
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSuppressing the formation of NOx and N2O in CO2/N2 dielectric barrier discharge plasma by adding CH4: scavenger chemistry at work†
Ramses
Snoeckx
 *ab,
Karen
Van Wesenbeeck
c,
Silvia
Lenaerts
c,
Min Suk
Cha
a and
Annemie
Bogaerts
b
*ab,
Karen
Van Wesenbeeck
c,
Silvia
Lenaerts
c,
Min Suk
Cha
a and
Annemie
Bogaerts
b
aKing Abdullah University of Science and Technology (KAUST), Clean Combustion Research Center (CCRC), Physical Science and Engineering Division (PSE), Thuwal 23955, Saudi Arabia. E-mail: Ramses.snoeckx@kaust.edu.sa
bResearch Group PLASMANT, Department of Chemistry, University of Antwerp, Universiteitsplein 1, BE-2610 Antwerp, Belgium
cResearch Group DuEL, Department of Bioscience Engineering, University of Antwerp, Antwerp, Belgium
First published on 20th February 2019
Abstract
The need for carbon negative technologies led to the development of a wide array of novel CO2 conversion techniques. Most of them either rely on high temperatures or generate highly reactive O species, which can lead to the undesirable formation of NOx and N2O when the CO2 feeds contain N2. Here, we show that, for plasma-based CO2 conversion, adding a hydrogen source, as a chemical oxygen scavenger, can suppress their formation, in situ. This allows the use of low-cost N2 containing (industrial and direct air capture) feeds, rather than expensive purified CO2. To demonstrate this, we add CH4 to a dielectric barrier discharge plasma used for converting impure CO2. We find that when adding a stoichiometric amount of CH4, 82% less NO2 and 51% less NO are formed. An even higher reduction (96 and 63%) can be obtained when doubling this amount. However, in that case the excess radicals promote the formation of by-products, such as HCN, NH3 and CH3OH. Thus, we believe that by using an appropriate amount of chemical scavengers, we can use impure CO2 feeds, which would bring us closer to ‘real world’ conditions and implementation.
1. Introduction
The global challenge of climate change and the need for carbon negative technologies have sparked research interest in a wide variety of techniques capable of converting CO2.1–6 This CO2 can be captured either from major emission sources or—preferably, in the long run— from air, through direct air capture (DAC).7 Numerous analyses and comparisons between different technologies have been made in the literature; however, they all overlook a key aspect that has major consequences, i.e. the fact that lab-scale studies generally use pure gases (99.999% purity), whereas industrial gases (with some exceptions) usually contain N2. Purification is one option, but an energy intensive, and thus costly one.8 Another—more practical—option is to directly use these impure gases.However, this option comes with an important obstacle. Most novel technologies under consideration for the conversion of CO2 into CO and O2 either require high temperatures (e.g., solar thermochemical and catalytic thermochemical conversion) or create highly reactive O species in situ (e.g., electrochemical, photochemical and plasmachemical conversion).1 As a result, the risk of producing nitrogen oxides (NOx) and nitrous oxide (N2O) is real.9,10 In combustion science, the formation of NOx and N2O is a well-understood phenomenon.11,12 Among the three major NOx formation mechanisms (i.e., thermal NOx (Zel'dovich), prompt NOx, and fuel NOx), the thermal mechanism consistently produces NOx, as long as O2 and N2 coexist under high temperature conditions (>1900 K).11 N2O, on the other hand, is not a major by-product in combustion processes, except for fluidized bed combustion.12 When released in the atmosphere, these compounds lead to severe air pollution, such as smog and acid rain, and they are responsible for the formation of tropospheric ozone.13 With respect to global warming, the production of N2O, in any CO2 conversion process, cancels out the carbon negative effect of any CO2 converted, since N2O is 298 times more potent as a greenhouse gas.13 This is why NOx and N2O emissions are so strictly regulated worldwide.
Despite the potential risk of producing unwanted NOx and N2O during the conversion of impure CO2 feeds containing N2, almost no research has been performed in this area, for the novel technologies that are being considered to convert CO2. It stands, without doubt, that this is an important issue, as additional deNOx post-treatment, or more severe CO2 pre-purification steps, will have a negative effect on the energy and cost balance of these CO2 conversion technologies. In previous studies, we reported that, for non-thermal plasma technology—one of the most promising technologies for the conversion of CO2 (ref. 1)—the presence of N2 indeed causes the aforementioned formation of NOx and N2O.9,10
Here, we explore a potential solution to prevent the formation of NOx and N2O, in situ, during the plasmachemical conversion of CO2. A well-known solution from combustion science has been the addition of more fuel (eq. to a higher fuel-to-air ratio).13 Despite the fact that we work under experimental conditions that are very different from those in combustion science, we can justify using a similar approach, based on the results obtained in our previous studies.9,14,15 We already know that the addition of a hydrogen source to non-thermal pure CO2 plasmas can trap free O species, in situ.14 And exactly these free O species are responsible for the NOx and N2O production pathways in non-thermal plasmas.9 Therefore, here we introduce the use of a hydrogen source, CH4, as a chemical oxygen scavenger to suppress the formation of NOx and N2O, in situ, during the conversion of CO2 mixtures containing N2, in a dielectric barrier discharge (DBD) plasma.
