
Toggling Geobacter sulfurreducens metabolic state reveals hidden behaviour and expanded applicability to sustainable energy applications†
 *ae
*ae
Abstract
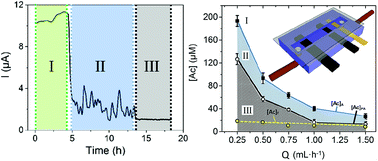
Electroactive biofilms are under intense scrutiny due to their potential to enable new sustainable technologies for energy production and bioremediation. However, severely reduced metabolic activity at low concentrations is a barrier to their implementation. A microfluidic approach was used for real-time respiration experiments on a Geobacter sulfurreducens biofilm to overcome these constraints. Precise changes to solution conditions enabled rapid and reversible switching between biofilm metabolic states, leading to the following discoveries. (i) Flow reactors can maintain biofilm activity at concentrations as low as 15 μM; (ii) a “pseudo-active” metabolic state separates active and inactive states; and (iii) acetate conversion can be as high as 90 percent for active biofilms at the pseudo-activity threshold.
 Please wait while we load your content...
Please wait while we load your content...

