
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMaking an ultralow platinum content bimetallic catalyst on carbon fibres for electro-oxidation of ammonia in wastewater†
A. M.
Pourrahimi
 *ad,
R. L.
Andersson
a,
K.
Tjus
b,
V.
Ström
c,
A.
Björk
*b and
R. T.
Olsson
*a
*ad,
R. L.
Andersson
a,
K.
Tjus
b,
V.
Ström
c,
A.
Björk
*b and
R. T.
Olsson
*a
aDepartment of Fibre and Polymer Technology, School of Chemical Science and Engineering, KTH Royal Institute of Technology, Stockholm, Sweden. E-mail: ampo@kth.se; rols@kth.se
bIVL Swedish Environmental Research Institute, Stockholm, Sweden. E-mail: anders.bjork@ivl.se
cKTH Royal Institute of Technology, School of Industrial Engineering and Management, Material Science and Engineering, SE-100 44 Stockholm, Sweden
dDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, 41296 Göteborg, Sweden
First published on 20th June 2019
Abstract
Electrocatalysis of wastewater containing ammonia is a promising alternative to chemical and biological water purification for several reasons, one being that energy-rich hydrogen gas is generated as a by-product while the reaction can be strictly controlled to meet demands. An objective has been to reduce the loading of expensive platinum (Pt) in the catalyst electrodes, and to reduce the poisoning of the metal surface during the electrolysis. Herein, the co-deposition of a copper–platinum (Cu–Pt) bimetallic alloy onto carbon filaments, stripped from their polymeric coating, is shown to give an electrocatalytic performance superior to that of pure Pt at a content of less than 3 wt% Pt. The key to the enhanced performance was to take advantage of micrometer-sized carbon filaments to distribute a very large bimetallic alloy surface uniformly over the filaments. The Cu–Pt-alloy-coated filaments also suffer less electrode poisoning than pure Pt, and are bonded more strongly to the carbon fibre due to better mechanical interlocking between the bimetallic alloy and the carbon filaments. High-resolution electron microscopy studies combined with a tuned electro-deposition process made it possible to tailor the catalyst micro/nano morphology to reach a uniform coverage, surrounding the entire carbon filaments. The results are promising steps towards large-scale wastewater treatment, combined with clean energy production from regenerated hydrogen.
1. Introduction
Ammonia enters our water resources via municipal effluent discharges and contributes to the eutrophication of rivers, lakes and coastal waters. Aquatic organisms cannot excrete high concentration of ammonia from wastewater leading to a toxic buildup in internal tissues, and ultimately death, i.e. contrary to other forms of nitrogen.1 The toxicity of ammonia increases with increasing pH and temperature.2 According to the Swedish Environmental Protection Agency (EPA), the highest concentration in effluents should be 10 ppm, whereas an acute criterion of 17 ppm total ammonia nitrogen (TAN) at pH 7 and 20 °C has been recommended by EPA in the United States.1,3 Ammonia removal is, therefore, a crucial process in large wastewater treatment plants (WWTPs), and methods to remove it from wastewater include; nitrification,4–7 ion-exchange8,9 and electrolysis.10–13 The most common method of removing ammonia from wastewaters is biological nitrification and denitrification, which removes 50–75% of the nitrogen species.3 A major challenge is the high cost of the ammonia oxidation, the long reaction times and the specific adjustment of the environmental conditions for bacteria growth required, i.e. pH and temperature.5 The nitrifying bacteria growth is sometimes inhibited by organic pollutants, and ion exchange can be used as an alternative method for removal of the ammonium ion.9 The estimated annual cost of these processes is ca. two billion US dollars worldwide, considering only the most concentrated water streams (500–2000 g m−3) of the 22 million tons of ammonia entering our wastewater treatment plants each year. The electrochemical decomposition of ammonia in wastewater plants would, therefore, be attractive since it could simultaneously be used to produce hydrogen using less thermodynamic energy than water electrolysis.10,12–15 The decomposition of ammonia in an alkaline electrolyte involves the oxidation of the ammonia to nitrogen and the reduction of water to hydrogen, according to the reactions:10,13(Anode)
| 2NH3 + 6OH− → N2(g) + 6H2O + 6e−, E = −0.77 V/SHE | (1) |
| 4OH− → O2(g) + 4H2O + 4e−, E = +0.40 V/SHE | (2) |
(Cathode)
| 2H2O(l) + 2e− → H2(g) + 2OH−, E = −0.83 V/SHE | (3) |
The oxidation of ammonia in an alkaline medium to generate hydrogen needs a cell potential of 0.06 V (reaction 1 and 3), while the electrolysis of water in the absence of ammonia needs a higher cell potential according to reactions 2 and 3; i.e. 1.23 V. The electrolysis of ammonia has the additional advantages of low operating temperatures, no hazardous emissions, and responsiveness to different concentrations of ammonia in the wastewater, while making use of hydrogen stored in the ammonia.12,13
So far, the noble metal platinum (Pt) has been used as the main component of electrodes for ammonia electro-oxidation and water splitting due to its high electrocatalytic activity,12,16–18 but the amount of Pt required to achieve a reasonable performance has restricted its use in large-scale applications (typically 0.05–34 mg cm−2).12 A large over-potential for ammonia electro-oxidation is also required, which leads to a loss in electrode efficiency.14 The challenge here is to develop electrodes at a reasonable cost, which show a minimum ohmic loss and long-term stability, while ensuring sufficient electro-catalytic activity.12 Two ways to solve this problem have been proposed; (i) using a high surface-to-volume support substrate that reduces the catalytic metal to a minimal amount of Pt while maintaining an extensive reactive surface;13,19,20 or (ii) alloying Pt with cheaper transition metals, e.g. copper (Cu).21 Pt-free or Pt-alloyed electrodes based on iridium, ruthenium, rhodium, and palladium metals have also been studied.12,22–30 These electrodes may perform better than pure Pt electrodes in the electro-oxidation of ammonia, but the preparation costs are even greater. A combination of the two approaches, using small amounts of expensive Pt while alloying more inexpensive compounds on high surface-to-volume support substrates has received less attention.31,32 This is partly due to the bottleneck of finding a useful conductive substrate ‘templates’ that can reliably hold the catalytic metal structures, which are in turn required to be on the micro/nanoscale to optimize its surface area to mass ratio.33
In this work, nano-structured catalytic Pt and bimetallic Cu–Pt coatings electroplated on carbon fibre filaments have been studied for inexpensive catalysis and electro-oxidation of ammonia in wastewater. The carbon fibres have a high specific-surface-area (ca. 0.4 m2 g−1),34 high stiffness (≈200 GPa), and higher mechanical strength (≈3.5 GPa normalized to mass) compared to the metallic high surface-to-volume substrate materials used in industrial electrolysis.35 These properties combined with low thermal expansion and high chemical resistance make the carbon fibres interesting as substrates for carrying electrocatalysts in both alkaline and acidic environments. The catalytic metals can be deposited on to carbon structures by wet precipitation using reducing agents,36,37 by deposition from the gas phase,38,39 and by electrochemical deposition (electroplating).13,40–42 The electroplating technique was chosen because of its inexpensive and versatile water chemistry, which show a potential to be up-scaled for industrial applications, as well as its unique ability to allow coating of complex surfaces. It is demonstrated that controlling the electrodeposition parameters (i.e. applied current, voltage and time) and surface chemistry of the carbon fibres, it is possible to grow morphologies with a high specific catalytic activity. The Pt electrocatalyst gave an electrochemically active surface area (ECSA) of 21 m2 g−1 for hydrogen generation, and a peak current density of ca. 6 mA mgPt−1 during the electro-oxidation of ammonia, although the filament electrodes showed poor mechanical stability. The conditions for Pt deposition were however further used to develop a novel bimetallic Cu–Pt alloy with an extremely low content of Pt (3 wt%), which had almost the same level of catalytic activity as the pure Pt system. In addition, the poisoning effect (the blockage of active surfaces by nitrogen species) was reduced due to the presence of a large amount of copper in the alloy. The presented design reduced the electrode cost dramatically, which allow for upscaling of active catalytic surfaces to be explored in a variety of applications.