2. Materials and methods
Experiments were carried out in a coaxial DBD plasma reactor operating at room temperature and atmospheric pressure. A stainless steel mesh (high voltage electrode) was wrapped over the outside of a quartz tube, and a stainless steel rod (ground electrode) was placed at its centre. Feed gases were composed of CO2, N2 and CH4 (Air Liquide, Alphagaz 1, 99.999%), and each flow rate was controlled using a mass-flow controller (Bronkhorst, EL-Flow select F-210CV). The DBD reactor was powered by an AC high-voltage power supply (AFS, custom made), and the applied voltage and electrical current were sampled using a four-channel digital oscilloscope (Picotech, PicoScope 64201). Finally, Fourier transform infrared spectroscopy (FTIR; Thermo Fischer Scientific, Nicolet 380) was used to study the effects of the addition of CH4 on the formation of N2O and NOx compounds (i.e., NO, NO2, N2O3 and N2O5). A detailed description of the set-up and experimental conditions can be found in Section 1 of the ESI.†2.1. CH4 as a chemical oxygen scavenger to suppress NOx production
Despite the many advantages offered by plasma technology for the conversion of CO2, two main challenges remain:1(1) Separation: the output of a plasma reactor consists of a homogeneous gas mixture; in the case of plasma-based CO2 conversion, it yields a mixture of CO and O2 (and any unreacted CO2) that is very difficult (and thus energy-intensive) to separate by conventional methods;
(2) Impurities: the presence of other gases (even those generally considered to be chemically inert) influences both the physical properties of the plasma and its chemistry; in the case of plasma-based CO2 conversion, the presence of N2 results in the undesired formation of NOx and N2O.
Here, we show how focussing on the plasma chemistry can help us to simultaneously find answers to both the separation and impurity issues, in the case of a DBD plasma reactor used for the conversion of an impure CO2 feed containing N2.
As a baseline case, we studied a DBD operating at a specific energy input (SEI) of 12 kJ L−1, for a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of CO2:N2. Detailed experimental and modelling results for a wide variety of CO2:N2 mixing ratios were discussed in a previous study,9 with the highest NOx production occurring for the 1:1 case, which is the main reason why we chose to further explore that condition first. A chemical analysis revealed that NOx species are formed through several pathways in the presence of N2, during plasma processing of CO2. The main formation mechanism, for all the different NOx species, starts with a reaction involving O (or O2) and N (or N2(A3)) (see also Section 2.2 below).9 This observation is complementary with that made in a previous study, which showed that it was possible to chemically trap oxygen species, in situ, by adding a hydrogen source.14 Additionally, another separate study showed that when O and H radicals are present in a plasma, their natural tendency is to form H2O.15 Therefore, by combining these three observations, it becomes apparent that we are presented with a ‘chemical opportunity’. We hypothesize, based on chemical analyses from these prior studies, that the addition of a small stoichiometric amount of a hydrogen source to a CO2:N2 mixture should be sufficient for trapping the O radicals with H species to form OH and H2O, before the N species can react with the O species and form NOx and N2O (Fig. 1).
:1 mixture of CO2:N2. Detailed experimental and modelling results for a wide variety of CO2:N2 mixing ratios were discussed in a previous study,9 with the highest NOx production occurring for the 1:1 case, which is the main reason why we chose to further explore that condition first. A chemical analysis revealed that NOx species are formed through several pathways in the presence of N2, during plasma processing of CO2. The main formation mechanism, for all the different NOx species, starts with a reaction involving O (or O2) and N (or N2(A3)) (see also Section 2.2 below).9 This observation is complementary with that made in a previous study, which showed that it was possible to chemically trap oxygen species, in situ, by adding a hydrogen source.14 Additionally, another separate study showed that when O and H radicals are present in a plasma, their natural tendency is to form H2O.15 Therefore, by combining these three observations, it becomes apparent that we are presented with a ‘chemical opportunity’. We hypothesize, based on chemical analyses from these prior studies, that the addition of a small stoichiometric amount of a hydrogen source to a CO2:N2 mixture should be sufficient for trapping the O radicals with H species to form OH and H2O, before the N species can react with the O species and form NOx and N2O (Fig. 1).
