
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA simple and general approach for in situ synthesis of sulfur–porous carbon composites for lithium–sulfur batteries†
Noel
Díez
 ,
Guillermo A.
Ferrero
,
Marta
Sevilla
and
Antonio B.
Fuertes
*
,
Guillermo A.
Ferrero
,
Marta
Sevilla
and
Antonio B.
Fuertes
*
Instituto Nacional del Carbón (CSIC), Fco. Pintado Fe 26, Oviedo 33011, Spain. E-mail: abefu@incar.csic.es
First published on 7th October 2019
Abstract
Porous carbon materials constitute an excellent conductive matrix for the immobilization of sulfur in the cathode of lithium–sulfur batteries. Herein, we provide a novel, easy, sustainable, general and scalable procedure for the preparation of such cathodes, which combines in the same process both the generation of a S-doped porous carbon and the incorporation of elemental sulfur within the pores. To achieve this, a biomass-based substance (i.e., tannic acid) is activated with sodium thiosulfate, yielding a carbonized solid that includes a high-surface area carbon (up to 2550 m2 g−1) and sodium polysulfides. Once this solid is immersed in an acid medium, the polysulfides generated as activation by-products are oxidized in situ to elemental sulfur that spontaneously diffuses into the pores of the carbon, which gives rise to the hybrid sulfur–carbon composite. When tested as cathodes in lithium–sulfur batteries, these hybrid materials exhibit high sulfur utilization (∼80%) even for the composites with a sulfur content as high as 82%, as well as an excellent rate performance (590 mA h g−1 S at 5C). The cycling stability of the batteries was confirmed by their low capacity fading (0.049% of capacity decay per cycle), maintaining 85% of their initial capacity after 300 charge–discharge cycles at 1C. Cathodes with a high sulfur loading (7.1 mg S per cm2) were steady cycled providing reversible areal capacities of 4.4 mA h cm−2 at 0.2C after 100 charge–discharge cycles.
1. Introduction
Lithium–sulfur batteries have generated a huge interest as energy storage systems because of the low cost of sulfur and its high theoretical capacity (1675 mA h g−1) that can provide a high specific energy (∼2500 W h kg−1).1–5 However, the commercial viability of such batteries is restricted by the rapid capacity decay of the sulfur cathode. This is caused by the combination of several factors related to the high volume expansion taking place during the reduction of S to Li2S, the poor electrical conductivity of the end-products (S and Li2S) and the loss of active material due to the dissolution and migration of intermediate lithium polysulfides (Li2Sx, 4 ≤ x ≤ 8) (shuttle effect).6–10 In an attempt to solve these problems, a great number of strategies aimed at the optimization of the design of the sulfur cathode have been proposed. Most of these efforts have been addressed towards the improvement of electrical conductivity and the suppression of polysulfide mobility.11–14 In this respect, the combination of elemental sulfur with several types of carbon materials (i.e., carbon nanotubes, carbon blacks, carbon nanofibers, graphene and porous carbons) has been thoroughly investigated.15–25 In particular, given their availability, low-cost and easy preparation, porous carbons constitute an ideal sulfur host because of their good electrical conductivity as well as their high pore volume, able to accommodate the volumetric changes and, in the narrow pores, trap and limit the diffusion of the polysulfides.Given the growing demand for carbon materials for electrochemical applications such as Li–S batteries, it can be envisaged that large amounts of these materials will be needed in the coming years. It is reasonable to assume that the porous materials should be produced following green criteria that mean the use of sustainable carbon precursors (i.e., biomass-based products) and less toxic chemical substances.26 On this subject, we recently reported a novel procedure for the production of high-surface area carbons based on these criteria.27,28 Indeed, as carbon precursors we investigated a variety of renewable biomass-based products and the activation was carried out using a harmless ingredient such as sodium thiosulfate. This activation process emerges as a general sustainable strategy for the production of highly porous carbons.
The development of simple approaches for the preparation of carbon-based Li–S cathodes is a significant challenge. Several authors have investigated straightforward procedures towards sulfur/carbon composites. In these synthesis strategies, elemental sulfur nanoparticles are generated in situ by means of a wet chemical process. Thus, in a pioneering work Wang et al. reported the fabrication of nanocomposites made up of graphene layers enveloping sulfur nanoparticles which were synthesized in situ by the disproportionation of sodium thiosulfate in the presence of hydrochloric acid (Na2S2O3 + 2HCl → S + SO2 + H2O + 2NaCl).16 These sulfur nanocomposites showed enhanced electrochemical performance. This approach has also been applied to other substrates such as activated carbons,29,30 carbon black,31 reduced graphene oxide24 or polypyrrole.32 In another approach, sulfur nanoparticles were also generated in situ by the decomposition of polysulfides in an acid medium [Sx2− + H+ → (x − 1)S + H2S] and then deposited onto carbon substrates such as carbon nanofibers33 or graphene sheets.34,35 Other alternatives to S/C nanocomposites based on the in situ production of sulfur nanoparticles include: (i) the Claus reaction [2H2S + SO2 → 3S + 2H2O] in the presence of carbon black,36 (ii) oxidation of H2S with graphene oxide that leads to S/RGO composites37 or (iii) electrolysis of H2S over a binder-free carbon monolith.38 Obviously, the above procedures require pre-synthesized carbon substrates, involving in many cases complicated and time-consuming synthesis procedures. On the other hand, the preparation of sulfur–carbon composites by co-processing the carbon and sulfur precursors has been investigated by several authors. For example, Roberts et al. proposed the preparation of S/C composites – although with low sulfur contents only up to 39% – by means of the thermal treatment of sodium poly(4-styrenesulfonate).39 Wang et al. reported the synthesis of carbon/sulfur composites by employing resorcinol–formaldehyde as the carbon precursor and zinc sulfide as the sulfur source via oxidation with iodine.40 Li et al. described the preparation of sulfur–carbon composites by using glucose as the carbon precursor and Na2S that acts as a template and the source of sulfur.41 The thermal treatment at 800 °C of the mixture gives rise to a composite made up of three-dimensional carbon and Na2S particles which are then oxidized to elemental sulfur with Fe3+. By using a similar approach, Luo et al. treated thermally at 900 °C a mixture of glucose and Na2SO4, obtaining a composite constituted by a porous carbon and sodium polysulfides that are converted into elemental sulfur with FeCl3.42 It is worth noting that in these examples, the porous carbons have a poor pore development (pore volume < 0.5 cm3 g−1), which means that only a small sulfur fraction can be accommodated within the pores, the rest being located at the outer surface of the carbon particles. Within this context, the development of a novel process that combines the synthesis of highly porous carbons with the generation and infiltration of elemental sulfur within the pore network constitutes a significant challenge.
Herein, we report an easy and general methodology for the fabrication of sulfur–carbon composites which is based on our recently reported green approach to producing high-surface carbons by the chemical activation of biomass-based substances with sodium thiosulfate.27 To illustrate this methodology we selected tannic acid, a type of polyphenol extracted from plants, as an example of biomass-derived carbon precursor. The solid obtained after the activation process consists of sodium sulfide/polysulfides intimately mixed with porous carbon particles. In an acid medium, the polysulfides suffer a disproportionation reaction that gives rise to elemental sulfur.33,34 This reaction gives us a direct way to produce sulfur–carbon composites via the incorporation of the in situ generated elemental sulfur into the pores of the carbon particles. In this way, the synthesis of highly porous carbon and the introduction of elemental sulfur within the pores can be combined in a simple process and sulfur–carbon composites with tunable sulfur content in the 50–90% range can be easily prepared. Importantly, because the synthesized porous carbons have high textural development with pore volumes over 2 cm3 g−1, they can accommodate a large amount of sulfur within their pores (>4 g of sulfur per gram of porous carbon). It constitutes a significant advance in relation to the single process approaches found in the literature that yield carbons with poor pore development. We tested these composites as cathodes in lithium–sulfur batteries and our results show that this material exhibits a high specific capacity of ca. 1300 mA h g−1 S even for the composites with sulfur content as high as 82%. Moreover, batteries cycled at 1C retained over 85% of their initial capacity after 300 cycles of charge–discharge, with a coulombic efficiency of above 99.5% over the whole cycling test. Cathodes with a high sulfur loading were also tested, showing high areal capacities of up to 6.50 mA h cm−2 (value superior to that of commercial Li-ion batteries) as well as steady cyclability, retaining 4.4 mA h cm−2 after 100 charge–discharge cycles at 0.2C.
2. Experimental
2.1. Preparation of the sulfur/porous carbon composites
In a typical synthesis, 3 g of tannic acid (Aldrich) was dissolved in 70 mL of deionized water together with potassium chloride (Aldrich) and sodium thiosulfate (Alfa-Aesar). The water solution was frozen in liquid nitrogen and then lyophilized (Telstar Cryodos) at −50 °C and 0.06 mbar. The dried powder was then heat treated under N2 up to 800 °C at a heating rate of 5 °C min−1 and held at this temperature for 1 h. The formed black monolith was then dispersed in 5 M HCl for 15 h under continuous stirring. In this step, the inorganic species (e.g. KCl and Na2S) are dissolved and elemental sulfur is generated due to the disproportionation of polysulfides. Finally, the sulfur/carbon composite was collected by filtration, washed with abundant deionized water and dried at 90 °C for several hours. In these experiments, we used different amounts of sodium thiosulfate (Na2S2O3/tannic acid weight ratio ranged between 1.5 and 3) while the Na2S2O3/KCl weight ratio was fixed at 0.45. The resulting sulfur/carbon samples are denoted as S/C-x, x being the Na2S2O3/tannic acid weight ratio (x = 1.5, 2.0, 2.3, 2.7 or 3). The porous carbon particles were collected from the S/C samples by removing the infiltrated sulfur, which was carried out by heat treatment of the composites up to 600 °C in a N2 atmosphere. These porous carbon samples are designated as C-x. For comparison purposes, the C-1.5/S composite (51.9 wt% S) was prepared by the melt-diffusion strategy. Thus, a certain amount of the porous carbon C-1.5 was ground together with elemental sulfur (Aldrich) and heated at 155 °C for 12 h in an argon-filled autoclave.2.2. Physicochemical characterization
Scanning electron microscopy (SEM) images were recorded by using a Quanta FEG650 (FEI) instrument, whereas transmission electron microscopy (TEM) images were acquired on a JEOL (JEM 2100-F) apparatus operating at 200 kV. The packing density and electrical conductivity of the carbon powders were determined in a homemade apparatus. 50–100 mg of carbon or sulfur–carbon composite was placed inside a hollow nylon cylinder with an inner diameter of 8 mm and pressed in between two stainless-steel plungers under a pressure of 7.1 MPa. The thickness of the disc formed by the pressed powdered carbon was determined by measuring the displacement of the upper plunger using a cathetometer. A certain current was passed through the plunger–sample–plunger system by using a DC power source (Promax, DC-405) and then, the voltage was measured with a multimeter (Fluke, 45 Dual display). The packing density of the carbon was determined in the same experiment. The N2 sorption isotherms of the carbon samples were measured at −196 °C using a Micromeritics ASAP 2020 sorptometer. The apparent surface area was calculated by the BET method. An appropriate relative pressure range was selected to ensure a positive line intersect of multipoint BET fitting (C > 0). The total pore volume was determined from the amount of nitrogen adsorbed at a relative pressure (p/po) of 0.99. The micropore volume and the pore size distributions (PSDs) were determined by means of the Quench Solid State Density Functional Theory (QSDFT) method for nitrogen. X-ray diffraction (XRD) patterns were obtained on a Siemens D5000 instrument operating at 40 kV and 20 mA, using a Cu-Kα radiation source. X-ray photoelectron spectroscopy (XPS) was carried out on a Specs spectrometer, using Mg KR (1253.6 eV) radiation from a double anode at 150 W. For the high resolution spectra, the calibration of the binding energies was performed by setting the C 1s signal to 284.5 eV. The elemental analysis of the samples was carried out on a LECO CHN-932 microanalyzer. The Raman spectra were recorded on a Horiba (LabRam HR-800) spectrometer. The source of radiation was a laser operating at a wavelength of 514 nm and at a power of 25 mW. Thermogravimetric analysis (TGA) curves were recorded on a TA Instruments Q6000 TGA system.2.3. Electrochemical measurements
160 mg of each sulfur/carbon composite, 20 mg of Super C65 carbon black and 20 mg of PVDF binder were mixed together and dispersed in N-methyl-2-pyrrolidone (NMP) by magnetic stirring to form thick slurries. The slurries were cast onto aluminium foil using the Doctor Blade technique and then dried at 50 °C under vacuum overnight. Once dried, the coated foil was cut into discs with a diameter of 10 mm. The areal sulfur loading of the cathodes was in the 1.0–1.5 mg cm−2 range, and over 7 mg cm−2 for the high S loading cathodes. CR2032 coin-type cells were assembled in an Ar filled glovebox using Li metal foil as the anode and a Celgard 2500 membrane as the separator. 100 μL of 1 M lithium bis(tri-fluoromethane) sulfonimide (LiTFSI) in a mixed solvent of 1,3-dioxolane and 1,2-dimethoxyethane (DOL/DME, vol/vol = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with 1 wt% LiNO3 was used as the electrolyte in all the batteries. The electrochemical performance of the cells was tested at room temperature in a computer-controlled potentiostat (Biologic VMP3 multichannel generator). Galvanostatic charge/discharge (CD) experiments were recorded over a potential range of 1.7–2.7 V (vs. Li+/Li) at different C rates, ranging from 0.05 to 5C (1C = 1675 mA g−1). Cyclic voltammetry (CV) experiments were recorded within the same potential window at a sweep rate of 0.05 mV s−1. Electrochemical impedance spectroscopy (EIS) curves were obtained at open-circuit potential within a frequency range from 100 kHz to 1 mHz using an amplitude of 10 mV.
:1) with 1 wt% LiNO3 was used as the electrolyte in all the batteries. The electrochemical performance of the cells was tested at room temperature in a computer-controlled potentiostat (Biologic VMP3 multichannel generator). Galvanostatic charge/discharge (CD) experiments were recorded over a potential range of 1.7–2.7 V (vs. Li+/Li) at different C rates, ranging from 0.05 to 5C (1C = 1675 mA g−1). Cyclic voltammetry (CV) experiments were recorded within the same potential window at a sweep rate of 0.05 mV s−1. Electrochemical impedance spectroscopy (EIS) curves were obtained at open-circuit potential within a frequency range from 100 kHz to 1 mHz using an amplitude of 10 mV.
3. Results and discussion
3.1. Synthesis of porous carbon and sulfur/carbon composites
The synthesis procedure is schematically illustrated in Fig. 1. In the first step, tannic acid, sodium thiosulfate and potassium chloride are dissolved in water and freeze-dried. Then the solid mixture is thermally treated under an inert atmosphere up to a high temperature (800 °C). During this thermal treatment, several consecutive processes take place: (i) the melting of tannic acid (∼200 °C) and decomposition of sodium thiosulfate to sodium sulfate (∼300 °C), (ii) the pyrolysis of the carbon precursor (220–400 °C) and (iii) the redox reaction between the pyrolyzed precursor and sodium sulfate (>500 °C) according to| Na2SO4 + [carbonaceous matter] → Na2Sx + [porous carbon] + CO/CO2 | (1) |
 | ||
| Fig. 1 Schematic illustration of the synthesis procedure. (a) Synthesis mixture (KCl, tannic acid and Na2S2O3), (b) melted KCl–tannic acid–Na2SO4 system, (c) carbonized monolith (carbon, Na2Sx and KCl) and (d) hybrid sulfur/carbon composite. | ||
As a result of this reaction, a black hard monolith constituting porous carbon particles, sodium sulfide/polysulfides and KCl is obtained. The inert salt (KCl) incorporated in the synthesis mixture has an important role because this substance melts at 770 °C providing a confined reaction medium that favours the activation reactions and therefore the development of porosity within the carbon. We have provided a detailed analysis of the reaction mechanism corresponding to this process elsewhere.27 When the carbonized solid is washed with water, the inorganic impurities (i.e., KCl and Na2Sx) are completely removed and, in this way, porous carbon particles can be easily collected. Taking another step forward, the polysulfides that are present in the washing liquor can be reused for the in situ generation of elemental sulfur. Thus, under acidic conditions the polysulfides suffer a disproportionation reaction that leads to the formation of elemental sulfur and hydrogen sulfide:33,34
| Na2Sx + 2HCl → (x − 1)S + H2S + 2NaCl | (2) |
An illustration of this process applied to the washing liquor is given in Fig. S1.† It can be seen that the filtrate obtained after washing the black monolith ((A) in Fig. S1†) with water exhibits a yellow colouring, which is indicative of the presence of sodium polysulfides ((B) in Fig. S1†). The addition of hydrochloric acid to this liquid provokes a sudden emission of H2S and the formation of a pale yellowish colloidal suspension ((C) in Fig. S1†), which slowly precipitates giving rise to a yellow solid ((D) in Fig. S1†). This solid product is identified as elemental sulfur by the EDX spectrum (Fig. S2a†) and also by the XRD pattern (Fig. S2b†), which reveals that the precipitated sulfur has an orthorhombic structure. This experiment clearly illustrates how the sulfur is generated in the procedure herein presented. In this way, when the carbonized monolith is immersed in HCl solution, there is a rapid dissolution of the inorganic species (i.e., KCl, sodium sulfide and sodium polysulfides) and the formation of elemental sulfur according to reaction (2). In the presence of the porous carbon particles, it can be envisaged that the generated sulfur nanoparticles will rapidly diffuse through the pore network as a consequence of capillary forces.41
In this approach, the formation of a sulfur/carbon composite takes place in an easy process that combines two consecutive steps: firstly, the production of a highly porous carbon and next, the generation and incorporation of elemental sulfur within the pores of the carbon material.
3.2. Structural, textural and chemical properties of the porous carbon host and the sulfur/carbon composites
The structure of the porous carbon host samples and the sulfur/carbon composites was examined by means of scanning electron microscopy (SEM) and transmission electron microscopy (TEM). It can be seen that the carbonized solids exhibit a monolithic structure where the porous carbon and Na2Sx particles are closely confined within a rigid framework built up by the KCl phase (Fig. 2a). In contrast, the porous carbon particles collected after the removal of the inorganic compounds exhibit a sponge-like structure with numerous macrovoids (size ∼ 5–10 μm) formed due to the templating effect of the inorganic particles (Fig. 2b). A close-up examination of the carbon particles reveals a microstructure consisting of interconnected carbon nanoparticles with numerous interparticle nanovoids (i.e., macropores and mesopores) (Fig. 2c and S3†). Further TEM inspection of the carbon samples confirms that the porous carbon particles have a very open porosity made up of mesopores with a size < 10 nm (Fig. 2d and e), which are separated by thin graphene-like carbon sheets (Fig. 2f). This hierarchical meso–macroporosity is advantageous to achieve a homogeneous deposition of the in situ generated sulfur, as well as to facilitate electrolyte infiltration and ion diffusion when used in a Li–S cell. Indeed, a comparative analysis of the morphological features of the porous carbon (Fig. 2b and c) with the sulfur/carbon composite (Fig. 3a–c) clearly shows that both samples exhibit an analogous microstructural appearance, suggesting that the incorporated sulfur is well-infiltrated into the pore network. Elemental mapping of carbon and sulfur obtained by SEM-EDX (energy dispersive X-ray spectroscopy) shows that sulfur is homogeneously distributed (Fig. 3d–f). | ||
| Fig. 2 SEM images of: (a) carbonized composite and (b and c) porous carbon sample C-1.5. (d–f) TEM images of porous carbon C-1.5. | ||
 | ||
| Fig. 3 Sulfur–carbon composites: (a–c) SEM images and (d–f) SEM-EDX elemental mappings of (d) carbon and (e) sulfur, which were collected from the area shown in (f). Sample: S/C-1.5. | ||
The physical properties of porous carbon hosts and S/C composites were examined by N2 physisorption, XRD, Raman and thermogravimetric analysis. The sorption isotherms and pore size distributions are shown in Fig. 4 and the main textural properties (i.e., BET surface area, pore volume and micropore volume) are listed in Table 1. The porous carbon samples produced by means of activation with sodium thiosulfate have high BET surface areas, in the 1900–2550 m2 g−1 range, and high pore volumes (2–2.9 cm3 g−1). The modification of the (Na2S2O3/tannic acid) weight ratio between 1.5 and 3 hardly modifies the textural properties but induces a significant diminution of the carbon yield from 18% for C-1.5 to 1.8% for C-3, which occurs concurrently with the increase of the amount of sulfur infiltrated in the corresponding S/C samples (see Fig. S4†). Interestingly, these carbons exhibit bimodal porosity with two pore systems centred at ∼0.8 nm and at ∼1.5–5 nm, as illustrated by the pore size distributions shown in Fig. 4b. Moreover, they have high micropore volumes (<2 nm) up to 0.76 cm3 g−1 in the case of the C-1.5 carbon, with the volume of narrower micropores (<1 nm) up to 0.33 cm3 g−1 (see Table 1). The incorporation of sulfur induces a drastic reduction of the textural properties as can be seen from the results in Fig. 4c and Table 1. In the case of the S/C composites with <82% S, sulfur is confined within the pores of the carbon, as can be inferred from the comparison of the measured and theoretically deduced unoccupied pore volumes (see Table 1). However, for the composites with sulfur content higher than 82% (i.e., S/C-2.7 and S/C-3), the pores are completely filled and it seems reasonable to assume that in these cases a certain amount of sulfur is located on the outer surface of the carbon particles.
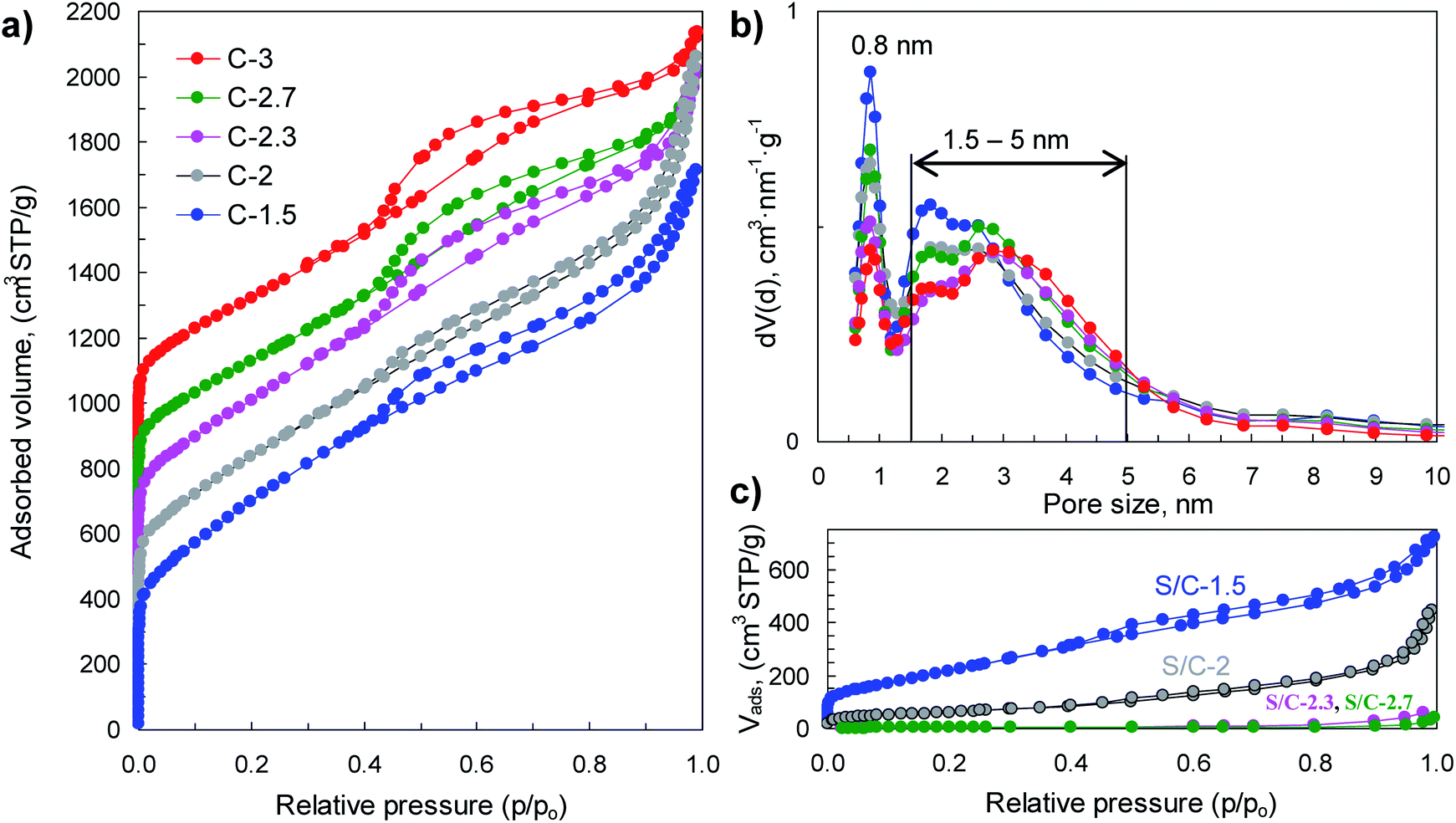 | ||
| Fig. 4 (a) Nitrogen sorption isotherms and (b) pore size distributions of the porous carbons, and (c) nitrogen sorption isotherms of the sulfur/carbon composites. In figure (a), the isotherms C-2, C-2.3, C-2.7 and C-3 are vertically shifted by 200, 400, 600 and 800 cm3 g−1 respectively. | ||
| Material | Sample code | S BET (m2 g−1) | V p (cm3 g−1) | V micro<2 nm (cm3 g−1) | Sinfiltratedc (wt%) | Sdopedd (wt%) | Carbon yield (%) | Electrical conductivity (S cm−1) | Packing density (g cm−3) |
|---|---|---|---|---|---|---|---|---|---|
| a The theoretical unoccupied pore volumes deduced by assuming that the sulfur is confined within the porosity of carbon are indicated in parentheses. This parameter was calculated by using the formula: [Vp × (1 − aS) − aS/2.07], aS being the fraction of sulfur and 2.07 its density. b The volume of micropores smaller than 1 nm are given in parenthesis. c Determined by thermogravimetric analysis. d Percentage of sulfur attached to the carbon framework and determined once the infiltrated sulfur has been removed by thermal treatment up to 600 °C (N2). | |||||||||
| S/carbon | S/C-1.5 | 790 | 1.12 (1.02) | 0.19 | 51.9 | 2.6 | 0.52 | ||
| S/C-2 | 230 | 0.69 (0.69) | 0.04 | 65.3 | 2.1 | 0.59 | |||
| S/C-2.3 | 14 | 0.10 (0.06) | — | 81.9 | 2.1 | 1.27 | |||
| S/C-2.7 | <10 | <0.05 | — | 86.1 | 8.5 × 10−1 | 1.55 | |||
| S/C-3 | <10 | <0.05 | — | 96.2 | 5.3 × 10−3 | 1.54 | |||
| Porous carbon | C-1.5 | 2550 | 2.65 | 0.76 (0.33) | 5.4 | 18.0 | 4.5 | 0.26 | |
| C-2 | 2310 | 2.88 | 0.65 (0.27) | 7.6 | 12.9 | 3.2 | 0.27 | ||
| C-2.3 | 2220 | 2.51 | 0.60 (0.25) | 7.2 | 8.3 | 4.3 | 0.23 | ||
| C-2.7 | 1920 | 2.18 | 0.50 (0.20) | 7.1 | 7.0 | 3.5 | 0.28 | ||
| C-3 | 1900 | 2.07 | 0.49 (0.18) | 9.7 | 1.8 | 3.9 | 0.27 | ||
The sulfur content in the sulfur/carbon composites was determined by TGA analysis (Fig. 5a). The amount of sulfur incorporated in the composites uniformly varies with the amount of sodium thiosulfate used in the activation process (see Fig. S4†). In that way, the percentage of sulfur can be precisely adjusted between ∼50% and ∼95% just by modifying the Na2S2O3/tannic acid weight ratio from 1.5 to 3. The X-ray diffraction patterns of the S/C composites are displayed in Fig. 5b. The samples with sulfur content of around 80% and lower (samples S/C-1.5, S/C-2 and S/C-2.3) only exhibit a broad band at 2θ ∼ 15–30°, which is typical of carbons with a disordered structure, no peak associated with elemental sulfur being detected. The absence of sharp diffraction peaks characteristic of crystalline sulfur clearly supports the idea that the sulfur infiltrated in the composites with <∼80% S is well dispersed and confined within the pores, probably as amorphous S8. In contrast, the XRD patterns of the composites with the highest sulfur content (e.g. S/C-2.7 and S/C-3) show diffraction peaks characteristic of orthorhombic sulfur, which indicates that a fraction of sulfur is located outside the pores as a crystalline phase. The structural characteristics of the infiltrated sulfur were further investigated by Raman spectroscopy (Fig. 5c). The spectra of the sulfur/carbon composites with <∼80% S only exhibit two broad bands at ∼1350 cm−1 (D band) and ∼1600 cm−1 (G band) that are typical of carbon materials with a disordered structure.43 However, in the case of a S/C composite with a very high sulfur content such as the S/C-3 sample, the Raman spectrum, in addition to the D and G bands characteristic of the carbon host, exhibits another three peaks at 154 cm−1, 219 cm−1 and 473 cm−1 distinctive of crystalline sulfur that are associated with S–S vibrational modes.44 These results prove that for the composites with sulfur content lower than around 80%, the sulfur is well embedded within the pores of the carbon host as an amorphous phase, which is in agreement with the data derived from the textural analysis.
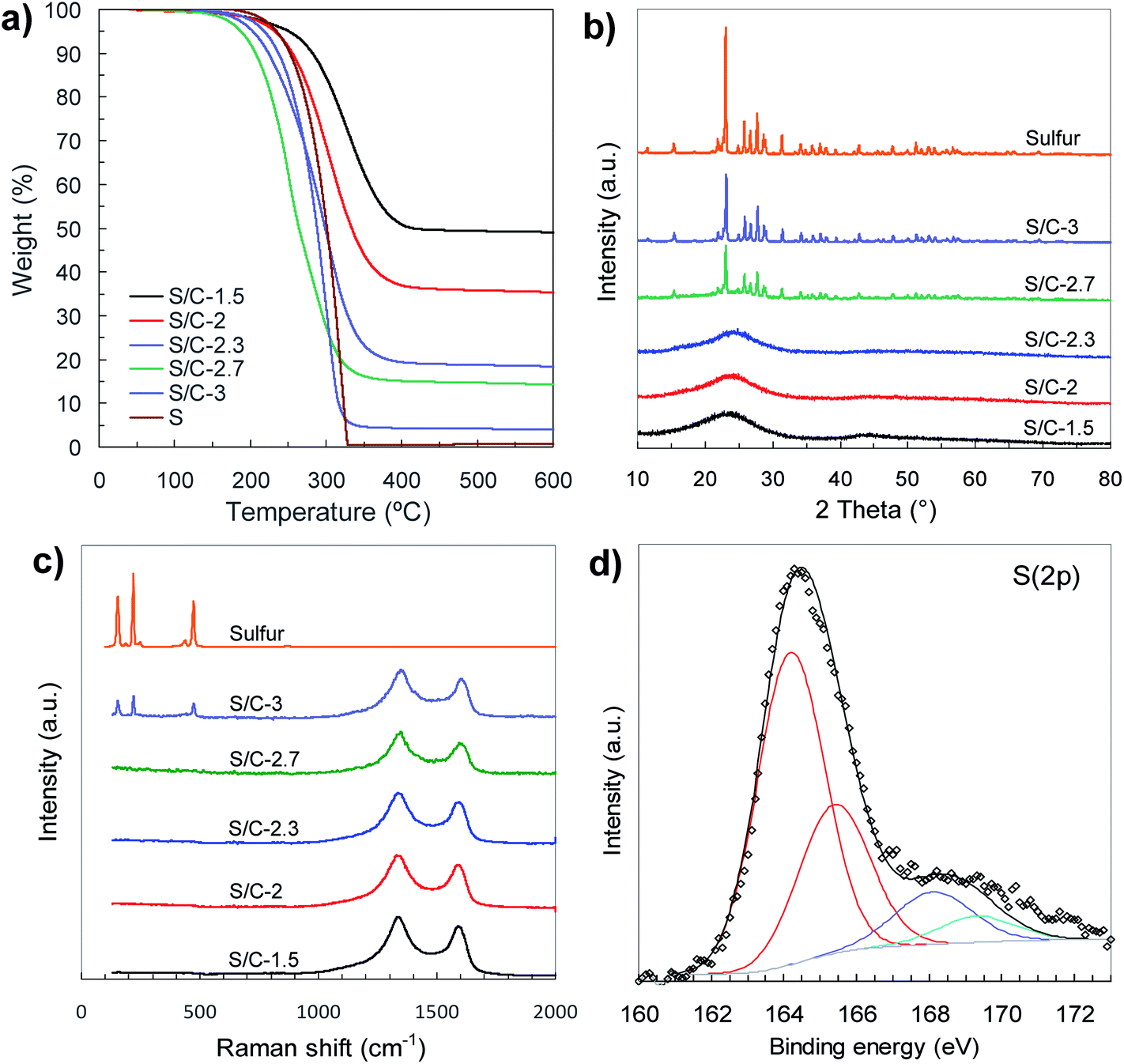 | ||
| Fig. 5 (a) TGA curves (N2, 5 °C min−1), (b) XRD patterns, (c) Raman spectra and (d) high-resolution S 2p XPS spectrum of C-1.5 sample. | ||
All of the carbon hosts, despite their highly porous structure and the absence of graphitic ordering, have good electronic conductivity, with values in the 3.2–4.5 S cm−1 range (see Table 1). Importantly, the S/C composites with sulfur content up to 82% have remarkable electrical conductivities, with values ≥2.1 S cm−1. This result corroborates the fact that the sulfur is mainly located inside the pores of the carbon host, and good electrical contact between the S/C particles is preserved. In contrast, higher sulfur contents led to a drastic decrease in the conductivity of the composites, due to the deposition of sulfur on the outer surface of the carbon. In addition, we observed that whereas the porous carbon samples have similar packing densities within the 0.23–0.28 g cm−3 range, the density of S/C composites gradually increases with the sulfur content from ∼0.5 g cm−3 to ∼1.5 g cm−3 (see Table 1).
A characteristic of our general synthesis strategy is that during the activation process the incorporation of sulfur heteroatoms within the carbon framework takes place. Notable sulfur contents between 5 wt% and 10 wt% were determined in the porous carbons (see Table 1). The nature of these S groups inserted in the carbon host was examined by X-ray photoelectron spectroscopy (XPS). The XPS survey spectra of porous carbons only contain three peaks ascribed to C 1s, O 1s and S 2p, thus excluding the presence of foreign elements (Fig. S5a†). Deconvolution of the high-resolution C 1s and S 2p XPS spectra corresponding to the C-1.5 sample reveals that sulfur is covalently attached to the carbon framework (Fig. 5d and S5b†). Thus, the high-resolution S 2p spectrum (Fig. 5d) shows two doublets; the most intense, at lower binding energies (164.2 and 165.4 eV), is assigned to thiophenic sulfur (C–S–C), whereas the higher energy doublet (at 168.1 and 169.3 eV) corresponds to oxidised sulfur (C–SOx–C). The presence of elemental sulfur can be discarded taking into account that the carbon material was heat-treated at 600 °C. Furthermore, the analysis of the C 1s XPS spectrum reveals that besides the typical peaks at 284.5 eV, 286.2 eV, 287.1 eV and 288.7 eV corresponding to sp2 carbon, C–O, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and O–CO, respectively, there is an additional peak at 285.4 eV that can be ascribed to the C–S bonds (see Fig. S5b†).41,45 These sulfur heteroatoms are uniformly distributed throughout the carbon particles as evidenced by the SEM-EDX mapping in Fig. S6.† The importance of the sulfur attached to the carbon lies in the fact that it can contribute to controlling the migration of polysulfide species and, in this way, improve the electrochemical performance of the Li–S battery.46–48
O and O–CO, respectively, there is an additional peak at 285.4 eV that can be ascribed to the C–S bonds (see Fig. S5b†).41,45 These sulfur heteroatoms are uniformly distributed throughout the carbon particles as evidenced by the SEM-EDX mapping in Fig. S6.† The importance of the sulfur attached to the carbon lies in the fact that it can contribute to controlling the migration of polysulfide species and, in this way, improve the electrochemical performance of the Li–S battery.46–48
3.3. Electrochemical performance of the sulfur/carbon composites in Li–S batteries
The S/C composites were tested as the cathode material in lithium–sulfur batteries by assembling coin cells using metallic lithium foil as the anode. Fig. 6a shows the cyclic voltammograms, recorded at a scan rate of 0.05 mV s−1, corresponding to the S/C-1.5 composite. In the cathodic sweep, the typical reduction peaks associated with the two-step reduction of sulfur appear at 2.29 V and 2.04 V. The peak registered at the highest voltage corresponds to the conversion of elemental sulfur to soluble long-chain lithium polysulfides (Li2Sx, 4 ≤ x ≤ 8), while the peak at lower voltage is related to further reduction of polysulfides to the solid products Li2S2 and Li2S.49 Another broad peak of low intensity was also identified at a voltage of ca. 1.80 V. This small peak vanishes during the first charge–discharge cycles and therefore can be ascribed to the irreversible reduction of the LiNO3 additive.41 Correspondingly, the oxidation sweep reveals two signals associated with the oxidation of Li2S and Li2S2 to high-order polysulfides, at 2.34 V, and to their oxidation to elemental sulfur, at 2.39 V. The overlap of the plots during cycling supports the reversibility of these reactions and the electrochemical stability of the cathode.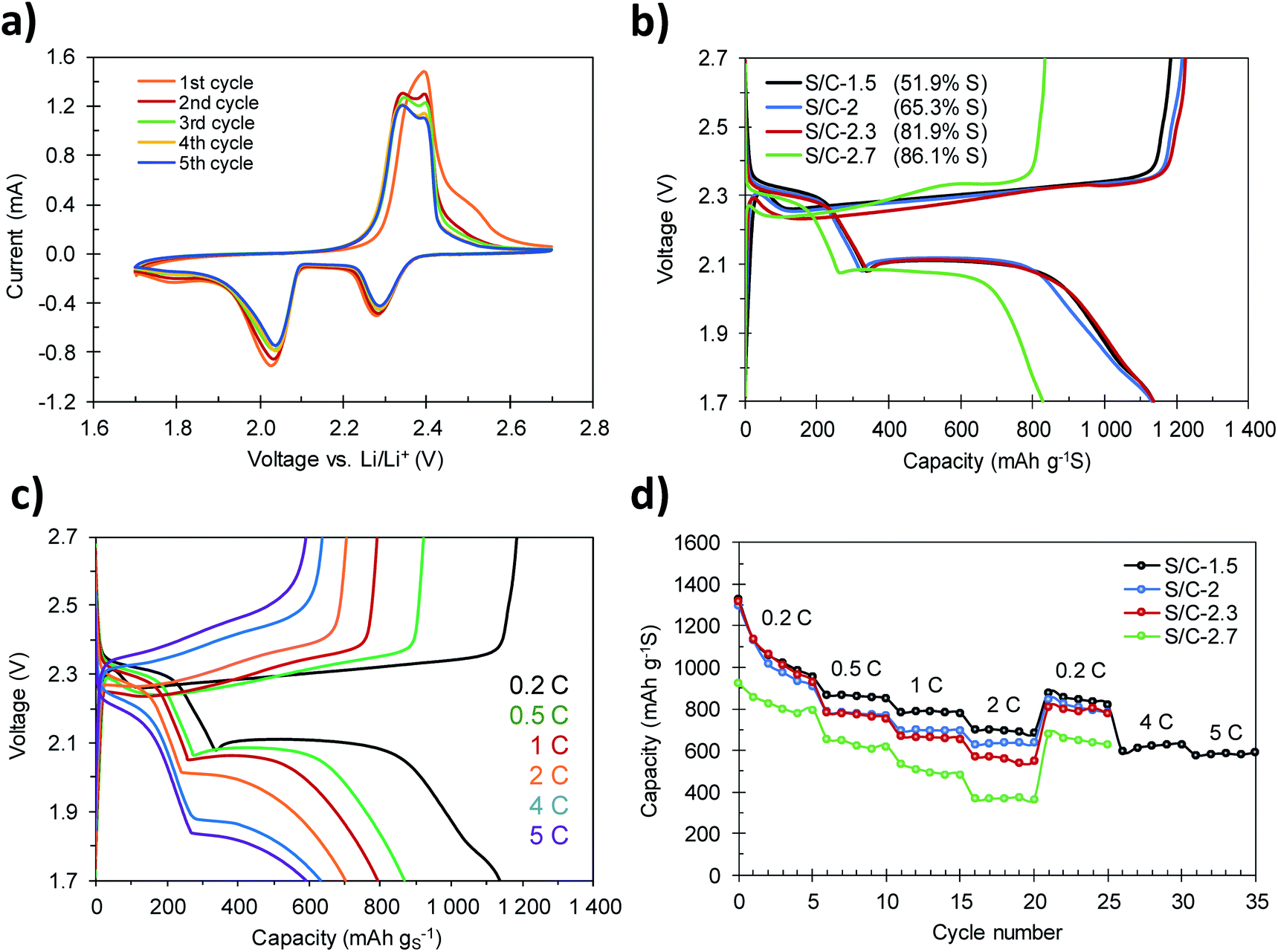 | ||
| Fig. 6 (a) CV profiles of the S/C-1.5 cathode recorded at a sweep rate of 0.05 mV s−1, (b) charge–discharge curves of the cathodes with different S contents at a current density of 0.2C, (c) galvanostatic charge–discharge curves corresponding to the S/C-1.5 cathode at different current rates, and (d) rate performance of the S/C cathodes with different S contents. | ||
Galvanostatic charge–discharge profiles of the cathodes with different S contents were acquired at different current densities in the 1.7–2.7 voltage range. Fig. 6b shows the voltage profiles of the first charge–discharge cycles recorded at a current density of 0.2C (1C = 1675 mA g−1). In accordance with the results obtained by cyclic voltammetry, a change in the slope at a voltage of ca. 1.8 V was recorded during the first cycles of charge–discharge (see Fig. S7†) as a consequence of the irreversible reduction of LiNO3. The discharge profiles show the two characteristic plateaus at voltages of ca. 2.3 V and ca. 2.1 V. Only when the sulfur content is higher than 82% (e.g. S/C-2.7, 86.1% S) a slight decrease of the voltage at the second plateau to 2.08 V can be observed. This polarization is more evident when the batteries are run at higher current rates (Fig. 6c and S8†), and is a consequence of the sluggish reaction kinetics due to the insulating nature of the end-products S8 (∼10−30 S cm−1) and Li2S (∼10−14 S cm−1).15 The voltage profiles corresponding to S/C-1.5 recorded at charge–discharge rates ranging from 0.2C to 5C are shown in Fig. 6c. The discharge plateaus can be clearly identified even at the highest charge–discharge rate of 5C. This proves that the hierarchically porous structure of the carbon host is able to correctly accommodate the discharge products (Li2S2 and Li2S) at high current loads, so that the electrochemical mechanisms occurring at high rates are similar to those taking place during slow charging and discharging.41
The rate capability of the S/C composites with different S contents is displayed in Fig. 6d. The cathodes with S content between 51 and 82% (S/C-1.5, S/C-2 and S/C-2.3) achieve initial discharge capacities of around 1300 mA h g−1, which, considering the theoretical capacity of elemental sulfur (1675 mA h g−1), corresponds to a high sulfur utilization of ∼80%. Among them, the S/C-1.5 cathode shows the best rate capability, retaining 870 mA h g−1, 791 mA h g−1 and 703 mA h g−1 at 0.5C, 1C and 2C, respectively. As expected, with the increase of the sulfur content to a value of ∼82% (S/C-2.3), the capacity retention of the cathodes decreases slightly. However, even so, the sample S/C-2.3 still retains high capacities of 832 mA h g−1, 709 mA h g−1 and 607 mA h g−1 at 0.5C, 1C and 2C, respectively. When the current rate is set again to 0.2C, the cathodes recover values of capacity of 880 mA h g−1 (S/C-1.5), 847 mA h g−1 (S/C-2) and 810 mA h g−1 (S/C-2.3), which suggests that the cathodes are able to tackle charge–discharge processes at high currents without detriment to their mechanical integrity. Remarkably, once the S/C-1.5 cathode is further cycled at 4C and 5C, outstanding capacities of 630 mA h g−1 and 590 mA h g−1 are still achieved (Fig. 6c and d). Such good capacity retention surpasses that of other high-performance sulfur–carbon composites previously reported.41,42,50–52 As previously shown, in the case of the samples with a sulfur content up to 82 wt%, the totality of the sulfur is confined within the porosity of the carbon host. Such high capacity values at high rates are explained by (i) the fact that the active material is effectively confined in the conductive carbon matrix, which enables a good carbon–sulfur electrical contact and (ii) the hierarchical pore structure of the carbon host, which guarantees short diffusion pathways for the transport of electrons.18,41 In contrast, in the composites with a S content of >82 wt% (e.g. S/C-2.7) the sulfur not only fills the porosity of the carbon, but also, a fraction covers the outer surface of the carbon particles. This sulfur fraction is less active electrochemically and reduces significantly the conductivity of the cathode (Table 1), which results in a lower degree of sulfur utilization (Fig. 6d).
The cycling stability of the batteries was evaluated by galvanostatic charge–discharge at 1C. As can be seen in Fig. 7a and e, the S/C-1.5 composite (51.9% S) exhibits very stable cycling. For this cathode, a capacity of 674 mA h g−1 is retained after 300 cycles of charge–discharge, which corresponds to a capacity retention of 85%. Furthermore, the average capacity decay per cycle is as low as 0.049%. It can be seen that the capacity fading stabilizes after the 100th cycle and, from this point, the average capacity loss per cycle is only 0.016%. The coulombic efficiency is higher than 99% during the whole cycling test (Fig. 7e), which proves that the shuttle effect is efficiently controlled. The evolution of the capacity values corresponding to the upper and lower voltage plateaus (QH and QL, respectively) at different cycling states is shown in Fig. S9.† Both parameters are highly stable and the QL/QH ratio remains constant over the whole cycling test, which evidences the good electronic accessibility of sulfur and the invariable polysulfide retention ability of the carbon host.53,54 In this point, it is important to indicate that several authors have observed that S-doped carbons exhibit an improved electrochemical behaviour, which agrees with the good cyclability observed in our experiments.46,47,55,56 In a recent work, Du et al. reported that S-doped carbon acts as a metal-free electrocatalyst that enhances the conversion kinetics of polysulfides, which leads to a superior performance.48 Remarkably, the porous carbon hosts presented in this work meet the requirements of a good metal-free electrocatalyst: (i) a high degree of heteroatom doping (>5.5% S) and (ii) good electrical conductivity (>3 S cm−1). The combination of both properties could explain the good electrochemical performance observed in our experiments. It may be noted that the doped sulfur heteroatoms did not participate in any faradaic process and remain stable upon cycling, as deduced from the electrochemical characterization of a battery assembled using the C-1.5 carbon as the cathode material (see Fig. S10†), ruling out any electrochemical activity from the carbon host. Fig. 7b and e compare the cycling stabilities of the cathodes with sulfur contents of 51.9% (S/C-1.5) and 81.9% (S/C-2.3). Noteworthy, despite the very high sulfur content in S/C-2.3, its cell has an initial capacity of 835 mA h g−1 and is successfully cycled for 200 cycles retaining 617 mA h g−1, the coulombic efficiency staying above 96% during the whole cycling test.
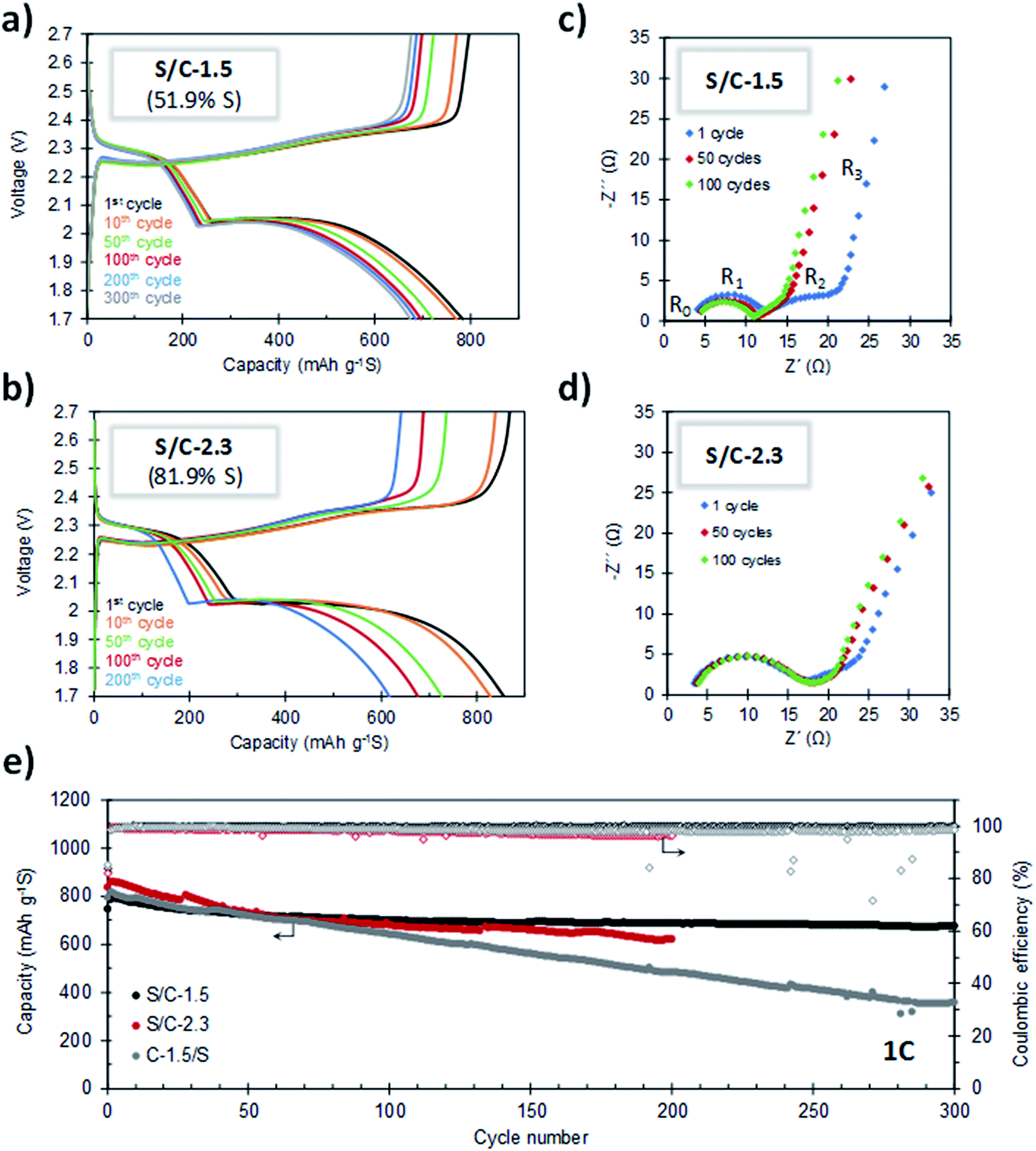 | ||
| Fig. 7 Galvanostatic charge–discharge curves upon cycling at 1C for: (a) S/C-1.5 and (b) S/C-2.3. Nyquist plots of (c) S/C-1.5 and (d) S/C-2.3 at different cycling states. (e) Cycling performance of the S/C-1.5 (51.9% S), C-1.5/S (51.9% S) and S/C-2.3 (81.9% S) cathodes at a charge–discharge rate of 1C. | ||
For the purpose of comparison, we investigated the electrochemical performance of a S/C composite prepared by the conventional melt-diffusion methodology with C-1.5 as the carbon host (C-1.5/S sample, sulfur content: 52% S). The cycling performance of this composite, measured at a rate of 1C, is included in Fig. 7e. It can be seen that after 300 cycles, whereas the S/C-1.5 sample still retains more than 85% of the initial capacity, the conventional C-1.5/S cathode undergoes a loss of around 60%. We found that the sulfur in the C-1.5/S composite is less homogeneously deposited within the porous system of the activated carbon and, in addition, it exhibits poorer electrical conductivity in relation to the S/C-1.5 sample (see Table S1, Fig. S11 and Supplementary Note 1 in the ESI†). The above results evidence that the methodology herein reported is advantageous in relation to the infiltration procedure conventionally used for the preparation of S/C cathodes.
