
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNuclear fuel cycle, with a liquid ore and fuel: toward renewable energy
Claude A.
Degueldre
 *,
Richard J.
Dawson
and
Vesna
Najdanovic-Visak
*,
Richard J.
Dawson
and
Vesna
Najdanovic-Visak
Engineering Department, Lancaster University, Lancaster LA1 4YW, UK. E-mail: c.degueldre@lancaster.ac.uk
First published on 27th March 2019
Abstract
To fulfill the conditions required for a nuclear renewable energy concept, one has to explore a combination of processes going from the front end of the nuclear fuel cycle to the fuel production and the energy conversion using specific fluid fuels and reactors. Extraction of uranium from a diluted fluid ore such as seawater has been studied in various countries worldwide. This extraction should be carried out parsimoniously. An extraction rate of 103 tons of U per year over centuries would not modify significantly the equilibrium concentration of uranium in the oceans (3.3 ppb). This equilibrium results from the input of 104 tons of U per year by river waters and its scavenging on the sea floor from the 1.37 × 1018 tons of water in the oceans. For a renewable uranium extraction, the use of a specific biomass material is suggested to adsorb uranium and subsequently other transition metals. The uranium loading on the biomass would be around 100 mg per kg. After contact time, the loaded material would be dried and burned (CO2 neutral) with heat conversion into electricity. The uranium ‘burning’ in a molten salt fast reactor helps to optimize the energy conversion by burning all actinide isotopes with an excellent yield for producing a maximum amount of thermal energy from fission and converting it into electricity. This optimisation can be reached by reducing the moderation and the fission product concentration in the liquid fuel/coolant. These effects can be achieved by using a maximum amount of actinides and a minimum amount of alkaline/earth alkaline elements yielding a harder neutron spectrum. Under these optimal conditions the consumption of natural uranium would be 7 tons per year and per gigawatt (GW) of produced electricity. The coupling of uranium extraction from the sea and its optimal utilisation in a molten salt fast reactor should allow nuclear energy to gain the label renewable. In addition, the amount of seawater used by a nuclear power plant to cool the last coolant fluid and the turbine would be ∼2.1 × 109 tons per year for a fast molten salt reactor, corresponding to 7 tons of natural uranium extractable per year. This practice justifies the label renewable.
1. Introduction
Nuclear fuel is traditionally obtained by extracting uranium from solid ores mined from uranium deposits or from rather uranium rich rocks. After extraction, concentration and specific solid fuel preparation, the fuel is ‘burnt’ in reactors for the production of heat and electricity. This mode of extraction of uranium is not renewable and current consumption levels indicate that the reserves in current mines will be depleted in 20 years (ref. 1) and all known economic reserves will be consumed in 80 years (ref. 2).Presently, nuclear fuel production is mainly driven by uranium mining from relatively rich deposits. The uranium extraction is, however, limited to approximately 100 ppm due to economic reasons as estimated using Mudd data.3 In reality, extraction may be carried out from less concentrated sources when their decontamination is required e.g. phosphates, or when the production of uranium oxide can be performed at very low costs. For a country like UK which imports all of its uranium from abroad with shipping distances of about 6000 km or more (e.g. Canada and Niger) the extraction of U from seawater should be the first priority.
It should also be noted that several radiotoxic isotopes are released during conventional uranium extraction from solid ores e.g. radon emanations and tailings. The protocol suggested here concerns seawater, a fluid in which the decay products are not retained.
Uranium extraction from seawater has been researched for approximately 30 years in Japan, focusing on amidoxime-based sorbents4,5 and recycling of the absorbent by desorption. Similar approaches have recently re-emerged in other countries, e.g. the USA,6 coupling sorption and electrochemical separation and even now in China7 mainly due to its strategic importance. Today the specific uranium sorption on amidoxime is well understood.8 The extraction of uranium from seawater is, however, limited by several criteria (e.g. strategy, costs, legal aspects, and ecological requirements) as recently discussed.9
This study explores the potential extraction of uranium from seawater using a versatile chemical approach based on combined chemisorption10 and potential reduction (as modelled earlier for neptunium11) onto an organic/bioorganic waste material.
After collection of the uranium loaded biomaterial, the wet phase must be dried and burnt (producing heat that can be converted into electricity). Uranium can then be extracted from the ashes following the classical hydrometallurgical route. It can finally be used in a fuel form in dedicated reactors.
Molten Salt Reactors (MSR) are liquid fuel reactors where, in the classical concept, the fuel is also the coolant. The use of molten salt reactors for example as a Small Modular Reactor (SMR) is suggested, together with optimisation of neutron economy and fissile materials management in the nuclear fuel cycle. The design of a SM-MS reactor deals with key questions such as: (i) how can one optimise the reactor design for flexible and transient utilisation? (ii) How can one upgrade the neutron economy? With the solubility increase of Xe and Kr in the M–U–37Cl (M: Na, K, Rb, Mg etc.) molten phase during temperature increase, the neutron economy is degraded because of the presence of strong neutron absorbers such as 135Xe. Some experimental and modelling research studies are still needed on the solubility of Kr, Xe and I, the Xe precursor, in molten salts.
The present study evaluates the potential of combining uranium extraction from seawater and its use in a liquid fuel reactor to make nuclear energy renewable.