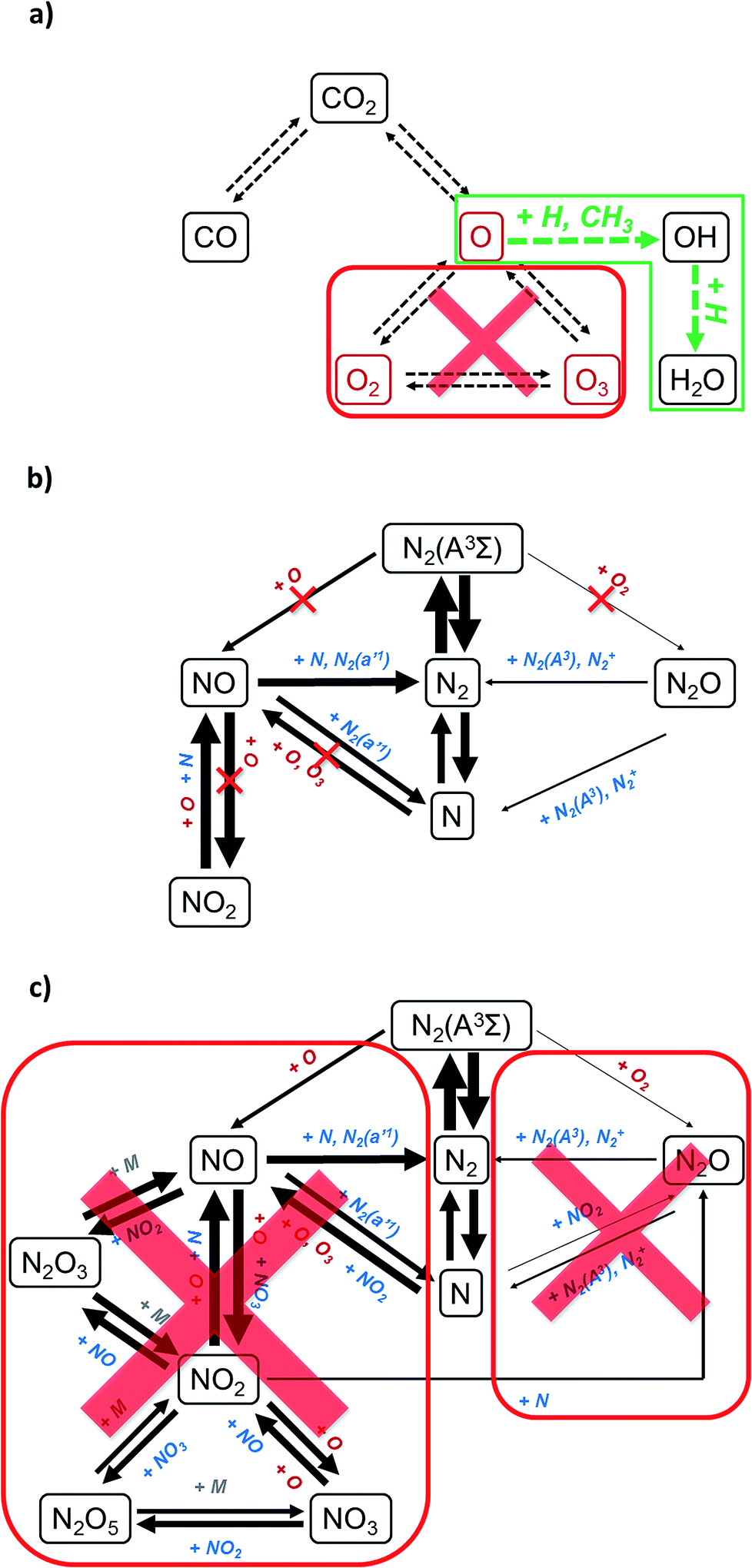 | ||
| Fig. 1 Simplified reaction scheme illustrating the suppression of the main pathways of the NOx and N2O chemistry through the addition of CH4. Reaction pathways starting from CO2 show the in situ trapping of O by H species (a); initiation of the NOx chemistry, indicating which pathways are eliminated by the in situ trapping of O (b); complete overview of the NOx and N2O chemistry to be suppressed by the scavenging of oxygen (c). Original reaction schemes have been adapted from ref. 9. | ||
To verify the validity of our hypothesis that an effective chemical oxygen scavenger can prevent the formation of NOx and N2O, we investigated the effects of using CH4 as a hydrogen source. Some of the most important plasmachemical reactions leading to the formation of the desired hydrogen radicals are the following electron impact dissociation reactions of CH4:
| e− + CH4 → CH3 + H + e− | (1) |
| e− + CH4 → CH2 + H + H + e− | (2) |
| e− + CH4 → CH2 + H2 + e− | (3) |
| e− + CH4 → CH + H2 + H + e− | (4) |
| e− + CH4 → C + H2 + H2 + e− | (5) |
These radicals react further through subsequent electron impact dissociation reactions:
| e− + CH3 → CH2 + H + e− | (6) |
| e− + CH3 → CH + H2 + e− | (7) |
| e− + CH2 → CH + H + e− | (8) |
| e− + CH → C + H + e− | (9) |
| e− + H2 → H + H + e− | (10) |
The most important electron impact dissociation and excitation reactions with CO2 and N2 are:
| e− + CO2 → CO + O + e− | (11) |
| e− + N2 → N2(A3) + e− | (12) |
| e− + N2 → N + N + e− | (13) |
For more details on these and other types of plasmachemical (electron impact) reactions refer to the existing literature and databases.1,9,16–19
We varied the CH4 addition from 0.1 up to 2.0 mol% of the total CO2:N2 mixture, for a DBD under operating conditions similar to those in the baseline case. It is important to note that the introduction of other components influences the physics of the plasma and its chemistry, especially with a species like CH4, which results in a cascade of reactive compounds, including H and CHx radicals. As a result, the electron density and temperature, which affect the conversions, can be altered significantly. Additionally, the conversion of CO2 can also decrease, due to additional back reactions to CO2, such as:
| HCO + O → CO2 + H, k = 5.00 × 10−11 cm3 per molecule per s at 300 K (ref. 20) | (14) |
| OH + CO → CO2 + H, k = 1.25 × 10−13 cm3 per molecule per s at 300 K (ref. 20) | (15) |
This effect was observed in a previous study, in which adding 2 mol% CH4 to pure CO2 yielded a drop in the relative conversion of CO2 by ∼10%.14
In Fig. 2, we can clearly see a decrease of both the NO (1875 cm−1) and NO2 (1599 cm−1) peaks, when adding CH4 to the mixture, with the NO peak showing the biggest initial decrease, and the NO2 peak showing a stronger overall response (see also Fig. 3a). The NO peak decreases by 42% upon adding 0.1 mol% CH4, by 51% with 1.0 mol%, and by 63% with 2.0 mol% CH4 added. The NO2 peak, on the other hand, decreases by 32% upon adding 0.1 mol% CH4, by 82% with 1.0 mol%, and by 96% with 2.0 mol% CH4 added.
