
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnravelling the practical solar charging performance limits of redox flow batteries based on a single photon device system†
Dowon
Bae‡
 *,
Gerrit M.
Faasse‡
,
Gerasimos
Kanellos
and
Wilson A.
Smith
*
*,
Gerrit M.
Faasse‡
,
Gerasimos
Kanellos
and
Wilson A.
Smith
*
Materials for Energy Conversion and Storage (MECS), Department of Chemical Engineering, Delft University of Technology, Van der Maasweg 9, 2629 HZ Delft, The Netherlands. E-mail: d.bae@tudelft.nl; w.smith@tudelft.nl
First published on 5th July 2019
Abstract
In recent years, solar redox flow batteries have attracted attention as a possible integrated technology for simultaneous conversion and storage of solar energy. Unlike solar water splitting technologies which require at least 1.8 V for meaningful performance, a lesson learned from previous studies on solar redox flow batteries (SRFBs) is that even single-photon devices can demonstrate unbiased photo-charging owing to the flexibility of redox couple selection. Thus, in this paper, we present a theoretical model reflecting experimental parameters, such that we can highlight important parameters that merit the most attention in further studies towards the practical development of SRFBs. Importantly, the results clearly show how to choose an optimum combination of semiconductors and redox couples under unavoidable conditions that a practical system would encounter, including, but not limited to, optical loss by the electrolyte, overpotential, device architecture and chemical potentials.
1. Introduction
In 2017 the contribution from renewable sources to the electricity production in OECD countries totalled 23.7%, of which 26.9% was from solar energy (i.e., photovoltaic (PV) and solar-thermal power).1 However, continued implementation is facing challenges concerning the security of supplies because of the intermittent nature of sunlight. In this context, storing solar energy directly in hydrogen or other chemicals via photoelectrochemical (PEC) water splitting or CO2 reduction has been regarded as a particularly attractive technology.2–5 However, the sluggish reaction kinetics (i.e., high overpotential) hampers their widespread implementation.2,3 In terms of technological readiness, the most feasible approach to store solar energy would be a photovoltaic (PV) panel integrated with Li-ion batteries as an energy storage system (ESS), but this method faces critical issues related to frequent thermal runaway6,7 and cost of the battery module.8 Alternatively, solar rechargeable redox flow batteries (SRFBs) are being studied as a means of simultaneous storage of solar energy into chemicals, which can be readily utilized to generate electricity via reversible redox reactions.9–11 Generally, redox flow batteries (RFBs) present facile reaction kinetics, which can be several orders of magnitude faster than water oxidation,12 and are deemed safer than other solid-state batteries owing to their wide discharging voltage range.13,14 Additionally, the system power rating and discharging time of RFBs surpass those of conventional batteries.15,16The architectural concept of the SRFB is a combination of a redox flow battery (RFB) and a PEC or PV-assisted device, which may be fully integrated into a single cell or separated into an RFB and a solar charging component. Since the initial pioneering work in 1976 by Hodes et al.,17 a lot of effort has been put into this field relatively recently9,10,18–22 in the wake of the maturation of PEC water splitting materials which offer practical utilization of PV materials for electrochemical applications.11 Unlike conventional PEC water splitting, SRFBs offer flexibility with respect to redox potential and solubility in a wide pH range. In recent studies, it is apparent that conventional inorganic redox chemicals combined with various organic chemicals allow an unprecedented wide selection of redox energy level matching with the photovoltage for an optimized charging reaction.11,23,24 Despite the above-mentioned advantages, one of the major drawbacks is the relatively low solar-to-chemical efficiency (i.e., charging efficiency, STC% hereinafter). An integrated system with a photo-anode and -cathode immersed in the PEC charging cell in a dual-bed or tandem device configuration (2-photon-device) leads to an increased photovoltage that is enough for the redox couples with a high cell voltage (≥0.8 V).10,18,20 Most recently, Urbain et al. reported a record-breaking STC% (12.3%) using a monolithic tandem a-Si/a-Si PV-assisted device which showed a high photovoltage (>2 V) owing to the wide bandgap of the two absorbing layers (ca. 1.95 eV).18
Nonetheless, the 2-photon-device approach is cost-advantageous only when both photo-absorbers have an ideal band-gap pairing and are made using technologies providing a similar cost per unit output power.25 Another critical challenge is the demonstration of a wide band-gap top cell with a cost below $100 per m2 and an efficiency above 20%, which is essential for having a lower LCOE (levelized cost of electricity) than a single-photon-device.25,26 Meanwhile, a lesson learned from previous SRFB studies is that even single-photon-devices can demonstrate unbiased photocharging owing to the flexibility of redox couple selection.
Previously published STC efficiencies for single photon devices are shown in Fig. 1, showing the efficiencies and the materials used. Initial studies in the eighties demonstrated meaningful charging efficiencies using n-WSe2.27,28 However, these references used a stationary redox chemical cell with a non-standard light source (e.g., He–Ne laser; 150 mW cm−2). Recently, McKone and co-workers22 have demonstrated an SRFB under standard light conditions (AM 1.5 irradiation). A crystalline WSe2 photocathode was used for charging an RFB with NaI and AQDS (2,7) redox couples with a cell voltage (Vcell) of 0.46 V with an STC% of 3.9%, which is one of the highest values among the SRFBs with an unbiased single PEC device.22 Interestingly, examples with already proven PEC materials, such as c-Si, GaAs, DSSC, etc.,19,29–31 exhibited quite low charging efficiencies. For the sake of consistency, all STC% data are calculated using the light-driven photocurrent measured at 0% state of charge (SOC%).
