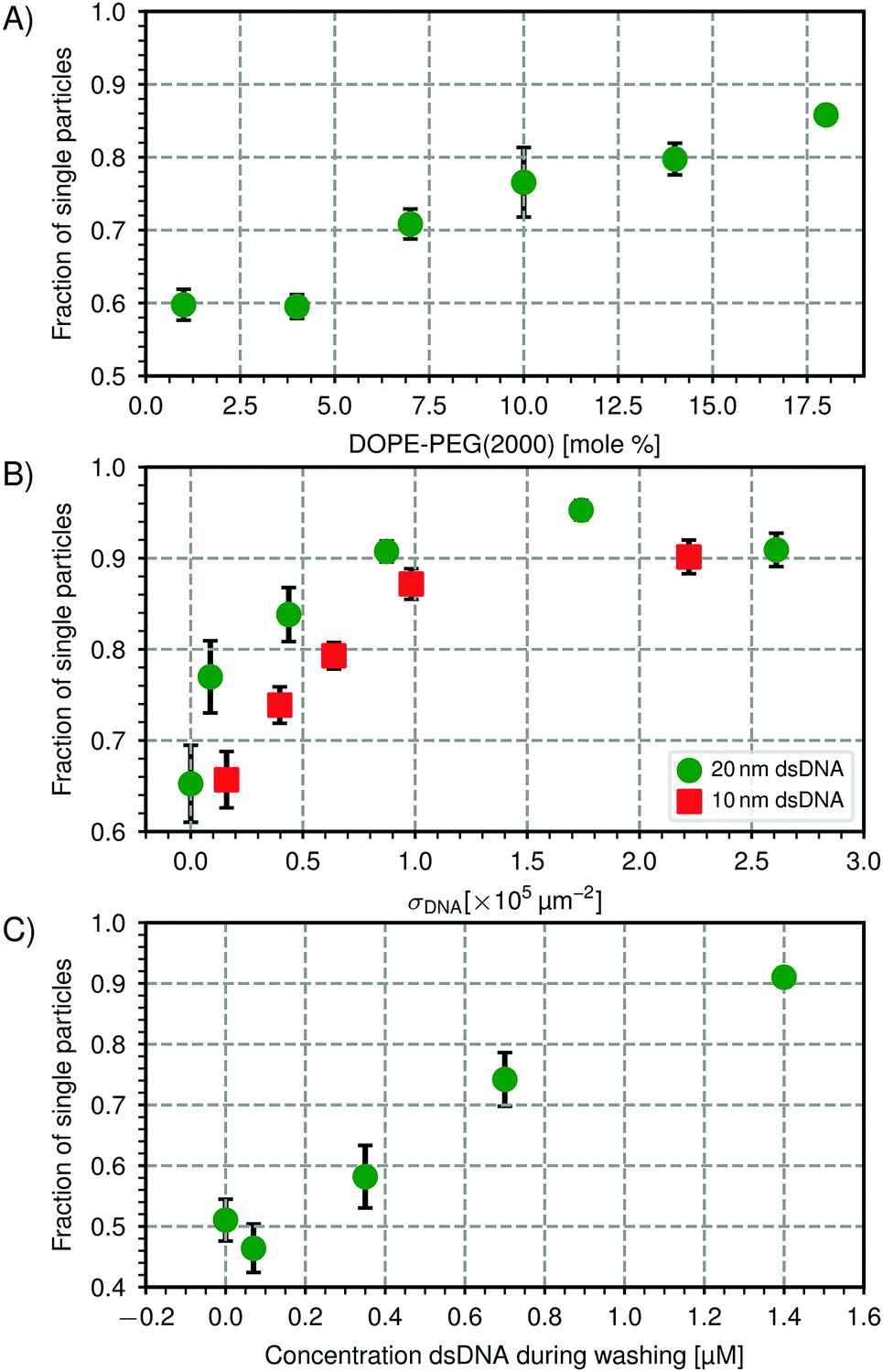 | ||
| Fig. 5 Steric stabilization of CSLBs. (A) Higher concentrations of DOPE-PEG(2000) lead to a higher fraction of single particles in the absence of linker DNA. Stability is shown after one washing cycle of 2 min at 43 rcf and overnight rotation. We hypothesize that above 8 mol%, the packing density of PEG on the surface of the membrane is high enough for the PEG to be in the brush state, making it more effective as a steric stabilizer. (B) Increasing the number of dsDNA strands on the particle surface increases the particle's stability for two different lengths of inert dsDNA (10 nm and 20 nm). (C) Centrifugation and redispersion with a solution containing inert dsDNA affects the fraction of single particles. After centrifuging particles that were initially stable (fsingle = 0.95, σDNA = 1.7 × 105 μm−2 dsDNA) 3× at 43 rcf for 2 min, we observed nonspecific aggregation (fsingle = 0.51) in the absence of dsDNA strands in solution while increasing the concentration of inert dsDNA (20 nm) in the washing solution could preserve stability. Particles of type 1 (see Table 1) was used in all experiments. | ||
While the colloidal stability can be improved by increasing the concentration of PEGylated lipids, the bilayer is not fluid at the concentrations required for colloidal stability as we showed earlier (see Fig. 4B). Therefore, embedded DNA linkers will also not be mobile in the bilayer and it is not possible to form reconfigurable structures from CSLBs stabilized by PEGylated lipids only.