 | ||
| Fig. 2 FTIR spectra of a 1:1 mixture of CO2:N2 with 0, 0.1, 0.5, 1.0 and 2.0 mol% CH4 added. For clarity, the CO2 peak has been removed. The negative absorbance of the CH4 bands is due to the subtraction of the blank spectra obtained before turning on the plasma. Original spectra are provided in Section 2.3 of the ESI.† | ||
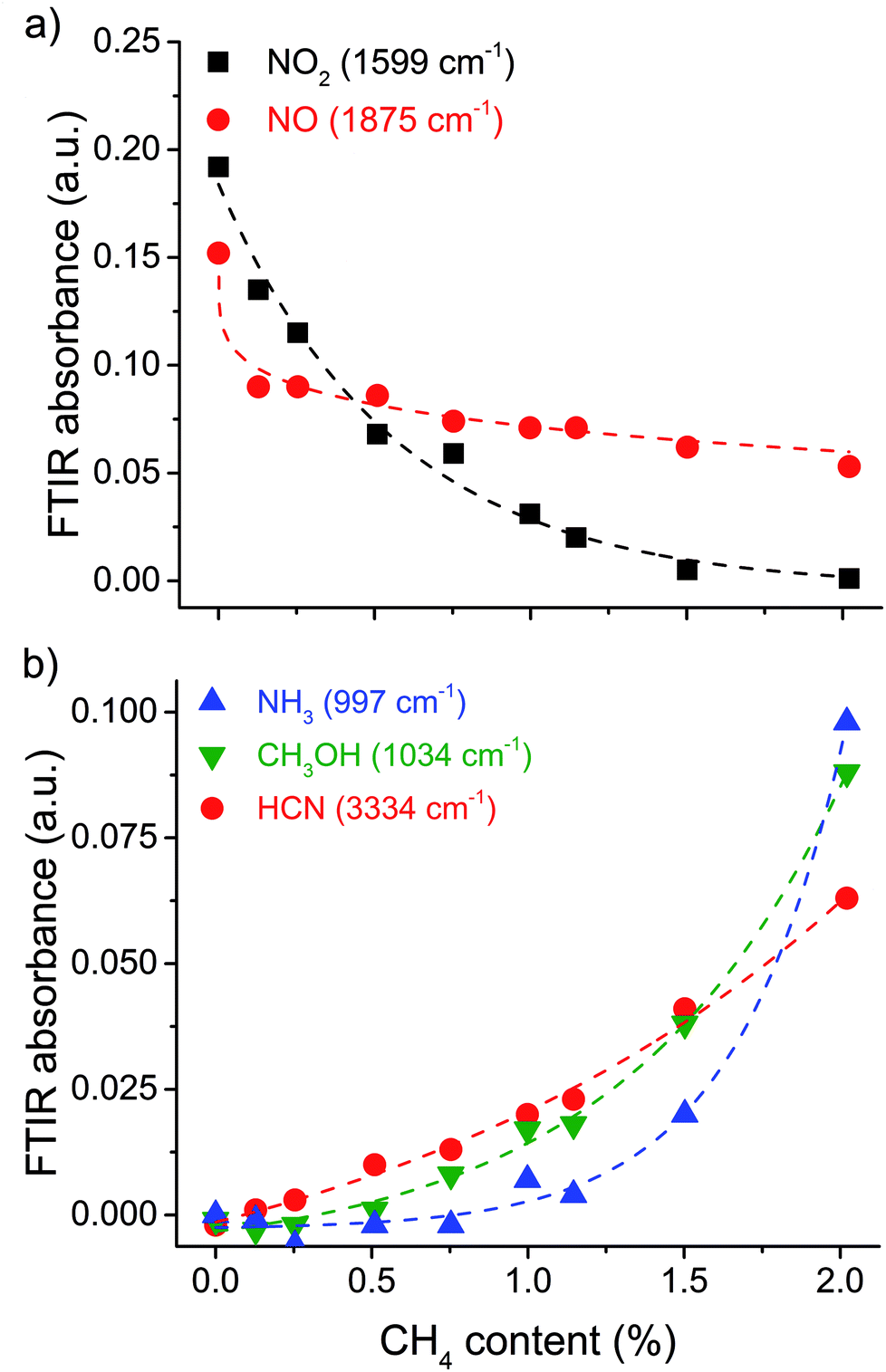 | ||
| Fig. 3 FTIR absorbance for NO and NO2 (a); and for NH3, CH3OH and HCN (b) as a function of the amount of CH4 added to a 1:1 mixture of CO2:N2 for a SEI of 12 kJ L−1. | ||
Due to a complete overlap of the CH4 peaks, we cannot determine whether the N2O3 (1309 cm−1) and/or N2O5 (1245 cm−1) peaks decrease, upon addition of CH4. Nevertheless, this would be a logical consequence, since N2O3 and N2O5 are secondary reaction products from NO and NO2 (Fig. 1).