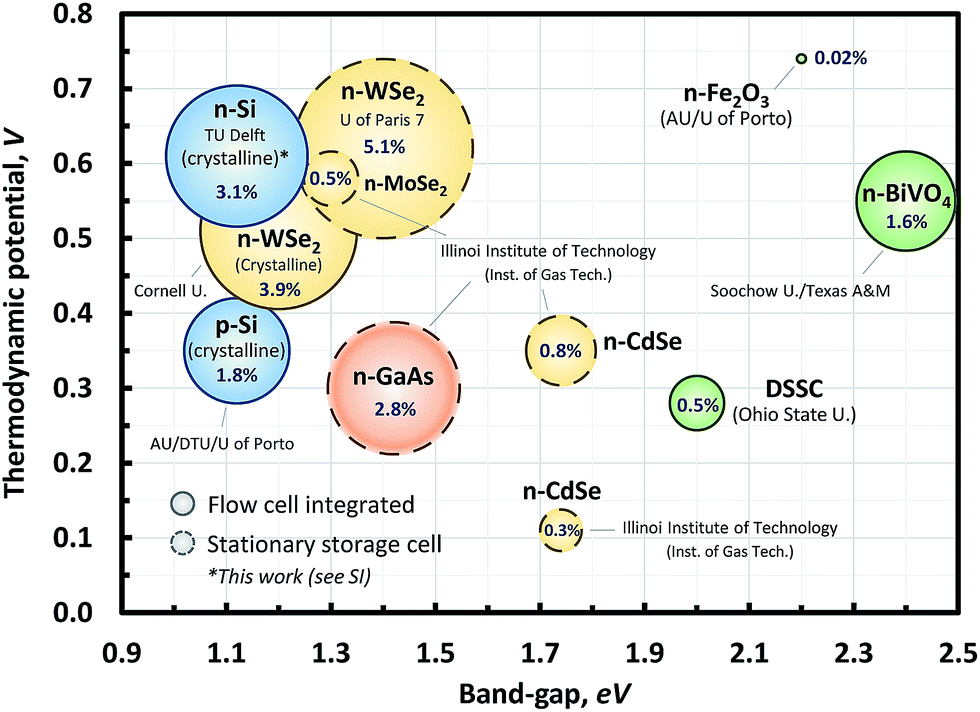 | ||
| Fig. 1 Experimental STC% of several unbiased single photo-absorber cells is shown with different combinations of the photo-absorber band-gap and thermodynamic potential (defined as the difference between the redox potential of the respective oxidation and reduction couples). For clarity, Si-based materials are colored in blue; chalcogenides in yellow; III–V in orange; and metal-oxides in green. Note that the data point with the dashed line was subtracted from the experimental results in this work (this will be addressed later in the Results and discussion section). Detailed working conditions and citation information for the references for the chart can be found in the ESI (Table S1†). | ||
For an ideal case, i.e., the Shockley–Queisser limit, one would expect a theoretical maximum efficiency of up to around 33%. However, Fig. 1 shows that the charging efficiencies are far from this maximum. There is even a precondition of sufficient solubility of the redox couples to balance the energy storage capacity, and thus there remains substantial room for improvement considering the gap between the performances of the SRFB with a single-photon-device and the state-of-the-art PV cell technology. The performance of the stationary redox cells in the bubble chart (Fig. 1) alone also highlights the experimental potential of SRFBs. In this respect, modeling the theoretical conversion efficiency for a PEC device containing SRFBs is useful as it can identify practical performance limits and aspects of material properties that need to be enhanced. A number of previous studies have addressed the theoretical performance limit for both single- and 2-photon PEC water splitting devices.32–35 However, to the best of our knowledge, no study on theoretical device performance has been done for SRFB applications.
In this work, we aim to provide a model for studying the charging performance of SRFBs by considering the inherent nature of SRFB operation, including, but not limited to, optical loss by the electrolyte and overpotential, to reveal the practical photo-charging performance limit of a single-device-based system. We emphasize that the calculations reflect experimental parameters from previous research studies and/or our own measurements (particularly, photocharging using c-Si with Fe(CN)63− and NH4Br; Fig. 1, circle with an asterisk) so that we can highlight which properties merit the most attention in further studies towards the practical development of SRFBs.
It should be noted that for any PEC or PV-assisted device, there needs to be a corresponding energy level matching between the photo-device and redox couples as described in a previous study.11 While understanding that these counter reactions is important, this work only focuses on the analysis of photo-charging performance with an assumption of appropriate energy matching. For readers interested in energy level matching and analysis on the electrochemical performance of RFBs we suggest previous studies by McCulloch et al.30 and Wedege et al.11 and technological review studies by Ye et al.36 and Park et al.23 It is also worthwhile to note that theoretical calculations are valid only if there is no positive photo-redox effect, such as modification of the redox properties owing to photo-excited states, as demonstrated in dye-sensitized PEC cells.37,38
2. Experimental
2.1. Theoretical modeling
In most instances in this work, photo-devices are assumed to possess the following properties: (I) photon absorptivity with an energy level lower than the band gap energy (Eg) is not considered (i.e., light utilization by intermediate states is zero). (II) The device has ideal contact with both the counter electrode and the conducting layer at the solid/liquid interface. (III) The current output is not limited by mass transport (e.g., flow or circulation rate of the electrolyte). (IV) Dynamics due to the state-of-charge ratio change is not considered. Under the given assumptions, the following diode equation is derived by Shockley and Queisser,39 in which the empirical ideality factor n was added later (e.g., described by Green40) to calculate the voltage output as a function of the current density: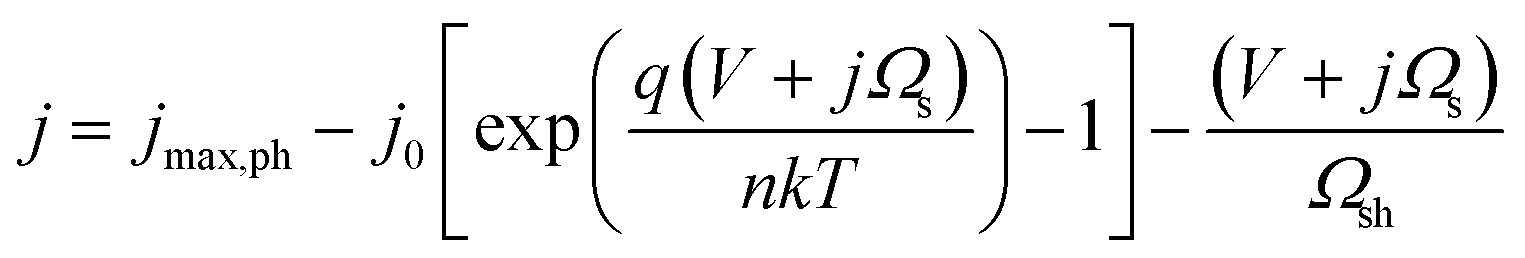 | (1) |
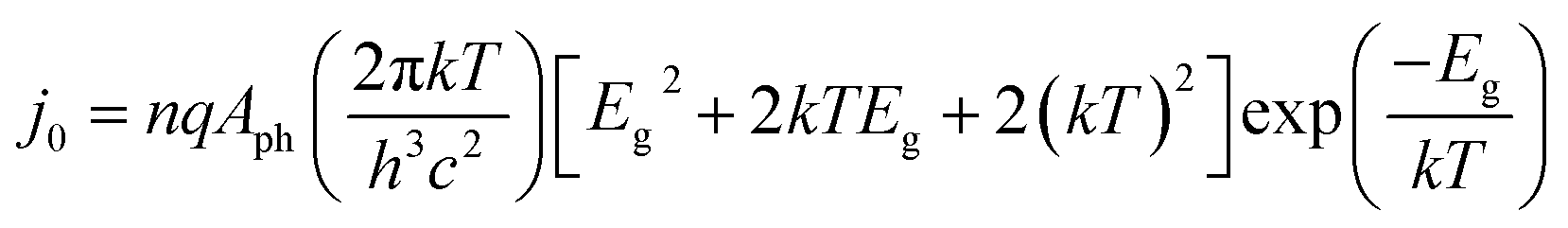 | (2) |
One important point to discuss is how jmax,ph can be defined. While conventional studies on PVs and solar-fuel use standard air mass 1.5 (i.e., AM 1.5) as an incident photon source, most SRFBs use electrolytes with distinctive colours which can lead to a significant loss in jmax,ph due to parasitic light absorption. So far, nearly all studies have employed front-side illumination despite the inevitable optical-loss by the electrolyte.10,19,20,42,43 Only a few studies have been performed using back-side illumination, which allows direct photon absorption on a dry surface.9,18,44 Döscher35 and Seger33 demonstrated a method to model solar water splitting technologies by using a detailed balance between the PEC device and optical losses by water with various overpotentials of the catalysts. However, light absorption by the electrolyte of SRFBs is incomparably high and has a strong dependence on the type of redox couples used. Unfortunately, experimentally measured absorbance data cannot be directly applied to the model, since all PEC systems have different electrolyte thicknesses and redox chemical concentrations. Hence, instead, the molar absorptivity (molar extinction coefficient, ε) data are employed for the modeling work. Simultaneously, practical effects, including reflection losses at the light-illuminated surface, are taken into account. According to the analysis by Frijnts et al.,45 the contribution of optical loss by direct reflection in the case of a flat c-Si device without an anti-reflection (ARF) layer reaches over 5.8 mA cm−2, whereas the textured device with an ARF layer showed a loss of only 3.4 mA cm−2. Therefore, losses due to both reflection and parasitic absorption are considered, by including their effects on the absorbed solar flux, and consequently on jmax,ph (detailed equations are shown in Section S2 of the ESI†).