2. Experimental section
2.1. Materials
Copper(II) sulphate pentahydrate (CuSO4·5H2O, ACS reagent, ≥99%, AnalaR NORMAPUR), sulfuric acid (H2SO4, ACS reagent, ≥95%, Sigma-Aldrich), hydrochloric acid (HCl, ACS reagent, 37% w/w, 12.2 M, Merck millipore), nitric acid (HNO3, analysis EMSURE® ISO, 65% w/w, 14.6 M, EMD millipore), platinum wire (Pt, 0.5 mm diameter, ≥99.9%, Metalor Technologies, Borås Sweden), potassium hydroxide (KOH, ACS reagent, ≥85%, Sigma-Aldrich), and ammonium hydroxide solution (ACS reagent, 28.0–30.0% NH3 basis, Sigma-Aldrich) were used as received. High resistivity MilliQ water, “ultrapure” water of “Type 1”, according to ISO 3696 and ASTM D1193-91 (18.2 Mohm cm at 25 °C) was used in all the reactions. Carbon fibres, Pyrofil TR30S 3K (200 mg m−1, density 1790 kg m−3, 3000 filaments per bundle, average filament diameter 7 μm) were bought from Easy Composites Ltd, UK. The surface area of the filaments inside the carbon fibres was estimated to 0.33 m2 g−1, but due to the irregular curvature it was experimentally determined by BET (Brunauer–Emmett–Teller) to be ca. 0.4 m2 g−1.342.2. Synthesis of chloroplatinic acid (H2PtCl6·6H2O)
Chloroplatinic acid was prepared by dissolving a Pt wire (5 g) in boiling aqua regia (50 ml), consisting of concentrated hydrochloric acid (37%) and nitric acid (65%) at a volume ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (HCl:HNO3), in a 250 ml glass container equipped with a thermometer, a magnetic stirrer, and a reflux condenser. The gases generated at the exit of the condenser (Cl2, NOCl, HCl, and NOx) were scrubbed into a cold-water container. The reactor was maintained at a temperature of 80 °C. During the dissolution time (12 h), it was necessary to add HCl to compensate for its depletion and evaporation. After dissolution, the acidic mixture was allowed to evaporate until the volume reached 10 ml and 30 ml of HCl was then added. This step was done twice to decompose and remove any remaining nitric acid, evident by the presence of brownish vapour. To avoid decomposition of the chloroplatinic acid into PtCl4, the last 10 ml of the Pt salt solution was allowed to crystallize and further dried and stored under vacuum in a desiccator.
:1 (HCl:HNO3), in a 250 ml glass container equipped with a thermometer, a magnetic stirrer, and a reflux condenser. The gases generated at the exit of the condenser (Cl2, NOCl, HCl, and NOx) were scrubbed into a cold-water container. The reactor was maintained at a temperature of 80 °C. During the dissolution time (12 h), it was necessary to add HCl to compensate for its depletion and evaporation. After dissolution, the acidic mixture was allowed to evaporate until the volume reached 10 ml and 30 ml of HCl was then added. This step was done twice to decompose and remove any remaining nitric acid, evident by the presence of brownish vapour. To avoid decomposition of the chloroplatinic acid into PtCl4, the last 10 ml of the Pt salt solution was allowed to crystallize and further dried and stored under vacuum in a desiccator.
2.3. Removal of surface polymer sizing from carbon fibres
Before the electrodeposition, the protective layer of polymer sizing applied as a non-conductive surface coating in the production of the carbon fibres was removed by a shock heat-treatment up to ca. 750–850 °C for 10 s. This was performed via resistive heating of the filament bundles by applying 130 Vrms (50 Hz) per meter of fibre length, with a variable autotransformer (METREL HSN0203, power: 1.82 kVA), Fig. 1 (top). The current was 7 A per fibre bundle, each consisting of 3000 filaments with a diameter of 7 μm resulting in a current density of 61 A mm−2. | ||
| Fig. 1 (top) The schematic experimental overview for removal of non-conductive polymer sizing and activation of the ca. 3000 filaments in one carbon fibre, followed by electrodeposition of Cu, Pt and Cu–Pt on carbon filaments; (below) the SEM images of (a) a pristine single filament of carbon fibre, and (b–d) electroplated copper on carbon filaments at different magnifications (from nano- to micro-structures). A 10 cent coin is shown to indicate the approx. width of a carbon fibre containing ca. 3000 filaments. | ||
2.4. Electroplating of metals (Cu and Pt) onto carbon fibre
The electrochemical deposition of metals onto the carbon filaments was carried out at room temperature using a Keithley 2450 Source Meter in a typical three-electrode setup. A 250 ml cylindrical beaker with an inner diameter of 60 mm and a height of 88 mm was used, in combination with an Ag/AgCl (RE-1S reference electrode, length: 52 mm, OD: 4.5 mm, electrolyte 3 M NaCl) as a reference electrode, Fig. 1 (top). The working electrode was 50 mm of carbon fibre consisting of the ca. 3000 carbon filaments placed vertically in the middle of the beaker. The counter electrode was 1 m of Pt wire (0.5 mm), spirally mounted on the inner surface of the beaker. The deposition process was carried out and optimized at room temperature by applying either a constant voltage (within a range +0.2 to −1 V) or a constant current (−5 to −100 mA). The concentrations of the metal-salt solutions were varied between 2 to 10 mM for Pt and Cu, respectively, and the pH was adjusted with sulfuric acid. The electroplated carbon filaments were washed with deionized water and dried at 80 °C overnight. The electroplating of the metals on the carbon filaments was assessed by cyclic voltammetry (scanning rate 20 mV s−1 within the range from +1 to −1 V) in the similar electrochemical set-up.2.5. Characterization
A field emission scanning electron microscope (SEM; Hitachi S-4800) connected to an energy dispersive spectrometer (EDS) (Oxford Instruments, UK) was used to assess the morphology and elemental composition of the coatings.X-ray diffractograms of the powder samples were recorded at room temperature using a PANalytical X'pert Pro MPD X-ray diffraction (XRD) diffractometer equipped with a Cu-Kα source (wavelength = 1.54178 Å), using a 2θ step size of 0.017° at a generator voltage of 45 kV and a current of 40 mA. The crystal size (D) and crystal interplanar distance (d) respectively were obtained from the Scherrer and Bragg equations:
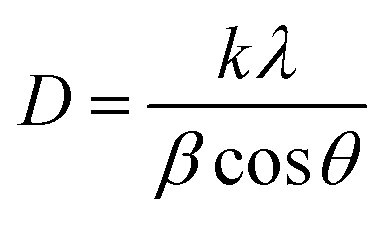 | (4) |
 | (5) |
X-ray photoelectron spectroscopy (XPS) spectra were collected with a Kratos Axis Ultra DLD electron spectrometer using a monochromatic Al Kα source operating at 150 W. An analyzer pass energy of 160 eV was used to acquire wide spectra and a pass energy of 20 eV was used for individual photoelectron lines. Since the carbon fibre samples were relatively large and heterogeneous in macrostructures and conductivity, the surface potential was stabilized using a spectrometer charge neutralization system.43 The spectra were processed with the Kratos software.
