
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlling the strength of interaction between carbon dioxide and nitrogen-rich carbon materials by molecular design†
Ralf
Walczak
a,
Aleksandr
Savateev
 a,
Julian
Heske
b,
Nadezda V.
Tarakina
a,
Sudhir
Sahoo
c,
Jan D.
Epping
c,
Thomas D.
Kühne
b,
Bogdan
Kurpil
a,
Markus
Antonietti
a and
Martin
Oschatz
*ad
a,
Julian
Heske
b,
Nadezda V.
Tarakina
a,
Sudhir
Sahoo
c,
Jan D.
Epping
c,
Thomas D.
Kühne
b,
Bogdan
Kurpil
a,
Markus
Antonietti
a and
Martin
Oschatz
*ad
aMax Planck Institute of Colloids and Interfaces, Colloid Chemistry, Research Campus Golm, Am Mühlenberg 1, D-14476 Potsdam, Germany. E-mail: martin.oschatz@mpikg.mpg.de
bChair of Theoretical Chemistry and Center for Sustainable Systems Design, University of Paderborn, Warburger Str. 100, D-33098 Paderborn, Germany
cFakultät II: Mathematik & Naturwissenschaften, Institut für Chemie, Technische Universität Berlin, Hardenbergstr. 40, 10623 Berlin, Germany
dInstitute of Chemistry, University of Potsdam, Karl-Liebknecht-Str. 24-25, D-14476 Potsdam, Germany
First published on 13th August 2019
Abstract
Thermal treatment of hexaazatriphenylene-hexacarbonitrile (HAT-CN) in the temperature range from 500 °C to 700 °C leads to precise control over the degree of condensation, and thus atomic construction and porosity of the resulting C2N-type materials. Depending on the condensation temperature of HAT-CN, nitrogen contents of more than 30 at% can be reached. In general, these carbons show adsorption properties which are comparable to those known for zeolites but their pore size can be adjusted over a wider range. At condensation temperatures of 525 °C and below, the uptake of nitrogen gas remains negligible due to size exclusion, but the internal pores are large and polarizing enough that CO2 can still adsorb on part of the internal surface. This leads to surprisingly high CO2 adsorption capacities and isosteric heat of adsorption of up to 52 kJ mol−1. Theoretical calculations show that this high binding enthalpy arises from collective stabilization effects from the nitrogen atoms in the C2N layers surrounding the carbon atom in the CO2 molecule and from the electron acceptor properties of the carbon atoms from C2N which are in close proximity to the oxygen atoms in CO2. A true CO2 molecular sieving effect is achieved for the first time in such a metal-free organic material with zeolite-like properties, showing an IAST CO2/N2 selectivity of up to 121 at 298 K and a N2/CO2 ratio of 90/10 without notable changes in the CO2 adsorption properities over 80 cycles.
Introduction
Due to the rapid increase of the CO2 level in the atmosphere and the problems associated with this, selective CO2 capture from air or from point sources of emission (e.g., coal-fired power plants) is intensively studied.1–4 Chemical absorption of CO2 in dissolved amines (“scrubbing”) is the industrially established technique for CO2 capture from power plant exhaust as it is very selective due to covalent carbamate bond formation. Chemisorption of CO2 on amines immobilized on solid surfaces can even be applied for selective CO2 capture from air, that is, at very low partial pressure.2 Due to the high binding enthalpy of CO2, these methods suffer from high energy penalty during regeneration.5–7 Physical adsorption (physisorption) of CO2 with lower binding enthalpy (in the range of van der Waals forces) is an alternative with less energy intensive regeneration. Great research efforts have been made recently and more and more nanomaterials for selective CO2 capture have been proposed.8–10Typical physical adsorbents applied are nanoporous materials such as zeolites or zeolitic imidazole frameworks,11–15 carbon-based materials,16–21 porous organic frameworks,22,23 and metal–organic-frameworks (MOFs).24–27 Common methods to increase their affinity towards CO2 (that is, the CO2 adsorption enthalpy) include the introduction of specific interaction sites (e.g., basic nitrogen sites/or extra framework cations in porous carbons or amine MOFs),28–33 as well as the tuning of the ultramicroporosity of the sorbents.18,34,35 Some thermodynamic sweet spots have been found for different families of materials, often inspired by mimicking nature's highly selective CO2 capture mechanism in the ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco).36 However, as long as high CO2/N2 selectivity is the target, these attempts suffer from one or more of the following problems: (i) the quadrupole moment (13.4 × 10−40 C m2) and polarizability (26.3 × 10−25 cm3) of CO2 are indeed higher than for N2 (4.7 × 10−40 C m2 and 17.6 × 10−25 cm3), but it should be kept in mind that any attempt to increase CO2 selectivity by introducing specific binding sites will also increase the binding enthalpy of the adsorbent towards N2; (ii) every porous material with a high porosity for nitrogen will always suffer from significant contribution of less discriminative van der Waals interaction; (iii) many of the record-holders in terms of selectivity for atmospheric CO2 capture suffer from limited stability under wet conditions, are difficult to be synthesized, and often contain metals.