For the back-illuminated case, total reflectance of the device is simply the reflectance spectrum of the photo-absorber (covered with any available ARF layer). In the case of front-side illumination, there are three reflection interfaces: the air–window, window–electrolyte and electrolyte–electrode interface. For each interface, the reflectance (R) is calculated using the Fresnel equation:
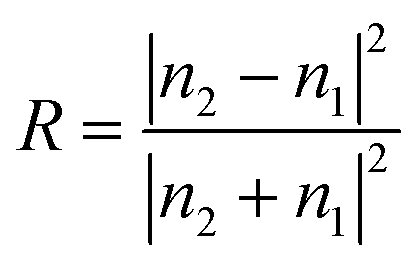 | (3) |
The effect of Ωsh is assumed to be negligible, since commercially available PV devices have Ωsh with triple digits (>2 kΩ cm−2).47,48 Typically, power losses caused by the presence of a shunt resistance are due to manufacturing defects, rather than poor device design. However, a fill factor loss due to the presence of Ωs cannot be eliminated. Unlike conventional PV systems with Ωs in the range of 3–10 Ω cm−2,45,49 PEC or PV-assisted systems have additional interfaces at the liquid side to protect devices from corrosion, and this interlayer with a conducting material causes an increased resistance. In addition, electrochemical losses due to redox reactions at the solid/liquid interfaces as well as ionic charge transport (i.e., solution resistance, Rsol) are taken into account as one of the contributing factors to the overpotential (i.e., resistance overpotential, ηr) to satisfy the following basic condition for unbiased photo-charging:
| Eph ≥ Eredox + Edev. loss + ηredox | (4) |
 | (5) |
The operating current jop can be obtained as the maximum possible current density in the current–voltage characteristic of eqn (1) at a voltage that also satisfies the condition in eqn (4). It is worth noting that the experimental jop (jop,exp) is defined by the photocurrent at zero bias voltage using a two-electrode setup. Knowing the operating current, the STC% can be calculated using the following equation:
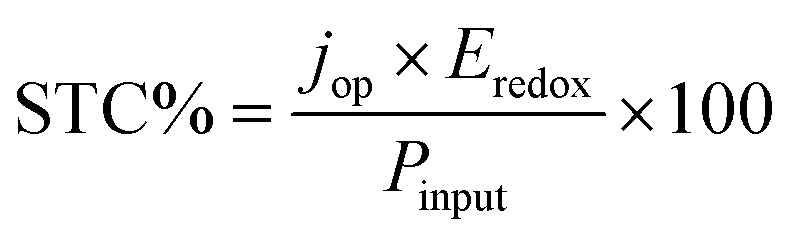 | (6) |
2.2. Sample preparation
Front-side illuminated photocathodes (pn+–Si) were prepared as described elsewhere19,50–52 and used with further optimization. In the case of the back-illuminated sample (Fig. 2a), a shallow np+-junction was formed at the surface of n-type c-Si wafers. Pt thin films were also sputtered at room temperature as a conducting layer with a Ti adhesion layer. Further experimental information can be found in the ESI (Section S3†). | ||
| Fig. 2 Schematic of solar charging compartments for the SRFB system. (a) Charging cell with a back-side illuminated device and (b) cell with a front-side illuminated device (i.e., illuminated through the window (2) and the electrolyte). The photocathode (1) in the catholyte (with redox couple A) and a polarizable counter electrode (4) in the anolyte (with redox couple B) are separated by an ion exchange membrane (3). In practical applications, an RFB stack (5) is connected in series with storage tanks and pumps (not shown) for discharging the solar charged electrolytes. Subscripts (Ox and Red) denote the oxidized form and reduced form of the redox couples. CB and VB correspond to the conduction and valence band edges of the semiconductor, respectively. Note that the illustration is not to scale. | ||
In addition to Pt, various materials, such as carbon, Ti, TiO2, and Au, have been used to provide experimental overpotentials and exchange current densities for the verification of the theoretical model. All conducting layers have been deposited using the same sputtering technique as previously mentioned at room temperature. Fabricating an SRFB requires careful selection of the conducting material since some redox potentials, such as V3+/2+, are favourable for the hydrogen evolution reaction (HER) that can lead to mechanical damage to the system.11
2.3. Characterization
The electrochemical measurements of the photoelectrodes were conducted using a three-electrode and a two-electrode configuration. Solar charging has been done under simulated AM1.5 solar irradiation (100 mW cm−2) for both back- and front-illuminated cases (Fig. 2a and b). During the measurements, 40 mL of the electrolyte was continuously circulated using a peristaltic pump at 70 mL min−1. Linear sweep voltammetry (LSV), chronovoltammetry measurements were performed with a carbon felt counter electrode. The solutions were purged with nitrogen before and during measurements. The absorbance and reflectance spectra were recorded by using a UV-vis spectrometer to estimate parasitic absorption by the electrolyte and reflection loss at the sample surface. Further experimental information can also be found in the ESI (S3†).3. Results and discussion
When varying the cell voltage (i.e., thermodynamic potential to drive the redox reaction) as well as the band-gap of the photo-absorber, a 2D contour-plot of the STC efficiency is obtained. This format will be used to assess several parameters. In Fig. 3a the ideal case is shown, in which no losses, for instance, due to parasitic light absorption, reflection or resistances, are taken into account. It is found that the theoretical maximum STC efficiency for a single-photo-absorber device is 32.4%, which concurs with literature estimations for PV solar cells.39,53 However, the SRFB system allows a wider operational range than the PEC water splitting system which has a fixed thermodynamic energy barrier for driving the redox reaction (i.e., water oxidation at 1.23 V). The top left of the plot shows no efficiency data, due to the limited photovoltage with respect to Eredox. When moving towards a higher bandgap energy, the STC% is limited by the number of incoming photons only with an energy higher than Eg. This figure indicates that there is an optimum thermodynamic potential depending on the band-gap of the absorber material, and finding this optimum could drastically enhance the solar charging performance of SRFBs. | ||
| Fig. 3 Efficiency plotted as a function of the thermodynamic potential and the bandgap of the photo-absorber. (a) The ideal case without any kinetic, resistance, or parasitic light absorption/reflection losses. The same plot in (b) and (c), but the reflection from the window and parasitic light absorption of 0.3 M ferricyanide and V3+ (with an optical path length of 1 cm) respectively are taken into account. (d) Experimental absorption spectra of several electrolytes overlapped with the photon flux of the AM1.5 spectrum (depicted by the shaded area). (e) The concentration dependence of the maximum efficiency for a 350 μm c-Si photo-absorber. The experimental LSV curve in (f) shows that in the case of a 0.4 M ferricyanide electrolyte, a front-side illuminated configuration results in a significantly lower saturation photo-current, which concurs with the modeled trends. The inset shows the LSV for a dual-electrolyte cell (i.e., 2-electrode configuration), resulting in a current density of 10.2 mA cm−2, corresponding to an estimated STC of 3.1% at a cell voltage of ∼300 mV. | ||
In Fig. 3b and c, the effect of parasitic light absorption by the electrolyte [Fe(CN)6]3− and V3+ (in 1 M NH4Cl and HCl, respectively) is shown for a concentration of 0.3 M and a path length of 1 cm. These figures mimic the working environment of a PEC compartment where the photoelectrode is directly immersed in the electrolyte. As shown in these figures, the maximum STC% decreases to 27.2% and 17.2% for [Fe(CN)6]3− and V3+ respectively. Moreover, the shape of the efficiency-landscape changes depending on the exact absorption spectrum of a given electrolyte (see also Fig. S1† for the AQS/AQSH2 and I−/I3− cases). This makes finding the optimum Eredox/Eg combination extensively more complex. In Fig. 3d, the experimental absorption spectra, based on the literature54–56 and experimental measurements, overlap with the photon flux (in current density) converted from the standard AM1.5 spectrum, showing the impact of electrolyte selection on the incoming solar spectrum. Note that this result does not take into account the individual effect of the supporting electrolyte since there is no significant loss up to 10 cm of water thickness in the case of a single-photon device.35 Overall, the reduced incoming-photon flux shifts the optimum Eredox/Eg combination towards a smaller value such that an electrolyte with a high concentration is required for balancing the discharging capacity.