2.6. Electrochemical measurements on ammonia solutions
The electrochemical measurements were performed with the electroplated carbon fibre filaments as working electrodes in an electrolyte ammonia solution (0.1 M NH3 + 1 M KOH) at room temperature in a typical three-electrode setup (250 ml cylindrical beaker, 60 mm inner diameter, 88 mm height), using a Keithley 2450 Source Meter. The Pt wire (1.5 g) was used as the counter-electrode. Both working and counter electrodes were immersed in the same electrolyte solution, and separated by open plastic cylinders. In this way, the gas bubbles produced at the electrodes were not accessible to each another. The reference electrode was of standard Hg/HgO type (KNO3 1 M inner electrolyte solution, XR440, Radiometer). The cyclic voltammetry measurements were performed in the presence and absence of ammonia at scanning rates of −2 to −200 mV s−1 within the range of −1 to 0.4 V.3. Results and discussion
3.1. Carbon fibre treatment and electrodeposition onto carbon filaments
Fig. 1 (top) shows a general scheme for the carbon fibre electrode preparation, and three separately coated carbon fibres electrodeposited with Cu–Pt, Pt, and with Cu alone. The pristine carbon fibres contained ca. 3000 filaments with an average diameter of 7 μm. These bundles of filaments were coated as hanging entities in the center of the electrodeposition cell (Fig. 1). Scanning electron micrographs of the carbon fibres prior to and after electrodeposition of pure Cu are shown in Fig. 1a–d. Before electroplating, an important pre-treatment step had to be carried out in order to activate the fibre surfaces, since commercial carbon fibres have a polymer coating (sizing), usually of epoxy resin on their surfaces. This is essential for the bundling and handling of the fibres during manufacture, but it also acts as an electrical insulator and chemical passivator.44 The surface conductivity is therefore considerably lower than that in the out-of-plane and in-plane graphite directions within the filaments (transverse and along the filaments), limiting their application in electrochemistry unless the sizing is effectively removed.45 The de-sizing of fibres had previously been carried out by thermal or chemical treatment.46 In addition to the physical removal of the sizing, however, these treatments were found to damage either the bulk or the surface of the carbon fibres and lowered the filament conductivity, possibly due to changes in the fibre microstructure. In contrast, an electrical shock heat-treatment by application of 61 A mm−2 current resulted in a power dissipation of ca. 0.3 W m−1 per carbon fibre filament, causing a quick decomposition and evaporation of the polymer sizing products at an estimated temperature of 750–850 °C. The conductivity along the filaments after removal of the polymer coating was 1.18 × 10−5 ohm m. For comparison, a perfect single crystal of graphite has been reported to show 1 × 10−6 ohm m parallel to, and 1 × 10−2 ohm m perpendicular to the basal plane.47 The high electrical conductivity thus appeared to stem from an undistorted dominant graphite crystal orientation along the filaments. The electrical shock treatment also opened up the carbon fibre filament bundles into separate filaments with the potential to be assessed in the electroplating reactions, demonstrated in Fig. 1. The deposited copper or platinum metal could be detected by their red-brown and grey-white colour, while the electroplated Cu–Pt alloy on the carbon fibre showed an intermediate colour (Fig. 1). Fig. 1b–d shows the marked changes in morphology of the copper-electroplated carbon filaments, used in the initial screening of the most useful electroplating condition. Lower magnifications revealed the ability of the copper to uniformly cover the filaments with an average thickness of 1–2 μm (Fig. 1c and d) when an excess of 10 mM metal solution was used during 2 hours of electroplating. The copper coverage consisted of polycrystalline and multi-faceted copper grains with sizes ranging from nano- to micrometer.Fig. 2a shows the multi-cyclic voltammograms measured in an aqueous CuSO4 solution (10 mM), which were scanned during deposition and dissolution onto the carbon filaments. The pH was adjusted to 1 and the scanning potential range was set from +1 to −1 V to ensure complete deposition and dissolution of copper metal. During the first cathodic scan, the copper deposition peak showed an onset potential of ca. −50 mV and a peak at ca. −300 mV vs. Ag/AgCl due to the reduction of copper ions. In the reversal sweeps, the cathodic current (absolute value) gradually decreased until it crossed 0.0 A at ca. −50 mV where the anodic reaction started due to the dissolution of the deposited copper. The corresponding anodic peak was observed at ca. +500 mV, and the current past the peak was not 0.0 A at the vertex +1 V, indicating that the oxidative dissolution of metallic copper at the carbon filament surface was not complete.48 Residual metallic copper on the carbon fibre surface could thus act as nuclei for further electrodeposition via layer-by-layer growth, which was indicated by an increase in the cathodic and anodic current peaks, and a shift in the potential to higher voltages during the subsequent cycles. For comparison, multi-cyclic voltammetry was carried out under the same conditions for electrodeposition of the copper onto a flat graphite substrate, Fig. S2a.† The cyclic voltammograms for the deposition and dissolution of copper on the graphite surface showed a more symmetrical pattern than the carbon filament substrate, indicating typical diffusion growth characteristics.40 The cathodic and anodic peaks for the graphite substrate were observed at lower values of respectively −220 mV and +185 mV than for the carbon filament substrates (cf.Fig. 2a and S2a†). In contrast to the carbon filament case, the subsequent voltammograms were almost superimposed on the first cycle in the case of the flat graphite substrate. It was suggested that these differences were due to different ion diffusion mechanisms onto substrates with the different surface areas and different curvatures40 and/or crystallographic substrate–metal misfits.48 It was therefore concluded that the high specific surface area and curvature of the carbon filament substrates affected and reduced the local ion diffusion, and the rates of electrodeposition and dissolution, the explanation being that the deposition coverage during the first cycle was incomplete, so that increased deposition could occur during the subsequent cycles due to the already nucleated metal species. Another possibility is that the carbon filaments were slightly rough and affected the layer-by-layer electrodeposition mechanism.
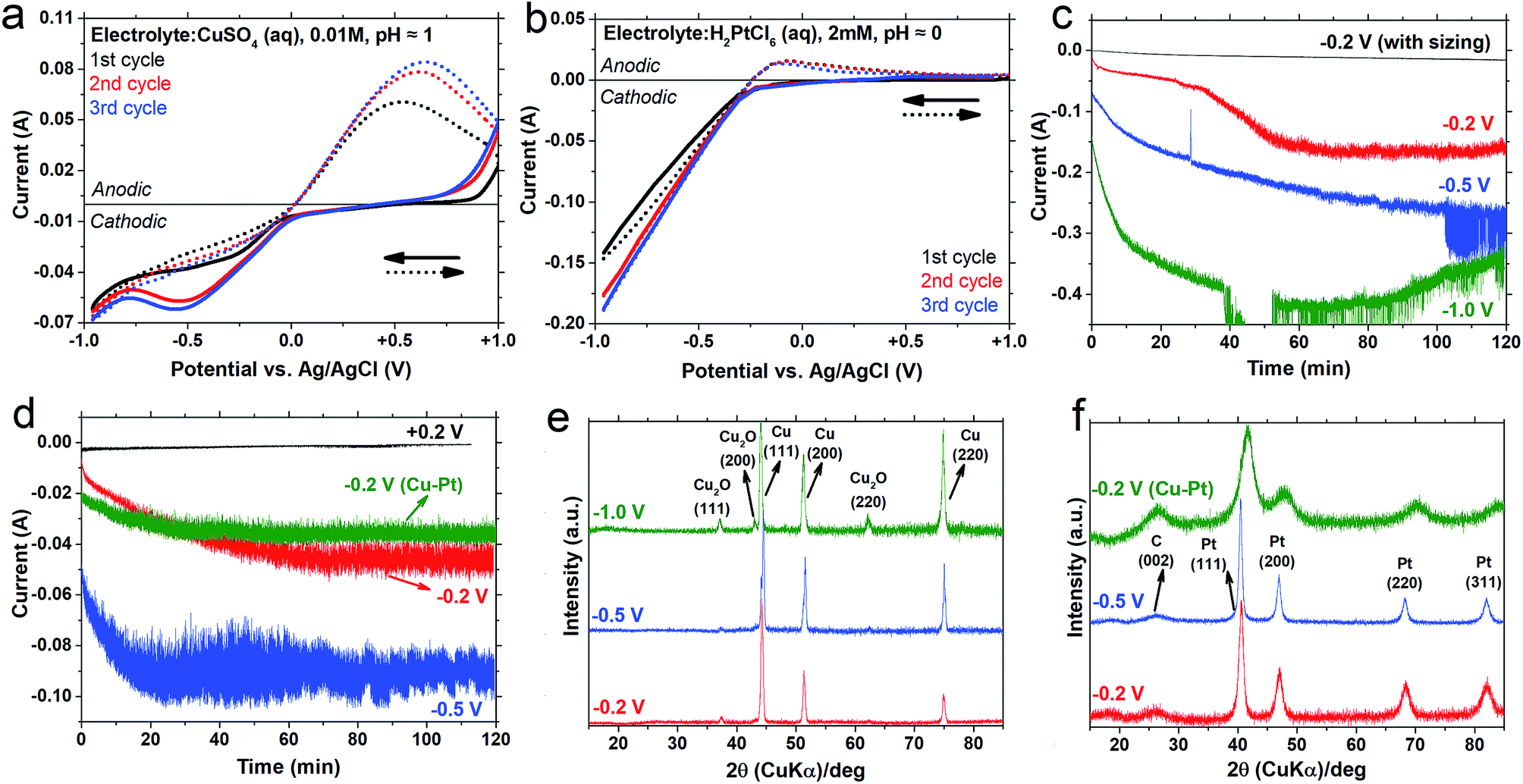 | ||
| Fig. 2 Electrochemical measurements of the deposition of copper, platinum and their alloy onto 50 mm carbon fibres filaments, together with the electroplated crystal structure; cyclic voltammogram scans without a polymer coating for Cu and Pt, with a sweep rate of 20 mV s−1 at room temperature (a and b); cathodic current–time profile during 2 h potentiostatic deposition of Cu and Pt (c and d), together with X-ray diffractograms of carbon fibres after 2 h potentiostatic deposition of Cu and Pt at different voltages vs. Ag/AgCl (e and f). The combined Cu–Pt deposition was made at −0.2 V from a solution of 0.01 M CuSO4 and 2 mM H2PtCl6, adjusted to pH ≈ 0, and marked as (Cu–Pt) in (d and f). | ||
Fig. 2b shows the multi-cyclic voltammograms for the electrodeposition of the aqueous chloroplatinic acid solution with the pH adjusted to about 0. A lower pH and a lower concentration were chosen for the platinum deposition due to its noble metal character. In the first negative sweep, the reduction of Pt ions on the carbon filament surfaces started at ca. −200 mV and the cathodic current increased linearly with the applied potential until vertex −1 V, but atomic hydrogen re-deposition49 occurred together with the Pt deposition. In the reversal sweep, the cathodic current decreased with the same slope to 0.0 A. The anodic peak at ca. −100 mV and its shoulder were thus related respectively to the ionisation of hydrogen adsorbed on the deposited Pt and to the slight formation of surface Pt-oxide species (PtO, PtO2, Pt(OH)2).49 In subsequent cycles, the cathodic current increased more sharply because the platinum deposited in the first cycle was not dissolved during the anodic sweep. In the case of copper, only small remains of Cu acted as a substrate for further deposition in subsequent cycles.
