
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding charge transfer, defects and surface states at hematite photoanodes
Jifang
Zhang
 and
Salvador
Eslava
*
and
Salvador
Eslava
*
Department of Chemical Engineering, University of Bath, Claverton Down, Bath, UK. E-mail: s.eslava@bath.ac.uk
First published on 11th April 2019
Abstract
Hematite (α-Fe2O3) has been widely investigated as a promising photoanode candidate in photoelectrochemical cells for solar water splitting. Although significant advances have been made to improve bulk charge properties as well as surface catalytic activity for oxygen evolution reaction, it still remains challenging to meet the standards for practical applications. As such, deeper understanding and analysis is necessary to guide efforts to achieve higher activities. This perspective reviews and analyzes the important progress on hematite photoanodes from multiple angles. We highlight the critical role of defect chemistry in terms of bulk properties and surface reaction kinetics. Careful manipulation of the quantity of oxygen vacancies and majority/minority charge carriers is shown to be essential for higher activity. One major type of surface recombination site, which can be readily removed, is identified to be an Fe2+ species based on multiple photoelectrochemical and spectroscopic observations. Analyzing X-ray absorption spectroscopy and electrochemical energy diagrams, we present a clear picture of water oxidation dynamics at different operating conditions, revealing the relationship between photo-generated holes and surface recombination states. Finally, we conclude that to make hematite photoanodes commercially viable, tuning the minority charge transport properties should be regarded as the priority.
 Jifang Zhang | Jifang Zhang received his B. Eng. from Beijing Institute of Technology in Chemical Engineering, followed by a MSc in Materials Chemistry at The University of Edinburgh. During this period, he studied the phonon dispersion behavior of thermosalient molecular crystals using density functional theory. He is currently a final year PhD student in the Department of Chemical Engineering at University of Bath under the supervision of Dr Salvador Eslava. His research interests cover mechanistic investigation of photoelectrodes for solar water splitting, including their interaction with electrocatalysts. He is also interested in the properties of low dimensional nanomaterials and optoelectronics. |
 Salvador Eslava | Dr Salvador Eslava is a Lecturer (Asst. Prof.) in Chemical Engineering at University of Bath, UK. He did his MEng in Chemical Engineering at Universitat Autònoma de Barcelona, Spain, and a taught master in Materials Science at Università degli Studi di Pavia, Italy. He completed his PhD in Leuven, Belgium, at the Centre for Surface Chemistry and Catalysis (COK) at Katholieke Universiteit Leuven and the international microelectronics center IMEC, in Prof. Johan Martens's group. He then held postdoctoral positions in the Department of Chemistry at The University of Cambridge working with Professors Dominic Wright and Richard Lambert and the Department of Materials at Imperial College London working with Prof. Eduardo Saiz. He has published 47 articles on materials for sustainable chemical technologies, including graphene composites, metal oxide semiconductors and porous dielectric materials. Eslava's lab currently focuses on finding greener routes for the production and optimization of semiconductor devices for solar energy conversion, with especial emphasis on solar fuels. |
Hydrogen production from solar water splitting has been an active research field in recent years with great promise to solve energy and environmental problems. It can be carried out in photoelectrochemical (PEC) cells with semiconductor materials, in which hydrogen and oxygen evolve at separate electrodes, hence making it convenient for their collection. The choice of these electrode materials is key to efficient water splitting devices. Of its two electrode reactions, the oxygen evolution reaction (OER) is more challenging than the hydrogen evolution reaction (HER), since the OER involves four electrons to be transferred to produce each O2 molecule. The OER with poor kinetics competes with much faster charge recombination processes in the semiconductor bulk material and at its surface.1–3 Hematite (α-Fe2O3) has been one of the most extensively investigated photoanode materials due to its relatively small band gap, remarkable stability and great elemental abundancy. However, it also comes with some notorious drawbacks such as low conductivity, small absorption coefficient, poor minority charge mobility (10−4 cm2 V−1 s−1), and slow OER kinetics at the semiconductor-liquid junction (SCLJ).4–6
A large number of attempts to tackle these issues, including defect engineering, band engineering and surface treatment, have led to better performance.7–10 Unfortunately, the underlying reasons for such improvements are not always well understood. For example, using perturbation-modulated techniques such as photoelectrochemical impedance spectroscopy (PEIS) and intensity modulated photocurrent spectroscopy (IMPS), one can obtain the rate constants for surface charge transfer and surface charge recombination.3,11,12 However, the physical implications of differences in these rate constants with and without modifications of interest, even if calculated correctly, are often not obvious. Band or defect engineering via doping, in spite of being one of the most commonly used methods to improve bulk charge transport properties, is also frequently found to significantly change OER kinetics.7,13–15 Therefore, PEC behavior must be interpreted by more advanced physical characterizations, such as X-ray absorption spectroscopies and infrared spectroscopy (ideally carried out in situ or in operando).16,17
This perspective aims to provide an insight into the effect of defect engineering of hematite photoanodes not only on bulk properties but also on surface reactions, by gathering information from all aspects of PEC research, both computational and experimental. We prove here that direct control of oxygen vacancies and tuning dopant level follow practically the same principle to change the photoresponse of hematite electrodes, while their differences are also discussed. We also show that the presence of defects is highly associated with two types of electronic bands for charge transfer and how they are influenced by overlayers. Moreover, we reveal one possible explanation for the chemical basis of recombination surface states and demonstrate the electrode dynamics at different voltages. Finally, we provide our opinion on future research directions.
Charge transfer in hematite
Upon irradiation, there are four types of transitions in hematite as shown in Fig. 1a: (a) single ligand field (LF) transitions; (b) pair LF transitions; (c) ligand to metal charge transfer (LMCT); (d) metal to metal charge transfer (MMCT). The first two transitions do not generate electron–hole pairs thus they will not be discussed here. Detailed description of them can be found elsewhere.4,18 The LMCT transition is prevalently recognized as the main, or sometimes, the only source of photocurrent.19,20 According to the reaction| Fe3+ + O2− → Fe2+ + O− | (1) |
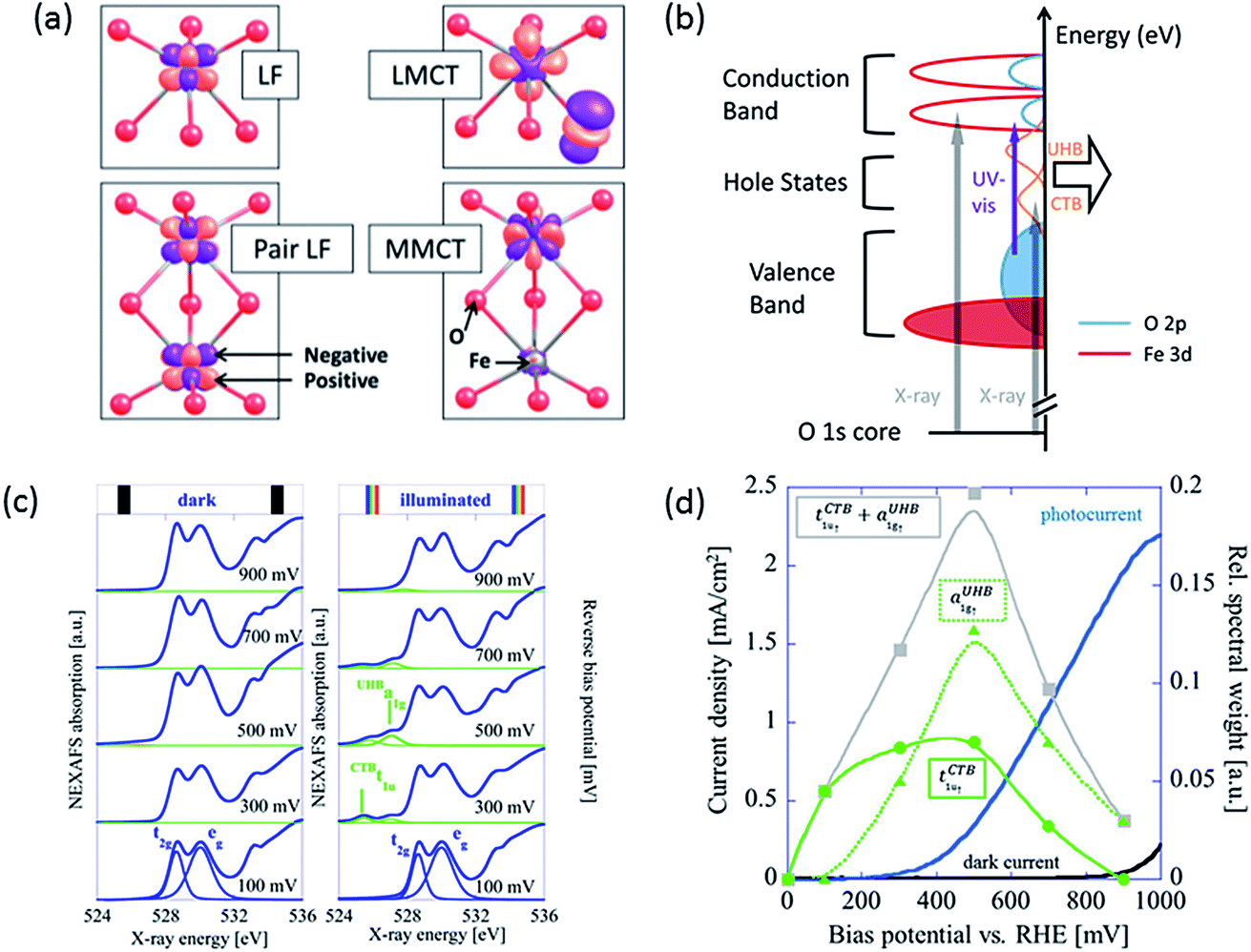 | ||
| Fig. 1 (a) Calculated charge density difference isosurfaces (purple) of four types of transitions in hematite upon irradiation: single ligand field (LF) transition, pair LF transition, ligand to metal charge transfer (LMCT) and metal to metal charge transfer (MMCT). Red and grey atoms represent O and Fe atoms. From ref. 4. Reprinted by permission of John Wiley & Sons, Inc. Also adapted with permission from ref. 18. Copyright 2014 American Chemical Society. (b) Schematic diagram of a hematite photoanode showing electronic transitions by UV-visible and X-ray excitation. Orange peaks indicate hole states of CTB and UHB. Reproduced from ref. 23 with permission from The Royal Society of Chemistry. (c) O 1s NEXAFS spectra recorded at bias from 100 to 900 mV in the dark (left) and under illumination (right) conditions. (d) The spectral weight of tCTB1u↑ (green squares) and aUHB1g↑ (green triangles) measured by NEXAFS under illumination in relation to photocurrent density of a hematite photoanode. Bias potential can be converted to applied potential vs. RHE by adding flat-band potential of roughly 0.5–0.6 V. Reprinted with permission from ref. 16. Copyright 2014 American Chemical Society. | ||
The excited electron resides at an Fe 3d orbital while the hole resides at an O 2p orbital, which is active for water oxidation. The MMCT indicates an electron transfer from one Fe site to another, resulting in split valences. This process is represented by
| 2Fe3+ → Fe2+ + Fe4+ | (2) |
In this case, a hole is generated in an Fe 3d orbital. These two transitions, the LMCT and the MMCT, are also responsible for the electron conduction of hematite; the former is known as O2−–Fe3+ electron hopping and the latter as charge disproportionation.16,21
Although MMCT has often been seen as not participating in solar water splitting by some early researchers,20,22 a more recent work by Braun et al. has found evidence that it does account for a significant proportion of photocurrent.16 They have used near-edge X-ray absorption fine structure (NEXAFS) spectroscopy to study the electronic structure of a hematite photoanode in a PEC cell in operando. They discovered that only under illumination did the NEXAFS spectra of the same film contain two pre-edge peaks that represent electron transitions from O 1s core level either to O 2p hole states through charge transfer band (tCTB1u↑) or to Fe 3d hole states through upper Hubbard band (aUHB1g↑), in addition to the main edge from transitions to CB levels (Fig. 1b).16,23 These two types of hole states, potentially active for charge transfer, are separated by about 1.3 eV (Fig. 1c). These peaks become apparent at applied potentials near and above the photocurrent onset potential only when irradiated. According to transient absorption spectroscopy (TAS) results, bulk recombination is found to be ultrafast and a high percentage of holes are lost within 1 ns,24 so only the rest which migrate to the surface can be detected by NEXAFS. The spectral weight distribution of CTB and UHB for illuminated samples versus applied bias is depicted in Fig. 1d. Interestingly, the spectral sum of these two has a peak located near the onset potential, which closely resembles surface capacitance (often referred to as trap states capacitance) measured using PEIS.7,11,25 The relationship between these two curves will be discussed in later sections.
