
Phenol–TiO2 complex photocatalysis: visible light-driven selective oxidation of amines into imines in air†
 *ab
*ab
Abstract
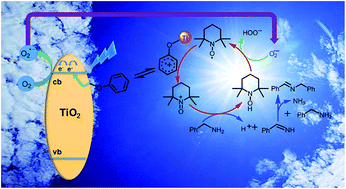
Strong interfacial charge-transfer (ICT) has been observed between phenol and TiO2. It was demonstrated that a single Ph–O–Ti linkage could induce ICT transitions in the visible light region. By utilizing the surface complexation of phenol with TiO2, we, herein, present a new photocatalytic protocol for the selective oxidation of amines to imines in air. Our success depended on merging the phenol–TiO2 complex photocatalyst with (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO), so called cooperative photocatalysis, which improved stability of the surface-complexed phenol under the oxidative circumstance and promoted the selective conversion of amines. With 5 mol% of TEMPO as a co-catalyst and phenol–TiO2 complex (containing 0.8 mol% of phenol) as a photocatalyst, amines could be efficiently oxidized into imines with atmospheric O2 under blue light-emitting diode (LED) irradiation. In addition, a mechanism is proposed to explain the visible-light photocatalytic performance of the present system.
- This article is part of the themed collections: 2019 Sustainable Energy and Fuels HOT Articles and 2018 Sustainable Energy and Fuels HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