Fig. 2c and d show the current–time profile during the potentiostatic (vs. Ag/AgCl) reduction of copper and platinum ions, respectively, as discussed in Fig. 2a and b. In order to eliminate the effect of ion diffusion, the electrolyte solutions were stirred during the deposition,50 and the cyclic voltammetry under stirring is shown in Fig. S2b and c.† According to Faraday's law, the weight of plating deposited on the cathode is a linear function of both time and current. In a typical carbon filament electroplating, however, the cathodic current for both electrolyte systems increased sharply during the first 20 min, which is indicative of a fast nucleation and growth; whereafter the current increased only gradually. The cathodic current for the combined aqueous chloroplatinic acid and copper sulphate solution at pH ≈ 0 was more monotonous than that for the chloroplatinic acid (see Cu–Pt in Fig. 2d), which was indicative of a more controlled nucleation and growth for the electroplating of the copper–platinum binary system. Fig. S1† shows the carbon filament coatings corresponding to the different deposition currents, and Fig. 2c shows that the carbon fibres with the intact polymer sizing showed a significantly lower cathodic current than the carbon fibre with removed polymer sizing, i.e. during deposition with the same applied potential of −0.2 V, and almost no Cu coverage occurred. In fact, the high-temperature removal of insulating polymer sizing not only extensively increased the out-of-plane conductivity of the carbon fibres, but was vital for the filaments to function as a deposition substrate. Reducing the thermal treatment only to an extent where the filaments were only partially glowing at lower temperature negatively affected the ability of the fibres to become entirely embedded in the catalytic coatings.
Fig. 2e and f show the X-ray diffractograms of the electrodeposited carbon fibre samples after 2 h potentiostatic deposition in aqueous solutions of respectively copper sulphate and chloroplatinic acid (with conditions stated in Fig. 2a and b). Both Cu and Pt electrodeposited on carbon fibres showed clear diffraction peaks corresponding to the cubic crystal structure lattice planes with a corresponding Miller index of (hkl). The electrodeposited samples from the Cu2+ electrolyte solution showed mainly a metal phase together with a small amount of copper(I) oxide (Fig. 2e). According to the Scherrer equation (eqn (4)), based on the peak at 44° corresponding to the (111) plane, the sizes of the copper crystals were 18.2, 26.2 and 16.9 nm for applied potentials of −0.2, −0.5 and −1.0 V, respectively. The oxide phase that formed on the surface of copper crystals in the acidic environment,51 was thus indicative of that the smaller copper crystals with a larger specific surface area were more prone to oxidize at the higher potential (−1.0 V). The electrodeposited samples from the chloroplatinic acid solution showed a pure platinum metal phase within the 3 nm detection limit (Fig. 2f).52,53 The X-ray diffractograms for samples containing Pt also showed a peak at 26° corresponding to the (002) crystal plane of carbon fibres. The relatively high intensity of this peak indicated a smaller Pt metal deposit after electroplating than the copper electrodeposition (cf.Fig. 2e and f). The calculated crystal sizes based on the peak at 44° corresponding to the Pt (111) plane were 10.0 and 13.4 nm respectively for applied potentials of −0.2 and −0.5 V.
Fig. 2f also shows that the simultaneous deposition of the two metals (Cu–Pt binary system at an applied electrodeposition voltage of −0.2 V) revealed a crystal size of 3.6 nm, which is much smaller than that of the pure Cu and Pt electrodeposited samples. A unique feature of the Cu–Pt binary diffractograms was that the diffraction peaks were located at diffraction angles between those of pure Cu and Pt systems, indicating that the Pt and Cu atoms were randomly distributed in a new cubic superlattice with a different intermediate interplanar distance according to the Bragg equation (eqn (5)).54
To gain more information regarding the surface composition of the prepared electrodes, the Pt and Cu–Pt electrodeposited carbon fibre samples were examined with high-resolution XPS (Fig. 3), which measures the kinetic energy and number of electrons that escape from up to 10 nm of the samples. Fig. 3a shows the Pt 4f7/2 spectrum of Pt electrodeposited carbon fibres deconvoluted into one intensive main peak at 71.2 eV assigned to metallic Pt, together with weaker peaks at 72.0 and 73.7 eV attributed to Pt in Pt(OH)2 and PtO oxidized states.55 The Cu–Pt electrodeposited samples showed a lower intensity for the peaks assigned to Pt species than the Pt electrodeposited samples, indicating that only 3 wt% of the platinum atoms were at the surface (Fig. 3b), although the EDS results indicated a higher Pt content in the bulk, which is discussed later in Fig. 7h. The peaks in the Cu 3p region suggested that metal atoms were doped into the copper lattice,56 and that a Cu–Pt superlattice was formed. The Cu 2p spectrum of the binary Cu–Pt sample also showed intense peaks from Cu2+ and only a very small fraction of metallic Cu was detected, Fig. 3c. The presence of a large amount of doped Cu2+ was confirmed by the presence of satellite peaks that suggested that platinum atoms were successfully alloyed with copper atoms. The absence of metallic Cu was further confirmed by multi-cyclic voltammetry, which was carried out by electrodepositing the Cu–Pt binary system on the carbon fibre in H2SO4 (aq), 0.5 M, pH ≈ 0, Fig. S3.† No sign of electro-dissolution or re-electrodeposition of pure Cu was observed.