2. Extraction of uranium from seawater on biowaste materials
A better economical and ecological way of extracting uranium from seawater by optimising the extraction process by studying surface complexation and its reduction on solid organic ligands is first required. The process has to be studied in a comprehensive and systematic way to provide key information on potential extraction routes from seawater. As the use of seawater is difficult at the ton level, a strategy is suggested using laboratory tests prior to on site tests in the final step. This extraction is a key issue for the scientists involved in achieving a sustainable nuclear fuel cycle with a renewable approach.Uranium extraction from seawater becomes renewable when the extraction rate does not exceed the input rate from rivers (i.e. 104 tons of soluble U per year (ref. 12)). Actually, if U is not extracted, it is discarded from the seawater by scavenging (sedimentation) on the seafloor and ultimately eliminated by subduction of the pelagic sediment layer in the Earth’s mantle. In addition, since the volume of water in the sea is extremely large the extraction over the next 1000 years would not be significant compared to the total amount e.g. ∼1012 kg. During extraction, the environmental flora and fauna need to be protected with respect to possible speciation changes of redox sensitive elements or dissolved traces from the sorbing phase; however, it could be anticipated that a careful extraction of U should not modify the natural environment.
The proposed extraction strategy using low cost bioorganic waste such as coffee-, black tea- and brewery-residues, seeds, fruit peels, juice waste, tree bark, straw, wheat bran etc. (as delivered or treated) as sorbents needs to be tested. The process would subsequently include burning of the dried sorbed material followed by extraction of U from the ashes such as in the case of U recovery from coal.13 A similar approach is followed for wastewater treatment.14
The thermodynamic modelling of the uranium surface complexes is performed using correlation methods15 applied to the reactions:
![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) So-H + UO22+ ⇔ So-UO2+ + H+ So-H + UO22+ ⇔ So-UO2+ + H+ |
| +4H+ + 2e− ⇕ 2H2O + ▪ + 4H+ + 2e− ⇕ 2H2O+ |
| So-H + U4+ ⇔ So-U3+ + H+ |
| So-H + [UO2(CO3)i](2−2i)+ ⇔ So-UO2(CO3)i](1−2i)+ + H+ |
| +4H+ + 2e− ⇕ 2H2O + (i − j)(CO3)2− + ▪ + 4H+ + 2e− ⇕ 2H2O + (i − j)(CO3)2−+ |
| So-H + [U(CO3)j](4−2j)+ ⇔ So-U(CO3)j](3−2j)+ + H+ |
So-H stands for the sorbing groups and ▪ displays the equation separation. The modelling of the thermodynamic properties of the sorbents and uranium in seawater using a dedicated thermodynamic approach is carried out to estimate if coupling of sorption and redox reaction takes place during the absorption process.
Presently, the formation of U aqueous complexes shall impact on their sorption onto the active groups (So-H). The complex properties are affected by water temperature (T10–20°C), pressure (1 bar), water acidity (pH around 8) and an ionic strength of 0.3 M. Sorbing functional groups are given in Table 1.16 The species concentrations based on the element composition of seawater are given by calculated output results using thermodynamic data from Ball & Nordstrom, 1991.17 The tricarbonate complex concentration is 1.25 × 10−8 M, and the bicarbonate complex is 1.81 × 10−9 M. All other species are well below 1.00 × 10−10 M.
| Nomenclature | Group name | Sorbing group formula |
|---|---|---|
| So-H |
Carboxylic | R–CO–OH |
| Alcohol or phenol | R–OH | |
| Thiol or thiophenol | R–SH | |
| Hydroxyl amine | R2N–OH | |
| So < Hn |
Amine or aminophenol | R–NH2 |
| Amide | R–CO–NH2 | |
| Amidoxime | R–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NOH)–NH2 NOH)–NH2 |
A relationship between a metal’s sorption coefficient and its complexation in solution has been derived, e.g. Degueldre et al. (1994),15 using surface complexation properties. The surface complexation constants may be evaluated on the basis of their correlation for each complex form with the hydroxo-complex formation constants. The impact of the redox is then evaluated using the Nernst equation considering surface complexation for all redox species e.g. see Degueldre (1997).11
In seawater (oxidative conditions) U(VI) should only be formed; its reduction would yield U(V) and U(IV). Reaction on specific substrates may stabilise one form versus another favouring sorption. The kinetics of these uranium species formation are also linked to the concentration of the species.
Uranium sorption from freshwater sources onto Citrus limetta peels18 and citrus reprocessing waste19 has been reported recently. With regard to the extraction of uranium from seawater, Chlorella dry cells and orange peels adsorb a great quantity of uranium; in contrast, some natural high polymers such as chitin, chitosan, cellulose and starch do not absorb uranium significantly from seawater. Phosphorylated polysaccharides (chitin phosphate, chitosan phosphate and cellulose phosphate) and acid polysaccharides (alginic acid and pectic acid) have also been found to strongly sorb uranium. The absorption of uranium by chitin phosphate as reported is rapid during the first 3 hours. It was observed that chitosan phosphate can recover uranium (2.6 μg per g adsorbent) from natural seawater.20 Among biological samples e.g. grasses, leaves, barks, roots and fruit bodies, Myrica cortex barks were found to be the most performing absorbents of U in seawater with >30% uranium absorbed.21
Clearly, uranyl sorption on a colloidal material should combine surface complexation, reduction and colloid aggregation/precipitation to be efficient.