Although the promise of kinetic size exclusion of N2 (kinetic diameter: 0.36 nm) over CO2 (kinetic diameter: 0.33 nm) to enhance CO2/N2 selectivity of physically adsorbing materials is generally accepted34,37 and although it is obvious that CO2/N2 selectivity increases significantly at lower available N2 surface area,8 a real molecular CO2 sieving adsorbent has not yet been reported. The likely reason is that a decrease of the “pore size” of the adsorbents down to the molecular dimensions of CO2 leads to a significant activation barrier for adsorption, or in other words, CO2 adsorption in such narrow spaces is kinetically hindered. The only way to overcome this limitation seems to be the development of adsorbent materials with higher chemical affinity towards CO2. It is not surprising that materials with “ionic character” (i.e., materials that contain Coulomb charges in their structure as does Rubisco) such as metal–organic materials and zeolites have already been successfully utilized for CO2/N2 separation at outstanding selectivity.38–40
In contrast to such crystalline materials with defined pore size, carbon-based materials instead show narrow pores with a distribution of sizes, but their mean size can in principle be sleeplessly adjusted. For the separation of small molecules like N2 and CO2, carbon materials still lack the combination of narrow porosity and high polarizability towards CO2. We have recently reported that simple thermal condensation of hexaazatriphenylene-hexacarbonitrile (HAT-CN) molecules results in the formation of nitrogen-rich HAT-CN-derived carbon materials with high structural microporosity and a near-perfect C2N-type composition. Porosity and nitrogen content can be adjusted by the synthesis temperature. The high heteroatom doping level with well-defined structure motives introduced from the pre-organized precursor and the narrow micropores lead to zeolite-like adsorption properties of these C2N materials.20,41
We further utilize this concept and realize a CO2/N2 molecular sieve effect by only partially condensing HAT-CN at temperatures below 550 °C. Synthesis at 525 °C leads to nitrogen-rich carbon materials with porosity that is still partly accessible for reversible CO2 adsorption but the material shows no notable uptake of N2. To the best of our knowledge, that is the first time, a metal-free carbon-based material with such narrow pores has been used for CO2 molecular sieving working on a reasonable timescale.
Experimental details
Materials synthesis
The synthesis of hexaazatriphenylene-hexacarbonitrile (HAT-CN) was carried out according to a previously described procedure.42 Hexaketocyclohexane octahydrate (4 g, 12.6 mmol) and diaminomaleonitrile (10.88 g, 100.8 mmol) were refluxed in acetic acid (AcOH, 150 mL) for 2 h. The black suspension was filtered off while hot and washed with hot AcOH (3 × 25 mL) resulting in a black solid. The solid was suspended in 30% HNO3 (60 mL) and heated at 100 °C for 3 h. The hot dark brown suspension was poured into ice water (200 mL) and cooled overnight. The suspension was filtered and the solid was refluxed in MeCN (400 mL) for 2 h and was filtered. The filtrate was dried under vacuum to give an orange solid (2.4 g, yield 50%). 13C NMR (DMSO-d6, 100 MHz): δ 142.0, 135.8, 114.5.Nitrogen-rich carbons (C-HAT-CN-X materials) were prepared using HAT-CN (500 mg) for carbonization in a horizontal tubular furnace at different temperatures for 1 h under N2 gas flow. The heating ramp was 2 °C min−1 from room temperature to 80 °C and 4 °C min−1 from 80 °C to the maximum temperature (500 °C, 525 °C, 550 °C, or 700 °C). The condensed materials are labelled as C-HAT-CN-X, where X represents the synthesis temperature.
Materials characterization
Prior to all physisorption measurements, the samples were degassed under vacuum at 200 °C for 20 h. Ar, CO2, N2, and H2O vapor physisorption were performed on a Autosorb IQ apparatus (Quantachrome Instruments). Low pressure physisorption measurements were performed using Ar at −186 °C with ∼20 mg of sample. The pore size distributions were calculated using the quenched solid density functional theory (QSDFT) method (adsorption branch kernel) for Ar adsorbed on carbon with cylindrical/sphere pore shape at 87 K, integrated into the ASiQwin 3.0 analysis software. The specific surface areas (SSAs) were calculated using the multi-point Brunauer–Emmett–Teller (BET) model (p p0−1 = 0.005–0.05). Total pore volumes (Vtotal) were determined at p p0−1 = 0.95. CO2 (0 °C and 25 °C), N2 (−196 °C, 0 °C, and 25 °C), and H2O (25 °C) physisorption measurements were carried out with 40–50 mg of sample at the same instrument. The isosteric heat of CO2 adsorption was obtained based on the Clausius–Clapeyron equation using an option integrated in the ASiQwin 3.0 analysis software (Quantachrome Instruments).The determination of CO2 over N2 selectivity (S) (at 298 K; for N2/CO2 ratio of 90/10) followed the ideal adsorption solution theory (IAST) method43 and was calculated using following equation:
| SCO2/N2 = XCO2/XN2 × YN2/YCO2 |
Thermal response measurements were performed at 1 bar and 298 K on an optical calorimeter (InfraSORP).44 For the CO2 adsorption/desorption measurements, ∼30 mg of the C-HAT-CN-X materials were placed in the sample holder, and purged under N2 flow of 72 sccm until a constant sample temperature was observed. Then, the sample was exposed to a CO2 flow of 72 sccm for 100 seconds. Finally, N2 flow was directed to the sample cell again for 200 s causing a decrease in temperature due to desorption of the test gas. The peak areas (A) of the thermal response curves have been integrated using OriginPro2015 software. The peak area was divided by the sample mass used for the measurements and is also normalized to the total pore volume.
The CO2 adsorption cycling experiments were performed using similar gas flow and time-programmed conditions.
All X-ray powder diffraction (XRPD) patterns were collected on a Bruker D8 Advanced diffractometer equipped with a scintillation counter detector using Cu Kα radiation (λ = 0.1518 nm) in the 2θ range from 5° to 70° with a step size of 0.03° and counting time 1 s per step. Possible impurity phases were checked by comparing XRPD patterns with those in the PDF4 database (powder diffraction file, ICDD, release 2016).
C/H/N Elemental analysis (EA) was accomplished as combustion analysis using a Vario Micro device. The oxygen content of the samples was calculated from the residual amount of the EA which has not been detected as C, H, or N.