 | ||
| Fig. 3 High-resolution XPS spectra of carbon fibres samples after 2 h potentiostatic deposition at an applied potential of −0.2 V vs. Ag/AgCl; (a and b) Pt 4f region of electrodeposited Pt and Cu–Pt samples, (c) Cu 2p region of the electrodeposited Cu–Pt sample. | ||
3.2. Morphology of copper electrodeposited on carbon fibres
The morphology of the electro-plated carbon fibres under various applied potentials (potentiostatic mode) and currents (galvanostatic mode) is shown in Fig. 4. Fig. 4a–c show the low magnification SEM images of Cu deposits on 7 μm single carbon filament for the three distinctive deposition potentials of −0.2, −0.5 and −1.0 V, respectively, after 2 h. The cathodic current–time profiles during 2 h and X-ray diffractograms are presented in Fig. 2b and c. An applied potential of −0.2 V gave the most uniform filament coverage with ca. 10 μm average thickness (Fig. 4a), whereas an applied potential of −0.5 V resulted in a non-uniform coverage almost twice as thick that contained smaller lump features with sizes ranging from 5 to 50 μm (Fig. 4b). An applied voltage of −0.5 V had a current density after 2 h electroplating twice that of −0.2 V (see Fig. 2b), which should indicate a twice as great electroplated Cu mass on carbon fibres. The greater coverage with the higher applied potential was thus in accordance with Faraday's law. Fig. S4† shows SEM micrographs of the electroplated layers initially formed at an applied voltage of −0.5 V, illustrating that the filaments were uniformly covered at the start of the electroplating reaction, as when the applied voltage was lower at −0.2 V. Accordingly, the micron-sized lump features were growing on top of an earlier formed uniform metal layer after longer reaction times. Song et al.40 suggested that this was a result of new nucleation sites and growth continuously activated when a more negative potential was applied. Although a higher applied voltage, i.e. −1.0 V (far beyond Cu/Cu2+ equilibrium potential), resulted in much larger current density and electrodeposition at a faster rate, it led to a cathodic current density (absolute value) that started to decrease after 1 h electrodeposition, Fig. 2b, due probably to the dissolution and/or detachment of Cu deposits during the later stages of the electrodeposition. Fig. S5† shows some detached and isolated copper deposits. It was evident that the higher applied voltage resulted in the growth of a nanostructured dendritic copper deposit, and the detachment occurred because the excessive deposition of nanostructured dendritic copper became sensitive to fragmentation from the surface of the carbon fibres. Fig. 4d and e show high magnification micrographs of the electrodeposited copper with a uniform coverage (Fig. 4d) and of the porous and radially grown leaf-like morphologies (Fig. 4e) at applied potentials of −0.2 and −1.0 V, respectively. | ||
| Fig. 4 SEM images of electrodeposited copper on carbon fibres in potentiostatic (applied constant potential vs. Ag/AgCl) and galvanostatic (applied constant current) modes after 2 h. | ||
It was reported previously that the electrodeposited copper on substrates (graphite and copper) showed mainly polycrystalline grains with sizes ranging from submicrons to microns.40,48 A uniform polycrystalline coverage was also observed in this work with the addition of small facetted morphologies ca. 10 nm in size for lower applied voltages (Fig. 4d), while dendritic superstructures were obtained at a higher applied voltage (Fig. 4e and S5†). These dendritic copper structures were also reported by Li et al.,57 as a result of hydrogen evolution together with the reduction of copper ions at the cathode surface at higher applied voltages. Although electrodeposition at a high voltage (close to hydrogen evolution) may seem to be a useful method to develop novel electrode surface with nano/hierarchical structures, these structures are a drawback because of their sensitive nature. A frequent detachment of the developed structures from highly curved carbon fibre surfaces occurred. In contrast, a more reliable and solid uniform copper coverage, surrounding the fibres was always obtained at lower voltages (−0.2 V).
Fig. 4f and g show SEM images of Cu samples electrodeposited in a galvanostatic mode (constant applied current) at respectively −20 and −50 mA after 2 h. The corresponding cathodic potential vs. Ag/AgCl-time profile during 2 h is shown in Fig. S6.† An abrupt decrease in the measured voltage (absolute value) occurred at an early stage (<200 s) and the voltage decreased slightly during the 2 h of electroplating. The lower applied current, −20 mA, resulted in a measured voltage of ca. −0.2 V at the earlier stages (<200 s), which finally levelled out at a voltage of ca. 0.0 V. This constant low applied current resulted in the most uniform Cu deposits with sizes less than 100 nm, Fig. 4f. It also served to avoid the formation of submicron polycrystalline grains, which were observed on the electrodeposited samples at a low applied voltage, i.e. −0.2 V (cf.Fig. 2d and f). Fig. 2g shows the presence of submicron cubic polycrystalline grains with truncated corners that were obtained using the higher applied current (−50 mA). A constant current (galvanostatic mode) during electroplating was thus the most effective way to control the microstructure morphology in the copper electroplating of the carbon filaments.
3.3. Morphology of platinum electrodeposited onto carbon fibres
Fig. 5a–c show SEM images of Pt deposits on carbon fibre surface for deposition potentials of +0.2, −0.2 and −0.5 V, respectively (vs. Ag/AgCl). The corresponding cathodic current–time profile during 2 h and X-ray diffractograms are shown in Fig. 2e and f. A positive applied voltage (+0.2 V) and a very low cathodic negative current (close to 0.0 A) led to a small mass of electroplated Pt formed as spherical nanoparticles on the carbon filament surfaces, Fig. 5a, but this low amount of Pt was could not be detected by X-ray diffraction,52 Fig. S7.† A negative voltage of −0.2 V resulted in larger micron-sized spherical particles forming inter-particle grain boundaries, Fig. 5b, and an even higher negative voltage, i.e. −0.5 V, yielded ca. 100 times smaller spherical Pt entities (Fig. 5c), but the growth conditions were then very heterogeneous and extended complex dendritic units with sizes close to 10 μm were formed (Fig. 5d and f). As in the case of the deposition of copper at −1 V (Fig. 4c, e and S4†), the platinum entities were composed of leaf-like units within micron-sized superstructures (Fig. 5d and f). Fig. 5e shows that the leaves were arranged as highly repetitive dendritic superstructures. In the spherical superstructures (Fig. 5f), the 20 nm thick leaf-like sheets were more randomly organized, as shown in Fig. 5g. Sheet formation occurred at a lower applied voltage in the Pt system because hydrogen was evolved at a lower voltage in the Pt system than in the case of copper electrodeposition since the platinum itself acts as an electrocatalyst for hydrogen evolution under acidic condition.58 These superstructures with specific nano-features could potentially provide more catalytic activity than spherical particles due to their high specific surface area and distinctive atomic termination,59–64 but the presence of hydrogen at the surface of the cathode causes some of the deposited metal to detach from the carbon fibre surface (as discussed earlier for the copper system). The application of a voltage close to the hydrogen evolution with a high rate of deposition also makes it difficult to maintain control of the deposited metal morphologies, e.g. when repetitive sample preparation is necessary. | ||
| Fig. 5 SEM images of platinum electrodeposited onto carbon fibres in potentiostatic mode for different applied potentials vs. Ag/AgCl after 2 h. | ||
Fig. 6a–c show SEM images of Pt electrodeposited samples in the galvanostatic mode (constant current) after 2 h for the three deposition currents of −5, −20 and −50 mA, respectively. The corresponding curves of cathodic potential vs. Ag/AgCl as a function of time during 2 h are shown in Fig. 6e. The lower deposition currents, i.e. −5 to −20 mA, resulted in spherical micron-sized particles, but the higher deposition current of −50 mA resulted in submicron Pt particles. In agreement with the earlier result, it was apparent that an increase in the deposition current density increased the number density of nucleation sites, and that this led to more and smaller Pt particles with a high specific surface area.59 The density of initial nucleation sites on the pristine carbon filament is shown for the −20 mA deposition current in Fig. 6f. In Fig. 6b, which shows a magnified region of Fig. 6f, it is evident that the deposition current also had a significant effect on the surface morphology of the Pt particles. A low deposition current gave smooth particle surfaces, but at −50 mA a dendritic nanostructure with spiky features was dominant. This nanostructure was discussed earlier in relation to the evolution of hydrogen during the electrodeposition (see the potential level in Fig. 6e). It was, however, apparent that already at a deposition current of −20 mA, smaller grain features favoured particle-to-particle nucleation before new nuclei were formed on the surface of the carbon filaments. This explains the preferred growth of particles on top of previously formed particles visible in Fig. 6f. The higher constant current, −50 mA, resulted in the most uniform dendritic Pt morphologies along the carbon filaments (Fig. 6c), when the measured potential levelled out at ca. −0.5 V vs. Ag/AgCl, Fig. 6e. These star-shaped morphologies can thus be compared with the mixed morphology (Fig. 5c–g) obtained in the potentiostatic mode at −0.5 V. Overall, it can be concluded that the carbon filaments dominantly supported a more radial growth of refined nanostructured platinum (and copper) deposits when a higher voltage was used, which agreed with and increased the nucleation on already deposited metal. At the same time, the application of a constant current was the easiest way to reach a more uniform and controlled particle morphology.
 | ||
| Fig. 6 (a–c and f) SEM images of carbon fibres electrodeposited with platinum in the galvanostatic mode for different applied currents (−5, −20 and −50 mA) after 2 h together with (e) voltage vs. Ag/AgCl-time profiles. (d) SEM image of Pt particles detaching from a carbon fibre surface. | ||
Fig. 6d shows the separation of three Pt particles from the carbon fibre surface (grown at −5 mA). The grooves in the filaments are clearly visible as an imprinted and replicated pattern on the released particles. These phase interface fractures were frequently observed and considered detrimental for the targeted catalytic application, although the Pt particles had successfully nucleated on the carbon filaments and grown into the filament grooves. Regardless of the electrodeposition mode, however, the Pt particles never resulted in a complete outer shell covering the high-curvature carbon fibre surfaces, and they always showed inter-particle boundaries due to preferential self-nucleation. This result was in contrast to the copper system and indicated that self-nucleation was greater for the Pt-ion than for the Cu-ion system.