Polyphenols are reported to be very efficient for extraction because of their strong complexation potential (see Fig. 1) and because of their antioxidant properties and their peptisation/flocculation potential.22 Quercetin, a widely found polyphenol e.g. in citrus fruits, wine tannin, etc., combines these complexation (Fig. 1a) and bridging properties (Fig. 1b) enhancing the extraction. These biomaterials are loaded within tannin which is mostly composed of polyphenols. The family of tannin-loaded biomaterials is being further investigated, by testing berries, barks, malt and tealeaf biomasses. A uranium loading on the biomass would be around 100 mg per kg.
 | ||
Fig. 1 Metal complex formation with polyphenols, example of the metal quercetin. (a) 3–1 and (b) 1–2 complexes, --- coordination bond,  H exchanged. H exchanged. | ||
After collection of the uranium loaded biomaterial, the wet phase must be dried, prior to burning in a green and clean way as suggested by Ashworth et al. (2013).23 Uranium can then be extracted from the ashes following the classical hydrometallurgical route.
3. Burning uranium in molten salt fast reactors
Molten salt reactors (MSR) are liquid fuel reactors in which, in the classical concept, the fuel is also the coolant.24 To run the reactor in low consumption mode, the MSR must consume a minimum amount of uranium for producing a maximum amount of thermal energy and hence electrical energy. To achieve this conversion in an optimal way, a strict neutron economy is required, achieved by using the actinide isotopes in an efficient way favoring fission instead of capture. This may be performed using fast neutrons. However, since the neutron spectra are never fully fast but include also epithermal and thermal components, some of the actinide isotopes called fertile isotopes undergo neutron capture up to the formation of fissile isotopes e.g.239Pu, 241Pu, 242Am, etc.Even in a fast MSR the neutron spectrum is a mix of fast, epithermal and thermal. Consequently, the following reactions occur:
| 235U + 1n ⇒ 92Kr + 141Ba + 3 1n takes place in a MSTR |
| 238U + 1n ⇒ 93Kr + 143Ba + 3 1n takes place in a MSFR |
The thermal part of the spectrum allows transmutation of the fertile isotopes into fissile isotopes
238U + 1n ⇒ 239U ⇒ β− + ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/char_e0ce.gif) + 239Np ⇒ β− + + 239Pu + 239Np ⇒ β− + + 239Pu |
| 239Pu + 1n ⇒ 94Rb + 143La + 3 1n |
During MSR operation, fission products are generated in the core of the reactor and in the primary loop. Some of them are strong poisons absorbing neutrons. The relative concentration of fission products follows the so called ‘kamel’ curve with the maxima located around Zr and Mo for the light, and around Xe, Cs, Ba and La for the heavy fission products. Some of them are not sustainable isotopes such as 135Xe (half-life 9.1 h), with a neutron capture cross-section of 2 × 106 barns, or its parent nuclide 135I with a half-life of 9.14 h.
| 135I ⇒ β− + + 135Xe(↑) ⇒ β− + + 135Cs |
They could be discarded from the core and loop by various strategies. A maximum elimination is required to optimise neutron economy. Once eliminated from the liquid fuel, they also need to be absorbed and fixed onto the absorber and embedded in waste matrices for disposition in a repository.
The behaviour and properties of Kr, Xe and its precursor I in fluoride and chloride molten salts have been addressed by Smirnov et al. (1988).25 Actually, isotopes of Xe and Kr can act as neutron poisons that must be countered by a high fuel loading. For iodine the case is evident, 135I decays into 135Xe, the stronger neutron absorber isotope.26 There are several strategies to eliminate an excess of 135Xe such as the decay process, degassing and ventilation. The evolution of 135I and 135Xe, which are entrained in the flowing salt, may be evaluated from their concentration change with the burn-up time.27 A fast circulation of the fuel salt could decrease the concentration of 135I and 135Xe, and the reduction can achieve purging of around 50 and 40% of 135I and 135Xe, respectively, at a small power level, e.g. 2 MW, when the core has the same fuel salt volume as that of the outer-loop. The second possibility is to use helium (less soluble) to remove the more soluble Xe.
The analysis of the fission product volatilisation and sorption is carried out by mass spectroscopy which is more sensitive than that reported earlier e.g.ref. 28. These analyses are done in-line (contamination free). Presently, thermodynamic codes are used for the description of the dissolution of metals in aqueous solution and the elimination of volatile phases from fluids.29 The codes have been used to predict the solubility of volatile species from their molten salts.30 To understand the rate of degassing the coupling of codes (e.g. with the code ‘fluent’) is suggested to examine bubble formation and full removal e.g.ref. 31.