Since holes from both CTB and UHB can directly contribute to photocurrent, it would be desirable to increase their density of states at PEC operational conditions. Judging from the relative positions and spectral weight of CTB and UHB, it is reasonable to hypothesize that CTB holes influence the low potential performance and even onset potential while UHB holes are associated with the high potential performance. As shown in Fig. 1c, the CTB peak is located at 525.8 eV, which is lower than the UHB peak at 527.1 eV. This means less X-ray energy is needed to inject a core electron into CTB than UHB, in turn proving that the energy level of the former is located lower. The CTB holes are found to be more reactive at lower potentials because their reaction is thermodynamically more favorable, as we will expand later. At higher potentials, more contribution must come from UHB holes as a majority of CTB holes have already been participating in OER. More specifically, CTB contribution has dropped to none above 900 mV bias while photocurrent density is still rapidly increasing, meaning UHB contribution is dominating at this stage. Depending on the characteristics of the measured film, the total contribution of UHB holes can be much less if most holes prefer to present themselves in the CTB band. As such, the task of improving photocurrent densities relies on finding the determinants that change the spectral weights of these two bands.
Oxygen vacancies
Two similar investigations in recent years have examined the effect of oxygen plasma on hematite photoanodes.26,27 This treatment, in both cases, leads to increased photocurrent densities but higher onset potential too. Hu et al. observes a decrease in the concentration of surface Fe2+ species upon oxygen-plasma treatment accompanied by an increase of OH− species by X-ray photoelectron spectroscopy (XPS).27 Pyeon et al. observes the same but also shows that after a posterior short annealing step the photocurrent is recovered and moreover enhanced (Fig. 2a).26 Both groups propose that oxygen plasma fills oxygen vacancies and attracts more OH− species as the oxidation state of Fe increases. After short-annealing, the newly formed oxygen vacancies are closer to the surface (Fig. 2b). Since both the oxygen vacancies and the CTB/UHB density have a direct influence on the photocurrent curves, we believe there could be a connection between the concentration of oxygen vacancies and the CTB/UHB density.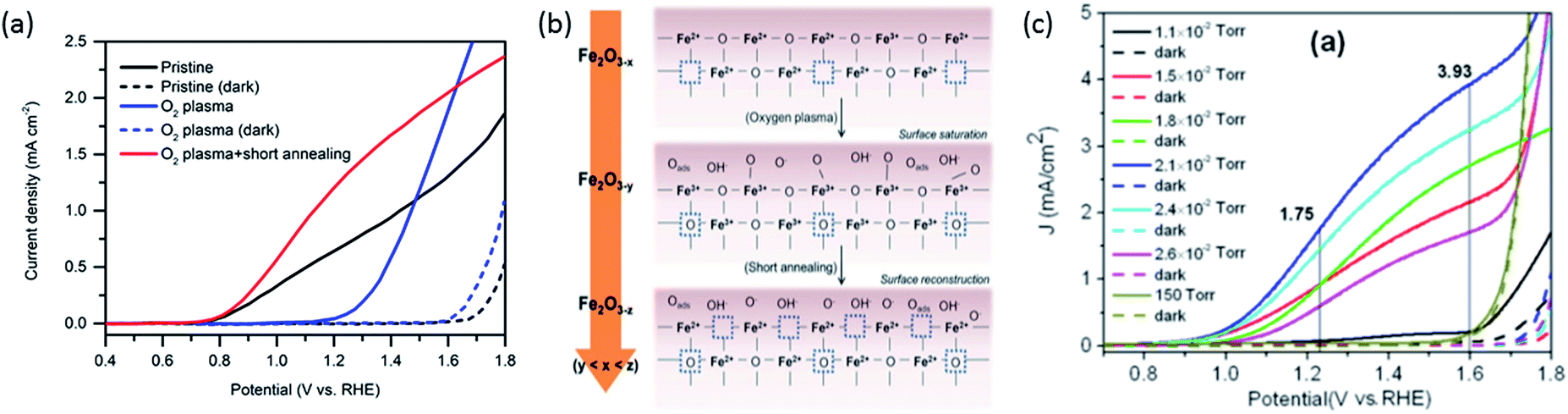 | ||
| Fig. 2 (a) Photocurrent density curves of pristine (black), oxygen-plasma treated (blue) and short-annealed after oxygen-plasma treated hematite photoanodes (red). (b) Scheme showing filling and reintroduction of oxygen vacancies after oxygen-plasma treatment and re-introduction after short-annealing. Adapted from ref. 26, reproduced with permission. (c) Current density curves of hematite photoanodes after annealing at different partial oxygen pressure. Dashed lines indicate measurements in the dark. Reproduced from ref. 13 with permission from The Royal Society of Chemistry. | ||
The link between photoelectrochemistry and the presence of oxygen vacancies can be explained by examining XPS results of hematite before and after annealing treatment.26 The binding energy of 530.10 eV for lattice oxygen O 1s electrons for untreated hematite is higher than the 529.40 eV for oxygen plasma-treated hematite, then returning to 530.00 eV after short annealing in air at 750 °C. These shifts indicate the possible presence of slightly more positively charged oxygen near oxygen vacancies. Such changes of O 1s binding energy, paired with the increased photocurrent density after oxygen plasma and post-annealing (Fig. 2a), are an indication that upon irradiation and LMCT transition (eqn (1)), transport of O 2p (CTB) holes near the surface is facilitated by virtue of oxygen vacancies. Conversely, the MMCT (eqn (2)) is favored upon irradiation if Fe2+ species from pristine hematite have been converted into Fe3+ by oxygen plasma, reducing the likelihood of recombination of a UHB hole (Fe4+) with Fe2+ (reversed eqn (2)). Although Fe2+ might also induce recombination with CTB holes (reversed eqn (1)), MMCT is more negatively affected because total Fe concentration is constant and Fe2+ ions directly suppress the generation of UHB holes (Fe4+).
Contrary to oxygen-plasma treatment, annealing in an oxygen deficient environment creates oxygen vacancies, as can be illustrated by the defect equilibrium of the standard oxygen reduction reaction for n-type oxides (Kröger–Vink notation is adopted in this article):28
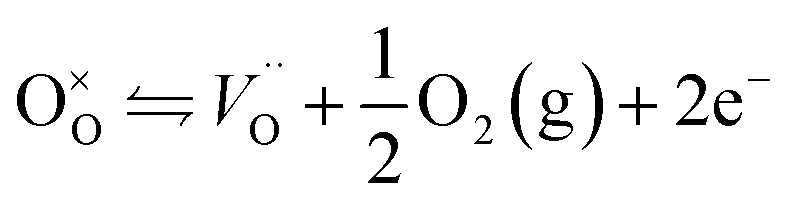 | (3) |
As the partial pressure of oxygen decreases, the reaction proceeds toward the right-hand side, creating more oxygen vacancies. Lower onset potentials have indeed been frequently measured after annealing in low partial O2 concentration (Fig. 2c) or in N2, or after air-plasma treatment, which gives the opposite effect of oxygen plasma treatment. However, the plateau photocurrent densities have also been improved.9,13,29 It can be noticed in eqn (3) that creating more 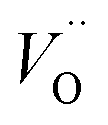 is accompanied by an increase in the concentration of electrons, leading to higher bulk conductivity. Therefore, plateau photocurrent still increases in spite of less contribution from UHB holes. However, excessive oxygen vacancies are detrimental, as evidenced by lower photocurrent densities.13,30 This has been associated with lattice distortion and crystallographic phase mixing of α-Fe2O3 and Fe3O4. A computational study has predicted an optimal concentration of surface oxygen vacancies of 1.26 nm−2 at (0001) surface, as a result of balancing its opposite effects of overpotential on hydroxyl terminated sites and oxygen terminated sites.31
is accompanied by an increase in the concentration of electrons, leading to higher bulk conductivity. Therefore, plateau photocurrent still increases in spite of less contribution from UHB holes. However, excessive oxygen vacancies are detrimental, as evidenced by lower photocurrent densities.13,30 This has been associated with lattice distortion and crystallographic phase mixing of α-Fe2O3 and Fe3O4. A computational study has predicted an optimal concentration of surface oxygen vacancies of 1.26 nm−2 at (0001) surface, as a result of balancing its opposite effects of overpotential on hydroxyl terminated sites and oxygen terminated sites.31
Cation doping
In addition to oxygen vacancies, doping also has an impact on bulk electronic properties as well as on surface kinetics. This perspective focuses on cation doping of hematite, to which a majority of studies are dedicated. We emphasize aliovalent doping (n-type and p-type) here but isovalent and dual-element doping will also be covered briefly.One of the most common n-type cation dopants for hematite photoanodes is titanium.7,13,32–37 Electronic (eqn (4)) and ionic (eqn (5)) compensation are competing processes upon Ti doping:28
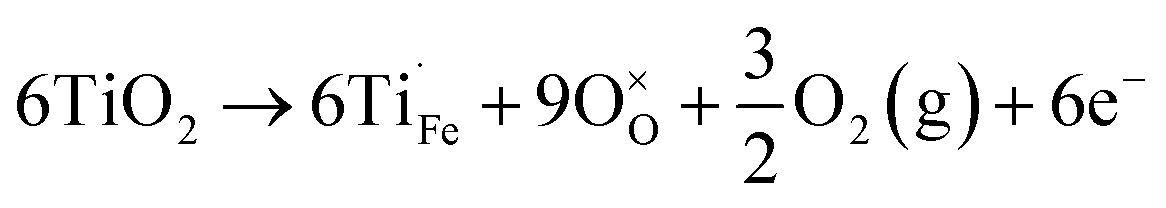 | (4) |
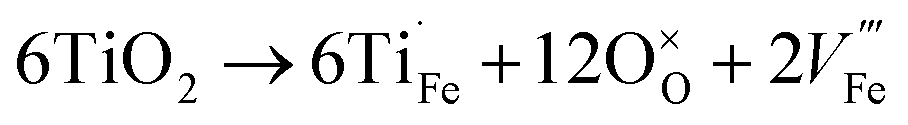 | (5) |
Each of them is irreversible but the resulting equilibrium obtained by subtracting one from the other can be seen as reversible:
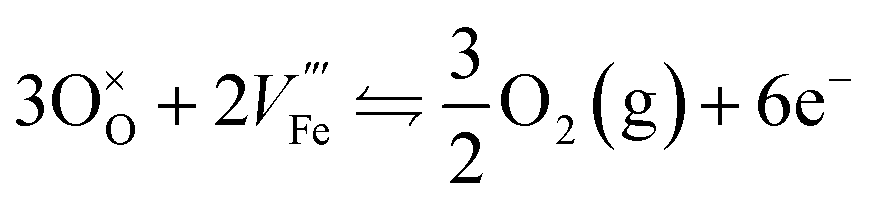 | (6) |
Here we only focus on substitutional rather than interstitial doping since the former was found to be more stable by simulation.5 The equations above deserve careful examination as they have implications in both bulk electronic properties and surface kinetics. The conductivity of Ti-doped hematite does not necessarily increase unless electronic compensation outweighs ionic compensation.28 Moreover, simulation by Liao et al. has compared the conductivities of several 4-valence cation doped hematite structures and has argued that electrons are more localized at Ti sites, which means they behave like traps.5 In contrast, dopants such as germanium and silicon form more covalent bonds with oxygen and free electrons occupy the anti-bonding orbitals, which become more mobile. However, we could not find experimental validation in the literature.