3.4. Morphology of copper–platinum (Cu–Pt) electrodeposited on carbon fibres
Since Cu could effectively stabilize a uniform filament coating, whereas Pt always displayed ‘island-like’ filaments due to extensive Pt–Pt nucleation instead of a uniform filament coverage, a bimetallic approach was considered based on inspiration by Stamenkovic et al.32Fig. 7a and b show the Cu–Pt coatings electrodeposited in the potentiostatic mode for the deposition potentials of respectively −0.2 and −0.5 V, after 2 h. The corresponding cathodic current–time profile during 2 h, and the X-ray diffractograms for deposition voltage of −0.2 V are shown in Fig. 2e and f. The high magnification micrographs from the −0.2 V deposition revealed a nano-sized polycrystalline phase together with submicron crystals (Fig. 7a and d). With an applied voltage of −0.5 V (Fig. 7b and e), only submicron crystals were formed and the X-ray diffractograms of the −0.5 V sample showed only the Cu/Cu2O phase with no sign of Pt species, Fig. S7.† It was therefore suggested that the facetted submicron crystals in the nanostructured phase obtained at −0.2 V consisted mostly of copper (Fig. 7d). Fig. 7c shows that a −20 mA electrodeposition current (in the galvanostatic mode) resulted in the aggregation of larger crystals in 1 μm lumps.Overall, the application of a lower voltage (−0.2 V) resulted in the most uniform filament coverage, completely surrounding the carbon filaments. An average thickness of ca. 1 μm for Cu–Pt deposits on single filaments was estimated from the SEM micrographs, Fig. 7a and f. A uniform coating coverage is considered to be important for the mechanical integrity of the coatings, preventing the release of catalytic metal as was observed with the pure Pt system (Fig. 6d). The cross-section of the bi-metallic coating on the carbon filament interface is shown in Fig. 7f–h. The Cu–Pt coating also showed an ability to orient and fit into carbon fibre surface grooves (observed in Fig. 7f and g), providing an interlock joint system similar to a “dovetail” joint (Fig. 7g inset), which hindered detachment of the deposited metal.
 | ||
| Fig. 7 SEM images of potentiostatic electrodeposited carbon fibres with layers of a binary alloy of copper–platinum after 2 h (a, b, d, and e); (c) low magnification of the galvanostatic electrodeposition mode; (f and g) low and high magnifications of the carbon fibre/deposit interface; (h) SEM-EDX element mapping of −0.2 V electrodeposited sample for carbon (C), oxygen (O), copper (Cu), and platinum (Pt) elements. | ||
EDX elemental mapping together with SEM was used to study the distribution of elements on the surface and bulk Cu–Pt coverage, Fig. 7h. The atomic percentages of C, O, Cu and Pt were calculated to be 77.1, 15.1, 5.7 and 2.1 respectively. That Cu and O were dominant on the surface of the deposits is in agreement with XPS surface analysis, Fig. 3, and these EDX results thus confirmed the ultralow Pt deposition of Pt in the binary system.
3.5. The catalytic performance of electrodeposited carbon fibres for electro-oxidation of ammonia solution
The cyclic voltammetry and stepwise chronoamperometry measurements were made for platinum and copper–platinum electrodeposited carbon fibre samples (2 h with the potential of −0.2 V vs. Ag/AgCl), Fig. 8. The behaviour of the Pt- and Cu–Pt-coated carbon fibres in 1 M KOH is shown in Fig. 8a. For the Pt-electrodeposited sample, the anodic current started on positive scan at −0.4 V and reached a peak at ca. −0.14 V, corresponding to the adsorption of OH on the catalyst surface together with the oxidation associated with the formation of an oxide layer on the platinum surface, occurring in the region from −0.20 to +0.4 V.55 In the Cu–Pt system, the anodic current started earlier at −0.65 V and reached a peak at −0.4 V, which was related to the formation of an oxide layer on copper (Cu2O) in an alkaline KOH electrolyte. The copper atoms may produce soluble copper hydroxide species at a voltage higher than 0.0 V during oxidation,65 but this did not occur in the Cu–Pt system. This supported the hypothesis that the copper atoms were alloyed with the Pt, and was in agreement with the X-ray, XPS and EDX results. This was considered to be important since metallic copper has the potential to contaminate wastewater systems. On negative scanning for the KOH electrolyte, the cathodic current for the Pt-electrodeposited sample started at +0.1 V, which was related to the reduction of Pt-oxide, whereas the cathodic current for the Cu–Pt system started at −0.3 V, Fig. 8a. The electrodeposited metals showed a peak between −0.6 and −0.8 V, which shifted to more negative potentials when the sweep rate was increased, Fig. S8.† Giri and Sarker65 carried out cyclic voltammetry for a copper monolayer on the platinum surface in a KOH electrolyte and compared the results with those for copper and platinum surfaces. The oxide phase on the monolayer copper and platinum monolayers was reduced at ca. −0.25 and −0.4 V, respectively,65 so that the peaks appearing on the negative scan in the −0.6 to −0.8 V range should be related to hydrogen adsorption/desorption, which is located in the −0.4 to −1.0 V range. The cyclic voltammograms for noble metals within basic electrolytes are characterized by hydrogen adsorption/desorption, double-layer and oxygen regions in potential ranges of respectively (−1.0 to −0.4 V), (−0.5 to −0.2 V) and (−0.2 to +0.6 V).55,66 The electrochemical active surface area (ECSA) of Pt electrodeposited on a carbon fibre was further derived from the integrated charge of the hydrogen adsorption/desorption peak (inset in Fig. 8a) divided by the scanning sweep rate and the constant 0.21 in mC cm−2 (the maximum surface charge transferred to Pt during adsorption of a monolayer of hydrogen).67,68 The calculated ECSA was ca. 21 m2 g−1, which is significantly higher than the value for ordinary electrodes of bulk Pt, Fig. S9a.†Fig. 8b shows the cyclic voltammogram of the electrodeposited carbon fibre catalysts in the presence of 0.1 M NH3. The Pt coated carbon fibres showed a broad irreversible anodic peak corresponding to NH3 oxidation with an onset and a maximum at respectively ca. −0.8 and +0.25 V. The maximum current density for electro-oxidation of ammonia was ca. 3 mA mgPt−1 at a sweep rate of −2 mV s−1, which shifted to a higher positive potential when the scanning rate was increased, see Fig. S8e.† This maximum current density was much higher than that of pure 220 mg Pt wire (0.5 mm in diameter). The Pt wire showed a peak current of 3.45 mA at a potential of −0.56 V, Fig. S9b,†i.e. ca. 0.02 mA mgPt−1, revealing the importance of the high active surface area achieved using carbon filaments as substrates for the catalytic metal. The onset of electro-oxidation of ammonia on Pt coated filaments (ca. −0.8 V) occurred earlier during cyclic voltammetry than previously reported for the electrodeposited Pt–carbon fibre systems,10,12,13,20 (ca. −0.5 V), and less energy was required for the electrolysis. The current density for electro-oxidation of ammonia based on Pt on carbon was previously reported to be 6.5 mA mgPt−1 for carbon fibre paper substrates at a potential of −0.05 V vs. Hg/HgO for 5 M KOH support electrolyte, but the ammonia concentration was not reported.20 Here, a similar current density was achieved at a much lower concentration of KOH (see Fig. 8b and S8†). The same group reported 2.7 mA mgPt−1 at a potential of +0.1 V vs. Hg/HgO for an electrolyte of 1 M KOH and 1 M ammonia at a sweep rate of 10 mV s−1.13 Our Pt–carbon electrodes showed ca. 3 mA mgPt−1 under 1 M KOH conditions, with a 10 times lower ammonia concentration, Fig. S8.† The anodic oxidation of ammonia and the cathodic evolution of hydrogen during cyclic voltammetry on the Pt surfaces is affected by electrolytic parameters and operation conditions,12 but ammonia is oxidized more easily at higher KOH concentrations, which suggests a very high catalytically performance of the Pt-coated filaments with regard to the current density and low onset potential.10 The electro-oxidation of ammonia with the binary Cu–Pt system showed an onset at −0.8 V and a double peak centred respectively at −0.31 and −0.06 V with a maximum current density of 2.5 mA mgCu–Pt(total loading)−1. The value suggests a slightly lower efficiency of ammonia oxidation than for Pt coated carbon filaments, but it should be noted that the surface-content of noble Pt in this binary system was only ca. 3 wt%.