The initial UCl4 can also be reduced to UCl3. The uranium redox buffer fixes the redox potential (see for example ref. 32 and 33). Reduction may be performed by doping H2 or metals e.g. Mg in the molten salt. The effect of redox in the molten salt may be modelled as performed earlier e.g.ref. 34. During burn-up, the U in UCl3 is substituted by fission products and the remaining 3Cl− modifies the redox.35 The thermodynamic properties of fission products need to be calculated by modelling. The iodine redox states (I(−I), I(0) or I(I)) and the species in M–U3/4+–Cl are subject to reactions such as:
| I0(M–U–Cl) + U3+(M–U–Cl) ⇔ I−(M–U–Cl) + U4+(M–U–Cl) |
| I0(M–U–Cl) ⇔ I(g) |
| Xe0(M–U–Cl) ⇔ Xe(g) |
Along with phase changes, the local distribution of I is also linked to the mobility or diffusion of I in the liquid fuel.36,37 The combination of thermodynamic modelling will help understand the impact of these factors on the distribution of I in the liquid fuel.
The reprocessing of non-volatile fission products can be performed in-line by precipitation and filtration of insolubles and by electro-refining.
A MSR offers high burn-up, breeding and improvement of neutron economy thanks to the potential elimination of fission products by volatilisation38 or electrolysis39 and for the MSFR, the consumption of actinides. Under these conditions the consumption of uranium is reduced by at least a factor of 20 in a thermal MSR and 200 in a fast MSR.
For economic and ecological reasons, small modular MSRs (SM-MS-R) are suggested. The design of the various components of the system must be optimised. A multi-modular system includes a central part with a core and primary loop and systems to optimise breeding and extraction of the volatile fission products.
The secondary loop coolant allows transfer of energy from the primary to the turbine. Other modules of the SM-MS-R system include a third cooling loop consisting of a classical heat exchanger using cold water as the cold source (seawater is suggested in this combined approach). An additional container includes a waste management unit with a related vessel. A fluid waste management module is foreseen in which standard nuclear waste management is performed. It includes absorbers that collect the Xe, Kr and I isotopes. At the end of its life the core/primary molten salt can be collected in a catcher in a waste package ingot and could be directly disposed. The core and primary could be refilled with fresh salt components. The SM-MS-R could be restarted finally with the input of fresh fissile salt.
Fig. 2 shows the various modules of the proposed version of the small modular-molten salt reactor adapted to 238U breeding in 239Pu. Separation of fissile and fertile molten salts in a decay tank is foreseen. The reactor itself is a 239Pu reactor in which the primary loop includes a heat exchanger. In the primary loop one also finds a reprocessing unit combined with a refuelling unit. The reprocessing unit is a simple degassing captor that allows fission gases to be purged out. The refuelling unit allows fresh plutonium to be reloaded in the fissile primary loop. The main issue at this level is to couple the decay tank and the refuelling unit. It is anticipated that 239U shall be radiochemically separated and fully decayed into 239Np. However, the decay of 239Np into 239Pu requires several days during which the U system has to rely on the fissile reactor loop prior to first refuelling the fissile loop with fresh plutonium. It would consequently be suggested that the refuelling would work intermittently with periods of at least 3 days of decay prior to refuelling. The other option would be an actinide separation in the decay tank but this would be difficult to operate in-line.
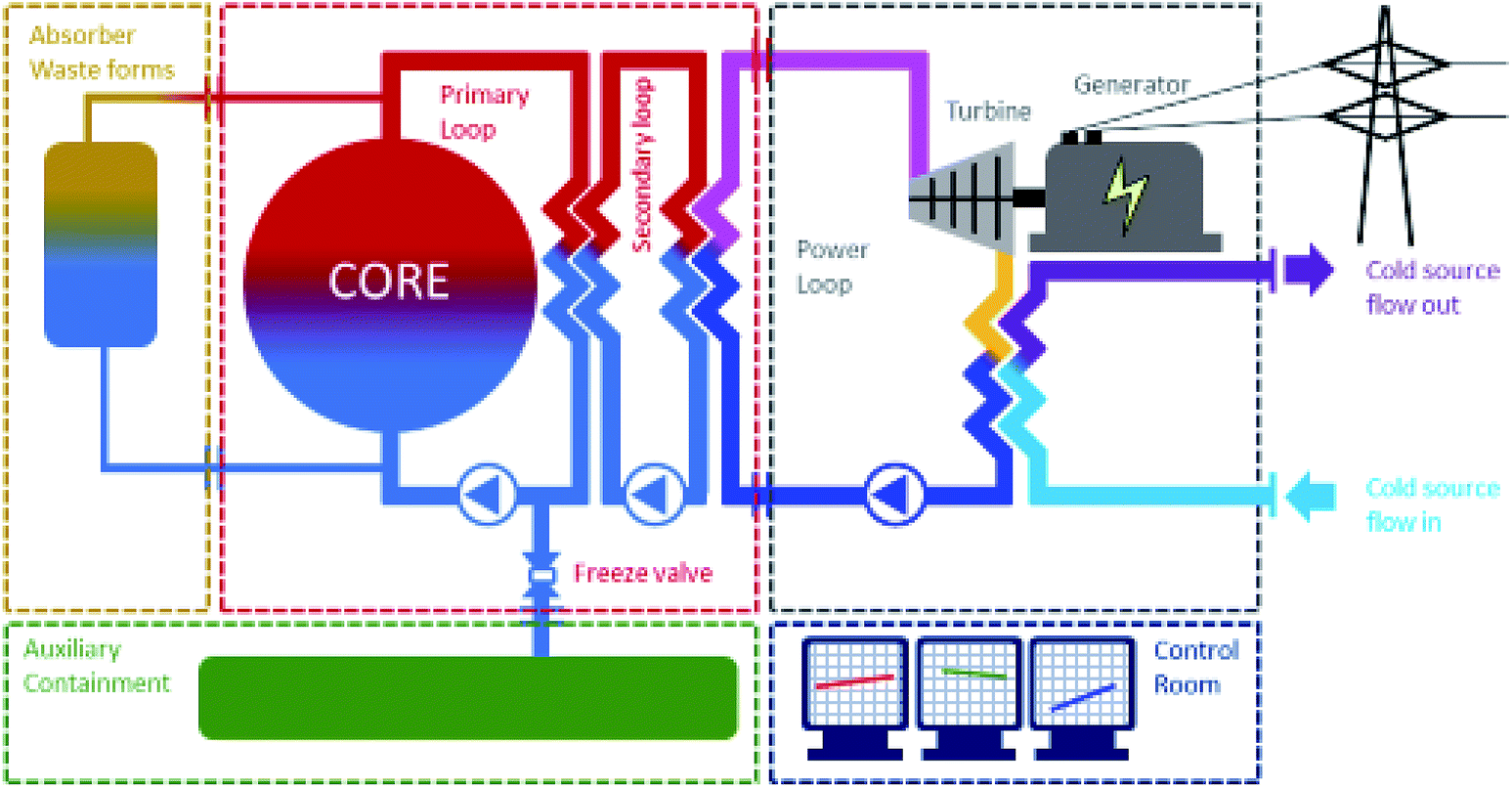 | ||