The N2O (2233 cm−1) peak, on the other hand, seems to increase when more CH4 is added (Fig. 2). This seems in contrast with a severe reduction of the formation of O2, which is necessary for the production of N2O from N2(A3) (Fig. 1). Therefore, there are two options: either the N2O concentration is indeed increasing or its decrease is masked in the FTIR spectra due to interference of other compounds with a similar absorption of the IR frequency (both options are further discussed in Section 2.2).
Besides the decrease in NO and NO2 peak intensities, some additional peaks started to emerge from the noise when we added 1 mol% CH4 to the mixture (Fig. 2); they became clearly visible as we increased the CH4 concentration to 2 mol%. The peak at 3334 cm−1 corresponds to HCN;21 the peak at 1034 cm−1 corresponds to CH3OH;21 and the peak at 997 cm−1 corresponds to NH3.21 The HCN peak increases almost linearly, starting from 0.1 mol% CH4, whereas the CH3OH and NH3 peaks only emerge clearly from the noise starting at 1.0 mol% of CH4 added, and exhibit an exponential increase with further addition of CH4, to 2 mol% (Fig. 3b).
The formation of these additional components indicates that adding more than 1 mol% CH4 generates an excess of the hydrogen source; most of the O species have been trapped into H2O and the excess radicals produce some of the typical products that can be expected in a CH4/N2 mixture (NH3 and HCN)17 and in a CH4/CO2 mixture (CH3OH).18,22 This result is not surprising and consistent with the stoichiometric balance for adding 1 and 2 mol% CH4.
By correcting the CO2 conversion of 3.8% of the baseline case9 to 3% for the lowered conversion, upon addition of CH4 (as well as to simplify the balance), and then adding 1 mol% CH4 (which is almost completely converted, see Table S3 and Fig. S3 in the ESI†), we can construct the following balance:
| 3% CO2 → 3% CO + 3% O | (16) |
| 1% CH4 → 1% C + 4% H | (17) |
| 4% CO + 2% H2O | (18) |
In this case, the O radicals will readily recombine with the C radicals and form CO, and with the H radicals and form H2O.
When adding 2 mol% CH4, (16), (17) and (18) become:
| 3% CO2 → 3% CO + 3% O | (19) |
| 2% CH4 → 2% C + 8% H | (20) |
| 4% CO + 2% H2O + 1% C + 4% H | (21) |
Hence, besides forming CO and H2O, the C and H radicals in excess will form other products, such as CH3OH, HCN, NH3 and HNCO, as revealed in Fig. 2 and 3. From these stoichiometric balances, it is also clear that the use of CH4 as a hydrogen source can lead to an increase in CO selectivity. Indeed, when increasing the CH4 content from 0.1 to 2 mol% the CO peak in the FTIR spectra increases by 42% (see Fig. 2).
In theory the formation of these additional components should not be a major problem, unlike the NOx formation we are aiming to inhibit, since CH3OH, HCN and HNCO can be condensed from the CO stream, and for NH3 efficient scrubbing systems exist.
2.2. Oxygen scavenging chemistry
The experimental results presented in Section 2.1 clearly show that the addition of CH4 as a chemical oxygen scavenger does indeed suppress the formation of NOx, in situ. The observed trends can be explained by looking at the different reaction rate coefficients of the most important reactions.Without a hydrogen source, the main components of the mixture are the following: the unreacted CO2 and N2, the CO2 electron impact dissociation products CO and O, and, to a very small extent, the N2 electron impact dissociation product N and the electron impact excited metastable N2(A3). However, due to its high dissociation energy threshold, the conversion of N2 and thus the concentration of N is very low (∼1017 cm−3), for a DBD plasma.9 In addition, although the concentration of N2(A3) is higher (∼2 × 1018 cm−3), only ∼2% (∼4 × 1016 cm−3) takes part in the formation of NOx, due to its fast quenching processes.9 For these main components, we can establish the following reaction chemistry, which recombines most of the O radicals to form O2:
| O + O (+M) → O2 (+M), k = 1.18 × 10−13 cm3 per molecule per s at 298 K (ref. 23) | (22) |
However, some of the O radicals, as well as O3, react with the few N radicals (see Fig. 1):
| O + N (+M) → NO (+M), k = 2.24 × 10−13 cm3 per molecule per s at 298 K (ref. 23) | (23) |
| O3 + N → NO + O2, k = 1.00 × 10−16 cm3 per molecule per s at 300 K (ref. 24) | (24) |
Subsequently, some of the O radicals react with the formed NO:
| O + NO → NO2, k = 2.42 × 10−12 cm3 per molecule per s at 300 K (ref. 25) | (25) |
Additionally, the metastable N2(A3) also reacts with the O radicals and O2:
| O + N2(A3) → NO + N, k = 7.00 × 10−12 cm3 per molecule per s at 300 K (ref. 9) | (26) |
| O2 + N2(A3) → N2O + O, k = 2.00 × 10−14 cm3 per molecule per s at 300 K (ref. 9) | (27) |
When a small amount (<2 mol%) of CH4 is added as a hydrogen source, the main components of the mixture are the following: the unreacted CO2 and N2 (and to a minor extent CH4), the CO2 electron impact dissociation products CO and O, the N2 electron impact dissociation product N (to a very small extent) and the electron impact excited metastable N2(A3), and the CH4 electron impact dissociation products CHx and H.17,18 Up to 0.5 mol% of CH4 added, the conversion of CH4 is close to 100%, for 1 mol% of CH4 added, the conversion is still 89%, but for 2 mol% of CH4 added, the conversion decreases to 59% (see ESI Table S3 and Fig. S3†). To effectively trap the O radicals and to suppress the formation of NOx and N2O, in situ, the scavenging reactions need to be faster than reactions 22 to 27 described above. It is important to note that the reaction rate coefficients can only give us an indication of the speed of reaction, so the information presented above needs to be put in perspective. In order to determine the real, exact reaction rates, we would also need to know the densities of all the species and the various chemical equilibria involved. Those can be obtained through the development of a complete and extensive chemical kinetics model.