 | ||
| Fig. 8 Electrochemical catalytic activity for different electrodeposited carbon fibre samples; (a and b) cyclic voltammogram scans in different electrolyte systems with sweep rate of 2 mV s−1 at room temperature, (c) polarization curves with stepwise applied potential with steps of 100 mV at 1 min intervals. The current was normalized with respect to the total mass of deposits including Pt and Cu by measurement of the weight of the carbon fibres before and after electrodeposition. Note: black refers to Pt, red refers to Cu–Pt in all graphs. | ||
Fig. 8c shows the current density versus time when the potential was increased stepwise in steps of 0.1 V from −0.6 V to +0.4 V with an interval of 1 min for each step. For Pt-coated samples, the current density level increased when the voltage level was increased to 0.0 V, but decreased slowly with time at each voltage step due to the poisoning effect. The electro-oxidation of ammonia in basic conditions is a multi-step process involving the successive dehydrogenation of nitrogen species and the formation of a variety of reaction intermediates,66,69 which accumulate at the catalyst surface and block the active noble metals sites, i.e. poisoning the main reaction.70 The poisoning effect became evident at potentials higher than 0.0 V for the Pt surface, where the current density decreased for higher potentials. For comparison, the Pt wire with a much lower active surface area was rapidly deactivated due to severe poisoning in the applied potential range, Fig. S9c.† The Cu–Pt binary system showed less poisoning than the pure Pt system at a voltage higher than ca. 0.0 V, Fig. 8c. The severe poisoning of Pt sites for ca. 10 min at the applied potential of 0.0 V was shown in chronoamperometry tests, Fig S10,† demonstrating both systems sensitivity to the formation of adsorbed reaction nitrogen intermediates. However, considering the possibility of reactivation of the catalytic surface applying a negative potential,66,69 the bimetallic alloy provided an almost similar level of electro-oxidation of wastewater ammonia as when pure Pt was used. It is therefore suggested that the copper hindered the poisoning effect while the low amount of Pt oxidized the ammonia to N2 and H2, since copper alone cannot dehydrogenate NH3.71
Electrodes containing noble metals doped with copper have shown useful activity for methanol electro-oxidation in acidic environments.56,67 Here, the successful alloying of the Cu and Pt in a superlattice formation can be regarded as beneficial from the perspective of a considerably retarded dissolution of copper during electro-oxidation.54
Conclusions
A novel concept of bimetallic carbon fibre electrodes to be prepared for the purification of wastewater from ammonia contaminant is presented. Only 3 wt% of platinum was required in a copper superlattice to generate a stable carbon fibre electrode surface with only 30% less capacity to oxidize ammonia, as compared to the pure platinum. A key component for the exploitation of this effect was to use commercial carbon fibres as an inexpensive electrodeposition material after a thermal shock treatment at ca. 800 °C. The rapid heating allowed their protective polymer coatings to rapidly decompose, while maintaining a high intrinsic filament conductivity of the carbon fibres at ca. 10−5 ohm m. Copper, platinum and a combination of copper and platinum ions were deposited on the filaments, and the nature of the metal coatings are corroborated with their methods of deposition as well as the type of metal ions used for similar electroplating conditions. The potentiostatic application of the coatings at higher voltages resulted in complex dendritic morphologies with a very high large surface area but a weak interface towards the filaments, whereas the galvanostatic preparation resulted in more uniform coatings. In contrast to platinum showing a strong preference for deposition on already nucleated surface areas, generating fragile ‘island-like’ filament coverage, copper–platinum coatings displayed uniform coating formation surrounding the entire filament cores. The use of carbon fibre filaments for the preparation of catalytic surfaces is expected to find a range of other uses where nanostructured morphologies with large surface area are required for catalytic performance.Conflicts of interest
No conflict of interest.Acknowledgements
The financial support from Vinnova, the Swedish Agency for Innovation Systems (Ref. 2016-03408), is acknowledged. We also would like to thank the engineering companies Polyproject Environment AB, Mercatus Engineering AB and the municipal wastewater treatment company SYVAB for their in-kind contributions to this project.Notes and references
- L. Huff, Aquatic Life Ambient Water Quality Criteria for Ammonia–Freshwater 2013, Report EPA 822-R-13-001, U.S. Environmental Protection Agency, Washington, DC, 2013 Search PubMed.
- W. A. Wurts, J. World Aquacult. Soc., 2003, 34, 20–21 Search PubMed.
- WasteWater treatment in Sweden, Swedish Environmental Protection Agency, Stockholm, Sweden, 2009, ISBN number is 978-91-620-8416-5, http://www.naturvardsverket.se/Om-Naturvardsverket/Publikationer/ISBN/8400/978-91-620-8416-5/ Search PubMed.
- M. A. H. J. van Kessel, D. R. Speth, M. Albertsen, P. H. Nielsen, H. J. M. Op den Camp, B. Kartal, M. S. M. Jetten and S. Lücker, Nature, 2015, 528, 555–559 CrossRef CAS PubMed.
- G. Ruiz, D. Jeison and R. Chamy, Water Res., 2003, 37, 1371–1377 CrossRef CAS PubMed.
- R. Hatzenpichler, Appl. Environ. Microbiol., 2012, 78, 7501–7510 CrossRef CAS PubMed.
- Y. Li, K. Ding, X. Wen, B. Zhang, B. Shen and Y. Yang, Sci. Rep., 2016, 6, 23747 CrossRef CAS PubMed.
- P. Ciambelli, P. Corbo, C. Porcelli and A. Rimoli, Zeolites, 1985, 5, 184–187 CrossRef CAS.
- T. C. Jorgensen and L. R. Weatherley, Water Res., 2003, 37, 1723–1728 CrossRef CAS PubMed.
- J. Gwak, M. Choun and J. Lee, ChemSusChem, 2016, 9, 403–408 CrossRef CAS PubMed.
- L. Li, J. Yao, X. Fang, Y. Huang and Y. Mu, Sci. Rep., 2017, 7, 41030 CrossRef CAS PubMed.
- C. Zhong, W. B. Hu and Y. F. Cheng, J. Mater. Chem. A, 2013, 1, 3216–3238 RSC.
- E. P. Bonnin, E. J. Biddinger and G. G. Botte, J. Power Sources, 2008, 182, 284–290 CrossRef CAS.
- D. J. Little, I. I. I. M. R. Smith and T. W. Hamann, Energy Environ. Sci., 2015, 8, 2775–2781 RSC.
- G. Zhang, Y. Zhou and F. Yang, Electrochim. Acta, 2019, 299, 672–681 CrossRef CAS.
- A. M. Pourrahimi and M. Pumera, Nanoscale, 2018, 10, 16398–16415 RSC.
- A. M. Pourrahimi, K. Villa, C. L. Manzanares Palenzuela, Y. Ying, Z. Sofer and M. Pumera, Adv. Funct. Mater., 2019, 1808678 CrossRef.
- A. M. Pourrahimi, K. Villa, Y. Ying, Z. Sofer and M. Pumera, ACS Appl. Mater. Interfaces, 2018, 10, 42688–42697 CrossRef CAS PubMed.
- B. K. Boggs and G. G. Botte, J. Power Sources, 2009, 192, 573–581 CrossRef CAS.
- B. K. Boggs and G. G. Botte, Electrochim. Acta, 2010, 55, 5287–5293 CrossRef CAS.
- K. Endo, Y. Katayama and T. Miura, Electrochim. Acta, 2004, 49, 1635–1638 CrossRef CAS.
- K. Siddharth, Y. Chan, L. Wang and M. Shao, Curr. Opin. Electrochem., 2018, 9, 151–157 CrossRef CAS.
- Y. Zhou, G. Zhang, M. Yu, J. Xu, S. Qiao, X. Cheng and F. Yang, ChemistrySelect, 2018, 3, 3433–3443 CrossRef CAS.