The good cycling stability of the cathodes was confirmed by impedance spectroscopy. Fig. 7c and d show the Nyquist plots for S/C-1.5 and S/C-2.3, respectively, after 1, 50 and 100 charge–discharge cycles. These plots consist of an ohmic resistance (R0, resistance of the electrolyte and current collectors) in series with three processes occurring at high, medium and low frequencies. The semicircle at high frequencies is ascribed to the charge transfer resistance at the anode (R1), while the depressed semicircle at medium frequencies is related to the charge transfer resistance of polysulfide anions in the cathode (R2). At the state of full discharge, the bent line at the lowest frequencies is attributed to the presence of an isolated layer of the discharge product, Li2S.57 The decrease of R2 with the first charge–discharge cycles is indicative of certain redistribution of the active material that becomes more accessible by electronic conduction. When the battery is further cycled for 100 cycles, the plot almost superimposes, which evidences the stable cyclability of the battery.
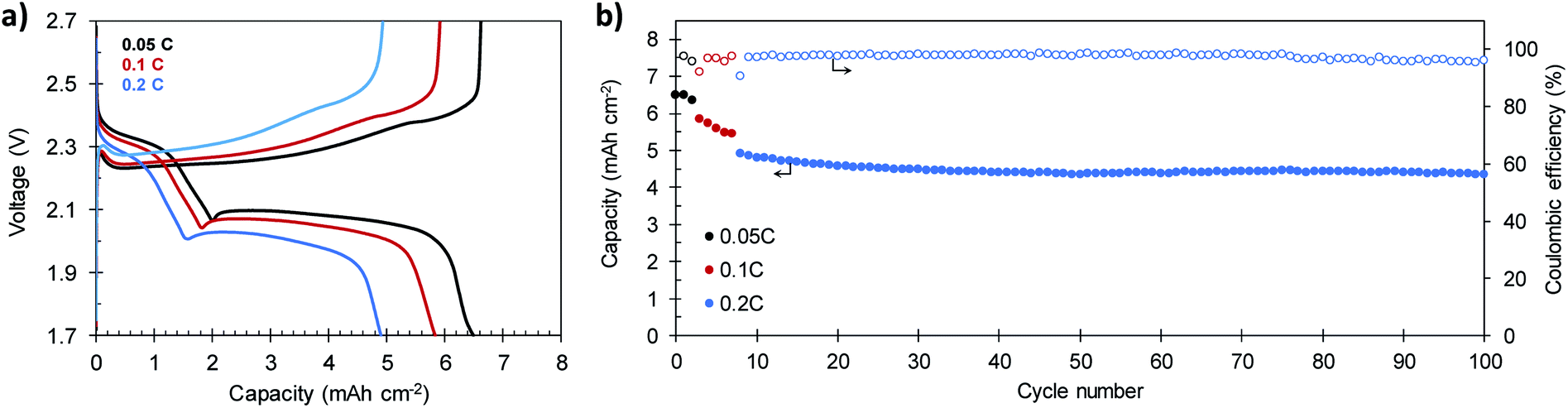
In order to verify the suitability of the composites for practical applications, the electrochemical performance of a S/C cathode that meets the specifications of high sulfur content (81.9%) and high sulfur loading (7.1 mg S per cm2) was investigated. The electrolyte-to-sulfur ratio was also reduced to 18 μL mg−1 S. Representative galvanostatic charge–discharge plots and the cycling stability of the battery built with this high load S/C-2.3 cathode are shown in Fig. 8a and b, respectively. As a consequence of its high areal loading, the battery reaches an initial capacity of 6.50 mA h cm−2 at 0.05C, which corresponds to a specific capacity of 921 mA h g−1 (see Fig. S12†). At higher current densities of 0.1C and 0.2C, the battery retains reversible capacities of 5.83 mA h cm−2 (826 mA h g−1) and 4.91 mA h cm−2 (696 mA h g−1), respectively. These values are comparable to those of other reported cathodes with similar or higher sulfur loading58,59 and superior to the 4 mA h cm−2 required to compete with the state-of-the-art Li-ion batteries.60 After 100 charge–discharge cycles, the battery still provides a capacity of 4.36 mA h cm−2 (618 mA h g−1) at a charge–discharge rate of 0.2C. The average capacity fading at 0.2C is as low as 0.12% per cycle, comparable to that of other high-performing cathodes with similar sulfur loading.61,62 Noteworthy, the battery becomes more stable with cycling, and the average capacity loss recorded from the 50th cycle is only 0.009% per cycle, while keeping the coulombic efficiency above 96% during the whole cycling test. Although very high areal capacities exceeding these values have been achieved by the use of 3D current collectors such as vertically aligned carbon nanotubes63 or carbonized cotton fibers,64,65 we believe our results prove the viability of our in situ synthetic procedure for the preparation of high sulfur loading cathodes for feasible lithium–sulfur batteries.
 | ||
| Fig. 8 (a) Galvanostatic charge–discharge plots and (b) cycling performance of a S/C-2.3 cathode (81.9% S) with a high sulfur loading of 7.1 mg cm−2. | ||
4. Conclusions
In summary, we have successfully synthesized sulfur–carbon composites through a general, easy, cost-effective and sustainable synthesis methodology that combines, in the same process, the production of a high surface area (up to 2550 m2 g−1) micro–mesoporous carbon with the incorporation of elemental sulfur within the pores of the carbon. In this way, hybrid sulfur–carbon materials with tunable sulfur contents in the ∼50–90% range can be obtained simply by adjusting the ratio between the activating agent (Na2S2O3) and the carbon precursor (tannic acid). We demonstrated that high performance can be attained when these sulfur–carbon hybrid composites are used as cathodes in lithium–sulfur batteries. Indeed, they displayed very good rate performance, achieving 590 mA h g−1 S at a high discharge current of 5C and excellent cycling stability, with a capacity fade of only 0.049% per cycle over 300 cycles at 1C. Even for a high sulfur loading (7.1 mg cm−2), the battery is able to deliver a high areal capacity of 6.50 mA h cm−2 at 0.05C, and retain 4.4 mA h cm−2 at 0.2C after 100 charge–discharge cycles. The synthesis strategy presented here has been demonstrated for tannic acid, but it could be further extended to other biomass-based substances. We believe that this work represents an important step forward towards the sustainable and scalable fabrication of high-performance cathode materials for lithium–sulfur batteries.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research work was supported by projects RTI2018-093712-B-I00 (MCIU/AEI/FEDER, UE) and IDI/2018/000148 (regional GRUPIN2018).References
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- R. Fang, S. Zhao, Z. Sun, D. W. Wang, H. M. Cheng and F. Li, Adv. Mater., 2017, 29, 1606823 CrossRef PubMed.
- Z. W. Seh, Y. Sun, Q. Zhang and Y. Cui, Chem. Soc. Rev., 2016, 45, 5605–5634 RSC.
- F. Wu and G. Yushin, Energy Environ. Sci., 2017, 10, 435–459 RSC.
- C. Dong, W. Gao, B. Jin and Q. Jiang, iScience, 2018, 6, 151–198 CrossRef CAS PubMed.
- A. Manthiram, Y. Fu, S.-H. Chung, C. Zu and Y.-S. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS PubMed.
- Y. X. Yin, S. Xin, Y. G. Guo and L. J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186–13200 CrossRef CAS PubMed.
- A. Manthiram, S. H. Chung and C. Zu, Adv. Mater., 2015, 27, 1980–2006 CrossRef CAS PubMed.
- Y. He, Z. Chang, S. Wu and H. Zhou, J. Mater. Chem. A, 2018, 6, 6155–6182 RSC.
- T. Ould Ely, D. Kamzabek, D. Chakraborty and M. F. Doherty, ACS Appl. Energy Mater., 2018, 1, 1783–1814 CrossRef CAS.
- Z. Zeng and X. Liu, Adv. Mater. Interfaces, 2018, 5, 1701274 CrossRef.
- Y. Zhu, S. Wang, Z. Miao, Y. Liu and S. L. Chou, Small, 2018, 14, 1801987 CrossRef PubMed.
- X. Li and X. Sun, Adv. Funct. Mater., 2018, 28, 1801323 CrossRef.
- J. Zhang, H. Huang, J. Bae, S. H. Chung, W. Zhang, A. Manthiram and G. Yu, Small Methods, 2018, 2, 1700279 CrossRef.
- X. Ji, K. T. Lee and L. F. Nazar, Nat. Mater., 2009, 8, 500 CrossRef CAS PubMed.
- H. Wang, Y. Yang, Y. Liang, J. T. Robinson, Y. Li, A. Jackson, Y. Cui and H. Dai, Nano Lett., 2011, 11, 2644–2647 CrossRef CAS PubMed.
- J. Guo, Y. Xu and C. Wang, Nano Lett., 2011, 11, 4288–4294 CrossRef CAS PubMed.
- S. Evers and L. F. Nazar, Acc. Chem. Res., 2012, 46, 1135–1143 CrossRef PubMed.