Fourier-transform infrared (FTIR) spectra were recorded on a Varian 1000 spectrometer with an attenuated total reflectance setup.
Thermogravimetric analysis (TGA) measurements were performed using a thermo microbalance TG 209 F1 Libra (Netzsch, Selb, Germany). A platinum crucible was used for the measurement of 10 ± 1 mg of samples in a N2 flow of 10 mL min−1 and a purge flow of 10 mL min−1. Additional 5 mL min−1 O2 flow has been used for the measurements in synthetic air. The samples were heated to 1000 °C with a heating rate of 5 °C min−1. The data was recorded and analysed by the Proteus (6.0.0) and Quadstar (7.03) software package.
Solid-state nuclear magnetic resonance (ssNMR) spectra were recorded with a Bruker Avance 400 MHz spectrometer operating at 100.56 MHz for 13C. 13C{1H} magic angle spinning (MAS) ssNMR experiments were carried out at a spinning rate of 10 kHz using 4 mm MAS HX double resonance probe. The 13C π/2 pulse length were 3.0 μs. Two pulse phase modulation (TPPM) heteronuclear dipolar decoupling was used during acquisition and a recycle delay of 20 s was implemented. All 13C{1H} spectra are referenced to external TMS at 0 ppm using solid adamantine as a secondary reference.
Details of the electron microscopy characterization of the samples and computational details are given in the ESI.†
Results and discussion
Structure of the HAT-CN-derived carbon materials
Thermal condensation of HAT-CN by elimination of the nitrile groups in the temperature range of 500–700 °C leads to the formation of nitrogen-rich carbon materials with a C2N-like stoichiometry (Fig. S1†). C-HAT-CN-500 and C-HAT-CN-525 show nitrogen contents of 31–34 at%, as determined by elemental analysis (EA) (Table 1). The significant amount of hydrogen and oxygen detected in these measurements likely arises from water adsorbed on the samples. The C/N atomic ratio of C-HAT-CN-500 (1.55) is only marginally higher than in HAT-CN (1.53). This indicates that no major condensation of the nitrile groups occurs at this temperature. After condensation at 525 °C and 550 °C, the C/N ratios increase to 1.60 and 1.67, respectively. Finally, at 700 °C, the ratio reached 1.96 and a nearly perfect C2N-type material is obtained. According to our previous findings, condensation of the HAT-CN precursor molecule with a composition of C18N12 releases cyanogen (C2N2) as the condensation byproduct. It can thus be assumed that the condensation proceeds according to the general equation C18N12(s) → C12N6(s) + 3C2N2(g). In agreement with thermal analysis of HAT-CN under inert atmosphere (Fig. S2†), there seems to be a sharp onset point of this condensation reaction located at 500 °C or slightly below. The yield of nitrogen-rich carbon material after condensation at 500 °C is 70% and drops significantly to 51% after thermal treatment at 525 °C and to 33% after reaction at 700 °C, which is in good accordance with the TGA experiment. These elemental compositions and yields do not only reveal that HAT-CN is a very well-suited precursor molecule for the synthesis of C2N materials, but also that the degree of condensation (and with it the chemical and textural properties of the resulting materials) is precisely adjustable via the condensation temperature because the nitrogen content decreases at higher condensation degree due to the release of cyanogen.| Material | CEA/at% (wt%) | NEA/at% (wt%) | HEA/at% (wt%) | OEA/at% (wt%) | Yield/% |
|---|---|---|---|---|---|
| HAT-CN | 53.9 (55.8) | 35.3 (42.6) | 10.3 (0.9) | 0.3 (0.6) | — |
| C-HAT-CN-500 | 52.5 (54.1) | 33.9 (40.8) | 10.6 (0.9) | 3.0 (4.2) | 70 |
| C-HAT-CN-525 | 50.4 (53.3) | 31.5 (38.9) | 13.4 (1.2) | 4.6 (6.5) | 51 |
| C-HAT-CN-550 | 43.7 (48.4) | 26.2 (33.7) | 19.3 (1.8) | 10.9 (16.1) | 50 |
| C-HAT-CN-700 | 42.5 (50.5) | 21.7 (30.1) | 25.0 (2.5) | 10.7 (16.9) | 33 |
FTIR (Fig. 1a) and XRPD (Fig. 1b) measurements further indicate a distinct structural change between 500 °C and 550 °C, resulting from condensation of the HAT-CN monomer. The XRD pattern of C-HAT-CN-500 is still in nearly perfect accordance to the pattern of the untreated HAT-CN. The crystal structure is still intact after heating to 525 °C, but there is already a notable contribution of defects in the HAT structure, as can be seen by the obvious distortion of the baseline underneath the characteristic peaks in the 2θ range from 10 to 30°. The contribution of amorphous scattering intensity at very low 2θ values can be indicative for the development of narrow pores. This is not observed for the HAT-CN and increases in intensity from C-HAT-CN-500 to C-HAT-CN-525. In accordance with our recent study, C-HAT-CN-550 and C-HAT-CN-700 have an amorphous structure as it is typical for high surface-area carbons. The (002) peak (representing graphitic stacking in the structure) has low intensity and can be found in the 2θ range from 25 to 30°. Its maximum shifts to a slightly lower angle with increasing condensation temperature.
 | ||
| Fig. 1 FTIR spectra (a), XRPD patterns (b), and 13C CP-MAS ssNMR spectra (c) of HAT-CN, C-HAT-CN-500, C-HAT-CN-525, C-HAT-CN-550, and C-HAT-CN-700. In (c), the green circle is marking aromatic carbon in the inner benzene ring, the orange circle marks the pyrazinic carbon, and blue circle corresponds to the nitrile carbon. | ||
FTIR spectra of the HAT-CN show sharp and narrow peaks, which disappear or get broader with increasing condensation temperature. The intense peak at 1339 cm−1 originates from C–N stretching vibrations. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and CC vibrations in the aromatic ring system appear at 1708 cm−1 and 1560 cm−1, respectively. The peak at 2240 cm−1 is attributed to the nitrile C
N and CC vibrations in the aromatic ring system appear at 1708 cm−1 and 1560 cm−1, respectively. The peak at 2240 cm−1 is attributed to the nitrile C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N groups. This peak does not disappear as the carbonization temperature increases. This might be related to the nitrile groups located on the edges of the carbon network. In accordance with EA and XRPD measurements, the spectra show that there is a continuous increase of the condensation degree with increasing synthesis temperature.