First, the rate coefficients for O radical scavenging reaction with H and CH3 radicals (see below) are clearly in the same order and higher than those for the above reactions (22), (23) and (26). Furthermore, the concentrations of H and CH3 radicals (∼2.5 × 1017 to 5 × 1018 cm−3, based on the (nearly) full conversion of CH4 at 0.1 to 2% CH4 added) are higher than those of the N radicals (∼1017 cm−3; see above) and available metastable N2(A3) (∼4 × 1016 cm−3; see above). Hence, these reactions are estimated to be faster, which means that H and CH3 radicals are indeed effective chemical oxygen scavengers:
| O + CH3 → CH2O + H, k = 1.40 × 10−10 cm3 per molecule per s at 300 K (ref. 26) | (28) |
| O + H (+M) → OH (+M), k = 1.06 × 10−12 cm3 per molecule per s at 300 K (ref. 27) | (29) |
The CH2O radical further reacts towards the formation of OH:
| O + CH2O → HCO + OH, k = 1.73 × 10−13 cm3 per molecule per s at 300 K (ref. 20) | (30) |
| O + HCO → CO + OH, k = 5.00 × 10−11 cm3 per molecule per s at 300 K (ref. 20) | (31) |
The formed OH radicals get rapidly trapped into H2O and CH3OH by subsequent reactions (32)–(35), some of them are even faster than the initial reactions (29)–(31) forming OH. This, in turn, enhances the formation of OH by Le Chatelier's principle, since these reactions rapidly remove the OH radicals from the mixture:
| OH + CH3 (+M) → CH3OH (+M), k = 1.00 × 10−10 cm3 per molecule per s at 300 K (ref. 28) | (32) |
| OH + H (+M) → H2O (+M), k = 1.65 × 10−11 cm3 per molecule per s at 300 K (ref. 20) | (33) |
| OH + CH2O → HCO + H2O, k = 9.37 × 10−12 cm3 per molecule per s at 300 K (ref. 29) | (34) |
| OH + CH3 → CH2 + H2O, k = 1.13 × 10−12 cm3 per molecule per s at 300 K (ref. 28) | (35) |
In general, all these chemical reactions ((28) to (35)) provide a clear indication of how the addition of CH4, as an oxygen scavenger, suppresses the formation of NOx and possibly N2O. As mentioned above, the increase in the N2O peak seems contradictory, at first, especially since the formation of O2 is severely suppressed. One possible explanation could be that the formation of N2O is effectively suppressed, and its concentration decreases, but this is masked in the FTIR spectra due to interferences from other compounds. Indeed, HNCO (2254–2268 cm−1),30,31 NCO (2175 cm−1)31 and NCO + OH interactions (2237 cm−1)31 have almost the same FTIR bands as N2O (2233 cm−1),21 making it likely that the increased peak in the range 2210–2250 cm−1 is the result of an increase of the (H)NCO concentration, which masks the decrease of the N2O concentration.
Another plausible explanation could be that, although the O2 formation is suppressed, N2O is being formed through new different pathways, as a result of the formation of HCN and NH3. For high temperature conditions, this has been detailed in numerous studies found in the literature describing the combustion chemistry of (de-)NOx (and fuel NOx).11,12 In the next section we analyse whether this chemistry is also relevant for the current low temperature plasma process under study.