Another factor used to differentiate between the efficiency of the back- and front-side illumination configurations is the possibility of introducing an antireflective treatment (e.g., AR coating). In the case of the electrode being immersed in the electrolyte, some corrosion protective layers for photoelectrodes (e.g., TiO2 with c-Si) show anti-reflective properties; however, typical AR treatments, such as MgF2 and SiO2 layers, are not applicable in this case due to their stability which is pH dependent.3 To account for this, the reflectance spectrum of c-Si with and without an AR coating is used for modeling the back-illuminated efficiency, while the (complex) refractive indices of air, glass, electrolyte and bare silicon are used to model the front-illuminated efficiencies. As shown by the dash-dot line in Fig. 3e, almost 8% efficiency loss is expected in the case of bare c-Si indicating the importance of having an AR treatment to minimize reflection losses. It is interesting to note that the back-illuminated bare c-Si case shows slightly lower charging efficiency than the front-illuminated cases in the low concentration range. According to the Fresnel equation (eqn (3)), the higher refractive index of the electrolyte with respect to air leads to reduced reflection of bare silicon for a front-illuminated configuration (also see Fig. S2 in the ESI†) with respect to back illumination, resulting in higher efficiencies in the cases where this effect is more dominant than electrolyte absorption.
The concentration dependency of the electrolyte on the STC% in Fig. 3e highlights that conventional vanadium-based redox couples with a front-illumination architecture cannot be considered as an efficient charging method for a high storage capacity system (i.e. high concentration) due to their high molar absorptivity in the high wavelength range (see the inset of Fig. 3d). On the other hand, the STC% with an iodide electrolyte is relatively less sensitive to the concentration but it showed drastic molar absorptivity in the short wavelength region (Fig. 3d). This study of the absorption spectra is particularly important for the wide band gap semiconductor-based device. For instance, GaP (2.24 eV) has a large absorption spectral overlap with the AQS electrolyte, leading to a drastic drop in Jph upon the addition of AQS into H2SO4 (Fig. S3 in the ESI†). The experimental LSVs in Fig. 3f support the modeled trends of the expected higher photo-currents (and thus efficiencies) for a back-illuminated configuration. The saturation current of the front-side illuminated device is significantly lower than that of the back-illuminated case, despite an otherwise almost identical setup (see Fig. S4 in the ESI† for detailed charging/discharging in RFB mode).
In a practical model, kinetic overpotentials should be considered, as the electron transfer between the photo-absorber and the electrolyte is never perfect. In general, an RFB presents facile kinetics, which can be several orders of magnitude faster than water oxidation that requires a high overpotential.12 In Fig. 4a and b, the effects of fixed kinetic overpotentials (ηk) of 0.2 V and 0.6 V on the efficiency-landscapes respectively are shown. First, it can be seen that the maximum STC%, as opposed to the previous electrolyte concentration effect case in Fig. 3a–c, shifts to higher Eg and lower Eredox, due to the kinetic overpotential effectively increasing the voltage needed to drive the reaction (eqn (4)).
 | ||
| Fig. 4 Efficiency plotted as a function of the thermodynamic potential and the bandgap of the photo-absorber. (a and b) The effect of 0.2 V and 0.6 V kinetic overpotential respectively. In (c) the STC% is plotted as a function of the bandgap-energy for various overpotentials. The dashed black line shows the maximum theoretical efficiency based on the AM1.5 spectrum. In (d), experimental LSV data are plotted for a single silicon photo absorber with various conducting layers, submerged in a [Fe(CN)6]3− electrolyte (in NH4Cl at pH 7), showing that the overpotential is strongly dependent on the type of conducting material. The inset depicts the LSV data for a carbon felt electrode, immersed in anolytes (NaI and TEMPO-sulfate at pH 0 and 7, respectively). | ||
Moreover, owing to the shift of the contour-plot to higher bandgap energies, the STC% decreases due to reduced absorbed solar flux (as shown more clearly in Fig. 4c). An important aspect is the selection of a conducting material at the solid/liquid interface. Conventionally, high overpotentials arise from the photoelectrode while the counter electrode shows quite fast kinetics. Experimental linear sweep voltammograms (LSVs) of n-type Si electrodes under dark conditions for the cathodic charging of Fe(CN)63− (Fig. 4d) show how the overpotential varies with the type of conducting layer. The inset shows the LSVs for a carbon felt electrode submerged in an anolyte (NaI and TEMPO-sulfate in this case) which exhibits a negligible overpotential owing to the sufficient active area of carbon felt. Unlike typical RFB systems in which the metallic electrodes or carbon is directly connected to a wire, SRFBs require integration of semiconductors with the conducting layer which may form an energy barrier with unfavourable band-bending at the semiconductor/metal junction. As shown in Fig. 4d, Pt shows outstanding kinetics; however, noble metals like Pt are not recommended for RFB applications since the formation of bubbles due to the concurrent hydrogen evolution reaction may lead to mechanical damage of the system. Addressing designing rules for efficient charge transfer at the solid/liquid interface is beyond the scope of this work.