The equilibrium of eqn (6) (applicable for other n-type dopants) is influenced by multiple factors including dopant concentration, partial pressure of oxygen during annealing, and annealing temperature.28 At high dopant levels, ionic compensation dominates electronic compensation, meaning a maximum of conductivity is only achieved at low dopant concentration. This phenomenon has been studied for Sn doping, where maximal conductance is achieved at 3 at%.14 Low oxygen pressure annealing is preferred (confirmed by DFT+U simulation) as more electrons can be generated.32 The transition from electronic compensation to ionic compensation has been theoretically illustrated with a Brower diagram as a function of oxygen partial pressure pO2.38 The authors demonstrate that at high oxygen level, negative charges are predominantly compensated by iron vacancies therefore free electron concentration falls. In the low pO2 region, this compensation is also present, causing the increase of majority charge carrier concentration to be dependent on the doping density by a power of 1/3. Hence, the authors appeal that researchers should take 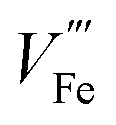 compensation into consideration when doping hematite. The roles of
compensation into consideration when doping hematite. The roles of 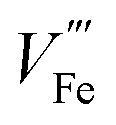 on hematite band structures and for OER have been computationally examined elsewhere but will not be elaborated here.39,40 It is worth noting that the optimal photocurrent has been achieved at a doping level of Sn different to 3%: 0.7 at% for 5 × 10−1 Torr pO2 and 0.1 at% for 5 × 10−5 Torr pO2 annealing.14 Therefore, doping requires fine tuning to obtain optimal photocurrent. Finally, high annealing temperature (above 700 °C for hematite electrodes in practice) drives eqn (6) to the right-hand side because of an increase in entropy, which means electronic compensation expressed by eqn (4) is more favored. Unintentional doping from diffusion of Sn from the fluorine-doped tin oxide (FTO) conductive support can be introduced to improve conductivity, only if the total dopant concentration is within the peak level. Furthermore, high annealing temperature also has an impact on surface properties, which will be discussed in more detail later.
on hematite band structures and for OER have been computationally examined elsewhere but will not be elaborated here.39,40 It is worth noting that the optimal photocurrent has been achieved at a doping level of Sn different to 3%: 0.7 at% for 5 × 10−1 Torr pO2 and 0.1 at% for 5 × 10−5 Torr pO2 annealing.14 Therefore, doping requires fine tuning to obtain optimal photocurrent. Finally, high annealing temperature (above 700 °C for hematite electrodes in practice) drives eqn (6) to the right-hand side because of an increase in entropy, which means electronic compensation expressed by eqn (4) is more favored. Unintentional doping from diffusion of Sn from the fluorine-doped tin oxide (FTO) conductive support can be introduced to improve conductivity, only if the total dopant concentration is within the peak level. Furthermore, high annealing temperature also has an impact on surface properties, which will be discussed in more detail later.
Due to the Schottky defects equilibrium as shown in eqn (7), iron vacancies created through the ionic compensation upon Ti doping (eqn (5)) inevitably reduce the amount of oxygen vacancies:
 | (7) |
Thus, Ti doping is expected to have a similar impact on surface kinetics to oxygen-plasma treatment that reduces oxygen vacancies. Indeed, it is frequently observed in photocurrent curves of Ti-doped samples (e.g., Fig. 3a) that the plateau photocurrent increases while onset potential also shifts anodically.
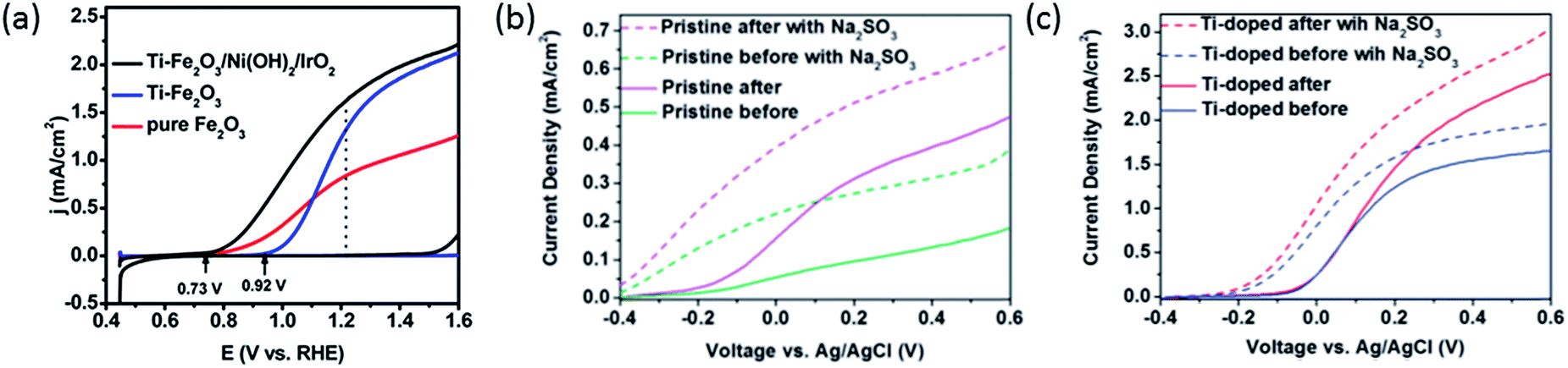 | ||
| Fig. 3 (a) (Photo)current density curves of pure (red) Ti doped (blue), and Ti doped Ni(OH)2–IrO2 coated (black) hematite photoanodes. Reprinted with permission from ref. 10. Copyright 2015 American Chemical Society. (b) Photocurrent density curves of pristine hematite photoanodes before (green) and after (purple) N2 annealing. (c) Photocurrent density curves of Ti doped hematite photoanodes before (blue) and after (red) N2 annealing. Dashed lines in (b) and (c) indicate measurements with Na2SO3 in the electrolyte. Reproduced from ref. 9 with permission from the PCCP Owner Societies. | ||
The plateau photocurrent is associated with a number of factors described above so the degree of improvements would vary. The onset potential, associated with surface kinetics, is more interesting to investigate. Often it is anodically shifted by 0.1 to 0.2 VRHE for films with n-type dopants, but only when annealed at high temperatures (700–800 °C).10,15,41 This is in good agreement with the distribution shift between CTB and UHB, and it is additionally proved by the fact that even with the presence of hole scavenger Na2SO3, the onset potential also has a noticeable shift (Fig. 3b and c).9 For films annealed at relatively lower temperatures, photocurrent is considerably lower or negligible for solution-processed electrodes.33,41,42 In contrast, films prepared by atmospheric-pressure chemical vapor deposition show appreciable photocurrent at the same annealing temperature.42 Given this, we believe that the consequences of high temperature annealing are two-fold: (a) enhancing the hematite crystallinity and (b) removing recombination surface states (r-SS). Zandi and Hamann have identified two types of surface states by rapid scan cyclic voltammetry and have shown that r-SS centered near 0.75 VRHE are removed upon 800 °C annealing (Fig. 4a).43 Notably, a bare hematite photoanode with a record low onset potential of 0.58 VRHE has been fabricated by 10 s of H2–O2 flame treatment at 1700 K.44 Coincidentally, Brillet and co-workers' study on functional annealing gives indirect evidence of r-SS oxidation.45 When their hematite electrode is calcined in a SiO2 template to preserve nanoporosity, the onset potential is 160 mV lower than without template, indicating unsuccessful removal of r-SS.