- J.-G. Wang, K. Xie and B. Wei, Nano Energy, 2015, 15, 413–444 CrossRef CAS.
- Z. Li, Y. Huang, L. Yuan, Z. Hao and Y. Huang, Carbon, 2015, 92, 41–63 CrossRef CAS.
- R. Sahore, B. D. Levin, M. Pan, D. A. Muller, F. J. DiSalvo and E. P. Giannelis, Adv. Energy Mater., 2016, 6, 1600134 CrossRef.
- J. Liang, Z.-H. Sun, F. Li and H.-M. Cheng, Energy Storage Materials, 2016, 2, 76–106 CrossRef.
- A. Eftekhari and D.-W. Kim, J. Mater. Chem. A, 2017, 5, 17734–17776 RSC.
- J. Gómez-Urbano, J. Gómez-Cámer, C. Botas, N. Díez, J. L. del Amo, L. Rodríguez-Martinez, D. Carriazo and T. Rojo, Carbon, 2018, 139, 226–233 CrossRef.
- S. Li, B. Jin, X. Zhai, H. Li and Q. Jiang, ChemistrySelect, 2018, 3, 2245–2260 CrossRef CAS.
- L. Borchardt, M. Oschatz and S. Kaskel, Chem.–Eur. J., 2016, 22, 7324–7351 CrossRef CAS PubMed.
- A. B. Fuertes, G. A. Ferrero, N. Diez and M. Sevilla, ACS Sustainable Chem. Eng., 2018, 6, 16323–16331 CrossRef CAS.
- M. Sevilla, N. Diez, G. A. Ferrero and A. B. Fuertes, Energy Storage Materials, 2019, 18, 356–365 CrossRef.
- C. Hernández-Rentero, R. Córdoba, N. Moreno, A. Caballero, J. Morales, M. Olivares-Marín and V. Gómez-Serrano, Nano Res., 2018, 11, 89–100 CrossRef.
- N. Moreno, A. Caballero, L. Hernan and J. Morales, Carbon, 2014, 70, 241–248 CrossRef CAS.
- Y.-S. Su and A. Manthiram, Electrochim. Acta, 2012, 77, 272–278 CrossRef CAS.
- Y. Fu and A. Manthiram, J. Phys. Chem. C, 2012, 116, 8910–8915 CrossRef CAS.
- L. Ji, M. Rao, S. Aloni, L. Wang, E. J. Cairns and Y. Zhang, Energy Environ. Sci., 2011, 4, 5053–5059 RSC.
- L. Ji, M. Rao, H. Zheng, L. Zhang, Y. Li, W. Duan, J. Guo, E. J. Cairns and Y. Zhang, J. Am. Chem. Soc., 2011, 133, 18522–18525 CrossRef CAS PubMed.
- S. Evers and L. F. Nazar, Chem. Commun., 2012, 48, 1233–1235 RSC.
- C. Wang, J.-j. Chen, Y.-n. Shi, M.-s. Zheng and Q.-f. Dong, Electrochim. Acta, 2010, 55, 7010–7015 CrossRef CAS.
- C. Zhang, W. Lv, W. Zhang, X. Zheng, M. B. Wu, W. Wei, Y. Tao, Z. Li and Q. H. Yang, Adv. Energy Mater., 2014, 4, 1301565 CrossRef.
- B. He, W.-C. Li, C. Yang, S.-Q. Wang and A.-H. Lu, ACS Nano, 2016, 10, 1633–1639 CrossRef CAS PubMed.
- A. D. Roberts, X. Li and H. Zhang, Carbon, 2015, 95, 268–278 CrossRef CAS.
- Q. Wang, Z.-B. Wang, C. Li and D.-M. Gu, J. Mater. Chem. A, 2017, 5, 6052–6059 RSC.
- G. Li, J. Sun, W. Hou, S. Jiang, Y. Huang and J. Geng, Nat. Commun., 2016, 7, 10601 CrossRef CAS PubMed.
- C. Luo, W. Lv, Y. Deng, G. Zhou, Z. Z. Pan, S. Niu, B. Li, F. Kang and Q. H. Yang, Small, 2017, 13, 1700358 CrossRef PubMed.
- A. Cuesta, P. Dhamelincourt, J. Laureyns, A. Martinez-Alonso and J. D. Tascón, Carbon, 1994, 32, 1523–1532 CrossRef CAS.
- A. T. Ward, J. Phys. Chem., 1968, 72, 4133–4139 CrossRef CAS.
- Z. Wang, Y. Dong, H. Li, Z. Zhao, H. B. Wu, C. Hao, S. Liu, J. Qiu and X. W. D. Lou, Nat. Commun., 2014, 5, 5002 CrossRef CAS PubMed.
- K. A. See, Y.-S. Jun, J. A. Gerbec, J. K. Sprafke, F. Wudl, G. D. Stucky and R. Seshadri, ACS Appl. Mater. Interfaces, 2014, 6, 10908–10916 CrossRef CAS PubMed.
- F. Nitze, K. Fossum, S. Xiong, A. Matic and A. E. Palmqvist, J. Power Sources, 2016, 317, 112–119 CrossRef CAS.
- L. Du, X. Cheng, F. Gao, Y. Li, Y. Bu, Z. Zhang, Q. Wu, L. Yang, X. Wang and Z. Hu, Chem. Commun., 2019, 55, 6365–6368 RSC.
- S. S. Zhang, J. Power Sources, 2013, 231, 153–162 CrossRef CAS.
- F. Wu, S. Chen, V. Srot, Y. Huang, S. K. Sinha, P. A. van Aken, J. Maier and Y. Yu, Adv. Mater., 2018, 30, 1706643 CrossRef PubMed.
- M. K. Rybarczyk, H.-J. Peng, C. Tang, M. Lieder, Q. Zhang and M.-M. Titirici, Green Chem., 2016, 18, 5169–5179 RSC.
- X. Yu, J. Zhao, R. Lv, Q. Liang, C. Zhan, Y. Bai, Z.-H. Huang, W. Shen and F. Kang, J. Mater. Chem. A, 2015, 3, 18400–18405 RSC.
- C.-H. Chang, S.-H. Chung and A. Manthiram, Sustainable Energy Fuels, 2017, 1, 444–449 RSC.
- S.-H. Chung and A. Manthiram, J. Phys. Chem. Lett., 2014, 5, 1978–1983 CrossRef CAS PubMed.
- J. Yang, F. Chen, C. Li, T. Bai, B. Long and X. Zhou, J. Mater. Chem. A, 2016, 4, 14324–14333 RSC.
- N. Li, F. Gan, P. Wang, K. Chen, S. Chen and X. He, J. Alloys Compd., 2018, 754, 64–71 CrossRef CAS.
- N. A. Canas, K. Hirose, B. Pascucci, N. Wagner, K. A. Friedrich and R. Hiesgen, Electrochim. Acta, 2013, 97, 42–51 CrossRef CAS.
- S. Lu, Y. Chen, X. Wu, Z. Wang and Y. Li, Sci. Rep., 2014, 4, 4629 CrossRef PubMed.
- M. Xiang, L. Yang, Y. Zheng, J. Huang, P. Jing, H. Wu, Y. Zhang and H. Liu, J. Mater. Chem. A, 2017, 5, 18020–18028 RSC.
- J. Song, T. Xu, M. L. Gordin, P. Zhu, D. Lv, Y. B. Jiang, Y. Chen, Y. Duan and D. Wang, Adv. Funct. Mater., 2014, 24, 1243–1250 CrossRef CAS.
- C. Hu, C. Kirk, Q. Cai, C. Cuadrado-Collados, J. Silvestre-Albero, F. Rodríguez-Reinoso and M. J. Biggs, Adv. Energy Mater., 2017, 7, 1701082 CrossRef.
- P. Ragupathy, S. A. Ahad, P. R. Kumar, H. W. Lee and D. K. Kim, Adv. Sustainable Syst., 2017, 1, 1700083 CrossRef.
- Z. Yuan, H. J. Peng, J. Q. Huang, X. Y. Liu, D. W. Wang, X. B. Cheng and Q. Zhang, Adv. Funct. Mater., 2014, 24, 6105–6112 CrossRef CAS.
- S.-H. Chung and A. Manthiram, Joule, 2018, 2, 710–724 CrossRef CAS.
- S.-H. Chung, C.-H. Chang and A. Manthiram, ACS Nano, 2016, 10, 10462–10470 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Evidence and characterization of the generation of sulfur. SEM images of carbons and sulfur–carbon composites, and XPS and EDX mappings of a S-doped carbon. Evolution of the amount of sulfur infiltrated and carbon yield in the composites using different Na2S2O3/tannic acid weight ratios. Comparison of different galvanostatic charge–discharge curves. Characterization of the melt-infiltrated carbon and discussion on the differences between this sample and its counterpart synthesized by our proposed in situ approach. See DOI: 10.1039/c9se00722a |
| This journal is © The Royal Society of Chemistry 2019 |