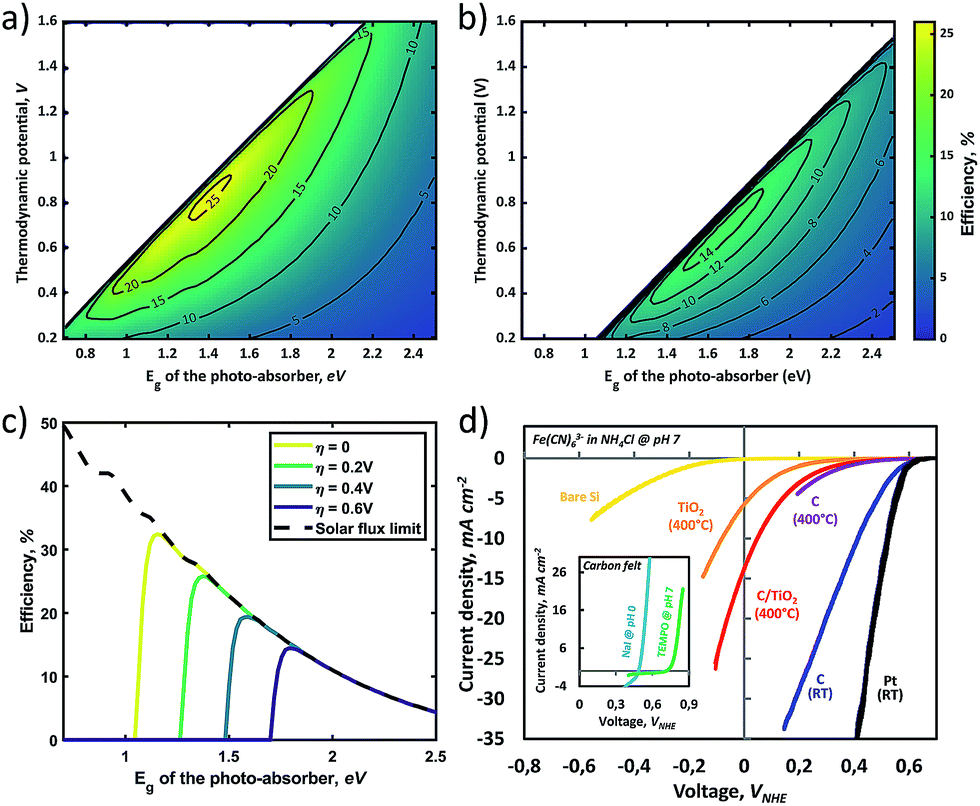
Fig. 5a and b display the effects of the overpotential and electrolyte resistance, respectively, on the photocharging efficiency landscape for the c-Si case (Eg = 1.12 eV). The dashed lines represent the efficiency curves for which surface reflection and shading by the front contact grid (i.e., a dead area of 4% of the total active area57) are taken into account. Interestingly, the efficiency loss due to the reflection and shadowing in the case of a photo-absorber with appropriate treatment (i.e., AR and optimized front grid) is limited. Fig. 5a shows the sensitivity analysis by plotting the charging efficiency against the thermodynamic potential. Naturally, it shows a similar trend to that found in Fig. 4c for overpotential variation; an increased overpotential greatly decreases the performance limit and threshold cell voltage (i.e., the potential differential between the catholyte and anolyte). As mentioned earlier, electrolyte resistance (Rsol) is one of the dominant contributors to the overpotential, and the plots in Fig. 5b were obtained from calculations with various resistance overpotentials at zero kinetic overpotential. Fig. 5b shows a relatively low sensitivity of the STC% to the electrolyte resistance variation. While a negative shift of the optimum thermodynamic potential is seen along with increasing solution resistance, the threshold potential below which achievable STC% is observed remains almost constant. The solution resistance is often a significant factor under practical working conditions where ionic transfer pathways are on the order of centimeters. Generally, the resistance of an ionic solution depends on the ionic concentration, type of ions, temperature, and geometry in which the current is carried. This is made evident in Fig. 5c, where the conductivity of the electrolytes is highly dependent on the electrolyte type and concentration. For example, for a system with a 1 M NH4Cl supporting electrolyte and an estimated conductivity of 75 mS cm−1 for the redox electrolyte, the total electrolyte resistance for a 2 cm path length (e.g., 1 cm each for catholyte and anolyte) is 11.35 Ω cm2 (see Section S5 in the ESI† for explicit calculations), implying that the impact of the electrolyte resistance should not be neglected in practical models. The plots for various electrolytes in Fig. 5c were calculated based on data found in the literature,58–61 where the measurements were conducted under precisely controlled conditions (e.g., temperature, electrode distance, purification treatment, etc.). As demonstrated in Fig. 5c, it is relatively easy to reduce the solution resistance simply via concentration control, whereas a reduction in the overpotential requires careful selection of conducting materials along with electrical and morphological design to provide sufficient conductivity and active sites over the whole surface.
 | ||
| Fig. 5 Sensitivity analysis for each type of loss. The charging efficiency is plotted as a function of the thermodynamic potential in the back-lit configuration for different kinetic overpotentials (a) and solution resistances (b), respectively. The dashed line represents the case in which surface reflection (by AR treated silicon) and contact shading are taken into account. Data found in the literature for the specific concentration are plotted versus the concentration of various electrolytes (c). In (d), a contour-plot of the efficiency landscape for an electrolyte resistance of 2.5 Ω cm2 is shown. The white circles indicate the optimum location (i.e., the optimum bandgap-thermodynamic potential combination) for electrolyte resistances of 0, 5, 10, 20 and 50 Ω cm2 respectively, showing a shift towards a higher bandgap and higher thermodynamic potential with increasing resistance. The same trend is observed in (e), where the STC efficiency is plotted versus the photo-absorber band-gap at a thermodynamic potential of 0.8 V, for various distances between the electrodes, effectively increasing the electrolyte resistance. | ||
As shown in Fig. 5d, the optimum point shifts towards lower Eg with decreasing solution resistance (white circles). In the extremely low solution resistance case, a lowered optimum point (Eg of 1.2–1.6 eV with an Ecell of ∼0.6–0.9 V) makes the use of commercial PV materials (e.g., c-Si, CIGSe, and GaAs)3,62 promising. However, the high ionic strength may potentially lead to corrosion of the materials, which will mostly likely require an additional protection layer. For instance, a solution resistance of 2.5 Ω cm−2 corresponds to highly acidic supporting electrolytes (e.g., a 3 M HCl solution). These extremely harsh conditions are not practical considering that most studies in the PEC water splitting field have been conducted in the pH range of 0–14, and still suffer from poor long term stability.3 Alternatively, minimizing the distance between the electrodes also can be an option to decrease solution resistance. Fig. 5e plots the STC efficiency versus the photo-absorber band-gap for which the distance between the electrodes decreases in magnitude, showing that a shorter electrode–electrode distance leads to a shift of the optimum band-gap towards lower values with a higher overall conversion efficiency. Again, we emphasize that this theoretical estimation has been conducted without the consideration of the possible photo-redox effect under the assumption that electrolytes are completely isolated from light due to the back-side illuminated architecture (e.g., Fig. 2a). In the front-illuminated design (e.g., Fig. 2b) case, reduction of the electrolyte thickness may increase the photo-redox effect of some specific redox couples, such as anthraquinones.37 Furthermore, the reduction of the electrode distance (i.e., channel size) may result in a notable mass transport phenomenon that can lead to a decrease in the obtainable STC% at a certain electrolyte flow rate,22,63 and this must be addressed using the Butler–Volmer model.