2.3. de-NOx chemistry
Despite scavenging the reactive O species to suppress the NOx an N2O formation, the presence of a hydrogen source also leads to a variety of reactants (such as HCN and NH3), leading, in turn, to the additional formation (or destruction) of NOx or N2O. Fig. 4 gives a visual representation of how, at low temperature, a general NOx reaction scheme of these interactions might look like, for CO2:N2 plasma with the addition of CH4. It is important to note that this reaction scheme is only of a general character. It is based on the products observed with FTIR and on the most important reactions, defined by their rate coefficients presented in Section 2.4 of the ESI.† To construct an accurate fully supported chemical pathway, it is necessary to build a complete plasma chemical kinetics model that includes a detailed description of the NOx and by-product chemistry, supported and validated by an extensive quantitative experimental study. For which the current analysis, together with the recent work of Wang et al.,16 can already provide a foundation.
 | ||
| Fig. 4 Basic visual representation of a low temperature NOx reaction scheme for CO2:N2 plasma with addition of CH4 (based on the most important reaction rate coefficients presented in Section 2.4 of the ESI†). For double-sided arrows, reactants above the arrows are those for reactions going from right to left, while reactants below the arrows are those for reactions going from left to right. NOx and N2O are marked in orange, whereas stable products without important loss processes are marked in green. | ||
We can summarize the reaction scheme as follows: HCN is formed from reactions of N and NO with CHx and its concentration increases linearly (Fig. 3b) due to the absence of important destruction reactions (contrary to what is found in combustion processes). The formation of NH3, on the other hand, is delayed until an excess of CH4 is added to the mixture (Fig. 3b), due to the consumption of the NHx precursors through a reaction with either NO (to form N2 and N2O) or O (to form HNO). The formation of CH3OH is also delayed until an excess of CH4 is added to the mixture (Fig. 3b), probably due to the consumption of CHx in the de-NOx chemistry. Upon addition of CH4, the NO2 concentration decreases more than the NO concentration (Fig. 3a) due to the interconversion of NO2 into NO through reactions with H and O, and due to the formation of NO through several reactions starting from NHx, HNO and NCO. Nevertheless, the NO concentration continues to decrease, due to destruction reactions with NHx, CH and H. Finally, N2O is formed from NO through reactions with NH and NCO, and destroyed by CH, whereas HNCO is formed by reaction of NCO with HCO, HNO and CH2O, indicating that HNCO is a stable end-product, and that N2O is converted into HCN as a stable end-product. As a result, the N2O concentration most likely decreases and the increased peak at 2233 cm−1 (Fig. 2) is, in fact, due to the formation of HNCO, rather than an increase in N2O concentration.
3. Discussion and outlook
We have demonstrated that it is possible to reduce the amount of NOx produced during plasma-based CO2 splitting in the presence of N2 simply by adding a hydrogen source such as CH4, leading to the almost stoichiometric in situ trapping of oxygen. Adding CH4 at 1 mol% of the total mixture yields NO2 and NO FTIR absorbance peaks that are 82% and 51% lower than those obtained without the addition of CH4. Even higher reductions, up to 96% and 63%, are possible when a stoichiometric excess of the hydrogen source is added, which was 2 mol% CH4 in our case. However, in that case, the excess hydrogen and carbon radicals will lead to the regular plasma-based reforming chemistry, creating several by-products in low concentrations, such as NH3, HCN, CH3OH and probably HNCO.From the data analysis it becomes clear that two processes are responsible for reducing the amount of NOx produced. The first one is—the process we were aiming for—the direct inhibition of NOx formation through the fast oxygen scavenging chemistry by the H and CHx radicals, arising from the introduced CH4. The second one is the known reduction of NOx to N2 in the presence of reducing agents, in this case occurring at room temperature.
These findings suggest that impure CO2 mixtures containing N2 may be used as a feedstock, which could have a significant positive impact on the implementation of plasma-based CO2 conversion research. As a result, there are several interesting follow-up questions. In the present study, we used the most convenient source of hydrogen, CH4, but it would be interesting to investigate other hydrogen sources.14 The most fundamental one would be H2, which could theoretically result in fewer by-products (cf. chemical analysis above). However, we could also look into greener and more sustainable hydrogen sources, such as glycerol.32 From the analysis side, an important challenge to be addressed in future studies is the issue of N2O and (H)NCO identification. Higher resolution FTIR, or separate N2O detection using a customized GC (with TCD, ECD, NPD or MS) or custom sensors, might offer a solution.
Additionally, to capture the complete complexity of the underlying mechanisms and to be able to fully analyse and comprehend all the chemical pathways, it will be necessary to build a complete plasma chemical kinetics model with a detailed NOx and by-product chemistry, supported and validated by a wide range of experiments. A good starting point for the development of such a model would be to expand the NOx chemistry from Wang et al.'s recent work on CO2/CH4/N2 mixtures.16
It would also be interesting to see whether the same effect can be found for different plasma types, especially for microwave (MW) and gliding arc (GA) plasmas. For these plasmas, the formation of NOx is much higher, and the dominant pathway proceeds through vibrationally excited N2 states, rather than through the metastable N2 state and N radicals.9,10
Finally, these results are a clear indication that the plasma chemistry can be controlled to a certain extent by adding small amounts of additives; a similar demonstration has been given by Snoeckx et al.33 in their work on the selective formation of methanol. Despite the seeming trivialness of this insight, directing more research towards simple chemical intervention steps—before turning to complex engineering or plasma-catalysis combinations—could lead to short-term promising advancements in the field of plasma-based CO2 conversion and hydrocarbon reforming.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research reported in this publication was supported by funding from the “Excellence of Science Program” (Fund for Scientific Research Flanders (FWO): grant no. G0F9618N; EOS ID: 30505023). The authors R. S. and M. S. C. acknowledge financial support from King Abdullah University of Science and Technology (KAUST), under award number BAS/1/1384-01-01.References
- R. Snoeckx and A. Bogaerts, Chem. Soc. Rev., 2017, 46, 5805–5863 RSC.