We varied the surface concentration σDNA (see eqn (1)) of two inert dsDNA strands with different lengths and measured its effect on the particle stability, which is shown in Fig. 5(B). The stability was determined after the particles were coated with inert dsDNA and rotated for one hour. For both the 10 nm inert dsDNA and the 20 nm inert dsDNA, we found that increasing the number of grafted dsDNA strands per particle led to an increase in particle stability from fsingle = 0.68 without dsDNA to fsingle = 0.90 to 0.95 above σDNA = 2 × 105 μm−2. We found that the 20 nm inert dsDNA is slightly more efficient as a steric stabilizer than the 10 nm inert dsDNA, as can be seen in Fig. 5(B). This is expected, because for the longer DNA, excluded volume interactions between particles start to become important already at larger particle separations than for the shorter DNA. Additionally, excluded volume interactions between DNA strands on the same particle force the DNA to extend outwards already at lower concentrations for the longer DNA strands as compared to the shorter DNA strands. Therefore, the repulsion between the particles also has a longer range, leading to better colloidal stability. However, at concentrations above σDNA = 2 × 105 μm−2, the difference between the 10 nm and 20 nm inert dsDNA is less pronounced, because the particles are so densely coated that adding longer or more DNA strands will not stabilize the particles any further.
To use these particles in self-assembly studies, specific interactions need to be present as well, which we here induce by adding dsDNA linkers, that is, dsDNA strands with a sticky end and double cholesterol anchors. After the particles are functionalised, any excess linker DNA left in the solvent needs to be removed to reduce unwanted aggregation or saturation of the complementarily functionalized particles. To remove excess linker DNA, we washed and redispersed the particles in buffer solution three times and measured the particle stability afterwards. Unexpectedly, it decreased from fsingle = 0.95 before washing to fsingle = 0.51 after washing. To detect whether the partial removal of the stabilizing inert dsDNA during washing had caused this aggregation, we washed the particles in a HEPES buffer that contained various concentrations of inert dsDNA. As shown in Fig. 5(C), increasing the concentration of inert dsDNA in the washing solution led to an increase in particle stability after washing, therefore confirming our hypothesis. Including 1.4 μM of inert DNA in the washing solution allowed us to preserve the particle stability (fsingle = 0.91).
However, washing the particles with such high concentrations of inert dsDNA proved detrimental to the bilayer and led to the formation of membrane tubes between 1 μm to 3 μm long (see Fig. S4 in the ESI†). These membrane tubes are highly curved surfaces that are only formed under specific conditions, for example a difference in spontaneous curvature between the inner and the outer membrane leaflets.77 In our case, since dsDNA is only added after the formation of the bilayer it is only present in the outer leaflet and hence induces a difference in spontaneous curvature which causes formation of tubes. If the DNA would be present on both leaflets, no tube formation is expected. We tested this by mixing the dsDNA with the SUVs before coating the particles but found no fluorescence signal on the particles' surface, implying the absence of a bilayer coating (see Fig. S5 in the ESI†). We believe that the dsDNA sterically stabilizes the SUVs and prevents spreading and fusion of SUVs on the particle surface.
To summarize, we found that inert double-stranded DNA can impart colloidal stability to the CSLBs. Asymmetric distribution of DNA causes the formation of membrane tubes at high coating concentrations (above approximately 1 × 105 μm−2), but is unavoidable if colloidal stability needs to be preserved during repeated washing cycles.
Linker functionalization for self-assembly
To be able to employ CSLBs in self-assembly experiments, we induce specific attractive interactions by using two sets of dsDNA linkers with complementary single-stranded sticky ends that can form bonds via hybridization. We use two hydrophobic anchors (either cholesterol or stearyl) per dsDNA complex to insert the dsDNA linker into the outer leaflet (see Fig. 1(2)), while at the same time adding inert dsDNA strands for steric stabilization. We use double hydrophobic anchors because dsDNA with a single hydrophobic anchor is less strongly confined to the bilayer.14 The dsDNA is attached to the double hydrophobic anchor with a tetraethylene glycol (TEG) or hexaethylene glycol (HEG) spacer to allow it to swivel with respect to the anchor. We label the dsDNA linkers with a fluorescent dye to image them using confocal microscopy. Full details on the DNA strands we used can be found in Table S1 of the ESI.†Previous experiments16 have shown that several interesting structures, such as flexible colloidal polymers and molecules, can be obtained via self-assembly of CSLBs. In order to form these reconfigurable structures, not only the lipids in the bilayer should be mobile but also the grafted linker DNA should be mobile on the surface of the membrane. We can quantify the mobility of dsDNA on the surface of the bilayer by measuring the FRAP of fluorescently labeled DNA. Note that for these experiments we did not employ fluorescent lipids. For a successful recovery after bleaching of the DNA linkers in the binding patch between two particles, two requirements need to be fulfilled: the DNA linkers outside of the binding patch have to be mobile and the bleached linkers inside the binding patch have to be able to unbind, to allow unbleached linkers to diffuse into the binding patch. We calculate the melting temperature of the sticky end using an approximate formula78 and find a melting temperature Tm = 30.4 °C, meaning that at 25 °C the probability for the sticky end to form a duplex is P(25 °C) = 0.68 based on melting temperature considerations only. Therefore, the sticky ends continuously bind and unbind in our experiments, making fluorescence recovery possible, while the sheer number of linkers in the patch area keeps the particles always bound.