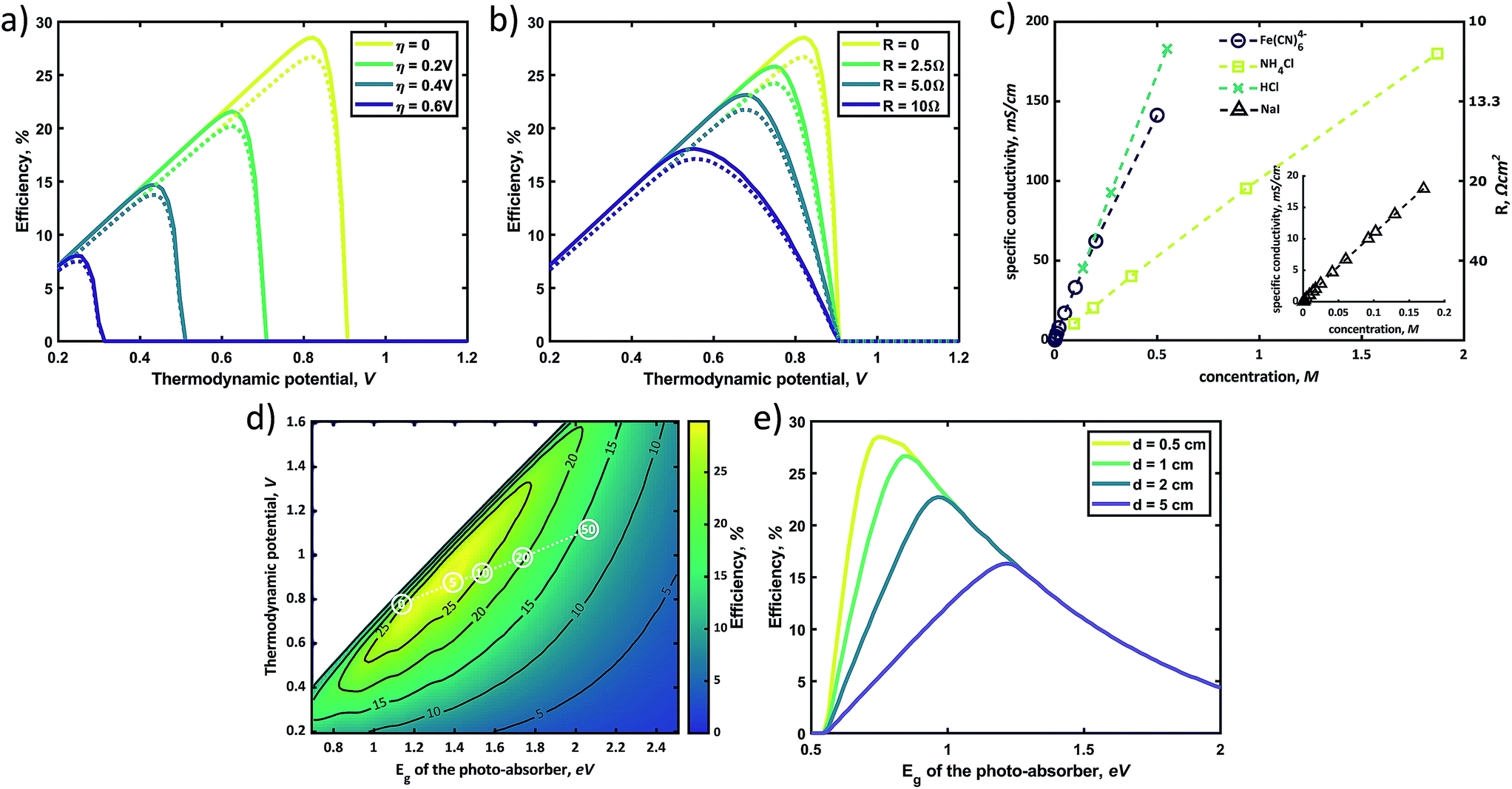
In general, the standard redox potential (E0redox) is defined as 50% state of charge (SOC), but this SOC-effect should be taken into account for optimizing an SRFB. Fig. 6 exhibits the maximum practical STC efficiency for a set of parameters: Rsol = 2.5 Ω cm2 (corresponding to a highly conductive, but useable electrolyte with an optimized path length of ∼ 1 cm), an internal series resistance of 3 Ω cm2, ηredox = 0.25 V with a constant parasitic light loss at the photo absorber of 5% and a contact shading of 4% of the active area. This is done for 10% and 90% SOC to show the effect of charging on the efficiency landscape (Fig. 6a and b, respectively). These plots highlight a shift of the entire STC shape towards a lower thermodynamic potential due to the potential polarization in accordance to the Nernst equation (eqn (5)).11
 | ||
| Fig. 6 The efficiency landscape for a realistic set of parameters: Rsol = 2.5 Ω cm2, ηk = 0.25 V with a constant parasitic light loss at the absorber of 5%, a contact shading of 4% of the active area, an internal series resistance of 3 Ω cm2 and 10% SOC (a) and the same plot with similar parameters, but with an SOC of 90% (b). Both plots assume a back-lit configuration. Several photo-absorbers are indicated in (a) to point out the possibilities of choosing an optimal photo-absorber. | ||
It is expected that an STC% of ∼16% can be obtained if the band-gap of the absorber material is within 1.6–1.8 eV and the thermodynamic cell voltage is around 0.9 V and 0.7 V for an SOC of 10% and 90%, respectively. To improve the STC, averaged over the entire charging cycle, this SOC-dependent shift should be taken into account when choosing optimal redox couples. Considering the fact that changing the redox couples during the operation is not unrealistic, these figures with SOC variance also imply that one should choose a photo-absorber material with a larger photovoltage than needed to allow operational flexibility. As discussed in a previous report, Wedege et al. demonstrated 95% SOC using a c-Si photocathode which exhibits a photovoltage exceeding the thermodynamic cell voltage by approximately 30%.19
When comparing Fig. 6 to Fig. 1, it can be seen that while practical parameters are taken into account for modeling the photo-charging performance, most experimental data are still far below the theoretical limit. It is partly because of the fact that most of these experimental studies have been demonstrated using a front-illuminated configuration showing a decrease of up to 34% in the theoretical photo-charging limit with respect to the back-illuminated configuration as shown earlier in Fig. 3. In this sense, the c-Si sample with ferricyanide/NH4Br (at pH 7) results in an STC% of around 3.1% with back-side illumination (Fig. 3f) which can be considered as a promising result. There is still, however, the possibility of degradation of bromine and ferricyanide molecules to form toxic side products, such as bromic acid and hydrogen cyanide gas, respectively,64,65 and this needs to be addressed for further development. Although the demonstrated theoretical model can be used to perform several sensitivity analyses and identify general trends, it is not yet suited to cover all specific experimental conditions in a single figure. As a remedy for this drawback, video clips showing dynamic contour plots with continuous parameter changes are presented in the ESI† to help the readers in tracking optimum combinations of thermodynamic potentials and band-gaps of the semiconductors.