| Fig. 2 SM-MS-R the modular concept. 1st module/container: core, primary and secondary. 2nd module/container: ternary, turbine/generator and cooling loop. 3rd module/container: waste management unit. 4th module/container: containment/accident MS catcher. 5th module/container: control room. | ||
For the fast MSR, the challenge is also at the beginning of fuel life, the initiation of the reaction. The chain reaction could be initiated by approaching externally a neutron source e.g. an actinide source (e.g. Cm) or by spiking in the molten salt the required amounts of fissile isotopes.
In all cases, molten salt composition, core volume and geometry, primary loop geometry, and molten salt flow rate have to be calculated together with secondary and ternary loop characteristics. Their adaptation to power production has to be studied in an analytical way.
For both thermal and fast MSRs, designs have to be adapted with the local requirement concerning nuclear waste management.
Nuclear waste management of the volatile products trapped during operation requires specific absorbers. Irreversible absorption of the fission products is required together with the preparation of the final waste package for the geological repository.
For the spent fuel, MS samples could be collected from the core – the primary system in the auxiliary container, forming ingots at the end of its life.
4. Discussion and proposed protocol
Today, nuclear energy conversion is still based on heat production, using steam turbines with condensation using a cold source. The cold source is linked to a heat exchanger using cold water, balancing the energy difference between turbine heat and electrical energy produced by the coupled generator. For a renewable nuclear fuel cycle, the use of seawater as the cooling source has to be promoted. To run in a sustainable way the conversion requires also parsimonious utilization of uranium. The loaded uranium must be produced by recovering it from seawater used as the reactor cooling source. For a 1 GW MSR running around 700 °C, the requirement of a nuclear power plant to run the condenser below the turbine would be 2.1 × 109 tons of seawater per year. This seawater has to be filtered prior to passing through a heat exchanger contributing to the collection of plastics in seawater. A maximum temperature increase of 10 °C of the water after heat exchange is currently the rule and 7 tons of natural uranium per year are anticipated to be extractable. Utilisation of a specific biomass absorber is required to extract pumped seawater uranium over weeks in pounds/pools downstream from the reactor. Each absorber (4 kg sorbing 400 mg U and other transition metals e.g. V, Mo, etc.) would be set in a 20 cm × 20 cm × 10 cm bag dipping in a pound/pool of 250 m × 250 m, a total of 8 × 107 bags made of (U non-sorbing) cellulose. Since the water is rejected in the sea, it is mandatory that the processes operate in an environmentally friendly way. The reinjected seawater plume must be oriented in a way to avoid mixing with the input seawater to separate in the local stream the depleted water from the input water.The sorbed biomass must be dried and burned in a carbon neutral way, converting the biomass into heat and then into electricity. The ash is then collected and treated for the production of UCl4. The nuclear fuel production must be performed in an optimal way using U3O8 and ash residues as starting materials. Its conversion to uranium tetrachloride is made using C37Cl4 around 500 °C. The purified U37Cl4 is obtained by sublimation. It is dissolved in a molten salt prior to nuclear initiation using a fissile source. The energy produced for 1 GW is obtained by burning 7 tons of uranium over a year. This corresponds to the amount of uranium present in the 2.1 × 109 tons of seawater used to cool down the reactor over a year. Fig. 3 depicts the processes required to run the nuclear fuel cycle though a MSR in a renewable way.
 | ||
| Fig. 3 Schematic view of the combination of processes including uranium extraction from seawater and burning in a molten salt fast reactor. Details of (a) cooling water pumping, (b) absorption on biomass in a pound downstream from the molten salt fast nuclear reactor, (c) biomass uranium recovery and fuel production plant and (d) the molten salt fast reactor needed for burning the recovered uranium in a renewable way with consumption rate compared to the recharge of uranium in the sea by rivers. | ||
The nuclear waste impact is also a key issue. Fast reactors are known for their optimal low ratios of nuclear waste amount for a fixed annual power. Actually, the potential of fission product recycling is very limited. Recently, Bourg and Poinssot (2017)40 investigated the potential of mining critical raw materials from fission products. They showed that the potential of reutilisation of fission products below the exemption limit after 50 years of cooling time is very limited. However, some radioactive fission products such as 99Tc could be specifically separated for medical applications such as radio-immuno-assays. The potential of the production of specific Xe radioisotopes for medical applications, such as 129Xe used in lung imaging studies41 and 133Xe used for brain imaging,42 must also be noted.
The impact of the MSR on the correction of the energy load on the grid is the next issue. A molten salt reactor is a liquid fuel reactor where, in the classical concept, the fuel is also the coolant. In this configuration the liquid fuel reactor can operate in fast transient mode such as that required when coupling a classical renewable and nuclear reactor on the grid. This was investigated by Dodson and Mccan (2013)43 for the thorium molten salt reactor and could also be the case for the uranium molten salt reactor. Clearly with the exception of metallic fuels all ceramic fuels are quite brittle and do not undergo transients such as those required to recover from wind turbines and photovoltaic cell production shortages. The photovoltaic transient may be of the order of 75% difference between full production for panel full illumination and reduction due to the clouding or shadowing of the panel in 30 s. An increase of production yield must be anticipated to meet the energy demand. This can be performed with the MSR by varying the molten salt flow rate.
The global impact of the renewable fuel cycle on the environment must be extended to the whole MSR park. A uranium extraction rate of 1000 tons per year from the sea would not modify significantly the equilibrium concentration of uranium in the oceans i.e. 3.3 ppb. This constant concentration results from an input of 10 kt of U per year by river water and its scavenging on the sea floor from the 1.37 × 1018 tons of water in the oceans corresponding to a total amount of tons of uranium. Since the volume of water in the sea is immense the extraction over the next 1000 years would not significantly affect the total uranium amount in the sea: ∼1012 kg. The strict renewable concept would correspond to a 1000 t uranium consumption which corresponds to 150 fast MSR units of 1 GW each. For a renewable uranium extraction, the use of a specific biomass material is suggested to fulfill the sorption of uranium and subsequently other transition metals (V and Mo) that could be extracted consequently. The annual consumption rate of a reactor park would even remain smaller than the natural uranium recovery rate by scavenging in seawater.
5. Conclusion and recommendations
The suggested conditions required to fulfill a nuclear renewable fuel cycle combine processes from the front end of the fuel cycle to the fuel production and the energy conversion using molten salt fast reactors.Extraction of uranium from seawater has been studied in various countries of the world. This extraction should be carried out parsimoniously. An extraction rate of 103 tons of U per year for a MSR park of 500 MSRs over centuries would not modify significantly the equilibrium uranium concentration (3.3 ppb) in the oceans, resulting from the input of 104 tons of U per year by river waters and its scavenging on the sea floor from the 1.37 × 1018 tons of water in the oceans.
The extraction of uranium from seawater is being studied on the laboratory scale. On the industrial scale it should be carried out utilising specific biowaste absorbents for performing the absorption of uranium from the rejected seawater from dedicated nuclear power plants’ cooling seawater, in large pounds/pools where the sorbent is in contact with seawater over several weeks. For a renewable uranium extraction, the use of a specific biomass material is suggested to fulfill the sorption of uranium and subsequently other transition metals with a loading on the biomass of around 100 mg per kg. At equilibrium, the loaded material shall be removed from the pound, dried, burned (CO2 neutral) with heat conversion into electricity.
The subsequent ashes are collected and treated by applying the classical hydrometallurgical route to produce UO2 then UCl4.
The uranium ‘burning’ with a molten salt fast reactor helps to optimize the energy conversion by burning all actinide isotopes with an excellent yield for producing a maximum amount of thermal energy from fission and converting it into electricity. This can be reached by reducing the moderation and the fission product concentration in the liquid fuel/coolant. This can be achieved by using a maximum amount of actinides and a minimum amount of alkaline/earth alkaline elements yielding a harder neutron spectrum. Under these optimal conditions the consumption of natural uranium would be 7 tons per year and per gigawatt (GW) of produced electricity.
The fuel utilized in molten salt fast reactors is in the form of Na/Rb/Mg–U–Pu–37Cl, the choice of 37Cl− being dictated by safety and neutronic reasons. The addition of Pu may be required to enhance the power at the beginning of its life.
During operation, the generation of fission products poisons the core of the reactor and the primary. However, some unsuitable isotopes such as 135Xe, or its parent nuclide 135I, could be discarded from the core/loop by various strategies. A maximum elimination is required and these gaseous/volatile phases need to be studied using a thermodynamics approach to better carry out their elimination. They also need to be absorbed and fixed (e.g. by specific sorption) onto waste matrices for disposal.
Coupling of uranium extraction from the sea and its optimal utilization in a molten salt fast reactor should allow nuclear energy to gain the label renewable. In addition the amount of seawater used by nuclear power plants to cool the last cooling fluid loop and the turbine would be ∼2 × 109 tons per year, corresponding to 7 tons of natural uranium per year. This practice justifies the label renewable.
Conflicts of interest
There are no conflicts to declare.References
- J. S. Henning, Uranium and thorium resource assessment, Encyclopedia of Energy, 2004, pp. 279–98 Search PubMed.
- NEA and IAEA, Uranium 2016 – resource, production and demand, OECD publishing, 2016, p. 548, available from http://www.oecd-nea.org/ndd/pubs/2016/7301-uranium-2016.pdf Search PubMed.
- G. M. Mudd, The environmental sustainability of mining in Australia: key megatrends and looming constraints, Resour. Pol., 2010, 35, 98–115 CrossRef.
- M. Tamada, N. Seko, N. Kasai and T. Shimizu, Cost Estimation of Uranium Recovery from Seawater with System of Braid type Adsorbent, J. At. Energy Soc. Jpn., 2006, 5, 358–363 CAS.
- K. Hara, T. Nishimoto, S. Fujiwara, T. Fujii, Y. Hidaka and H. Okabe, Attempts at capturing ppb-level elements from sea water with hydrogels, Prog. Nucl. Energy, 2016, 92, 228–233 CrossRef CAS.
- C. Liu, P.-C. Hsu, J. Xie, J. Zhao, T. Wu, H. Wang, S. Chu and Y. Cui, A half-wave rectified alternating current electrochemical method for uranium extraction from seawater, Nat. Energy, 2017, 2, 17007 CrossRef CAS.
- L. Chen, Z. Bai, L. Zhu, L. Zhang, Y. Cai, Y. Li, W. Liu, Y. Wang, L. Chen, J. Diwu, J. Wang, Z. Chai and S. Wang, Ultrafast and Efficient Ch.Extraction of Uranium from Seawater Using an Amidoxime Appended Metal–Organic Framework, ACS Appl. Mater. Interfaces, 2017, 9(38), 32446–32451 CrossRef CAS PubMed.
- S. Kung, Uranium in seawater, Ind. Eng. Chem. Res., 2016, 55, 4101–4361 CrossRef.
- K. Dungan, G. Butler, F. R. Livens and L. M. Warren, Uranium from seawater – Infinite resource or improbable aspiration?, Prog. Nucl. Energy, 2017, 99, 81–85 CrossRef.
- C. Degueldre, Uranium as a renewable for nuclear energy, Prog. Nucl. Energy, 2017, 94, 174–186 CrossRef CAS.
- C. Degueldre, Retention of redox sensitive elements in aquifers—the case of neptunium, J. Environ. Radioact., 1997, 34, 211–214 CrossRef CAS.
- M. R. Palmer and J. M. Edmond, Uranium in river water, Geochim. Cosmochim. Acta, 1993, 57, 4947–4955 CrossRef CAS.
- O. D. Maslov, S. Tserenpil, N. Norov, M. V. Gustova, M. F. Filipov, A. G. Belov, M. Altangerel and N. Enhbat, Uranium recovery from coal ash dumps of Mongolia, Solid Fuel Chem., 2010, 44, 433–438 CrossRef.
- G. Lofrano, M. Grassi and M. Notarnicola, Characteristics and adsorption capacities of low-cost sorbents for wastewater treatment: a review, Sustainable Mater. Technol., 2016, 9, 10–40 CrossRef.
- C. Degueldre, H.-J. Ulrich and H. Silby, Sorption of Am onto montmorillonite, illite and hematite colloids, Radiochim. Acta, 1994, 65, 173–179 CAS.
- B. B. Mathew, M. Jaishankar, V. G. Biju and K. N. Beeregowda, Role of Bioadsorbents in Reducing Toxic Metals, J. Toxicol., 2016, 2016, 4369604 Search PubMed.
- J. W. Ball and D. K. Nordstrom, User's manual for WATEQ4F, with revised thermodynamic data base and test cases for calculating speciation of major, trace, and redox elements in natural waters: U.S. Geological Survey Open-File Report 91-183, 1991 Search PubMed.
- S. C. Gondhalekar and S. R. Shukla, Equilibrium and kinetics study of uranium(VI) from aqueous solution by Citrus limetta peels, J. Radioanal. Nucl. Chem., 2014, 302, 451–457 CrossRef CAS.
- B. Satari and K. Karimi, Citrus processing wastes: Environmental impacts, recent advances, and future perspectives in total valorization, Resour., Conserv. Recycl., 2018, 129, 153–167 CrossRef.
- T. Sakaguchi, A. Nakajima and T. Horikoshi, Absorption of uranium from sea water by biological substances, Nippon Nogeikagaku Kaishi, 1979, 53, 149–156 CrossRef CAS.
- A. Nakajima and T. Sakaguchi, Adsorption of Uranium by Vegetable Crude Drugs, Agric. Biol. Chem., 1989, 53, 2853–2859 CAS.
- T. Sakagushi and A. Nakajima, Recovery of uranium from seawater by immobilized tannin, Sep. Sci. Technol., 1987, 22, 1609–1623 CrossRef.