- N. Sacré, M. Duca, S. Garbarino, R. Imbeault, A. Wang, A. Hadj Youssef, J. Galipaud, G. Hufnagel, A. Ruediger, L. Roué and D. Guay, ACS Catal., 2018, 8, 2508–2518 CrossRef.
- Y. Liu, B. Huang and Z. Xie, Appl. Surf. Sci., 2018, 427, 693–701 CrossRef CAS.
- B. Huang, Y. Liu and Z. Xie, J. Mater. Chem. A, 2017, 5, 23481–23488 RSC.
- B. Huang, Y. Liu, X. Huang and Z. Xie, J. Mater. Chem. A, 2018, 6, 22277–22286 RSC.
- B. Huang, X. Hu, Y. Liu, W. Qi and Z. Xie, J. Power Sources, 2019, 413, 408–417 CrossRef CAS.
- M. Wu, J. Du, C. Tao, Z. Liu and Y. Li, J. Colloid Interface Sci., 2019, 542, 451–459 CrossRef CAS PubMed.
- I. Lucentini, A. Casanovas and J. Llorca, Int. J. Hydrogen Energy, 2019, 44, 12693–12707 CrossRef CAS.
- Y. Liu, B. Huang, X. Zhang, X. Huang and Z. Xie, J. Power Sources, 2019, 412, 125–133 CrossRef CAS.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241 CrossRef CAS PubMed.
- Y. Liu, B. Huang, X. Hu and Z. Xie, Int. J. Hydrogen Energy, 2019, 44, 3702–3710 CrossRef CAS.
- J. Hagberg, S. Leijonmarck and G. Lindbergh, J. Electrochem. Soc., 2016, 163, A1790–A1797 CrossRef CAS.
- M. MInus and S. Kumar, JOM, 2005, 57, 52–58 CrossRef CAS.
- R. Yu, L. Chen, Q. Liu, J. Lin, K.-L. Tan, S. C. Ng, H. S. O. Chan, G.-Q. Xu and T. S. A. Hor, Chem. Mater., 1998, 10, 718–722 CrossRef CAS.
- S. H. Joo, S. J. Choi, I. Oh, J. Kwak, Z. Liu, O. Terasaki and R. Ryoo, Nature, 2001, 412, 169–172 CrossRef CAS PubMed.
- H. Yang-Chih, W. Chih-Chieh, L. Chueh, K. Chi-Chung and P. Tsong-Pyng, Nanotechnology, 2012, 23, 405603 CrossRef PubMed.
- R. D. Sanderson and E. R. Sadiku, J. Appl. Polym. Sci., 2003, 87, 1051–1058 CrossRef CAS.
- S. Song, C. M. Ortega, Z. Liu, J. Du, X. Wu, Z. Cai and L. Sun, J. Electroanal. Chem., 2013, 690, 53–59 CrossRef CAS.
- B. Pierozynski, Croat. Chem. Acta, 2012, 85, 1–8 CrossRef CAS.
- A. Subrenat, L. M. Le Leuch and P. Le Cloirec, Environ. Technol., 2008, 29, 993–1000 CrossRef CAS PubMed.
- P. E. Larson and M. A. Kelly, J. Vac. Sci. Technol., A, 1998, 16, 3483–3489 CrossRef CAS.
- A. Sumida and S. Watanabe, US Pat. 4555446 A, 1985.
- T. R. Pozegic, J. V. Anguita, I. Hamerton, K. D. G. I. Jayawardena, J. S. Chen, V. Stolojan, P. Ballocchi, R. Walsh and S. R. P. Silva, Sci. Rep., 2016, 6, 37334 CrossRef CAS PubMed.
- P. Mitschang, M. Blinzler and A. Wöginger, Compos. Sci. Technol., 2003, 63, 2099–2110 CrossRef CAS.
- A. K. Dutta, Phys. Rev., 1953, 90, 187–192 CrossRef CAS.
- D. Grujicic and B. Pesic, Electrochim. Acta, 2002, 47, 2901–2912 CrossRef CAS.
- B. E. Conway, H. Angerstein-Kozlowska, W. B. A. Sharp and E. E. Criddle, Anal. Chem., 1973, 45, 1331–1336 CrossRef CAS.
- A. Mallik and B. C. Ray, Mater. Res., 2013, 16, 539–545 CrossRef CAS.
- A. Lalitha, S. Ramesh and S. Rajeswari, Electrochim. Acta, 2005, 51, 47–55 CrossRef CAS.
- A. M. Pourrahimi, D. Liu, V. Ström, M. S. Hedenqvist, R. T. Olsson and U. W. Gedde, J. Mater. Chem. A, 2015, 3, 17190–17200 RSC.
- S. Akhlaghi, A. M. Pourrahimi, C. Sjöstedt, M. Bellander, M. S. Hedenqvist and U. W. Gedde, Polym. Degrad. Stab., 2017, 138, 27–39 CrossRef CAS.
- N. Jung, Y. Sohn, J. H. Park, K. S. Nahm, P. Kim and S. J. Yoo, Appl. Catal., B, 2016, 196, 199–206 CrossRef CAS.
- M. H. M. T. Assumpção, R. M. Piasentin, P. Hammer, R. F. B. De Souza, G. S. Buzzo, M. C. Santos, E. V. Spinacé, A. O. Neto and J. C. M. Silva, Appl. Catal., B, 2015, 174–175, 136–144 CrossRef.
- X. Du, S. Luo, H. Du, M. Tang, X. Huang and P. K. Shen, J. Mater. Chem. A, 2016, 4, 1579–1585 RSC.
- S. Li, R. Furberg, M. S. Toprak, B. Palm and M. Muhammed, Adv. Funct. Mater., 2008, 18, 2215–2220 CrossRef CAS.
- L. Zheng, D. Yang, R. Chang, C. Wang, G. Zhang and S. Sun, Nanoscale, 2017, 9, 8918–8924 RSC.
- J. Liu, C. Zhong, Y. Yang, Y. T. Wu, A. K. Jiang, Y. D. Deng, Z. Zhang and W. B. Hu, Int. J. Hydrogen Energy, 2012, 37, 8981–8987 CrossRef CAS.
- J. L. Castro-Mayorga, M. J. Fabra, A. M. Pourrahimi, R. T. Olsson and J. M. Lagaron, Food Bioprod. Process., 2017, 101, 32–44 CrossRef CAS.
- A. M. Pourrahimi, T. A. Hoang, D. Liu, L. K. Pallon, S. Gubanski, R. T. Olsson, U. W. Gedde and M. S. Hedenqvist, Adv. Mater., 2016, 28, 8651–8657 CrossRef CAS PubMed.
- A. M. Pourrahimi, D. Liu, R. L. Andersson, V. Ström, U. W. Gedde and R. T. Olsson, Langmuir, 2016, 32, 11002–11013 CrossRef CAS PubMed.
- F. J. Vidal-Iglesias, N. García-Aráez, V. Montiel, J. M. Feliu and A. Aldaz, Electrochem. Commun., 2003, 5, 22–26 CrossRef CAS.
- A. M. Pourrahimi, R. T. Olsson and M. S. Hedenqvist, Adv. Mater., 2018, 30, 1703624 CrossRef PubMed.
- S. D. Giri and A. Sarkar, J. Electrochem. Soc., 2016, 163, H252–H259 CrossRef CAS.
- S. Le Vot, D. Reyter, L. Roué and D. Bélanger, J. Electrochem. Soc., 2012, 159, F91–F96 CrossRef CAS.
- C. Wang, F. Ren, C. Zhai, K. Zhang, B. Yang, D. Bin, H. Wang, P. Yang and Y. Du, RSC Adv., 2014, 4, 57600–57607 RSC.
- Y. Holade, T. W. Napporn, C. Morais, K. Servat and K. B. Kokoh, ChemElectroChem, 2015, 2, 592–599 CrossRef CAS.
- H. Gerischer and A. Mauerer, J. Electroanal. Chem. Interfacial Electrochem., 1970, 25, 421–433 CrossRef CAS.
- F. Vitse, M. Cooper and G. G. Botte, J. Power Sources, 2005, 142, 18–26 CrossRef CAS.
- A. C. A. de Vooys, M. T. M. Koper, R. A. van Santen and J. A. R. van Veen, J. Electroanal. Chem., 2001, 506, 127–137 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9se00161a |
| This journal is © The Royal Society of Chemistry 2019 |