N groups. This peak does not disappear as the carbonization temperature increases. This might be related to the nitrile groups located on the edges of the carbon network. In accordance with EA and XRPD measurements, the spectra show that there is a continuous increase of the condensation degree with increasing synthesis temperature.
The chemical environment of carbon atoms in HAT-CN and HAT-CN-derived carbon materials is investigated by 13C{1H} magic angle spinning (MAS) solid-state (ss) NMR experiments (Fig. 1c). Three characteristic peaks for HAT-CN are present. The peak at ∼132 ppm (marked with an orange circle) corresponds to aromatic carbon atoms in the inner benzene ring of the HAT-CN molecule. The peak at ∼139 ppm (green circle) corresponds to the pyrazinic carbon, and the one at ∼110 ppm (blue circle) to the nitrile carbon.45 As expected for a well crystallized organic molecule, the peaks for HAT-CN are sharp and well separated. The condensation of the organic molecule towards a porous amorphous network clearly leads to broadening of the peaks which gets more pronounced at increasing temperature. The peak of the nitrile carbons remains for the C-HAT-CN-700, but weakens. This is due to the surface terminations at grain boundaries of the porous network particles, as it was already shown in the FTIR spectra.
Besides the atomic construction, the porosity of the HAT-derived carbon materials is also controllable by the synthesis temperature. In our recent study on HAT-CN-derived C2N-materials, it has been shown that (despite the absence of any additional porogen) condensation at 550 °C or above leads to the formation of structural microporosity.41 The Ar physisorption (−186 °C) isotherm (Fig. 2a) shows that the C-HAT-CN-700 material with near-ideal C2N-stoichiometry has indeed a high specific BET surface area and a significant total pore volume of 785 m2 g−1 and 0.31 cm3 g−1, respectively. Starting from the HAT-CN with no notable surface area, there is a step-wise increase of the porosity detected with Ar at −186![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C with increasing condensation temperature. Whereas the samples C-HAT-CN-500 and C-HAT-CN-525 still do not show any notable uptake of Ar, condensation at 550 °C forms a material with a BET surface area of 627 m2 g−1 and a total pore volume of 0.24 cm3 g−1. The lager pore volume of C-HAT-CN-700 is the consequence of further increasing pore size with increasing degree of condensation (Fig. S3†). The micropore volume detected for C-HAT-CN-700 with Ar physisorption at −186 °C (0.30 cm3 g−1) is comparable to the one detected with N2 physisorption at −196 °C (0.27 cm3 g−1, Fig. 2b). Notably, the difference for C-HAT-CN-550 is much larger. In this material, Ar physisorption analysis detects a micropore volume of 0.24 cm3 g−1, whereas nitrogen physisorption analysis only results in a micropore volume of 0.20 cm3 g−1. This indicates accessibility limitations for the slightly larger N2 molecules with higher quadrupole moment at lower measurement temperature into the very small micropores of C-HAT-CN-550. In other words, these materials are possible molecular sieves, not only for CO2/N2 adsorption, but also for Ar/N2 adsorption. The preferred adsorption is based on a kinetic size exclusion on the expense of the (even stronger adsorbing) nitrogen molecules, that is, a (kinetic) molecular sieving effect. The uptakes of argon and nitrogen at relative pressures close to 1 bar are due to adsorption in the larger pores, which is in line with the TEM investigations as discussed below.
°C with increasing condensation temperature. Whereas the samples C-HAT-CN-500 and C-HAT-CN-525 still do not show any notable uptake of Ar, condensation at 550 °C forms a material with a BET surface area of 627 m2 g−1 and a total pore volume of 0.24 cm3 g−1. The lager pore volume of C-HAT-CN-700 is the consequence of further increasing pore size with increasing degree of condensation (Fig. S3†). The micropore volume detected for C-HAT-CN-700 with Ar physisorption at −186 °C (0.30 cm3 g−1) is comparable to the one detected with N2 physisorption at −196 °C (0.27 cm3 g−1, Fig. 2b). Notably, the difference for C-HAT-CN-550 is much larger. In this material, Ar physisorption analysis detects a micropore volume of 0.24 cm3 g−1, whereas nitrogen physisorption analysis only results in a micropore volume of 0.20 cm3 g−1. This indicates accessibility limitations for the slightly larger N2 molecules with higher quadrupole moment at lower measurement temperature into the very small micropores of C-HAT-CN-550. In other words, these materials are possible molecular sieves, not only for CO2/N2 adsorption, but also for Ar/N2 adsorption. The preferred adsorption is based on a kinetic size exclusion on the expense of the (even stronger adsorbing) nitrogen molecules, that is, a (kinetic) molecular sieving effect. The uptakes of argon and nitrogen at relative pressures close to 1 bar are due to adsorption in the larger pores, which is in line with the TEM investigations as discussed below.