4. Conclusions
In this work, the necessity of accurately matching the photo-absorber bandgap-energy with the thermodynamic potential of the respective redox reaction in an SRFB is emphasized along with supporting experimental evidence. The key advancement of the present work is the incorporation of realistic losses and assumptions based on the experimental studies in the field. The optimum solar-to-chemical efficiency is shown to shift significantly in the efficiency landscape depending on the kinetic overpotential, electrolyte resistance, state-of-charge and compartment architecture (i.e., front- or backside illumination).The model used in this work identifies these trends and shows that in addition to reducing the respective losses in an SRFB, photo-absorber/redox-couple matching should be carried out carefully for maximizing the obtainable STC efficiency for a single absorber system. A baseline single absorber SRFB system can achieve a maximum STC efficiency of ∼18%, which decreases with increasing SOC at a certain Eg and cell voltage (i.e., thermodynamic potential). This value surpasses the theoretical conversion efficiency for PEC water splitting (∼11% for the single-absorber case) owing to the flexibility of the redox potential design and relatively fast redox kinetics. As the field explores new and higher performance materials (e.g., additives for increasing the solution conductivity, non-Pt conducting materials with higher conductivity, etc.), our model indicates that higher STC efficiencies (>20%) can be achieved.
Although this model is a strong tool to identify general trends and sensitivities, the accuracy and predicted value could be increased by including certain effects, for instance, the photo-redox effect which has not been considered in this study. Furthermore, implementation of the dynamic overpotential as a function of the current density and mass-transport phenomena, which are governed by the Butler–Volmer model, can enhance the accuracy of the model further.
To aid further development of the field, we suggest here a range of recommendations that can be undertaken to make SRFB development competitive with other solar utilization systems:
• Back-side illumination is highly recommended unless there is no significant overlap between the light absorption spectra of the chosen electrolyte and photo-absorber, and no anti-reflective layer is needed.
• It is necessary that the electrolyte has a sufficiently low resistance (<2.5 Ω cm2) such that implementation of commercially available, mature, but low band-gap PV materials, such as Si and CIGSe, are possible.
• In the same manner, the overall distance between the electrodes should be minimized. For example, a reduction of 1 cm in distance corresponds to a reduction of ∼50% of the solution resistance.
• Albeit forming a porous conducting layer with an extremely high surface area at the surface of a photoelectrode is technically possible, individual evaluation of intrinsic activity of the conducting layer should be conducted in order to minimize the kinetic overpotential.
• In the case of wide band-gap materials, such as a-SiC and Fe2O3, which require redox couples with a quite high potential gap (>1.3 V) to obtain a feasible STC%, care must be taken to avoid the risk of having a competitive reaction (e.g., water splitting).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the LEaDing Fellowship grant from the European Union's Horizon 2020 research and Innovation Programme under the Marie Sklodowska-Curie grant agreement no. 707404. The authors also thank Dr Kristina Wedege and Dr D. N. Østedgaard-Munck at Aarhus University for assistance in preparing the carbon felt electrodes and redox flow cell operation, and Joost Middelkoop and Herman Schreuders at the MECS group of TU Delft for preparation of the flow cell and assistance with training for use of the vacuum deposition system.References
- Key electricity trends 2017 based on monthly data, 2018.
- P. C. K. Vesborg and B. Seger, Chem. Mater., 2016, 28, 8844–8850 CrossRef CAS.
- D. Bae, B. Seger, P. C. K. Vesborg, O. Hansen and I. Chorkendorff, Chem. Soc. Rev., 2017, 46, 1933–1954 RSC.
- I. A. Digdaya, G. W. P. Adhyaksa, B. J. Trześniewski, E. C. Garnett and W. A. Smith, Nat. Commun., 2017, 8, 15968 CrossRef CAS.
- S. Ardo, D. Fernandez Rivas, M. A. Modestino, V. Schulze Greiving, F. F. Abdi, E. Alarcon Llado, V. Artero, K. Ayers, C. Battaglia, J. P. Becker, D. Bederak, A. Berger, F. Buda, E. Chinello, B. Dam, V. Di Palma, T. Edvinsson, K. Fujii, H. Gardeniers, H. Geerlings, S. M. Hashemi, S. Haussener, F. Houle, J. Huskens, B. D. James, K. Konrad, A. Kudo, P. P. Kunturu, D. Lohse, B. Mei, E. L. Miller, G. F. Moore, J. Muller, K. L. Orchard, T. E. Rosser, F. H. Saadi, J. W. Schüttauf, B. Seger, S. W. Sheehan, W. A. Smith, J. Spurgeon, M. H. Tang, R. Van De Krol, P. C. K. Vesborg and P. Westerik, Energy Environ. Sci., 2018, 11, 2768–2783 RSC.
- H.-W. Nam, The Korean Times, 2018 Search PubMed.
- X. Liu, D. Ren, H. Hsu, X. Feng, G. L. Xu, M. Zhuang, H. Gao, L. Lu, X. Han, Z. Chu, J. Li, X. He, K. Amine and M. Ouyang, Joule, 2018, 2, 2047–2064 CrossRef CAS.
- N. S. Lewis, Science, 2016, 351, aad1920 CrossRef.
- W. Li, H.-C. Fu, L. Li, M. Cabán-Acevedo, J.-H. He and S. Jin, Angew. Chem., Int. Ed., 2016, 55, 13104–13108 CrossRef CAS.
- Q. Cheng, W. Fan, Y. He, P. Ma, S. Vanka, S. Fan, Z. Mi and D. Wang, Adv. Mater., 2017, 29, 1700312 CrossRef.
- K. Wedege, D. Bae, W. A. Smith, A. Mendes and A. Bentien, J. Phys. Chem. C, 2018, 122, 25729–25740 CrossRef CAS.
- E. H. Calderon, A. Katsaounis, R. Wüthrich, P. Mandin, G. Foti and C. Comninellis, J. Appl. Electrochem., 2009, 39, 1827–1833 CrossRef CAS.
- M. Uhrig, S. Koenig, M. R. Suriyah and T. Leibfried, Energy Procedia, 2016, 99, 35–43 CrossRef CAS.
- M. Paiss, Energy Storage System Safety, 2017 Search PubMed.
- W. Wang, Q. Luo, B. Li, X. Wei, L. Li and Z. Yang, Adv. Funct. Mater., 2013, 23, 970–986 CrossRef CAS.
- J. Wang, K. Lu, L. Ma, J. Wang, M. Dooner, S. Miao, J. Li and D. Wang, Energies, 2017, 10, 991 CrossRef.
- G. Hodes, J. Manassen and D. Cahen, Nature, 1976, 261, 403–404 CrossRef CAS.
- F. Urbain, S. Murcia-López, N. Nembhard, J. Vázquez-Galván, C. Flox, V. Smirnov, K. Welter, T. Andreu, F. Finger and J. R. J. R. Morante, J. Phys. D: Appl. Phys., 2019, 52, 044001 CrossRef.
- K. Wedege, D. Bae, E. Dražević, A. Mendes, P. C. K. Vesborg and A. Bentien, RSC Adv., 2018, 8, 6331–6340 RSC.
- S. Liao, X. Zong, B. Seger, T. Pedersen, T. Yao, C. Ding, J. Shi, J. Chen and C. Li, Nat. Commun., 2016, 7, 11474 CrossRef CAS.
- G. Kim, M. Oh and Y. Park, Sci. Rep., 2016, 1–9 Search PubMed.
- J. R. McKone, F. J. DiSalvo and H. D. Abruña, J. Mater. Chem. A, 2017, 5, 5362–5372 RSC.