We confirmed the mobility of linker DNA on the particle surface using FRAP experiments, shown in Fig. 6. Note that the whole cluster is immobilized on the (non-passivated) glass coverslip to enable FRAP on these micrometer-sized particles. Therefore these clusters do not show translational diffusion or cluster flexibility. In Fig. 6(A), a representative FRAP experiment on a single sphere is shown. We measured an average recovery time of (8 ± 2) s from independent measurements on 16 different particles. Outside the bond area (Fig. 6B), we measured an average recovery time of (6 ± 2) s from independent measurements on 15 different clusters. Within the error, the diffusion of DNA outside of the bond area is the same as on free particles, as expected. Inside the bond area (Fig. 6C), we measured recovery times of 5 s to 62 s with an average recovery time of (17 ± 16) s from independent measurements on 12 different clusters. This is longer than the recovery times we measured outside the bond area and on single particles, which indicates the diffusion is slower in the bond area. However, the recovery time inside the bond area varied greatly between different clusters indicating that diffusion into and out of the bond area can be different. A likely cause is the spread in the DNA concentration between individual particles in a batch. Higher DNA concentrations imply a higher concentration of bonded linkers in the bond area. This will sterically hinder the diffusion of unbonded linkers inside the patch area and will thus lead to longer recovery times. Furthermore, we hypothesize that the linker concentration in the patch area slowly increases as a function of time after binding, so that the recovery time depends on the time that has elapsed after the formation of the cluster, which we did not control. In summary, we have shown here that the dsDNA linkers are mobile on each part of the (un)bound particle, which is a prerequisite for creating flexibly linked clusters.
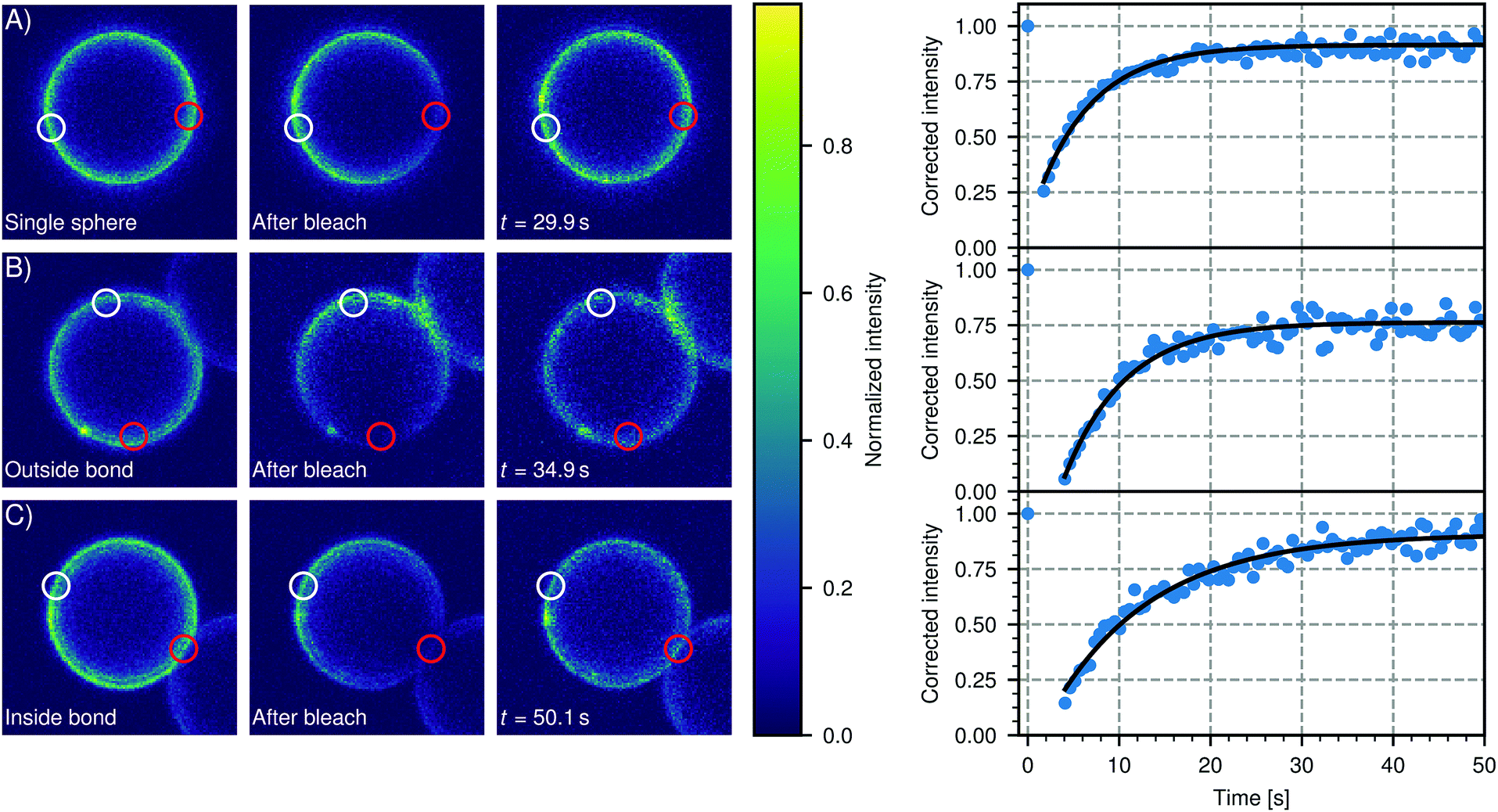 | ||
| Fig. 6 DNA linker mobility on CSLBs. Three representative FRAP experiments are shown that probe the mobility of DNA linkers on the surface of a 7 μm CSLB (particle type 1, Table 1). In the intensity color plots, a red circle indicates the bleached area and a white circle shows the reference area. For all experiments, the DNA linkers are mobile. (A) FRAP on a single sphere. We measured an average recovery time of (8 ± 2) s from independent measurements on 16 different particles. (B) FRAP on a sphere in a cluster, taken outside the bond area. We measured an average recovery time of (6 ± 2) s from independent measurements on 15 different clusters. Note that, within the error, this is the same as the recovery time we measured for single particles in (A). (C) FRAP on a sphere in a cluster, inside the bond area. We measured recovery times of 5 s to 62 s with an average recovery time of (17 ± 16) s from independent measurements on 12 different clusters. Note that this is longer than the recovery times we measured outside the bond area in (A) and (B), which indicates the diffusion is slower in the bond area. However, the spread in recovery times was also larger for bleached areas inside the bond area. | ||
Mobility of self-assembled structures
The mobility of individual dsDNA linkers on the surface does not necessarily imply that bonded clusters of DNA functionalized CSLBs are also reconfigurable. For example, for emulsion droplets functionalized with DNA linkers, the large linker patch that is formed between particles can slow down the motion when the supporting fluid is inhomogeneous61 and colloidal joints lose their flexibility with an increasing concentration of dsDNA linkers in the bond area.16To measure the flexibility of larger structures, we assembled CSLBs with complementary dsDNA linkers and imaged chains of three CSLBs, so called trimers, over time. We extracted the position of the individual particles and the opening angle θ (see inset Fig. 7A) for all frames and calculated the mean squared angular displacement (MSAD) to characterize the flexibility.16 Because of the high density of the silica spheres (particle type 1, Table 1), the particles sedimented to the glass substrate and were therefore confined to move in a quasi-2D plane. To investigate the influence of the membrane homogeneity on the structural flexibility, we compared trimers assembled from CSLBs with homogeneous, fluid bilayer to trimers with bilayers that had spontaneously formed membrane tubes. In the following, we will only show the free energy landscape for θ from 60° to 180° due to the symmetry of a trimer.
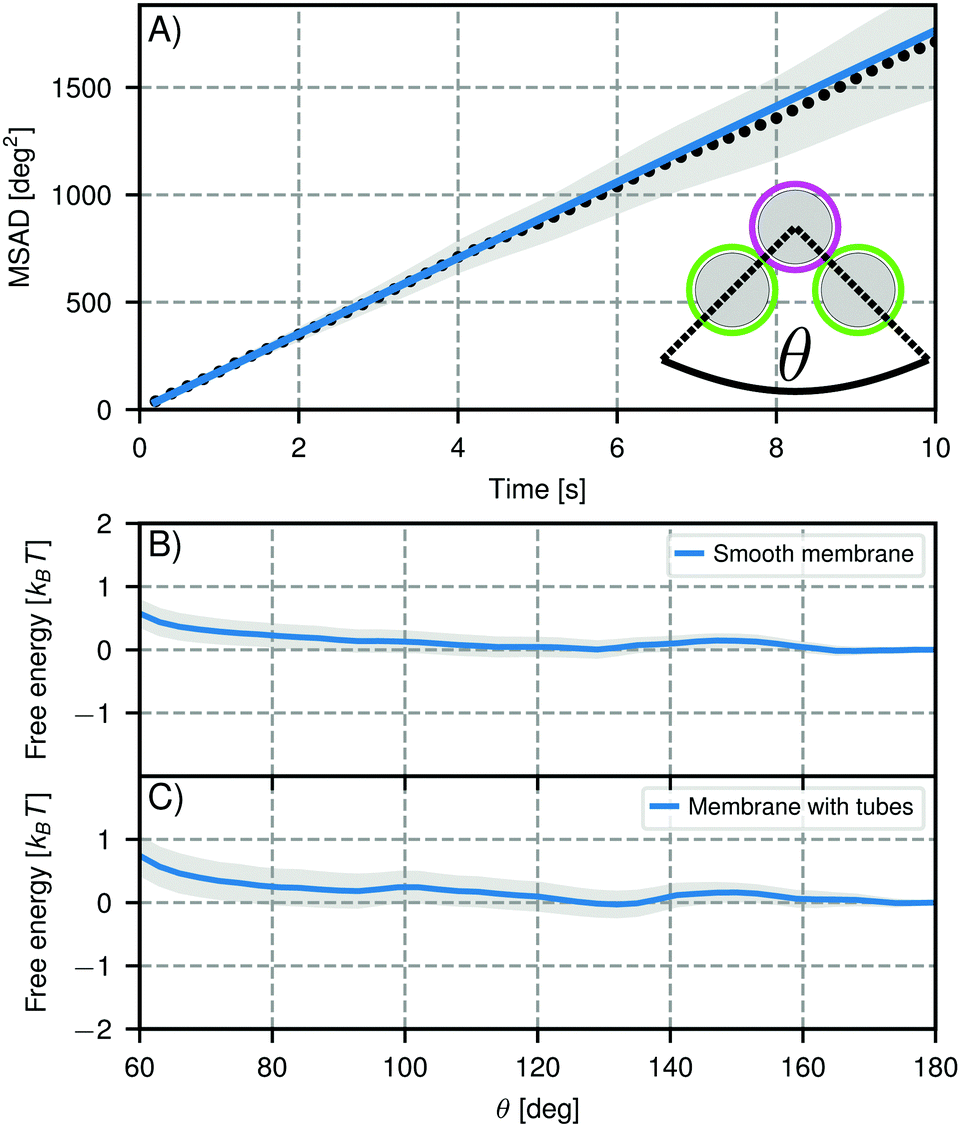 | ||
| Fig. 7 Mobility of self-assembled trimers. (A) Mean squared angular displacement (MSAD) of the opening angle θ for a mobile trimer assembled from particles of type 1, Table 1. The MSAD is linear and we find a joint flexibility J = (176 ± 12) deg2 s−1 (J = (184 ± 101) deg2 s−1 when averaging over all experiments). (B and C) Free energy as function of opening angle θ for particles with (B) smooth bilayers and (C) bilayers that have membrane tubes. The grey shaded area marks one standard deviation confidence interval. We analyzed 53 clusters with smooth bilayers and 18 clusters with membrane tubes. In both cases, we find no preference for specific opening angles within the experimental error, meaning the particles are freely-jointed. Note that the slight repulsion at small angles is caused by boundary effects inherent to our analysis method. | ||
For trimers made from CSLBs with smooth lipid bilayers, we found that the particles (D = 2 μm) move with respect to each other over the full angular range. We analyzed the opening angle θ for 53 different clusters by tracking the individual particles and calculating θ for all frames (see inset Fig. 7A). The average value of the flexibility of the trimers is J = (184 ± 101) deg2 s−1 (or (0.03 ± 0.02) μm2 s−1) and agrees well with previous experiments.16 The spread in the flexibility that we observe is likely caused by the spread in dsDNA linker density on the particles. We then determined the free energy using the maximum likelihood estimation of angular displacements method (see Experimental section, as shown in Fig. 7B). We found no preference in the opening angle that is significant with respect to the thermal energy kBT, indicating that the surface is smooth enough to allow the particles to move over one another without restrictions. We observed a boundary effect inherent to the analysis for angles smaller than 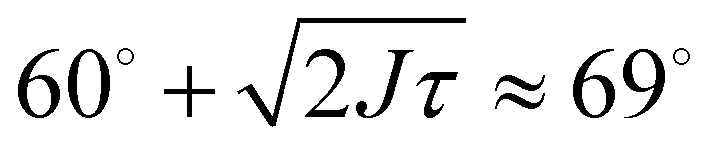 (where J is the joint flexibility) leading to a slight overestimation of the free energy for those angles. In earlier experiments, we found a small preference (0.9kBT free energy difference) for opening angles around 140°.16 However, at that time we could not accurately determine the confidence interval of the calculated free energy. When re-analyzing the data from ref. 16 using our maximum likelihood method, we noted that the observed free energy difference is within the experimental error (see Fig. S3 in the ESI†). This is in line with the free energy reported here, which we calculated from a more extensive dataset, and with recent experiments on flexibly linked emulsion droplets, which are freely-jointed like the CSLBs we present here.18
(where J is the joint flexibility) leading to a slight overestimation of the free energy for those angles. In earlier experiments, we found a small preference (0.9kBT free energy difference) for opening angles around 140°.16 However, at that time we could not accurately determine the confidence interval of the calculated free energy. When re-analyzing the data from ref. 16 using our maximum likelihood method, we noted that the observed free energy difference is within the experimental error (see Fig. S3 in the ESI†). This is in line with the free energy reported here, which we calculated from a more extensive dataset, and with recent experiments on flexibly linked emulsion droplets, which are freely-jointed like the CSLBs we present here.18
To test the robustness of this structural flexibility in the presence of membrane inhomogeneities, we prepared particles with a large number of membrane tubes by washing with inert dsDNA. As shown in Fig. 7(C), this did not alter the flexibility of the clusters: they still explore the full angular range and do not show a significant preferred opening angle, similar to the particles coated with a smooth membrane. Surprisingly, this means that particles stabilized by dsDNA can in principle be used for self-assembly studies despite the fact that the high concentration of dsDNA causes membrane tubes to form, because the tubes do not significantly alter the relative motion of clustered particles.
Summary and conclusions
We investigated various factors in the preparation protocol of colloid supported lipid bilayers (CSLBs) in view of their emerging use in self-assembly and model membrane studies. Specifically, we focused on realizing a homogeneous and fluid bilayer, while achieving colloidal stability and functionalization with DNA linkers at the same time.Similar to what has been reported for flat supported lipid bilayers, we found that the quality of the lipid bilayer on colloidal particles critically depends on the material of the particle's surface. The bilayer was not fluid on particles made from polystyrene (with or without carboxyl groups), hematite and TPM particles (with or without carboxyl or amino groups). Colloids featuring a silica surface, on the other hand, were able to host a fluid and homogenous bilayer, at least in the absence of any polymer residues from the synthesis. We furthermore observed that the variation in the substrate curvature does not affect the bilayer formation if it is sufficiently gentle, while excessive surface roughness can hinder the spreading and fusion of SUVs.
Use of PEGylated lipids in the bilayer increased the colloidal stability, but affected the bilayer homogeneity and mobility negatively. Addition of the amphiphilic surfactant SDS led to a disintegration of the bilayer. A better way to provide colloidal stability is by steric stabilisation by excluded volume effects through the insertion of double-stranded inert DNA. Increasing the concentration of dsDNA leads to an increase in colloidal stability.
Finally, we demonstrated that these CSLBs can be functionalized with surface-mobile DNA linkers and assembled them into flexible structures of freely-jointed particles. We found that local bilayer inhomogeneities in the form of membrane tubes do not affect the free energy landscape of the connected particles.
CSLBs with fluid, homogeneous membranes and surface-mobile binding groups have great promise in a wide range of applications and fundamental studies. The fact that the bonded particles can flexibly move with respect to each other opens the door to overcoming equilibration issues previously encountered in hit-and-stick processes and assembling structures with internal deformation modes. This enables studying the impact of structural flexibility on the phase behaviour, such as the formation of crystals with new lattices or properties,19–22 and the experimental realization of information elements for wet computing.25 CSLBs with increased membrane fluidity also mimic biological membranes more closely which may be advantageous for model membrane and cell biology studies8–12 smart drug delivery2–4 and bio-sensing applications.5,6
Author contributions
MR and RWV contributed equally to the work. MR and RWV performed the experiments and the data analysis. IC performed exploratory experiments. MR, RWV and DJK conceived the experiments and wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Rachel Doherty and Vera Meester for help with the colloidal syntheses and electron microscopy imaging, and Piermarco Fonda and Luca Giomi for useful discussions. This work was supported by the Netherlands Organization for Scientific Research (NWO/OCW), as part of the Frontiers of Nanoscience program and VENI grant 680-47-431. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement no. 758383).References
- A. L. Troutier and C. Ladavière, An overview of lipid membrane supported by colloidal particles, Adv. Colloid Interface Sci., 2007, 133(1), 1–21 CrossRef CAS PubMed.
- A. M. Carmona-Ribeiro, Preparation and characterization of biomimetic nanoparticles for drug delivery, Nanoparticles in Biology and Medicine: Methods and Protocols, Humana Press, Totowa, NJ, 2012, pp. 283–294 Search PubMed.
- J. Li, et al., A review on phospholipids and their main applications in drug delivery systems, Asian J. Pharm. Sci., 2015, 10(2), 81–98 CrossRef.
- S. Savarala, S. Ahmed, M. A. Ilies and S. L. Wunder, Formation and Colloidal Stability of DMPC Supported Lipid Bilayers on SiO2 Nanobeads, Langmuir, 2010, 26(14), 12081–12088 CrossRef CAS PubMed.
- E. T. Castellana and P. S. Cremer, Solid supported lipid bilayers: From biophysical studies to sensor design, Surf. Sci. Rep., 2006, 61(10), 429–444 CrossRef CAS.
- S. Chemburu, K. Fenton, G. P. Lopez and R. Zeineldin, Biomimetic Silica Microspheres in Biosensing, Molecules, 2010, 15(12), 1932–1957 CrossRef CAS PubMed.
- I. Brouwer, et al., Direct quantitative detection of Doc2b-induced hemifusion in optically trapped membranes, Nat. Commun., 2015, 6, 8387 CrossRef CAS PubMed.
- E. Sackmann, Supported membranes: scientific and practical applications, Science, 2007, 271(5245), 43–48 CrossRef.
- C. Madwar, G. Gopalakrishnan and R. B. Lennox, Interfacing living cells and spherically supported bilayer lipid membranes, Langmuir, 2015, 31(16), 4704–4712 CrossRef CAS PubMed.
- S. Mashaghi, T. Jadidi, G. Koenderink and A. Mashaghi, Lipid Nanotechnology, Int. J. Mol. Sci., 2013, 14, 4242–4282 CrossRef CAS PubMed.
- M. Rinaldin, P. Fonda, L. Giomi and D. J. Kraft, Geometric pinning and antimixing in scaffolded lipid vesicles, 2018, arXiv e-prints.
- P. Fonda, M. Rinaldin, D. J. Kraft and L. Giomi, Interface geometry of binary mixtures on curved substrates, Phys. Rev. E, 2018, 98, 032801 CrossRef.
- S. A. Van Der Meulen and M. E. Leunissen, Solid colloids with surface-mobile DNA linkers, J. Am. Chem. Soc., 2013, 135(40), 15129–15134 CrossRef CAS PubMed.
- S. A. J. Van Der Meulen, G. V. Dubacheva, M. Dogterom, R. P. Richter and M. E. Leunissen, Quartz crystal microbalance with dissipation monitoring and spectroscopic ellipsometry measurements of the phospholipid bilayer anchoring stability and kinetics of hydrophobically modified DNA oligonucleotides, Langmuir, 2014, 30(22), 6525–6533 CrossRef CAS PubMed.
- S. A. J. Van der Meulen, G. Helms and M. Dogterom, Solid colloids with surface-mobile linkers, J. Phys.: Condens. Matter, 2015, 27(23), 233101 CrossRef PubMed.
- I. Chakraborty, V. Meester, C. Van der Wel and D. J. Kraft, Colloidal joints with designed motion range and tunable joint flexibility, Nanoscale, 2017, 9, 7814–7821 RSC.
- L. Feng, L.-L. Pontani, R. Dreyfus, P. Chaikin and J. Brujic, Specificity, flexibility and valence of DNA bonds guide emulsion architecture, Soft Matter, 2013, 9(41), 9816–9823 RSC.
- A. McMullen, M. Holmes-Cerfon, F. Sciortino, A. Y. Grosberg and J. Brujic, Freely jointed polymers made of droplets, Phys. Rev. Lett., 2018, 121, 138002 CrossRef PubMed.
- K. L. Kohlstedt and S. C. Glotzer, Self-assembly and tunable mechanics of reconfigurable colloidal crystals, Phys. Rev. E – Statistical, Nonlinear, and Soft Matter Physics, 2013, 87(3), 032305 CrossRef.
- D. Ortiz, K. L. Kohlstedt, T. D. Nguyen and S. C. Glotzer, Self-assembly of reconfigurable colloidal molecules, Soft Matter, 2014, 10(20), 3541–3552 RSC.
- F. Smallenburg, L. Filion and F. Sciortino, Erasing no-man's land by thermodynamically stabilizing the liquid-liquid transition in tetrahedral particles, Nat. Phys., 2014, 10(9), 653–657 Search PubMed.
- H. Hu, P. S. Ruiz and R. Ni, Entropy Stabilizes Floppy Crystals of Mobile DNA-Coated Colloids, Phys. Rev. Lett., 2018, 120(4), 048003 CrossRef PubMed.
- J. D. Joannopoulos, P. R. Villeneuve and S. Fan, Photonic crystals: putting a new twist on light, Nature, 1997, 386(6621), 143–149 CrossRef CAS.
- Y. Lin, et al., Photonic band structure of colloidal crystal self-assembled in hollow core optical fiber, Appl. Phys. Lett., 2005, 86(12), 121106 CrossRef.
- C. L. Phillips, et al., Digital colloids: reconfigurable clusters as high information density elements, Soft Matter, 2014, 10(38), 7468–7479 RSC.
- M. Hadorn and P. Eggenberger Hotz, DNA-Mediated Self-Assembly of Artificial Vesicles, PLoS One, 2010, 5(3), e9886 CrossRef PubMed.
- P. A. Beales, J. Nam and T. K. Vanderlick, Specific adhesion between dna-functionalized janus vesicles: size-limited clusters, Soft Matter, 2011, 7, 1747–1755 RSC.
- L. Parolini, et al., Volume and porosity thermal regulation in lipid mesophases by coupling mobile ligands to soft membranes, Nat. Commun., 2015, 6, 5948 CrossRef CAS PubMed.
- V. T. Milam, Oligonucleotide-based recognition in colloidal systems – opportunities and challenges, Curr. Opin. Colloid & In. Sci., 2016, 26, 75–83 Search PubMed.
- Y. Wang, et al., Colloids with valence and specific directional bonding, Nature, 2012, 491(7422), 51–55 CrossRef CAS PubMed.
- W. B. Rogers and V. N. Manoharan, Programming colloidal phase transitions with DNA strand displacement, Science, 2015, 347(6222), 639–642 CrossRef CAS PubMed.
- Y. Wang, et al., Crystallization of DNA-coated colloids, Nat. Commun., 2015, 6, 7253 CrossRef CAS PubMed.
- N. B. Schade, et al., Tetrahedral Colloidal Clusters from Random Parking of Bidisperse Spheres, Phys. Rev. Lett., 2013, 110(14), 148303 CrossRef PubMed.
- Y. Zhang, et al., Sequential self-assembly of DNA functionalized droplets, Nat. Commun., 2017, 8(1), 21 CrossRef PubMed.
- T. Sugimoto and K. Sakata, Preparation of monodisperse pseudocubic α-Fe2O3 particles from condensed ferric hydroxide gel, J. Colloid Interface Sci., 1992, 152(2), 587–590 CrossRef CAS.
- L. Rossi, et al., Cubic crystals from cubic colloids, Soft Matter, 2011, 7(9), 4139–4142 RSC.
- V. Meester and D. J. Kraft, Spherical, Dimpled, and Crumpled Hybrid Colloids with Tunable Surface Morphology, Langmuir, 2016, 32(41), 10668–10677 CrossRef CAS PubMed.
- C. Van der Wel, et al., Preparation of colloidal organosilica spheres through spontaneous emulsification, Langmuir, 2017, 33(33), 8174–8180 CrossRef CAS PubMed.
- R. P. Doherty and D. J. Kraft, One-pot surfactant-free synthesis of organosilica colloids with various surface functional groups, in preparation.
- J. Appel, S. Akerboom, R. G. Fokkink and J. Sprakel, Facile One-Step Synthesis of Monodisperse Micron-Sized Latex Particles with Highly Carboxylated Surfaces, Macromol. Rapid Commun., 2013, 34(16), 1284–1288 CrossRef CAS PubMed.
- P. S. Cremer and S. G. Boxer, Formation and Spreading of Lipid Bilayers on Planar Glass Supports, J. Phys. Chem. B, 1999, 103(13), 2554–2559 CrossRef CAS.
- A. Y. Antipina and A. A. Gurtovenko, Molecular-level insight into the interactions of DNA with phospholipid bilayers: barriers and triggers, RSC Adv., 2016, 6, 36425–36432 RSC.
- C. Van der Wel, et al., Lipid membrane-mediated attraction between curvature inducing objects, Sci. Rep., 2016, 6, 32825 CrossRef CAS PubMed.
- D. Axelrod, D. Koppel, J. Schlessinger, E. Elson and W. Webb, Mobility measurement by analysis of fluorescence photobleaching recovery kinetics, Biophys. J., 1976, 16(9), 1055–1069 CrossRef CAS PubMed.
- D. B. Allan, T. Caswell, N. C. Keim and C. M. Van der Wel, trackpy: Trackpy v0.4.1, 2018, URL https://doi.org/10.5281/zenodo.1226458.
- C. M. Van der Wel, Lipid Mediated Colloidal Interactions, Casimir PhD Series, Leiden University, The Netherlands, 2017.
- R. Sarfati, J. Bławzdziewicz and E. R. Dufresne, Maximum likelihood estimations of force and mobility from single short Brownian trajectories, Soft Matter, 2017, 13(11), 2174–2180 RSC.
- D. Foreman-Mackey, D. W. Hogg, D. Lang and J. Goodman, emcee: The MCMC Hammer, 2013, 125, 306, ArXiv e-prints.
- G. Gopalakrishnan, I. Rouiller, D. R. Colman and R. B. Lennox, Supported bilayers formed from different phospholipids on spherical silica substrates, Langmuir, 2009, 25(10), 5455–5458 CrossRef CAS PubMed.
- R. P. Richter, R. Bérat and A. R. Brisson, Formation of solid-supported lipid bilayers: an integrated view, Langmuir, 2006, 22(8), 3497–3505 CrossRef CAS PubMed.
- E. Sackmann, Supported Membranes: Scientific and Practical Applications, Science, 1996, 271(5245), 43–48 CrossRef CAS PubMed.
- J. Rädler, H. Strey and E. Sackmann, Phenomenology and Kinetics of Lipid Bilayer Spreading on Hydrophilic Surfaces, Langmuir, 1995, 11(11), 4539–4548 CrossRef.
- I. Gözen and A. Jesorka, Instrumental methods to characterize molecular phospholipid films on solid supports, Anal. Chem., 2012, 84(2), 822–838 CrossRef PubMed.
- R. Macháň and M. Hof, Lipid diffusion in planar membranes investigated by fluorescence correlation spectroscopy, Biochim. Biophys. Acta, 2010, 1798(7), 1377–1391 CrossRef PubMed.