- K. Ashworth, O. Wild and C. N. Hewitt, Impact of biofuel cultivation on mortality and crop yield, Nat. Clim. Change, 2013, 3, 492–496 CrossRef CAS.
- J. Serp, M. Allibert, O. Beneš, S. Delpech, O. Feynberg, V. Ghetta, D. Heuer, D. Holcomb and V. Ignatiev, The molten salt reactor (MSR) in generation IV: Overview and perspectives, Prog. Nucl. Energy, 2014, 77, 308–319 CrossRef CAS.
- M. V. Smirnov, I. V. Korzun and V. A. Olegnikeva, Hydrolysis of molten alkali chlorides, bromides and iodides, Electrochim. Acta, 1988, 33, 781–788 CrossRef CAS.
- M. J. Eades, E. S. Chaleff, P. F. Venneri and T. Blue, The influence of Xe-135m on steady-state xenon worth in thermal molten salt reactors, Prog. Nucl. Energy, 2016, 93, 397–405 CrossRef CAS.
- J. Wu, C. Guo, X. Cai, C. Yu and J. Chen, Flow effect on 135I and 135Xe evolution behavior in a molten salt reactor, Nucl. Eng. Des., 2017, 314, 318–325 CrossRef CAS.
- D. S. Akerib, H. M. Araújo, X. Bai, A. J. Bailey and C. Zhang, Chromatographic separation of radioactive noble gases from xenon, Astropart. Phys., 2018, 97, 80–87 CrossRef.
- R. Dawson and G. J. Kelsall, Recovery of platinum group metals from secondary materials: I palladium dissolution in iodide solutions, J. Appl. Electrochem., 2007, 37, 3, DOI:10.1007/s10800-006-9218-8.
- I. Scott, Gaseous and volatile fission product release from moltensalt nuclear reactor, Proceedings Thorium Energy Conference 2015, ThEC15, Mumbai, India, Oct. 19–22, 2015 Search PubMed.
- R. Dawson, Electrochemical Recovery of platinum and Palladium from Alumina Supported Catalysts, PhD thesis, Imperial College, London, UK, 2006.
- T. Nagai, A. Uehara, T. Fujii, O. Shirai, N. Sato and H. Yamana, Redox equilibrium of U4+/U3+ in Molten NaCl-2CsCl by UV-Vis and cyclic voltammetry, J. Nucl. Sci. Technol., 2005, 42, 1025–1031 CrossRef CAS.
- T. Nagai, A. Uehara, T. Fujii, O. Shirai, N. Sato and H. Yamana, Electrochemical behaviour and electronic absorption spectra of uranium trivalent in molten LiCl-CsCl mixtures, J. Nucl. Mater., 2011, 414, 226–231 CrossRef CAS.
- Y.-J. Cho, H.-C. Yang, H.-C. Eun, E.-H. Kim and I.-T. Kim, Characteristics of Oxidation Reaction of Rare-earth Chlorides for Precipitation in LiCl-KCl Molten Salt by Oxygen Sparging, J. Nucl. Sci. Technol., 2006, 43, 1280–1286 CrossRef CAS.
- V. Singh, M. R. Lish, O. Chvála and B. R. Upadhyaya, Dynamics and control of molten-salt breeder reactor, Nucl. Eng. Technol., 2017, 49, 887–895 CrossRef CAS.
- M. V. Smirnov, I. V. Chebykia and L. A. Tsiovkina, Thermodynamic properties of sodium and potassium in molten chlorides, bromides and iodides, Electrochim. Acta, 1981, 26, 1275–1288 CrossRef CAS.
- M. V. Smirnov and O. Tkacheva, Interaction of oxygen with molten alkali chlorides, Electrochim. Acta, 1992, 37, 2681–2690 CrossRef CAS.
- E. L. Compere, S. S. Kirslis, E. G. Bohlmann, F. F. Blankenship and W. R. Grimes, Fission product behaviour in the molten salt reactor experiment, ORNL-4865, 1975, p. 143 Search PubMed.
- V. M. Oversby, C. C. McPheeters, C. Degueldre and J. M. Paratte, Control of civilian plutonium inventories using burning in a non-fertile fuel, J. Nucl. Mater., 1997, 245, 17–26 CrossRef CAS.
- S. Bourg and C. Poinssot, Could spent nuclear fuel be considered as a non conventional mine of critical row material, Prog. Nucl. Energy, 2017, 94, 222–228 CrossRef CAS.
- F. W. Hersman, I. C. Ruset, S. Ketel, I. Muradian and S. Patz, Large Production System for Hyperpolarized 129Xe for Human Lung Imaging Studies, Acad. Radiol., 2008, 15, 683–692 CrossRef PubMed.
- R. Wirestam, E. Ryding, A. Lindgren, B. Geijer and F. Ståhlberg, Absolute cerebral blood flow measured by dynamic susceptibility contrast MRI: a direct comparison with Xe-133 SPECT, Magn. Reson. Mater. Phys., Biol. Med., 2000, 11, 96–103 CrossRef CAS.
- A. M. Dodson and R. A. McCann, Investigation of thermal feedback design for improved load following capability of thorium molten salt reactor, Proc. IEEE 2013 green technology, pp. 215–219, DOI:10.1109/Green Technol. 2013 40.
| This journal is © The Royal Society of Chemistry 2019 |