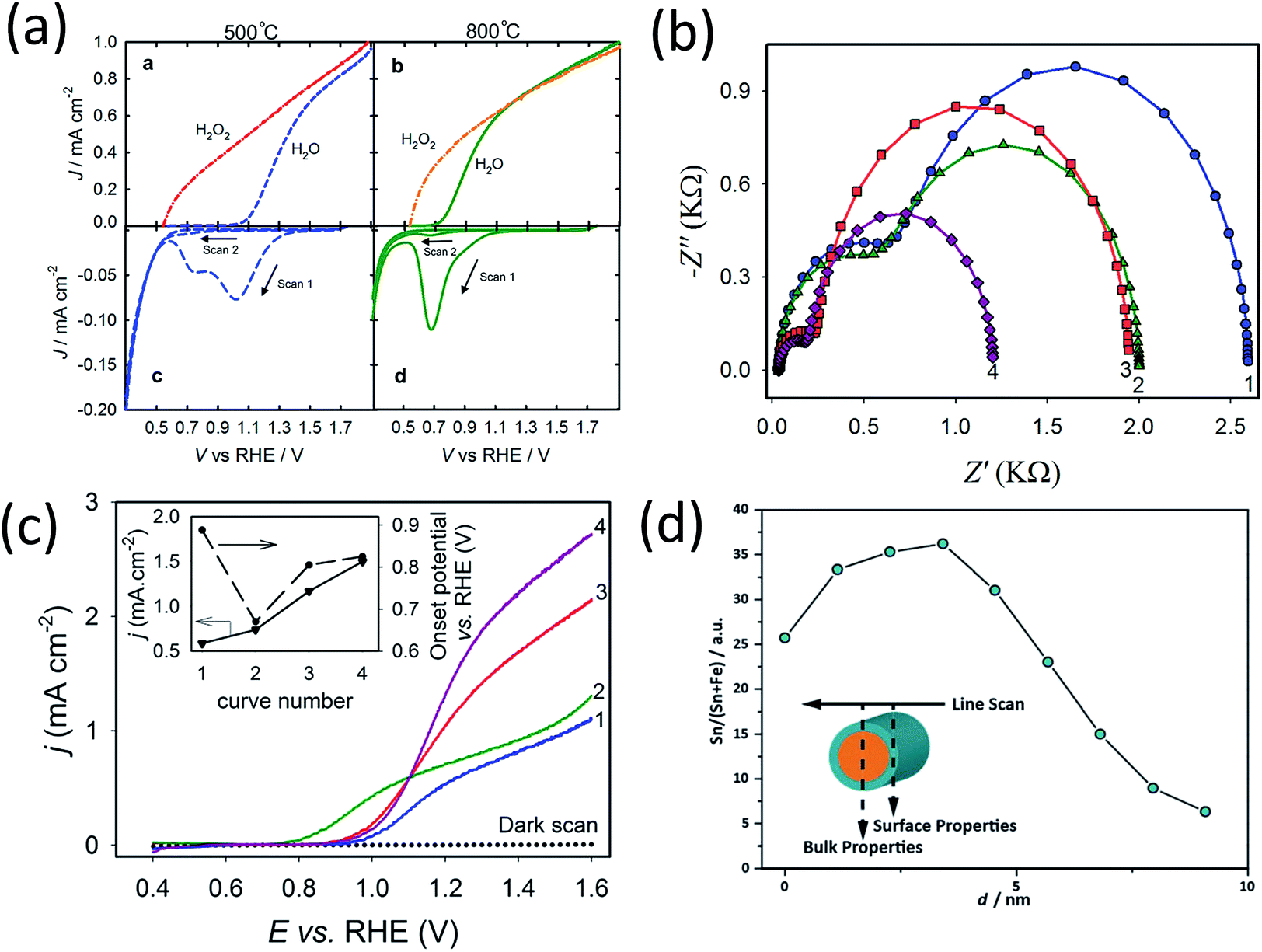 | ||
| Fig. 4 (a) Linear sweep voltammograms for hematite electrodes annealed at (top left) 500 °C and (top right) 800 °C in air measured in H2O and H2O2. Cyclic voltammograms scanned at 1 V s−1 in the dark of the electrodes annealed at 500 °C (bottom left) and 800 °C (bottom right). Reprinted with permission from ref. 69. Copyright 2014 American Chemical Society. (b) Nyquist plots for and (c) photocurrent density curves of (1) pristine hematite, (2) 10.71% Zn, (3) 1.96% Ti, and (4) 1.40% Ti + 5.58% Zn-modified hematite electrodes. Reprinted with permission from ref. 35. Copyright 2014 American Chemical Society. (d) Atomic concentration of Sn as a function of position for a 20% Sn–Fe2O3 photoanode. Reproduced from ref. 50 with permission from the PCCP Owner Societies. | ||
Amongst the n-type dopants, manganese is an exception that negatively shifts onset potential. In spite of an increase of predicted reaction energy (related to overpotential),46 the surface charge transfer accelerates and charge recombination slows down according to IMPS results.3 It has been postulated that Mn forms multivalent oxidation states that leads to a low (O–Mn–O) energy barrier for hole transfer.47 Vibronic superexchange effect is also possibly playing a part, since it has been recently found to enhance electrocatalytic performance of La2NiMnO6.48
Acceptor-type (p-type) dopants such as Zn and Cu create more oxygen vacancies as a result of the dissolution reaction:28
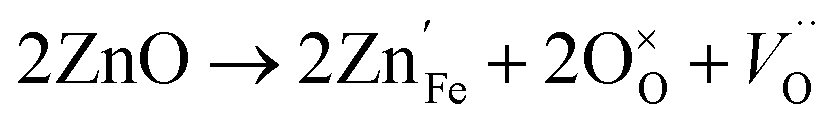 | (8) |
If oxygen is present, it also proceeds via
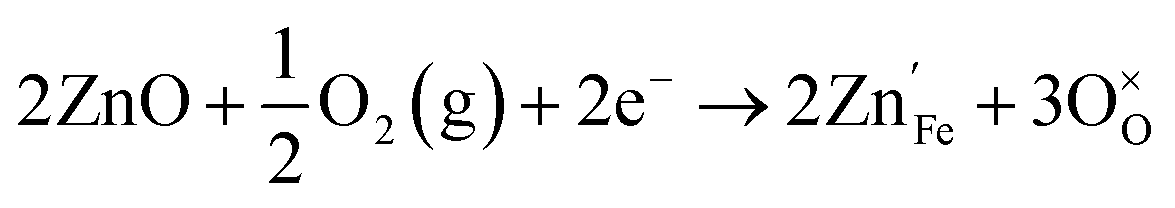 | (9) |
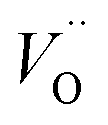 introduced by doping indirectly assist OER, the downside is the drop of bulk conductivity (eqn (3) and (9)). For example, doping of Be at 6% can reduce the plateau photocurrent by nearly 30% at 1.6 VRHE,15 and therefore the dopant concentration should be limited, preferably close to the surface.
introduced by doping indirectly assist OER, the downside is the drop of bulk conductivity (eqn (3) and (9)). For example, doping of Be at 6% can reduce the plateau photocurrent by nearly 30% at 1.6 VRHE,15 and therefore the dopant concentration should be limited, preferably close to the surface.
It can be seen now that overall improvement of photocurrent cannot be easily achieved by using only one treatment. Although annealing in an oxygen deficient atmosphere seems to be one possibility as we have mentioned, the improvement in plateau photocurrent is often quite limited compared to n-type doping (Fig. 3b and c).9 Dual-element doping has been attempted by adding a mixture of n-type and p-type dopants. Mirbagheri et al. has co-doped Ti and Zn into hematite and characterized films with PEIS.35 The advantages of each individual doping, i.e., decreased bulk charge transport resistance for Ti–Fe2O3 and decreased surface charge transfer resistance for Zn–Fe2O3, are both obtained in Ti/Zn–Fe2O3 film at 1.0 VRHE (Fig. 4b). The plateau photocurrent exceeds films doped with either Ti or Zn, which can be attributed to released lattice strain (Fig. 4c).15 Interestingly, its onset potential is maintained at 0.9 VRHE as opposed to shifting toward 0.8 VRHE for Zn–Fe2O3. This result implies that dopant or oxygen vacancy concentration is not simply linearly related to onset potential. In another work in which Sn and Be is co-doped, a similar behavior is recorded. Enhancement in plateau photocurrent density exceeds that of individual doping but onset potential (Eon) is also unchanged compared with Sn–Fe2O3.15 A plausible explanation is that n-type dopants prefer to dominate electrode surface; this has been confirmed in works on Sn and Mn doping, which reveal a gradient of dopant concentration across the film as well as in individual particles (Fig. 4d).3,50
A more desirable configuration combining both n-type and p-type doping has been fabricated by Kay et al. to produce layered single-junctions (Fig. 5a).51 Photocurrents improve at both potential ends when hematite is divided into three layers where top layer is doped with Zn and bottom layer with Ti, although Eon is not as low as with individual Zn doping (Fig. 5b). An interesting feature to note here is that Eon drops with increasing thickness of Zn doped layer. This means that p-type doping should not merely be placed at the outermost layer but also somewhat deeper so that O 2p holes transport more easily through slightly positively charged oxygen anions. On the other hand, this penetration depth should not be too high because, as seen before with Ti/Zn–Fe2O3, bulk co-doping does not reduce Eon. Therefore, control of surface doping of lower valence elements appears to be intricate and challenging.
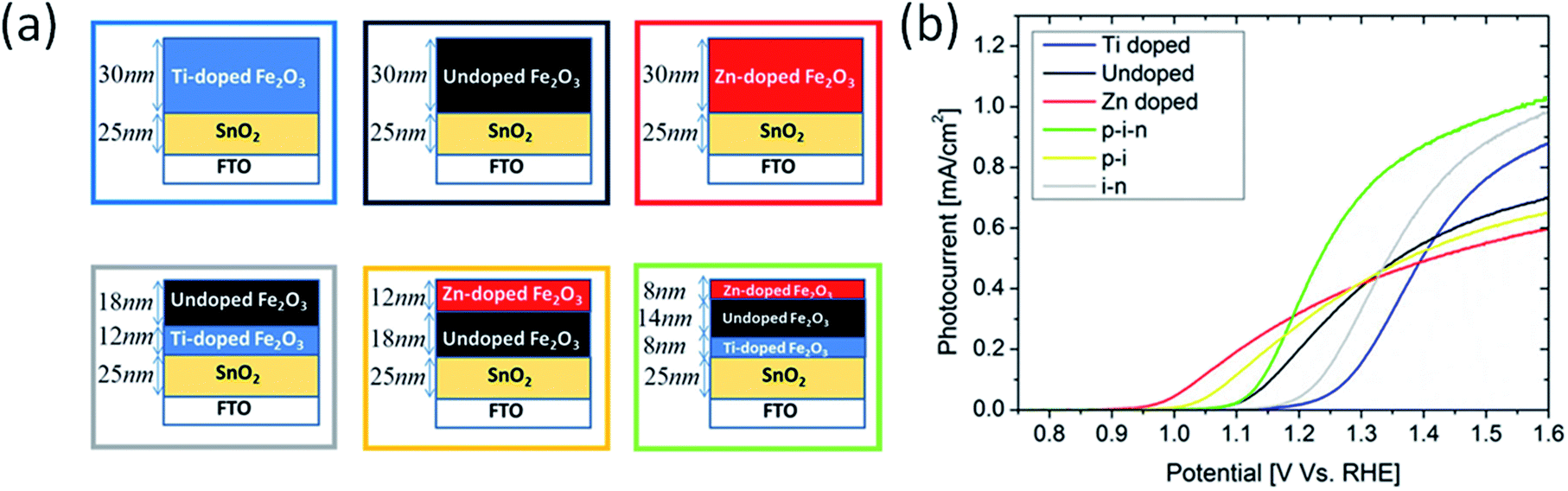 | ||
| Fig. 5 (a) homogeneously Ti-doped (∼1 cation%) hematite film (blue), undoped hematite film (black), homogeneously Zn-doped (∼1 cation%) hematite film (red), heterogeneously doped i–n stack (grey) heterogeneously doped p–i stack (yellow), and heterogeneously doped p–i–n stack (green). (b) Photocurrent density curves of films in (a), following color scheme of boxes in (a). Reprinted with permission from ref. 51. Copyright 2016 American Chemical Society. | ||
The less researched dopants are isovalent elements such as Al and Ga. They also show some promise of improving plateau photocurrent density which is attributed to better conductivity due to small polaron migration.52,53 When doped with Al, there is no apparent change in defect chemistry other than cation replacement and hence no change in Eon.
Surface states
The origin of surface states as intermediate species or as recombination centers has been under heavy debate. In this perspective, we attempt to combine several existing theories and construct a clearer picture. Durrant group has previously summarized some of their pioneering TAS research and has suggested that recombination centers exist and lie a few hundred millivolts below CB of a Si–Fe2O3 (annealed at 500 °C).1 Their dynamic model based on TAS is shown in Fig. 6a.1 At low potentials, when Fermi level is above r-SS level, electrons are intrinsically trapped. Thus, further trapping becomes more difficult (μs) compared to recombination from VB holes (ps–ns). As applied potential becomes more anodic, band bending depletes these states near the surface, and electron trapping (ps–ns) becomes faster than hole trapping (μs–ms). The population of depleted r-SS is reflected by the bleach signal detected at 580 nm (electronic transition illustrated in Fig. 6b and signal in Fig. 6c). Notably, its intensity follows the square root relationship with applied potential, meaning the r-SS are formed within space charge layer. These states are tentatively assigned to Fe2+–Vo that turn into Fe3+–Vo when become depleted.1 They have also been previously regarded as chromophores in Fe–TiO2.54 We have found further evidence in the literature from recent years supporting this assignment, as we shall illustrate below. | ||
| Fig. 6 Energy diagrams and processes involving photogenerated charge carriers for a hematite photoanode at various applied potentials at (i) 0.5, (ii) 1.1 and (iii) 1.6 VRHE. Circles indicate electron trap states. (b) Scheme of electronic transitions responsible for the Vis-NIR transient absorption spectra of hematite photoanodes. Reproduced from ref. 1 with permission from The Royal Society of Chemistry. (c) Transient absorption decay dynamics of 650 nm. (d) Transient absorption decay dynamics of 580 nm. Reproduced from ref. 65 with permission from The Royal Society of Chemistry. | ||
The positive signal above 650 nm on TAS results indicates the population and lifetime of holes (electronic transition illustrated in Fig. 6b and signal in Fig. 6d).1,26 Its intensity at short time domains doubles after oxygen-plasma treatment, likely due to the reduction in the quantity of oxygen vacancies, which are ultrafast recombination centers.26 This is supported by a simulation demonstrating that oxygen vacancies are even faster charge recombination centers than direct VB–CB recombination by 10 and 30 times for charged and neutral forms, respectively.55 The timescale (ps–ns) of electron trapping from CB to r-SS measured by another work of Durrant group24 is also computationally supported by Zhou et al.55 Consequently, oxygen vacancies are very likely to be an integral component of r-SS.
The other part of r-SS, Fe2+, can be more confidently confirmed with several approaches. Hu et al. has found a numeric match between the density of states and surface Fe2+ species concentration, thus confirming that r-SS are highly associated with Fe2+.56 As previously mentioned, high temperature annealing is capable of removing this type of r-SS (Fe2+–Vo), presumably by oxidation of Fe2+ to Fe3+. For a 500 °C treated sample, the empty form of electron traps can live up to 10 ms indicated by 580 nm bleach signals, whereas the signal disappears for a 750 °C treated sample for a wide range of applied potentials.26 Although a feature emerges at 1.5 VRHE, the timescale is 100 μs as opposed to 10 ms, so it must originate from recombination surface states of a different nature.26 Hence, high temperature treatment can successfully reduce the population of Fe2+, not to mention the improvement in crystallinity. Here, we should note that although high temperature introduces more oxygen vacancies, they are not as influential as Fe2+, which act as redox centers. Although oxidation potential of Fe2+ at hematite surface is undefined, we notice that the standard redox potential of Fe3+/Fe2+ (0.77 VRHE) is located several hundred millivolts below CB edge (0.3–0.4 VRHE) and extremely close to that observed by Zandi et al. (0.75 VRHE).43 Moreover, the computationally predicted position of 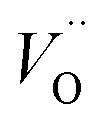 at 400 mV below CB by Zhou et al.55 suggests possible interaction between Vo and Fe2+ due to their close proximity in energy alignment.
at 400 mV below CB by Zhou et al.55 suggests possible interaction between Vo and Fe2+ due to their close proximity in energy alignment.
The spectral weight distribution of CTB, interestingly, follows a square root dependence on bias potential, in line with the development of 580 nm signal in TAS.1 It suggests strengthened band bending with applied potential, while r-SS is consequently being depleted (Fig. 6a). However, it is hard to judge which of the two types of holes is performing the depletion. Although we believe it is more likely to be the UHB holes due to their closer proximity in energy and the absence of its spectral weight at low bias, possibility of recombination between CTB and r-SS cannot be ruled out. This question could be readily answered by NEXAFS measurements on the same samples after r-SS removal by high temperature annealing, although it remains unexplored in literature.
The surface states directly detected by TAS suggest that they are not only at the top surface layer but extend into the bulk as the space charge region builds up as a function of applied bias. However, since the space charge region is only a few nanometers wide, these states can be still termed as “surface states”. This point has been previously mentioned by Barroso et al.42
Knowing the chemical origin of r-SS, we now focus on the crucial different effects that annealing and cation doping can have on the presence of oxygen vacancies and the relative amounts of CTB and UHB holes. N-type dopants (for example, Ti, Si and Sn) increase the concentration of nearby Fe2+.15,57,58 On the other hand, filling oxygen vacancies by oxygen plasma treatment or likewise reduces Fe2+ concentration.27 Although the increase of Fe2+ is known to assist polaron hopping and subsequently electrical conductivity,59 its existence near the surface (forming r-SS) would quench both CTB and UHB holes. An amorphous layer of FexSn1−xO4 of 1–2 nm reduces the charge injection efficiency below ca. 1.1 VRHE.60 But more efficient charge injection was recorded at higher potentials, where Fe2+ (r-SS) species are oxidized by the bias, so that UHB can be effectively used for OER. Such response is a firm proof of our theory on the role of UHB and CTB holes as there are no bulk effects involved in this case. Conversely, p-type doping would reduce Fe2+ concentration, therefore expected to enhance both CTB and UHB hole fluxes. At lower potentials, in particular, the removal of r-SS allows for a much higher generation of effective CTB holes for water oxidation. Hence, lowered onset potential is often observed.8,35,61 Direct evidence can be found in a work where an Ni-doped overlayer of NixFe2−xO3 cathodically shifts onset potential of hematite by 100 mV while surprisingly enhancing photocurrent density by 2–3 fold.61 Creating a p-type α-Fe2O3 layer by 3% Mg doping on the top 20 nm layer has reduced onset potential by more than 200 mV, while slightly raising plateau saturation too.8 Although the establishment of internal fields by homojunctions is claimed responsible for the improvement in the latter case, we believe changes in the contribution of holes are also playing a role here since uniform bulk p-type doping sometimes produces similar effects (Fig. 4c).35
In contrast to r-SS, the forms and chemistry of intermediate surface states (i-SS) that mediate OER are far more complicated and are still under heavy debate. Multiple oxidation states of Fe, such as IV, V, and even VI have been proposed to be present during OER, some of which have been experimentally observed.17,62–64 It is also possible for a variety of OER mechanisms to occur simultaneously with either single or multiple oxidation states of Fe.
Regardless of the nature of i-SS, a procedural scheme can be demonstrated to show the fate of CTB and UHB holes in hematite photoanodes when r-SS are present. Fig. 7 shows a schematic illustration of mixed energy diagrams of hematite photoanodes with CTB (red) and UHB (blue) and with the presence of r-SS at three applied potentials (0.55, 0.9, and 1.3 VRHE). The X-ray energy scale is also displayed, where peak edge is simply set to be at half-height (528.2 eV), matching the CB minimum at electrode surface. A few basic assumptions were made in Fig. 7 for a typical hematite photoanode: (a) a band gap of 2.2 eV;65 (b) CB minimum at 0.3 VRHE,66 (c) a flat band potential of 0.5 VRHE,67 and (d) r-SS at 0.75 VRHE. As shown in Fig. 7a at EF = 0.55 VRHE, which is close to the flat-band potential, the holes at the surface are very limited because they come from only a short diffusion layer of about 2 nm into the surface with little assistance from band bending.16 Holes can easily recombine with r-SS since they lie below Fermi level and will be immediately refilled with electrons (Fig. 7a). As potential is swept anodically, r-SS near the surface begin to be depleted primarily by UHB holes because of smaller energy differences (marked by thin purple arrow in Fig. 7b). Dare-Edwards and co-workers has pointed out that VB holes arrive at surface in Fe3+ orbitals; these Fe3+ have negligible rate constants for water oxidation.67 Meanwhile, CTB holes do not generate photocurrent as well because of their low surface concentration. They mostly undergo surface recombination (illustrated by a thick purple arrow, Fig. 7b). Therefore, even if r-SS are removed by high temperature annealing, photocurrent onset still cannot approach the same value in H2O2 (c.f. green curve, Fig. 7d). When potential increases further, r-SS near the surface soon become fully depleted above 0.9 VRHE according to constant 650 nm signal intensity below 10−3 s from TAS results shown in Fig. 6c.1 At this point, CTB holes reach a critical concentration, leading to photocurrent onset as holes arriving in O 2p have a facile faradaic route to O2 formation (thin red arrow in Fig. 7b).67 A small amount of UHB holes also exist but do not yet contribute to photocurrent.16,68 As bias is further swept anodically, rapid water splitting kinetics is capable of dissipating the CTB (O 2p) accumulated holes, reducing its spectral weight observed with NEXAFS (Fig. 7c). Likewise, this trend appears for UHB holes but approximately 0.1–0.2 V more positive (Fig. 7d). Note that the peak intensity is much higher than that of CTB, which is possibly due to the lower overpotential of UHB holes compared to 1.23 VRHE. Information about surface OER kinetics of excessive long-lived holes can be extracted from the second decay phase of 650 nm signal (Fig. 6c).1Fig. 7d shows the J–V responses of a typical hematite electrode (produced at 500 °C) marking out the points where each scenario projects to. The presence of r-SS is partly responsible for the strong contrast between J–V curves measured in H2O and H2O2.
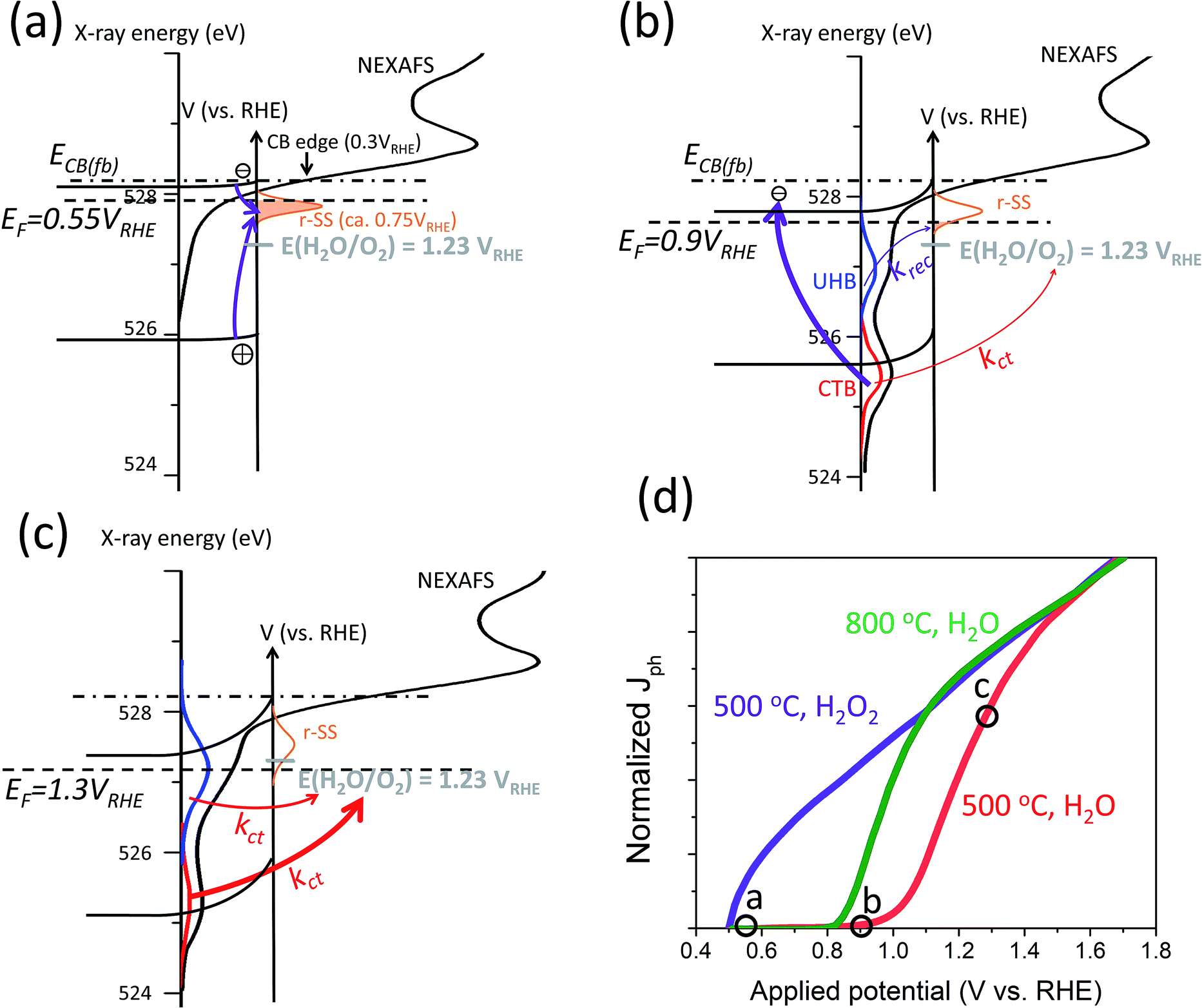 | ||
| Fig. 7 Schematic illustration of mixed energy diagrams of hematite photoanodes at operating conditions with CTB (red) and UHB (blue) and with the presence of r-SS at applied potentials of (a) 0.55, (b) 0.9, and (c) 1.3 VRHE. Arrows of different thicknesses indicate relative rates of important charge transfer and surface recombination processes. Purple arrows indicate surface recombination processes; red arrows indicate charge transfer processes. Grey dashes indicate OER potential of 1.23 VRHE. NEXAFS data obtained from ref. 16. (d) Normalized typical J–V responses of a hematite photoanode calcined at 500 °C in H2O (red) and H2O2 (purple) and calcined at 800 °C in H2O (green). The position of the scenarios presented in (a), (b) and (c) are marked out in the J–V red curve of (d). | ||
A better understanding of the photoelectrochemistry of a hematite photoanode in operation from spectroscopic findings allows for more rational investigation of PEIS outcomes. It is apparent that the overall spectral weight curve from NEXAFS closely resembles surface capacitance measured by PEIS. We believe that the surface capacitance is a reflection of CTB and UHB holes, in the form of both intermediate species and possibly positively charged r-SS but further investigation on this topic remains worthwhile.
At present, PEIS data is commonly interpreted following different models. In one model, charge transfer takes place directly from VB holes and surface states only account for recombination (Fig. 8a). In an alternative model, both charge transfer and recombination take place via the same surface states (Fig. 8b). Deciding which model is more appropriate depends on the relative quantities of i-SS and r-SS as well as applied potential. Equivalent circuits of these two models are mathematically indistinguishable, and calculation of rate constants by either method produces same values.68 However, Klahr et al. points out that the latter model is more physically meaningful because a peak maximum corresponds to a dip in charge transfer resistance.25 We agree with this statement but moreover, we believe that the other model is possibly not suitable at all for hematite on the basis of more experimental evidence, as follows. For hematite films calcined at intermediate temperatures (around 500 °C), surface capacitance Css peaks were not observed to start from 0.6 V, which would be the case if charged r-SS were oxidized by CTB holes according to NEXAFS spectral weights. Instead, Css only starts near the onset potential of 0.9 to 1 V.2,25 In contrast, for 800 °C calcined samples, density of surface states Nss (derived directly from Css) does rise from 0.6 V as extrapolated from Fig. 8c,7 which demonstrates the appearance of CTB holes for OER. This is also in line with the intense peak centered at 0.75 V in Fig. 4a(d) recorded using CV in the dark.69 Furthermore, Ti-doped films heated at 800 °C that are known to have more Fe2+ show depressed Css at low potentials (Fig. 8c), which further proves that PEIS does not detect r-SS. Note at high potentials, Css increases substantially, peaking at 0.2 V above non-doped samples, indicating a much higher maximum surface concentration of UHB.
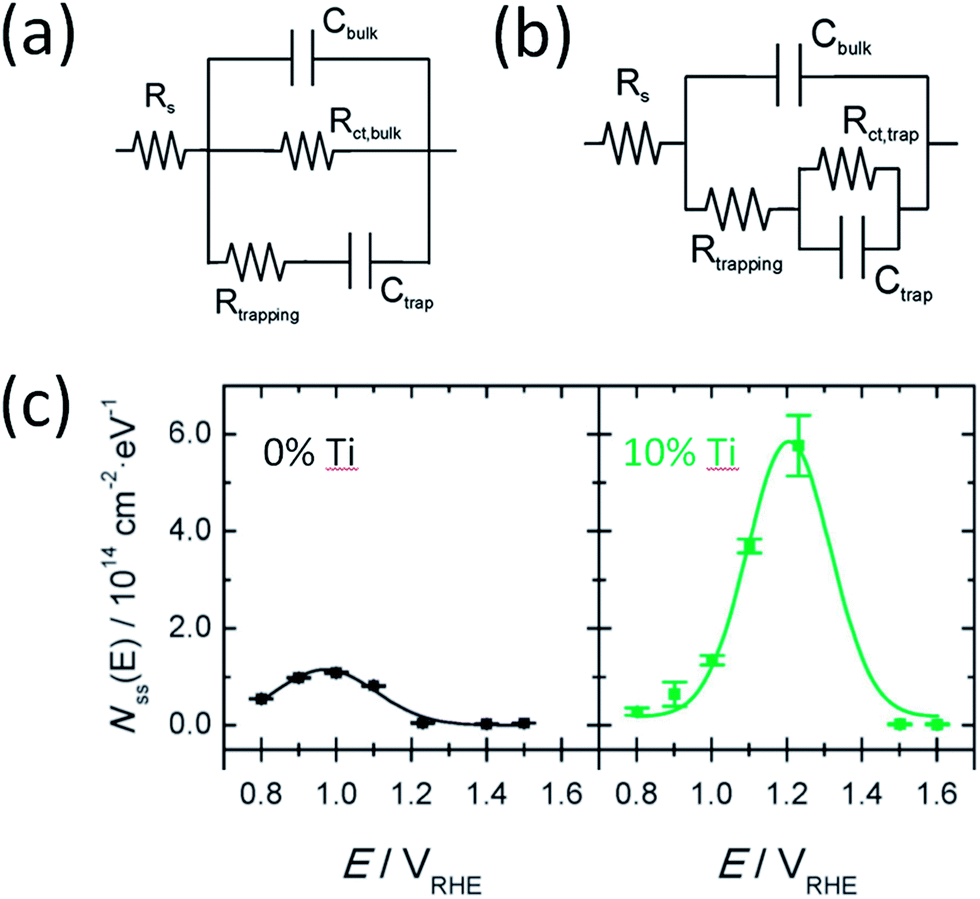 | ||
| Fig. 8 Two types of equivalent circuit from PEIS to represent hematite photoanodes under illumination. Model in (a) suggests charge transfer through valence band and recombination through trap states. Model (b) suggests charge transfer and recombination both occur through surface trap states. Reprinted with permission from ref. 25. Copyright 2012 American Chemical Society. (c) Density of surface states calculated from surface capacitance measured using PEIS with model (b) for pristine and Ti doped hematite photoanodes. Reproduced from ref. 7 with permission from The Royal Society of Chemistry. | ||
Knowing the physical origin of r-SS, we are able to summarize the mixed effects of oxygen vacancies in hematite photoanodes. First, they discharge the surrounding oxygen atoms to yield more O− species that are beneficial for charge transfer of CTB holes. Second, they act as fast recombination sites either on their own or coupled with Fe2+. Consequently, although the distribution of surface capacitance is located more cathodically, its intensity is lower than when there are less oxygen vacancies, as evidenced again in Fig. 8c. This trade-off is probably the source of difficulties when trying to improve the activity of a hematite photoanode, especially at low applied potentials. Apart from these roles already discussed, oxygen vacancies may also reduce the adsorption energy of H2O molecules, and subsequently reduce OER overpotential and Tafel slope, as observed for NiCo2O4.39,70
Deposition of overlayers
It would be also helpful to consider the effect of overlayers on surface states, which is still under heavy debate. As Barroso has mentioned in an early report, there is some ambiguity regarding the definition of “surface state”.71 A majority of publications use this term for states that exist only at the top molecular layer, whereas Fe2+–Vo can spread deeper into the surface, for example, in the space charge region. This notion clarified, the roles of surface layers including co-catalysts, compact non-catalytic layers, and surface doped layers become clearer.The deposition of an overlayer can partly or entirely cover the defects of the outermost hematite, including a fraction of Fe2+–Vo and other possible types of surface states. Here, we emphasize the difference between a co-catalyst (e.g., transition metal oxides and (oxy)hydroxides) and a compact non-catalytic layer (e.g., Ga2O3 and Al2O3). It has often been reported that the plateau photocurrent density drops with a Ga2O3 overlayer.53,72 A possible justification is that when Ga2O3 covers Fe2+ sites, it influences O 2p (CTB) and UHB holes differently: (a) more O 2p (CTB) holes will be transferred to O atoms of Ga2O3 rather than used for oxidizing r-SS, improving the low potential performance; (b) as more Fe sites are covered, charge transfer through UHB holes is inhibited. In the case of cobalt-based co-catalysts such as CoPi, the increase in O 2p (CTB) hole flux toward Co sites does not lead to acceleration in catalysis.2,71 Instead, recombination is retarded, which is associated with the low electrocatalytic activity of Co by itself compared with Fe sites, as reported by Boettcher group.73 For instance, the measured turnover frequency was 0.035 s−1 for Co3O4 at η = 325 mV,74 while 12 s−1 for CoFeOx at η = 350 mV.75 Our group has recently reported a moderate improvement of charge transfer rate with CoFeOx coating only when the loading is extremely low;11 and we have observed the same for thin NiFeOx coating. Thus, it can be said that the only part of the co-catalyst that assists OER is the layer where it connects to the hematite structure.