 | ||
| Fig. 2 Ar physisorption isotherms (−186 °C) (a) and N2 physisorption isotherms (−196 °C) (b) of C-HAT-CN-500, C-HAT-CN-525, C-HAT-CN-550, and C-HAT-CN-700. | ||
In order to utilize these materials for selective CO2/N2 capture based on molecular sieving, a pore size close to the dimensions of the CO2 molecules, but smaller than for N2, is required. On the other hand, the internal surface of the materials should be highly polarizing in order to have a high adsorption affinity towards CO2. H2O vapor (a molecule with even higher adsorption enthalpy than CO2 and thus a well-suited probe for the presence of polar adsorption sites) physisorption isotherms of the samples show an extremely polar character of the pore walls in all HAT-CN-derived materials, as indicated by the significant water uptake at low relative pressures (Fig. S4†). The adsorbed volumes follow the available pore volumes detected with Ar physisorption. It should be noted that the strong interaction with H2O will of course remain a problem for CO2 separation with these materials under practical conditions.
High-resolution transmission electron microscopy (HRTEM) as well as annular dark-field scanning transmission electron microscopy (ADF-STEM) images of the condensed HAT-CN materials (Fig. 3 and S5†) show the presence of an amorphous, covalent, carbonaceous microstructure without distinct stacking of layers in the C-HAT-CN-550 and C-HAT-CN-700, which is typical for highly microporous carbon materials and in line with our previous findings.41 This highly disordered structure likely results from the rapid condensation due to elimination of the nitrile groups of HAT-CN. C-HAT-CN-500 and C-HAT-CN-525 show an amorphous carbon microstructure in the TEM images. In combination with the XRPD results (Fig. 1b), this indicates that the condensed HAT-CN materials consist of crystalline and amorphous constituents. The C-HAT-CN materials seem to have a core-shell-like structure with a distinct difference between the constructions of the inner and outer areas of the individual disk-shaped particles (Fig. S5†). While the inner areas consist of a 3-dimensional network of agglomerated small grains with larger pores in between, the double-walled shell has a rather dense appearance.
 | ||
| Fig. 3 HRTEM images of C-HAT-CN-700 (a) and (b), C-HAT-CN-550 (c) and (d), C-HAT-CN-525 (e) and (f), and C-HAT-CN-500 (g) and (h). Constituents of core and shell areas are labelled in the images. | ||
CO2/N2 molecular sieving with HAT-CN-derived carbon materials
The narrow pore size of the HAT-CN-derived carbons can be rather precisely adjusted by the synthesis temperature, and the materials generally have a very polar surface resulting from their high nitrogen content. On the one site, these pyrazinic nitrogen sites have basic and electron-donating properties which are particularly strong in the C2N structure and able to strongly bind to the electrophilic carbon atom in CO2. A synergistic effect can be achieved by the similar presence of electron deficient carbon atoms in the C2N which can strongly rebind to the nucleophilic oxygen atoms in CO2. These properties render the materials attractive candidates for selective adsorption of CO2 in the presence of N2.CO2 and N2 physisorption isotherms of the HAT-CN-derived carbon materials at 0 °C and 25 °C show a maximum IAST CO2/N2 selectivity of 121 for the C-HAT-CN-525 (Table 2 and Fig. 4). In this material, the pores are already large enough to adsorb a significant amount of CO2, but are not yet too large to take up high volumes of N2. At higher synthesis temperatures, CO2 uptakes increase further as a result of the increasing pore volume, but the CO2/N2 selectivity decreases. At both temperatures, the selectivity of C-HAT-CN-550 with smaller pores is still higher than for C-HAT-CN-700. This confirms that the use of materials with a high N2 surface area is not suitable for selective CO2 capture, as long as high selectivity is required because of the increasing contribution of less discriminative van der Waals interactions.8 In accordance to the nanostructure and nitrogen content, C-HAT-CN-525 shows a significantly higher isosteric heat (Qst) of CO2 adsorption (Table 2 and Fig. S6†) of ∼52 kJ mol−1 as compared to C-HAT-CN-550 (∼36 kJ mol−1) and C-HAT-CN-700 (∼42 kJ mol−1). It should be mentioned that these numbers may have a relatively high experimental error but such a high Qst is the result of the high nitrogen content, as well as the pore size, which is perfectly tuned for the molecular encapsulation of CO2. Typically, isosteric heats of adsorption, as well as IAST CO2/N2 selectivity in this range are only achievable with metal-containing materials including coulomb charges.8,24,33 In this way, C-HAT-CN-525 indeed combines the necessary requirements for highly selective CO2 capture, which are molecular sieving and an atomic construction tuned for strong CO2 binding (i.e., separation of the electron density between carbon and nitrogen atoms) without the need for any metal.