- M. Park, J. Ryu, W. Wang and J. Cho, Nat. Rev. Mater., 2016, 2, 1–18 Search PubMed.
- K. Wedege, J. Azevedo, A. Khataee, A. Bentien and A. Mendes, Angew. Chem., Int. Ed., 2016, 55, 7142–7147 CrossRef CAS.
- I. M. Peters, S. Sofia, J. Mailoa and T. Buonassisi, RSC Adv., 2016, 6, 66911–66923 RSC.
- Z. J. Yu, J. V. Carpenter and Z. C. Holman, Nat. Energy, 2018, 3, 747–753 CrossRef CAS.
- B. Keita and L. Nadjo, J. Electroanal. Chem., 1984, 163, 171–188 CrossRef CAS.
- B. Keita and L. Nadjo, J. Electroanal. Chem., 1983, 151, 283–288 CrossRef CAS.
- Y. Zhou, S. Zhang, Y. Ding, L. Zhang, C. Zhang, X. Zhang, Y. Zhao and G. Yu, Adv. Mater., 2018, 30, 1–7 Search PubMed.
- W. D. McCulloch, M. Yu and Y. Wu, ACS Energy Lett., 2016, 1, 578–582 CrossRef CAS.
- P. G. P. Ang and A. F. Sammells, Faraday Discuss. Chem. Soc., 1980, 70, 207–222 RSC.
- M. F. Weber, J. Electrochem. Soc., 1984, 131, 1258 CrossRef CAS.
- B. Seger, O. Hansen and P. C. K. Vesborg, Sol. RRL, 2017, 1, e201600013 CrossRef.
- J. D. Benck, L. C. Seitz, B. A. Pinaud, T. F. Jaramillo, A. J. Forman and Z. Chen, ChemSusChem, 2014, 7, 1372–1385 CrossRef.
- H. Döscher, J. F. Geisz, T. G. Deutsch and J. a. Turner, Energy Environ. Sci., 2014, 7, 2951–2956 RSC.
- R. Ye, D. Henkensmeier, S. J. Yoon, Z. Huang, D. K. Kim, Z. Chang, S. Kim and R. Chen, J. Electrochem. Energy Convers. Storage, 2017, 15, 010801 CrossRef.
- G. O. Phillips, N. W. Worthington, J. F. McKeller and R. R. Sharpe, J. Chem. Soc. A, 1969, 767–773 RSC.
- E. A. Gibson, L. Le Pleux, J. Fortage, Y. Pellegrin, E. Blart, F. Odobel, A. Hagfeldt and G. Boschloo, Langmuir, 2012, 28, 6485–6493 CrossRef CAS.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- M. A. Green, Third Generation Photovoltaics – Advanced Solar Energy Conversion, Springer, Heidelberg, 2008 Search PubMed.
- P. M. Cuce and E. Cuce, Int. J. Low-Carbon Technol., 2012, 7, 159–165 CrossRef CAS.
- C. Flox, S. Murcia-López, N. M. Carretero, C. Ros, J. R. Morante and T. Andreu, ChemSusChem, 2018, 11, 125–129 CrossRef CAS.
- K. Wedege, J. Azevedo, A. Khataee, A. Bentien and A. Mendes, Angew. Chem., Int. Ed., 2016, 55, 7142–7147 CrossRef CAS.
- D. Liu, Z. Wei, C. J. Hsu, Y. Shen and F. Liu, Electrochim. Acta, 2014, 136, 435–441 CrossRef CAS.
- T. Frijnts, S. Kühnapfel, S. Ring, O. Gabriel, S. Calnan, J. Haschke, B. Stannowski, B. Rech and R. Schlatmann, Sol. Energy Mater. Sol. Cells, 2015, 143, 457–466 CrossRef CAS.
- M. A. Green, Sol. Energy Mater. Sol. Cells, 2008, 92, 1305–1310 CrossRef CAS.
- S. Correiaa, J. Lossena, M. Bährb and E. S. S. E. Ag, Sol. Cells, 2006, 2, 1–4 Search PubMed.
- L. Ma, L. Xu, K. Zhang, W. Wu and Z. Ma, 2011 Int. Conf. Electr. Control Eng. ICECE 2011 – Proc., 2011, pp. 2711–2714 Search PubMed.
- D. Bae, J. Gho, M. Shin and S. Kwon, Thin Solid Films, 2013, 535, 162–165 CrossRef CAS.
- D. Bae, T. Pedersen, B. Seger, M. Malizia, A. Kuznetsov, O. Hansen, I. Chorkendorff and P. C. K. Vesborg, Energy Environ. Sci., 2015, 8, 650–660 RSC.
- D. Bae, T. Pedersen, B. Seger, B. Iandolo, O. Hansen, P. C. K. Vesborg and I. Chorkendorff, Catal. Today, 2017, 290, 59–64 CrossRef CAS.
- D. Bae, B. Seger, O. Hansen, P. C. K. Vesborg and I. Chorkendorff, ChemElectroChem, 2019, 6, 106–109 CrossRef CAS.
- A. Polman, M. Knight, E. C. Garnett, B. Ehrler and W. C. Sinke, Science, 2016, 352, aad4424 CrossRef.
- N. H. Choi, S. Kwon and H. Kim, J. Electrochem. Soc., 2013, 160, A973–A979 CrossRef CAS.
- X. T. Yang, X. S. Chai, Q. X. Hou, J. Y. Zhu and L. G. Danielsson, Anal. Chim. Acta, 2002, 474, 69–76 CrossRef CAS.
- J. M. Gardner, M. Abrahamsson, B. H. Farnum and G. J. Meyer, J. Am. Chem. Soc., 2009, 131, 16206–16214 CrossRef CAS.
- M. F. Stuckings and A. W. Blakers, Sol. Energy Mater. Sol. Cells, 1999, 59, 233–242 CrossRef CAS.
- P. A. Lasselle and J. G. Aston, J. Am. Chem. Soc., 1933, 55, 3067–3071 CrossRef CAS.
- C. R. C. Handbook, C. R. C. Press, B. Raton, A. Solutions and B. Fluids, Handb. Chem. Physics, CRC Press, 95th edn, 1989, p. 178 Search PubMed.
- G. Jones and F. C. Jelen, J. Am. Chem. Soc., 1936, 58, 2561–2565 CrossRef CAS.
- W. M. Haynes, CRC Handbook of Chemistry and Physics, 95th edn, 2014–2015, CRC Press, 95th edn, 2014 Search PubMed.
- M. G. Mali, H. Yoon, B. N. Joshi, H. Park, S. S. Al-Deyab, D. C. Lim, S. Ahn, C. Nervi and S. S. Yoon, ACS Appl. Mater. Interfaces, 2015, 7, 21619–21625 CrossRef CAS.
- T. Sawant and J. McKone, ChemRxiv, DOI:10.26434/chemrxiv.6203297.v1.
- S. Mubeen, J. Lee, N. Singh, M. Moskovits and E. W. McFarland, Energy Environ. Sci., 2013, 6, 1633 RSC.
- O. Scialdone, C. Guarisco, S. Grispo, A. D. Angelo and A. Galia, J. Electroanal. Chem., 2012, 681, 66–75 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9se00333a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |