
Nitrogen and fluorine co-doped holey graphene hydrogel as a binder-free electrode material for flexible solid-state supercapacitors†

Abstract
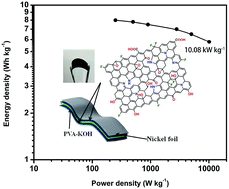
Two-dimensional (2D) graphene-based materials are widely applied as electrodes for supercapacitors due to their vast surface area and excellent charge mobility. However, the pursuit of the theoretical capacitance of graphene has lasted besides the improvements of charge/discharge rate and cycling life. In this work, a nitrogen and fluorine co-doped holey graphene hydrogel (NF-HGH) was synthesized by using consecutive solvothermal treatments for creating vacancies on the graphene basal plane and introducing heteroatoms into the graphene framework. The NF-HGH as an electrode for supercapacitors exhibited a remarkable electrochemical performance of 345.4 F g−1 at 1 A g−1 in 6 M KOH electrolyte. In addition, the specific capacitance of the assembled all-solid-state flexible symmetric supercapacitor (SSC) was 67.3 F g−1 at 1 A g−1, with an outstanding long-term cycling stability over 10 000 cycles. The delivered maximum energy and power densities were 7.99 W h kg−1 and 10.08 kW kg−1, respectively. The abundant pyrrolic-N doped at the edge of etched vacancies and the formed semi-ionic C–F bonds around pyrrolic-N were critical for electrochemical developments, which provided a sensational method to construct carbonaceous frameworks for energy storage systems.
 Please wait while we load your content...
Please wait while we load your content...