In contrast, if Co or Ni atoms are dispersed into hematite near the surface and interact with Fe atoms inside hematite structure, a far more significant benefit will be harnessed, in addition to reducing the number of Fe2+. This comparison of surface doping and deposition has been made clear by Cheng et al. with Co;61 the same prediction was also made through simulation in the same year by Liao et al.46 Unfortunately, this approach has been considerably outnumbered in the literature by surface deposition methods to reduce onset potential.
Charge generation and transport
As described in Fig. 1b, single and double ligand field transitions do not generate charge pairs upon irradiation. For this reason, Kennedy and Frese's calculation of the absorption coefficient based on J–V curves was found lower than the real value.20 A few more theoretical and experimental approaches confirmed this major efficiency loss mechanism that hampers high photogeneration yields.76,77 This is indeed another major curtailment to the expected photocurrent output. The low photogeneration yield has a strong wavelength dependence, which has been associated to polaronic trapping23,78 but different interpretations coexist.79 Therefore, better understanding of charge carrier dynamics must be first attained before research moves on to minimizing these unwanted transitions.Currently, the best hematite-based photoanodes reported in the literature can only output photocurrents around 5 mA cm−2 at 1.23 VRHE under 1-sun illumination (AM1.5G), which are much lower than the theoretical limit of 12.6 mA cm−2.80–83 It has frequently been reported that while charge injection efficiency can reach 90–100% with strong bias, charge separation efficiency is far lower regardless of applied potential.9,60 Therefore, we and other researchers3 believe that to further improve the performance of hematite as photoanodes and make them industrially viable, research should be mainly aiming at enhancing the bulk charge transport properties rather than surface OER kinetics, since researchers have established multiple tools to achieve this, e.g., by surface doping or controlled annealing.
To increase the electronic transport, or conductivity, the most obvious way is n-type doping. In Engel and Tuller's work, the conductivity of 1% Ti-doped hematite shows around four orders of magnitude higher conductivity at room temperature if calcined in air, which even reaches nine orders of magnitude higher in low O2 concentration (0.1% and 0.01%).38 In another work with the same doping level, photocurrent density for OER is indeed substantially increased. However, it only increases by less than three times at 1.23 VRHE in the presence of 0.5 M H2O2, which indicates that the hole flux increases by no more than one order of magnitude. The authors also confirmed that the surface hole concentration for water splitting reaction increases by only about four times using IMPS. The most probable reason for such low improvements in hole current as opposed to significant improvements in conductivity is the strengthened band bending due to the higher concentration of free electrons.
For hematite, conductivity along crystal orientation (110) has been reported to be four orders of magnitude higher than along (001).84 However, these two orientations offer the same hole flux,85 presumably because there is no difference in free electron density, giving a similar chance of bulk recombination.
Improving the hole (minority carrier) transport, i.e., mobility and lifetime, is a much more significant issue. For example, Peter et al. discovered that in stark contrast to the Gartner model, recombination is an apparent hindrance even within the space charge region.86 Applied bias has been recognized as a way to retard ultrafast recombination in the bulk hematite, but not much progress has been made by means of film fabrication.24 Another notable measure to enhance minority carrier mobility is by thermal energy, which can be achieved by concentrated solar illumination. Moreover, higher light intensity can also increase photovoltage and fill factor.87 Future research in this direction would be highly meaningful.
Conclusion and outlook
In summary, we have discussed the presence in hematite of charge transfer band (CTB) O 2p holes and upper Hubbard band (UHB) Fe 3d holes upon illumination and critically analyzed the role of oxygen vacancies and doping, as well as the origin of recombination surface states. Our main conclusion is that introducing oxygen vacancies at surfaces, by controlled annealing or p-type doping, gives rise to more positively charged oxygen anions, which assist the transport of CTB holes; whereas reducing the amount of oxygen vacancies, by oxygen-plasma treatment or n-type doping, decreases the degree of Fe–O hybridization and facilitates charge transfer via UHB.26 Additionally, concentration of near-surface Fe2+ species is influenced differently upon annealing or doping. Among the methodologies covered here, the most promising way is to n-type dope in the bulk plus p-type dope near the surface. We then define one type of recombination surface states to be Fe2+–Vo that mainly consumes CTB holes and prevents early onset potential. These states are strongly affected by heating conditions: a short high temperature can readily remove them. On the basis of TAS findings, integration of NEXAFS results into photoelectrochemistry has led us to a far more clarified energy picture at hematite electrodes. Our theory is in agreement with a majority of theoretical and experimental data in the literature. The impacts of high temperature treatment, overlayer deposition and surface doping have also been discussed. We note that the key limiting factor of hematite as photoanodes is its poor hole transport properties instead of electronic transport (conductivity). If this issue is overcome, and given its remarkable stability, hematite would undoubtedly secure its progress toward commercial PEC application.Finally, this perspective has deepened the understanding of the PEC performance of hematite photoanodes by various surface or bulk modifications and provided a useful guide to more efficient photoanodes with other semiconductors for solar water splitting. More importantly, this methodology of interdisciplinary literature review has rarely been applied in materials science, but would be valuable in other fields to gain novel and original understanding from present knowledge.
Conflicts of interest
The authors declare no conflicts of interest.References
- M. Barroso, S. R. Pendlebury, A. J. Cowan and J. R. Durrant, Chem. Sci., 2013, 4, 2724–2734 RSC.
- G. M. Carroll and D. R. Gamelin, J. Mater. Chem. A, 2016, 4, 2986–2994 RSC.
- Gurudayal, L. M. Peter, L. H. Wong and F. F. Abdi, ACS Appl. Mater. Interfaces, 2017, 9, 41265–41272 CrossRef CAS PubMed.
- D. A. Grave, N. Yatom, D. S. Ellis, M. C. Toroker and A. Rothschild, Adv. Mater., 2018, 1706577, 1–10 Search PubMed.
- P. Liao, M. C. Toroker and E. A. Carter, Nano Lett., 2011, 11, 1775–1781 CrossRef CAS PubMed.
- R. F. G. Gardner, R. L. Moss and D. W. Tanner, Br. J. Appl. Phys., 1966, 17, 55–61 CrossRef CAS.
- D. Monllor-Satoca, M. Bärtsch, C. Fàbrega, A. Genç, S. Reinhard, T. Andreu, J. Arbiol, M. Niederberger and J. R. Morante, Energy Environ. Sci., 2015, 8, 3242–3254 RSC.
- Y. Lin, Y. Xu, M. T. Mayer, Z. I. Simpson, G. McMahon, S. Zhou and D. Wang, J. Am. Chem. Soc., 2012, 134, 5508–5511 CrossRef CAS PubMed.
- X. Zhao, J. Feng, S. Chen, Y. Huang, T. C. Sum and Z. Chen, Phys. Chem. Chem. Phys., 2017, 19, 1074–1082 RSC.
- Z. Wang, G. Liu, C. Ding, Z. Chen, F. Zhang, J. Shi and C. Li, J. Phys. Chem. C, 2015, 119, 19607–19612 CrossRef CAS.
- J. Zhang, R. García-Rodríguez, P. Cameron and S. Eslava, Energy Environ. Sci., 2018, 11, 2972 RSC.
- C. Zachäus, F. F. Abdi, L. M. Peter and R. van de Krol, Chem. Sci., 2017, 8, 3712–3719 RSC.
- A. Pu, J. Deng, M. Li, J. Gao, H. Zhang, Y. Hao, J. Zhong and X. Sun, J. Mater. Chem. A, 2014, 2, 2491–2497 RSC.
- T. Y. Yang, H. Y. Kang, U. Sim, Y. J. Lee, J. H. Lee, B. Koo, K. T. Nam and Y. C. Joo, Phys. Chem. Chem. Phys., 2013, 15, 2117–2124 RSC.
- A. Annamalai, H. H. Lee, S. H. Choi, S. Y. Lee, E. Gracia-Espino, A. Subramanian, J. Park, K. J. Kong and J. S. Jang, Sci. Rep., 2016, 6, 23183 CrossRef CAS PubMed.
- A. Braun, K. Sivula, D. K. Bora, J. Zhu, L. Zhang, M. Grätzel, J. Guo and E. C. Constable, J. Phys. Chem. C, 2012, 116, 16870–16875 CrossRef CAS.
- O. Zandi and T. W. Hamann, Nat. Chem., 2016, 8, 778–783 CrossRef CAS PubMed.
- P. Liao and E. A. Carter, J. Phys. Chem. C, 2011, 115, 20795–20805 CrossRef CAS.
- R. F. G. Gardner, F. Sweett and D. W. Tanner, J. Phys. Chem. Solids, 1963, 24, 1183–1196 CrossRef CAS.
- J. H. Kennedy and K. W. Frese, J. Electrochem. Soc., 1978, 125, 709–714 CrossRef CAS.
- J. C. Papaioannou, G. S. Patermarakis and H. S. Karayianni, J. Phys. Chem. Solids, 2005, 66, 839–844 CrossRef CAS.
- R. F. G. Gardner, F. Sweett and D. W. Tanner, J. Phys. Chem. Solids, 1963, 24, 1183–1196 CrossRef CAS.
- J. Husek, A. Cirri, S. Biswas and L. R. Baker, Chem. Sci., 2017, 8, 8170–8178 RSC.
- S. R. Pendlebury, X. Wang, F. Le Formal, M. Cornuz, A. Kafizas, S. D. Tilley, M. Grätzel and J. R. Durrant, J. Am. Chem. Soc., 2014, 136, 9854–9857 CrossRef CAS PubMed.
- B. Klahr, S. Gimenez, F. Fabregat-Santiago, T. Hamann and J. Bisquert, J. Am. Chem. Soc., 2012, 134, 4294–4302 CrossRef CAS PubMed.
- M. Pyeon, T. P. Ruoko, J. Leduc, Y. Gönüllü, M. Deo, N. V. Tkachenko and S. Mathur, J. Mater. Res., 2018, 33, 455–466 CrossRef CAS.
- Y. Hu, F. Boudoire, I. Hermann-Geppert, P. Bogdanoff, G. Tsekouras, B. S. Mun, G. Fortunato, M. Graetzel and A. Braun, J. Phys. Chem. C, 2016, 120, 3250–3258 CrossRef CAS.