| Material | SSABET/m2 g−1 | SSADFT/m2 g−1 | V Total/cm3 g−1 | Selectivity/CO2/N2 | Q st/kJ mol−1 | |
|---|---|---|---|---|---|---|
| 273 K | 298 K | |||||
| C-HAT-CN-500 | 13 | 13 | 0.01 | — | — | — |
| C-HAT-CN-525 | 82 | 172 | 0.03 | 72 | 121 | 52 |
| C-HAT-CN-550 | 627 | 1013 | 0.24 | 34 | 65 | 36 |
| C-HAT-CN-700 | 785 | 1182 | 0.31 | 23 | 53 | 42 |
 | ||
| Fig. 4 CO2 (a) and (b), as well as N2 (c) and (d) physisorption isotherms of C-HAT-CN-500, C-HAT-CN-525, C-HAT-CN-550, and C-HAT-CN-700 measured at 0 °C (a) and (c) and at 25 °C (b) and (d). | ||
Thermal response measurements of CO2 adsorption with the InfraSORP technology44 (Fig. 5a) provide further information about CO2 adsorption capacities and kinetics of the HAT-CN-derived materials. In line with the volumetric measurements, the highest temperature peak areas are present for C-HAT-CN-700 and C-HAT-CN-550 due to their large micropores, and thus highest CO2 uptake at 1 bar. Regarding their very low Ar and N2 accessible microporosity, the significant temperature signals of C-HAT-CN-525 and C-HAT-CN-500 are remarkable. According to their higher Qst values their normalized peak areas (divided by mass and the total pore volumes determined with Ar physisorption) are significantly higher as compared to the materials obtained by HAT-CN condensation at higher temperatures (Fig. 5b). The mass-related peak areas for adsorption and desorption are nearly similar for one and the same material, indicating no occurrence of irreversible binding of CO2. Compared to a microporous carbide-derived carbon material (TiC-CDC, synthesized at 800 °C),44 the presence of specific CO2 binding sites is further indicated by the minor asymmetry in the adsorption and desorption temperature signals in the cases of the C-HAT-CN materials (Fig. 5c and 5d). The CO2 desorption process by flushing with N2 causes a slightly lower peak temperature decrease than the adsorption of CO2. This is typical for materials with narrow pores and abundant nitrogen sites, and thus not observable for the heteroatom-free TiC-CDC with slightly larger micropores. The latter is showing rather symmetrical and “sharp” temperature signals in CO2 adsorption.
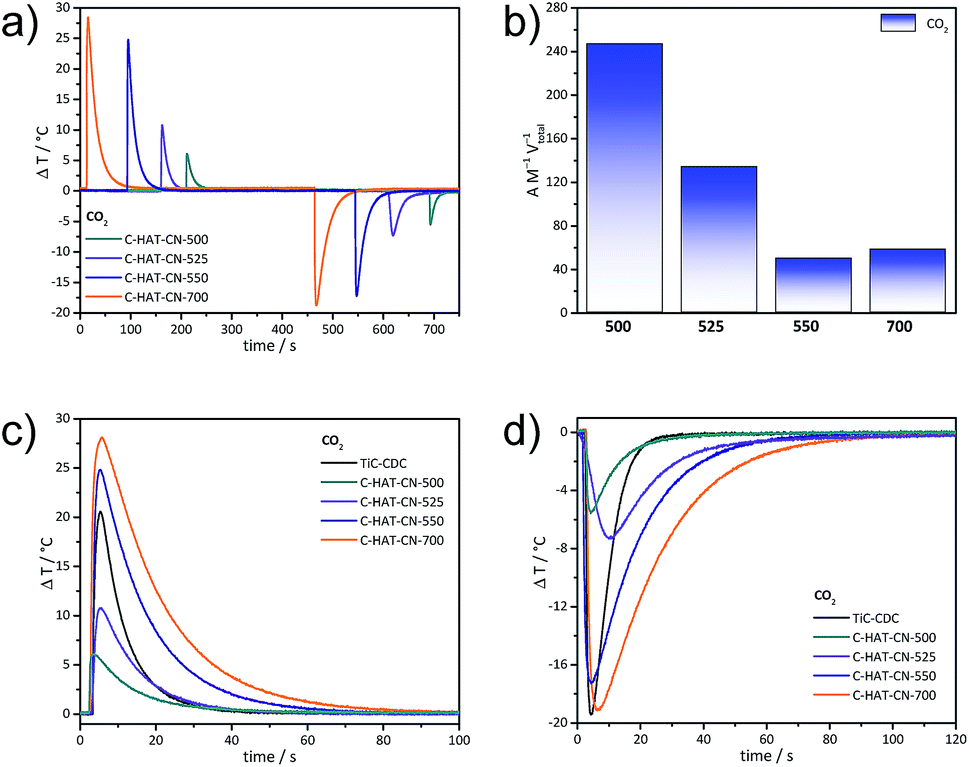 | ||
| Fig. 5 CO2 thermal response adsorption and desorption measurements at 1 bar and 25 °C (a), integrated adsorption temperature peak area normalized by mass and total pore volume (b) of C-HAT-CN-500, C-HAT-CN-525, C-HAT-CN-550, and C-HAT-CN-700, as well as their adsorption (c) and desorption (d) thermal response curves at higher magnifications together with a microporous non-nitrogen-doped carbon material (TiC-CDC) for comparison. | ||
Despite the high CO2 adsorption enthalpy of C-HAT-CN-525, the binding is fully reversible for 80 cycles by simple flushing with N2 (Fig. S7†). This means, that this molecular sieving concept is applicable to simple pressure swing adsorption without loss of CO2 separation efficiency over time.
Calculation of the CO2 adsorption state on C2N
An atomistic model of the adsorption state of CO2 on an idealized C-HAT-CN material with perfect C2N-type stoichiometry and AAA-type layer stacking (Fig. 6a) has been obtained by dynamical simulated annealing46,47 using the second-generation Car-Parrinello molecular dynamics approach of Kühne et al. (computational details are described in the ESI†).48,49 We find that the CO2 molecules are oriented orthogonal to the C2N-planes occupying every second vacancy in a checkerboard-like fashion. This energy minimization arises from collective stabilization effects from the nitrogen atoms in the C2N layers surrounding the carbon atom in the CO2 molecule and due to the electron acceptor properties of the carbon atoms from C2N in close proximity to the oxygen atoms in CO2. In simple words, all atoms of the gas molecule are surrounded in their electronically favourable environment. In the present supercell, which contains of 4 layers with 4 holes each, the insertion of 8 CO2 molecules entails an adsorption energy of −231.5 kJ mol−1 that equals to −29 kJ mol−1 per molecule. This Qst value is slightly lower as compared to the experimental value determined for the C-HAT-CN-700 material. In accordance with our gas physisorption data, this indicates that the pore size in the experimental sample is lower than in the idealized C2N material used for the theoretical calculations with pores which would result from perfect AAA stacking. Another possible reason for the higher experimental Qst value could be the overlapping of the adsorption potentials from two pore walls in narrow cavities which is not taken into consideration by the theoretical calculations. In the corresponding nitrogen-free control system with C3H stoichiometry, all nitrogen atoms have been substituted by C–H groups. In this case, the adsorption energy is strictly positive being as high as +206 kJ mol−1 (Fig. 6b). No energetically favourable coordination of CO2 takes place in this environment. | ||
| Fig. 6 Lowest energy structure of C2N with maximum amount of CO2 molecules adsorbed (top). For comparison the considered control system of C3H stoichiometry is also shown (bottom). Left: top views (a); right: side views (b). | ||
Conclusions
We have shown that the thermal condensation of HAT-CN is a process that produces extremely nitrogen-rich carbon materials under precise control over the molecular composition and nanostructure/porosity. At a condensation temperature of 525 °C, pores start forming with a size that allows for CO2/N2 molecular sieving. To the best of our knowledge, this is the first time that a metal-free and non-organic material with an isosteric heat of CO2 adsorption of up to 52 kJ mol−1 and a CO2/N2 selectivity of more than 100 has been prepared.These properties are mainly related to the combination of strong binding of CO2 (provided by negatively polarized nitrogen atoms and positively polarized carbon atoms) as a result of the combination of the atomic construction with the narrow pore size of the C-HAT-CN materials. A defined pore size alone for CO2 adsorption is useless as the capture will become too slow and a designed chemical construction combined with too large pores is also not beneficial, as then the selectivity is limited by non-discriminative van der Waals interactions.
As it can be assumed that only a small part of the bulk volume present in C-HAT-CN-525 is really used for CO2 binding, the logical next step would be the synthesis of a hierarchical C2N-type material in order to shorted mass transport pathways and thus to enhance the accessibility of the internal pore volume. This approach is currently applied in zeolite research50,51 and will also help in case of such “organic zeolites”. On the other hand, the insertion of extra-framework cations in these materials may be an approach to further increase the CO2 adsorption enthalpy. The main focus of this work was to illuminate the origin of the strong interaction between CO2 and nitrogen-rich carbon materials prepared by molecular design. After the general possibility of CO2 molecular sieving with metal-free materials has been shown in this work, the applicability of such materials under real-world conditions, as well as the kinetics of CO2 adsorption, has to be further investigated with mixed-gas breakthrough experiments under wet conditions.
Finally, this synthetic concept towards such zeolite-like carbon materials is potentially also applicable for the separation of other relevant gas mixtures.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. W. and M. O. thank the German Chemical Industry Fund (Fonds der Chemischen Industrie, FCI) for financial support with a Liebig Fellowship. J.H, S.S. and T.D.K acknowledges funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme (grant agreement No 716142). The authors would like to thank the Paderborn Center for Parallel Computing (PC2) for the generous allocation of computing time on the FPGA-based supercomputer “Noctua”. The authors would like to thank Ms. Bolortuya Badamdorj for help with collecting TEM images. Open Access funding provided by the Max Planck Society.References
- F. A. Rahman, M. M. A. Aziz, R. Saidur, W. A. W. A. Bakar, M. R. Hainin, R. Putrajaya and N. A. Hassan, Renewable Sustainable Energy Rev., 2017, 71, 112–126 CrossRef.
- E. S. Sanz-Perez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef CAS PubMed.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- R. S. Hazeldine, Science, 2009, 325, 1647–1652 CrossRef PubMed.
- B. Dutcher, M. Fan and A. G. Russell, ACS Appl. Mater. Interfaces, 2015, 7, 2137–2148 CrossRef CAS PubMed.
- C. Gouedard, D. Picq, F. Launay and P. L. Carrette, Int. J. Greenhouse Gas Control, 2012, 10, 244–270 CrossRef CAS.
- F. Inagaki, C. Matsumoto, T. Iwata and C. Mukai, J. Am. Chem. Soc., 2017, 139, 4639–4642 CrossRef CAS PubMed.
- M. Oschatz and M. Antonietti, Energy Environ. Sci., 2018, 11, 57–70 RSC.
- Y. S. Bae and R. Q. Snurr, Angew. Chem., Int. Ed., 2011, 50, 11586–11596 CrossRef CAS PubMed.
- X. Lu, D. Jin, S. Wei, Z. Wang, C. An and W. Guo, J. Mater. Chem. A, 2015, 3, 12118–12132 RSC.
- O. Cheung and N. Hedin, RSC Adv., 2014, 4, 14480–14494 RSC.
- M. Abu Ghalia and Y. Dahman, Energy Technol., 2017, 5, 356–372 CrossRef CAS.
- F. Akhtar, Q. Liu, N. Hedin and L. Bergström, Energy Environ. Sci., 2012, 5, 7664–7673 RSC.
- R. Banerjee, H. Furukawa, D. Britt, C. Knobler, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 3875–3877 CrossRef CAS PubMed.
- F. Gao, Y. Li, Z. Bian, J. Hu and H. Liu, J. Mater. Chem. A, 2015, 3, 8091–8097 RSC.
- Y. Oh, V.-D. Le, U. N. Maiti, J. O. Hwang, W. J. Park, J. Lim, K. E. Lee, Y.-S. Bae, Y.-H. Kim and S. O. Kim, ACS Nano, 2015, 9, 9148–9157 CrossRef CAS PubMed.