- Y. Jing, H. Trefna, M. Persson, B. Kasemo and S. Svedhem, Formation of supported lipid bilayers on silica: relation to lipid phase transition temperature and liposome size, Soft Matter, 2014, 10(1), 187–195 RSC.
- S. I. R. Castillo, et al., Silica cubes with tunable coating thickness and porosity: From hematite filled silica boxes to hollow silica bubbles, Microporous Mesoporous Mater., 2014, 195, 75–86 CrossRef CAS.
- T. Cha, A. Guo and X.-Y. Zhu, Formation of supported phospholipid bilayers on molecular surfaces: Role of surface charge density and electrostatic interaction, Biophys. J., 2006, 90(4), 1270–1274 CrossRef CAS PubMed.
- P. G. De Gennes, Polymers at an interface; a simplified view, Adv. Colloid Interface Sci., 1987, 27(3–4), 189–209 CrossRef CAS.
- S. Upadhyayula, et al., Coatings of polyethylene glycol for suppressing adhesion between solid microspheres and flat surfaces, Langmuir, 2012, 28(11), 5059–5069 CrossRef CAS PubMed.
- C. Van der Wel, et al., Surfactant-free colloidal particles with specific binding affinity, Langmuir, 2017, 33(38), 9803–9810 CrossRef CAS PubMed.
- C. Van Der Wel, G. L. Van De Stolpe, R. W. Verweij and D. J. Kraft, Micrometer-sized TPM emulsion droplets with surface-mobile binding groups, J. Phys.: Condensed Matter, 2018, 30(9), 094005 CrossRef PubMed.
- C. A. Naumann, et al., The polymer-supported phospholipid bilayer: Tethering as a new approach to substrate- membrane stabilization, Biomacromolecules, 2002, 3(1), 27–35 CrossRef CAS PubMed.
- M. Tanaka and E. Sackmann, Polymer-supported membranes as models of the cell surface, Nature, 2005, 437(7059), 656–663 CrossRef CAS PubMed.
- M. Tanaka, Polymer-Supported Membranes: Physical Models of Cell Surfaces, MRS Bull., 2006, 31, 513–520 CrossRef CAS.
- M. L. Wagner and L. K. Tamm, Tethered Polymer-Supported Planar Lipid Bilayers for Reconstitution of Integral Membrane Proteins: Silane-Polyethyleneglycol-Lipid as a Cushion and Covalent Linker, Biophys. J., 2000, 79(3), 1400–1414 CrossRef CAS PubMed.
- M. A. Deverall, et al., Transbilayer coupling of obstructed lipid diffusion in polymer-tethered phospholipid bilayers, Soft Matter, 2008, 4, 1899–1908 RSC.
- R. Lipowsky, Bending of membranes by anchored polymers, Europhys. Lett., 1995, 30(4), 197 CrossRef CAS.
- N. Geerts and E. Eiser, DNA-functionalized colloids: physical properties and applications, Soft Matter, 2010, 6(19), 4647–4660 RSC.
- M.-P. Valignat, O. Theodoly, J. C. Crocker, W. B. Russel and P. M. Chaikin, Reversible self-assembly and directed assembly of DNA-linked micrometer-sized colloids, Proc. Natl. Acad. Sci. U. S. A., 2005, 102(12), 4225–4229 CrossRef CAS PubMed.
- P. L. Biancaniello, J. C. Crocker, D. A. Hammer and V. T. Milam, DNA-mediated phase behavior of microsphere suspensions, Langmuir, 2007, 23(5), 2688–2693 CrossRef CAS PubMed.
- G. Nordlund, R. Lönneborg and P. Brzezinski, Formation of Supported Lipid Bilayers on Silica Particles Studied Using Flow Cytometry, Langmuir, 2009, 25(8), 4601–4606 CrossRef CAS PubMed.
- A. Tan, A. Ziegler, B. Steinbauer and J. Seelig, Thermodynamics of sodium dodecyl sulfate partitioning into lipid membranes, Biophys. J., 2002, 83(3), 1547–1556 CrossRef CAS PubMed.
- T. Drobek, N. D. Spencer and M. Heuberger, Compressing PEG brushes, Macromolecules, 2005, 38(12), 5254–5259 CrossRef CAS.
- F. Meng, G. H. M. Engbers and J. Feijen, Polyethylene glycol-grafted polystyrene particles, J. Biomed. Mater. Res., Part A, 2004, 70(1), 49–58 CrossRef PubMed.
- O. Garbuzenko, Y. Barenholz and A. Priev, Effect of grafted PEG on liposome size and on compressibility and packing of lipid bilayer, Chem. Phys. Lipids, 2005, 135(2), 117–129 CrossRef CAS PubMed.
- S. Angioletti-Uberti, P. Varilly, B. M. Mognetti and D. Frenkel, Mobile linkers on DNA-coated colloids: Valency without patches, Phys. Rev. Lett., 2014, 113(12), 128303 CrossRef PubMed.
- R. Lipowsky, Spontaneous tubulation of membranes and vesicles reveals membrane tension generated by spontaneous curvature, Faraday Discuss., 2013, 161, 305–331 RSC.
- S. I. Nakano, M. Fujimoto, H. Hara and N. Sugimoto, Nucleic acid duplex stability: Influence of base composition on cation effects, Nucleic Acids Res., 1999, 27(14), 2957–2965 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: The supporting information consists of 5 figures and 1 table: large fields of view of microscopy images; a table listing all employed DNA strands; a schematic representation of the particle tracking algorithm; a graph showing data from a previous paper re-analyzed using a different method; a microscopy picture of three CSLBs that feature membrane tubes; a microscopy picture of particles coated with DNA-stabilized SUVs. See DOI: 10.1039/c8sm01661e |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2019 |