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC.
- A. Goeppert, M. Czaun, J.-P. Jones, G. K. Surya Prakash and G. A. Olah, Chem. Soc. Rev., 2014, 43, 7995–8048 RSC.
- P. Lanzafame, G. Centi and S. Perathoner, Chem. Soc. Rev., 2014, 43, 7562–7580 RSC.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- P. Furler, J. R. Scheffe and A. Steinfeld, Energy Environ. Sci., 2012, 5, 6098–6103 RSC.
- D. W. Keith, Science, 2009, 325, 1654–1655 CrossRef CAS PubMed.
- C. Kolster, E. Mechleri, S. Krevor and N. Mac Dowell, Int. J. Greenhouse Gas Control, 2017, 58, 127–141 CrossRef CAS.
- R. Snoeckx, S. Heijkers, K. Van Wesenbeeck, S. Lenaerts and A. Bogaerts, Energy Environ. Sci., 2016, 9, 999–1011 RSC.
- S. Heijkers, R. Snoeckx, T. Kozák, T. Silva, T. Godfroid, N. Britun, R. Snyders and A. Bogaerts, J. Phys. Chem. C, 2015, 119, 12815–12828 CrossRef CAS.
- J. A. Miller and C. T. Bowmans, Prog. Energy Combust. Sci., 1989, 15, 287–338 CrossRef CAS.
- A. N. Hayhurst and A. D. Lawrence, Prog. Energy Combust. Sci., 1992, 18, 529–552 CrossRef CAS.
- C. Baird and M. Cann, Environmental Chemistry, W. H. Freeman, 4th edn, 2008 Search PubMed.
- R. Aerts, R. Snoeckx and A. Bogaerts, Plasma Processes Polym., 2014, 11, 985–992 CrossRef CAS.
- R. Snoeckx, A. Ozkan, F. Reniers and A. Bogaerts, ChemSusChem, 2017, 10, 409–424 CrossRef CAS PubMed.
- W. Wang, R. Snoeckx, X. Zhang, M. S. Cha and A. Bogaerts, J. Phys. Chem. C, 2018, 122, 8704–8723 CrossRef CAS.
- R. Snoeckx, M. Setareh, R. Aerts, P. Simon, A. Maghari and A. Bogaerts, Int. J. Hydrogen Energy, 2013, 38, 16098–16120 CrossRef CAS.
- R. Snoeckx, R. Aerts, X. Tu and A. Bogaerts, J. Phys. Chem. C, 2013, 117, 4957–4970 CrossRef CAS.
- LXCAT database, http://www.lxcat.net Search PubMed.
- D. L. Baulch, C. J. Cobos, R. A. Cox, C. Esser, P. Frank, T. Just, J. A. Kerr, M. J. Pilling, J. Troe, R. W. Walker and J. Warnatz, J. Phys. Chem. Ref. Data, 1992, 21, 411–429 CrossRef CAS.
- NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg MD, 2018 Search PubMed.
- R. Snoeckx, A. Rabinovich, D. Dobrynin, A. Bogaerts and A. Fridman, Plasma Processes Polym., 2017, 14, 1600115 CrossRef.
- I. M. Campbell and C. N. Gray, Chem. Phys. Lett., 1973, 18, 607–609 CrossRef CAS.
- A. J. Barnett, G. Marston and R. P. Wayne, J. Chem. Soc., Faraday Trans. 2, 1987, 83, 1453 RSC.
- R. Atkinson, D. L. Baulch, R. A. Cox, R. F. Hampson, J. A. Kerr, M. J. Rossi and J. Troe, J. Phys. Chem. Ref. Data, 1997, 26, 1329–1499 CrossRef CAS.
- R. Atkinson, D. L. Baulch, R. A. Cox, R. F. Hampson, J. A. Kerr and J. Troe, J. Phys. Chem. Ref. Data, 1992, 21, 1125–1568 CrossRef CAS.
- W. Tsang and R. F. Hampson, J. Phys. Chem. Ref. Data, 1986, 15, 1087–1279 CrossRef CAS.
- D. L. Baulch, C. J. Cobos, R. A. Cox, P. Frank, G. Hayman, T. Just, J. A. Kerr, T. Murrells, M. J. Pilling, J. Troe, R. W. Walker and J. Warnatz, J. Phys. Chem. Ref. Data, 1994, 23, 847–1033 CrossRef CAS.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, J. A. Kerr, M. J. Rossi and J. Troe, Summary of Evaluated Kinetic and Photochemical Data for Atmospheric Chemistry, 2001 Search PubMed.
- M. S. Lowenthal, R. K. Khanna and M. H. Moore, Spectrochim. Acta, Part A, 2002, 58, 73–78 CrossRef CAS.
- I. Czekaj, J. Wambach and O. Kröcher, Int. J. Mol. Sci., 2009, 10, 4310–4329 CrossRef CAS PubMed.
- X. Zhu, T. Hoang, L. L. Lobban and R. G. Mallinson, Chem. Commun., 2009, 2908 RSC.
- R. Snoeckx, W. Wang, X. Zhang, M. S. Cha and A. Bogaerts, Sci. Rep., 2018, 8, 15929 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8se00584b |
| This journal is © The Royal Society of Chemistry 2019 |