- R. Van de Krol and M. Gratzel, Photoelectrochemical Hydrogen Production, Springer, 2012 Search PubMed.
- C. Zhu, C. Li, M. Zheng and J. J. Delaunay, ACS Appl. Mater. Interfaces, 2015, 7, 22355–22363 CrossRef CAS PubMed.
- M. Rioult, D. Stanescu, E. Fonda, A. Barbier and H. Magnan, J. Phys. Chem. C, 2016, 120, 7482–7490 CrossRef CAS.
- X. Zhang, P. Klaver, R. Van Santen, M. C. M. Van De Sanden and A. Bieberle-Hütter, J. Phys. Chem. C, 2016, 120, 18201–18208 CrossRef CAS.
- Z. Zhou, P. Huo, L. Guo and O. V. Prezhdo, J. Phys. Chem. C, 2015, 119, 26303–26310 CrossRef CAS.
- K. D. Malviya, D. Klotz, H. Dotan, D. Shlenkevich, A. Tsyganok, H. Mor and A. Rothschild, J. Phys. Chem. C, 2017, 121, 4206–4213 CrossRef CAS.
- M. C. Toroker, J. Phys. Chem. C, 2014, 118, 23162–23167 CrossRef CAS.
- N. Mirbagheri, D. Wang, C. Peng, J. Wang, Q. Huang, C. Fan and E. E. Ferapontova, ACS Catal., 2014, 4, 2006–2015 CrossRef CAS.
- R. Franking, L. Li, M. A. Lukowski, F. Meng, Y. Tan, R. J. Hamers and S. Jin, Energy Environ. Sci., 2013, 6, 500–512 RSC.
- M. Rioult, H. Magnan, D. Stanescu and A. Barbier, J. Phys. Chem. C, 2014, 118, 3007–3014 CrossRef CAS.
- J. Engel and H. L. Tuller, Phys. Chem. Chem. Phys., 2014, 16, 11374–11380 RSC.
- M. T. Nguyen, S. Piccinin, N. Seriani and R. Gebauer, ACS Catal., 2015, 5, 715–721 CrossRef CAS.
- J. Lee and S. Han, Phys. Chem. Chem. Phys., 2013, 15, 18906–18914 RSC.
- Y. Ling and Y. Li, Nano Lett., 2011, 11, 2119–2125 CrossRef CAS PubMed.
- M. Barroso, C. a Mesa, S. R. Pendlebury, A. J. Cowan, T. Hisatomi and K. Sivula, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15640–15645 CrossRef CAS PubMed.
- O. Zandi and T. W. Hamann, J. Phys. Chem. Lett., 2014, 5, 1522–1526 CrossRef CAS PubMed.
- J. Han, X. Zong, Z. Wang and C. Li, Phys. Chem. Chem. Phys., 2014, 16, 23544–23548 RSC.
- J. Brillet, M. Grätzel and K. Sivula, Nano Lett., 2010, 10, 4155–4160 CrossRef CAS PubMed.
- P. Liao, J. A. Keith and E. A. Carter, J. Am. Chem. Soc., 2012, 134, 13296–13309 CrossRef CAS PubMed.
- P. Liao and E. A. Carter, J. Appl. Phys., 2012, 112, 1–13 CrossRef PubMed.
- Y. Tong, J. Wu, P. Chen, H. Liu, W. Chu, C. Wu and Y. Xie, J. Am. Chem. Soc., 2017, 139, 5242 CrossRef PubMed.
- J. Simfukwe, R. E. Mapasha, A. Braun and M. Diale, MRS Adv., 2018, 3, 669–678 CrossRef CAS.
- H. K. Dunn, J. M. Feckl, A. Müller, D. Fattakhova-Rohlfing, S. G. Morehead, J. Roos, L. M. Peter, C. Scheu and T. Bein, Phys. Chem. Chem. Phys., 2014, 16, 24610–24620 RSC.
- A. Kay, D. A. Grave, D. S. Ellis, H. Dotan and A. Rothschild, ACS Energy Lett., 2016, 1, 827–833 CrossRef CAS.
- A. Kleiman-Shwarsctein, M. N. Huda, A. Walsh, Y. Yan, G. D. Stuckyst, Y. S. Hu, M. M. Al-Jassim and E. W. McMland, Chem. Mater., 2010, 22, 510–517 CrossRef CAS.
- L. Steier, I. Herraiz-Cardona, S. Gimenez, F. Fabregat-Santiago, J. Bisquert, S. D. Tilley and M. Grätzel, Adv. Funct. Mater., 2014, 24, 7681–7688 CrossRef CAS.
- W. Clark and P. Broadhead, J. Phys. C: Solid State Phys., 1970, 3, 1047–1054 CrossRef CAS.
- Z. Zhou, J. Liu, R. Long, L. Li, L. Guo and O. V. Prezhdo, J. Am. Chem. Soc., 2017, 139, 6707–6717 CrossRef CAS PubMed.
- Y. Hu, F. Boudoire, I. Hermann-Geppert, P. Bogdanoff, G. Tsekouras, B. S. Mun, G. Fortunato, M. Graetzel and A. Braun, J. Phys. Chem. C, 2016, 120, 3250–3258 CrossRef CAS.
- A. Bandyopadhyay, J. Velev, W. H. Butler, S. K. Sarker and O. Bengone, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 174429 CrossRef.
- I. Cesar, A. Kay, J. A. Gonzalez Martinez and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 4582–4583 CrossRef CAS PubMed.
- G. S. Parkinson, Surf. Sci. Rep., 2016, 71, 272–365 CrossRef CAS.
- L. Xi, S. Y. Chiam, W. F. Mak, P. D. Tran, J. Barber, S. C. J. Loo and L. H. Wong, Chem. Sci., 2013, 4, 164–169 RSC.
- W. Cheng, J. He, Z. Sun, Y. Peng, T. Yao, Q. Liu, Y. Jiang, F. Hu, Z. Xie, B. He and S. Wei, J. Phys. Chem. C, 2012, 116, 24060–24067 CrossRef CAS.
- L. M. Peter, K. G. U. Wijayantha and A. A. Tahir, Faraday Discuss., 2012, 155, 309–322 RSC.
- N. Yatom, O. Neufeld and M. Caspary Toroker, J. Phys. Chem. C, 2015, 119, 24789–24795 CrossRef CAS.
- B. M. Hunter, N. B. Thompson, A. M. Müller, G. R. Rossman, M. G. Hill, J. R. Winkler and H. B. Gray, Joule, 2018, 2, 747–763 CrossRef CAS.
- S. R. Pendlebury, A. J. Cowan, M. Barroso, K. Sivula, J. Ye, M. Grätzel, D. R. Klug, J. Tang and J. R. Durrant, Energy Environ. Sci., 2012, 5, 6304–6312 RSC.
- M. Gratzel, Nature, 2001, 414, 338–344 CrossRef CAS PubMed.
- M. P. Dare-Edwards, J. B. Goodenough, A. Hamnett and P. R. Trevellick, J. Chem. Soc., Faraday Trans. 1, 1983, 79, 2027 RSC.
- F. Le Formal, S. R. Pendlebury, M. Cornuz, S. D. Tilley, M. Grätzel and J. R. Durrant, J. Am. Chem. Soc., 2014, 136, 2564–2574 CrossRef CAS PubMed.
- O. Zandi and T. W. Hamann, J. Phys. Chem. Lett., 2014, 5, 1522–1526 CrossRef CAS PubMed.
- J. Bao, X. Zhang, B. Fan, J. Zhang, M. Zhou, W. Yang, X. Hu, H. Wang, B. Pan and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 7399–7404 CrossRef CAS PubMed.
- M. Barroso, C. a Mesa, S. R. Pendlebury, A. J. Cowan, T. Hisatomi and K. Sivula, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15640–15645 CrossRef CAS PubMed.
- T. Hisatomi, F. Le Formal, M. Cornuz, J. Brillet, N. Tétreault, K. Sivula and M. Grätzel, Energy Environ. Sci., 2011, 4, 2512–2515 RSC.
- M. S. Burke, S. Zou, L. J. Enman, J. E. Kellon, C. A. Gabor, E. Pledger and S. W. Boettcher, J. Phys. Chem. Lett., 2015, 6, 3737–3742 CrossRef CAS PubMed.
- F. Jiao and H. Frei, Energy Environ. Sci., 2010, 3, 1018–1027 RSC.
- C. G. Morales-Guio, L. Liardet and X. Hu, J. Am. Chem. Soc., 2016, 138, 8946–8957 CrossRef CAS PubMed.
- D. Hayes, R. G. Hadt, J. D. Emery, A. A. Cordones, A. B. F. Martinson, M. L. Shelby, K. A. Fransted, P. D. Dahlberg, J. Hong, X. Zhang, Q. Kong, R. W. Schoenlein and L. X. Chen, Energy Environ. Sci., 2016, 9, 3754–3769 RSC.
- G. Segev, H. Dotan, D. S. Ellis, Y. Piekner, D. Klotz, J. W. Beeman, J. K. Cooper, D. A. Grave, I. D. Sharp and A. Rothschild, Joule, 2018, 2, 210–224 CrossRef CAS.
- L. M. Carneiro, S. K. Cushing, C. Liu, Y. Su, P. Yang, A. P. Alivisatos and S. R. Leone, Nat. Mater., 2017, 16, 819–825 CrossRef CAS PubMed.
- Z. Su, J. S. Baskin, W. Zhou, J. M. Thomas and A. H. Zewail, J. Am. Chem. Soc., 2017, 139, 4916–4922 CrossRef CAS PubMed.
- J. Y. Kim, G. Magesh, D. H. Youn, J. W. Jang, J. Kubota, K. Domen and J. S. Lee, Sci. Rep., 2013, 3, 1–8 Search PubMed.
- P. Peerakiatkhajohn, J. H. Yun, H. Chen, M. Lyu, T. Butburee and L. Wang, Adv. Mater., 2016, 28, 6405–6410 CrossRef CAS PubMed.
- K. Sivula, F. Le Formal and M. Grätzel, ChemSusChem, 2011, 4, 432–449 CrossRef CAS PubMed.
- T. H. Jeon, G. Moon, H. Park and W. Choi, Nano Energy, 2017, 39, 211–218 CrossRef CAS.
- N. Iordanova, M. Dupuis and K. M. Rosso, J. Chem. Phys., 2005, 122, 144305 CrossRef CAS PubMed.
- D. A. Grave, D. Klotz, A. Kay, H. Dotan, B. Gupta, I. Visoly-Fisher and A. Rothschild, J. Phys. Chem. C, 2016, 120, 28961–28970 CrossRef CAS.
- L. M. Peter, Gurudayal, L. H. Wong and F. F. Abdi, J. Electroanal. Chem., 2018, 819, 447–458 CrossRef CAS.
- X. Ye, J. Yang, M. Boloor, N. A. Melosh and W. C. Chueh, J. Mater. Chem. A, 2015, 3, 10801–10810 RSC.
| This journal is © The Royal Society of Chemistry 2019 |