- G. P. Hao, W. C. Li, D. Qian and A. H. Lu, Adv. Mater., 2010, 22, 853–857 CrossRef CAS PubMed.
- M. Sevilla, P. Valle-Vigón and A. B. Fuertes, Adv. Funct. Mater., 2011, 21, 2781–2787 CrossRef CAS.
- J. Wang, A. Heerwig, M. R. Lohe, M. Oschatz, L. Borchardt and S. Kaskel, J. Mater. Chem., 2012, 22, 13911–13913 RSC.
- Z. Tian, N. Fechler, M. Oschatz, T. Heil, J. Schmidt, S. Yuan and M. Antonietti, J. Mater. Chem. A, 2018, 6, 19013–19019 RSC.
- Y. Zhao, X. Liu and Y. Han, RSC Adv., 2015, 5, 30310–30330 RSC.
- X. Zhu, C. Tian, G. M. Veith, C. W. Abney, J. Dehaudt and S. Dai, J. Am. Chem. Soc., 2016, 138, 11497–11500 CrossRef CAS PubMed.
- Y. Zhao, K. X. Yao, B. Teng, T. Zhang and Y. Han, Energy Environ. Sci., 2013, 6, 3684–3692 RSC.
- Y. Belmabkhout, V. Guillerm and M. Eddaoudi, Chem. Eng. J., 2016, 296, 386–397 CrossRef CAS.
- J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41, 2308–2322 RSC.
- R. Luebke, J. F. Eubank, A. J. Cairns, Y. Belmabkhout, L. Wojtas and M. Eddaoudi, Chem. Commun., 2012, 48, 1455–1457 RSC.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- Y. K. Kim, G. M. Kim and J. W. Lee, J. Mater. Chem. A, 2015, 3, 10919–10927 RSC.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocella, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303–308 CrossRef CAS PubMed.
- X. Ren, H. Li, J. Chen, L. Wei, A. Modak, H. Yang and Q. Yang, Carbon, 2017, 114, 473–481 CrossRef CAS.
- J. W. To, J. He, J. Mei, R. Haghpanah, Z. Chen, T. Kurosawa, S. Chen, W. G. Bae, L. Pan, J. B. Tok, J. Wilcox and Z. Bao, J. Am. Chem. Soc., 2016, 138, 1001–1009 CrossRef CAS PubMed.
- R. Vaidhyanathan, S. S. Iremonger, G. K. H. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650–653 CrossRef CAS PubMed.
- Y. Zhao, X. Liu, K. X. Yao, L. Zhao and Y. Han, Chem. Mater., 2012, 24, 4725–4734 CrossRef CAS.
- J. R. Li, J. Yu, W. Lu, L. B. Sun, J. Sculley, P. B. Balbuena and H. C. Zhou, Nat. Commun., 2013, 4, 1538 CrossRef PubMed.
- N. P. Wickramaratne and M. Jaroniec, J. Mater. Chem. A, 2013, 1, 112–116 RSC.
- G. Zhu, R. G. Jensen, R. B. Hallick and G. F. Wildner, Plant Physiol., 1992, 98, 764–768 CrossRef CAS PubMed.
- A. Silvestre-Albero, S. Rico-Francés, F. Rodríguez-Reinoso, A. M. Kern, M. Klumpp, B. J. M. Etzold and J. Silvestre-Albero, Carbon, 2013, 59, 221–228 CrossRef CAS.
- Q. Liu, A. Mace, Z. Bacsik, J. Sun, A. Laaksonen and N. Hedin, Chem. Commun., 2010, 46, 4502–4504 RSC.
- P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80–84 CrossRef CAS PubMed.
- O. Shekhah, Y. Belmabkhout, Z. Chen, V. Guillerm, A. Cairns, K. Adil and M. Eddaoudi, Nat. Commun., 2014, 5, 4228 CrossRef CAS PubMed.
- R. Walczak, B. Kurpil, A. Savateev, T. Heil, J. Schmidt, Q. Qin, M. Antonietti and M. Oschatz, Angew. Chem., Int. Ed., 2018, 57, 10765–10770 CrossRef CAS PubMed.
- B. Kurpil, A. Savateev, V. Papaefthimiou, S. Zafeiratos, T. Heil, S. Özenler, D. Dontsova and M. Antonietti, Appl. Catal., B, 2017, 217, 622–628 CrossRef CAS.
- A. L. Myers and J. M. Prausnitz, AIChE J., 1965, 11, 121–127 CrossRef CAS.
- M. Oschatz, M. Leistner, W. Nickel and S. Kaskel, Langmuir, 2015, 31, 4040–4047 CrossRef CAS PubMed.
- H. T. Chifotides, B. L. Schottel and K. R. Dunbar, Angew. Chem., Int. Ed., 2010, 49, 7202–7207 CrossRef CAS PubMed.
- S. Kirkpatrick, C. D. Gelatt and M. P. Vecchi, Science, 1983, 220, 671–680 CrossRef CAS PubMed.
- R. Car and M. Parrinello, Phys. Rev. Lett., 1985, 55, 2471–2474 CrossRef CAS PubMed.
- T. D. Kühne, M. Krack, F. R. Mohamed and M. Parrinello, Phys. Rev. Lett., 2007, 98, 066401 CrossRef PubMed.
- T. D. Kühne, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 391–406 Search PubMed.
- R. Srivastava, M. Choi and R. Ryoo, Chem. Commun., 2006, 4489–4491 RSC.
- D. Verboekend and J. Pérez-Ramírez, Catal. Sci. Technol., 2011, 1, 879–890 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9se00486f |
| This journal is © The Royal Society of Chemistry 2019 |