
Free-standing protective films for enhancing the cyclability of organic batteries†
Shaolong
Zhu
,
Mi
Tang
,
Yanchao
Wu
,
Yuan
Chen
,
Cheng
Jiang
,
Cong
Xia
,
Shuming
Zhuo
,
Bo
Wang
and
Chengliang
Wang
 *
*
School of Optical and Electronic Information, Wuhan National Laboratory for Optoelectronics (WNLO), Huazhong University of Science and Technology, Wuhan 430074, PR China. E-mail: clwang@hust.edu.cn
First published on 14th November 2018
Abstract
Organic sodium-ion batteries (OSIBs) are regarded as potential alternatives to traditional inorganic lithium-ion batteries. But organic active materials always suffer from dissolution in organic electrolytes. The dissolution significantly reduces the capacity, coulombic efficiency and cyclability, and subsequently limits the possible large-scale applications of organic batteries. Herein, inspired by the success of poly(3,4-ethylenedioxythiophene)-poly(styrene sulfonate) (PEDOT:PSS) film in alleviating the shuttle of organic electrodes to a certain extent and the good mechanical properties of polyethylene oxide (PEO) which has been extensively studied as a solid-state electrolyte (SSEs), we develop a free-standing protective film (only ∼11 μm) with blended PEDOT:PSS and PEO for inhibiting the shuttle of organic electrode materials. The application of free-standing protective films significantly improves the capacity, coulombic efficiency and cyclability of organic active electrodes, by using 5,7,12,14-pentacenetetrone (PT) as a typical demonstration, much better than the previously reported protective films.
Introduction
The rapid development of electronic products (especially wearable electronics) and electric vehicles requires large-scale multifunctional rechargeable energy storage systems with high electrochemical performance.1–8 Organic sodium-ion batteries (OSIBs) have attracted great attention to meet large-scale multifunctional applications and hence become one of the most promising alternatives to current commercial inorganic lithium-ion batteries, due to the advantages of abundant resources, environmental benignity, flexibility and convenient molecular design for tuning the electrochemical performance.9–18However, organic active materials are usually soluble in organic electrolytes. The dissolved active materials diffuse into the electrolytes and may react with (or electrodeposit on) the counter electrode.19–21 Consequently, the active materials continue to dissolve in the electrolytes, due to the consumption of the dissolved active materials. The vicious cycle of dissolution–reaction–dissolution will continue till the active materials are exhausted. The dissolution and the shuttle of organic active materials will lead to performance degradation of batteries (decrease of capacity, poor cyclability and low coulombic efficiency).19 The solution to solve the dissolution problem generally includes two strategies: (i) using a protective layer with selective permeability of Na-ions to isolate the electrode materials;19,22–30 (ii) using insoluble molecules31,32 or polymers33–38 as active materials. The second method can efficiently inhibit the dissolution but always encounters the challenge of complex syntheses and purification of the materials. Complex and high-cost processes are therefore inevitable. The first method seems much more convenient, by mixing the active materials with protective carbon materials or polymeric binders, or simply applying a protective layer. Among them, the utilization of protective films is more efficient and can encapsulate the active materials thoroughly. Moreover, this method will extremely simplify the process if the protective layer is combined with a separator.
In one of our pioneering studies, inspired by the pinhole-free polymeric dielectric layer in organic electronics,39 poly(3,4-ethylenedioxythiophene)-poly(styrene sulfonate) (PEDOT:PSS, which is widely used as an electrode in electronics through spin coating) was used to fabricate a selectively permeable membrane to enhance the capacity and cyclability of 5,7,12,14-pentacenetetrone (PT).19 PEDOT:PSS was cast directly on a porous separator, and the obtained selectively permeable membrane allows the passage of the Na-ion, but blocks the active materials preventing their direct reaction with Na electrodes. The enhanced capacity and cyclability of batteries, together with the reduced rate performance, proved its effectiveness. However, this method has some shortcomings. For example, the PEDOT:PSS films show poor mechanical properties and may break during drying the films or assembly of the batteries, leading to capacity fading. The obtained capacity (73 mA h g−1) is also much lower than the theoretical capacity (317 mA h g−1).
Herein, inspired by our previous work and the good mechanical properties and moderate ionic conductivity of polyethylene oxide (PEO) which has been extensively studied as a solid-state electrolyte (SSE),40–42 we developed a free-standing protective film (only ∼11 μm) which consisted of PEDOT:PSS and PEO (PPF, Fig. 1) for suppressing the shuttle effect and enhancing the electrochemical performance of organic electrodes. We found that the single component of PEDOT:PSS (difficult to form a free-standing film)43–45 or PEO (crystallization and relatively low ionic conductivity) is not sufficient to protect the active materials. Our work presents a facile method for inhibiting the shuttle effect and thereby enhancing the capacity and cyclability of organic sodium-ion batteries. What's more, the utilization of SSE materials and free-standing films would pave a way to simplify the processing technology by combining the protective layer with the separator through further optimizing the materials and mixing ratio.
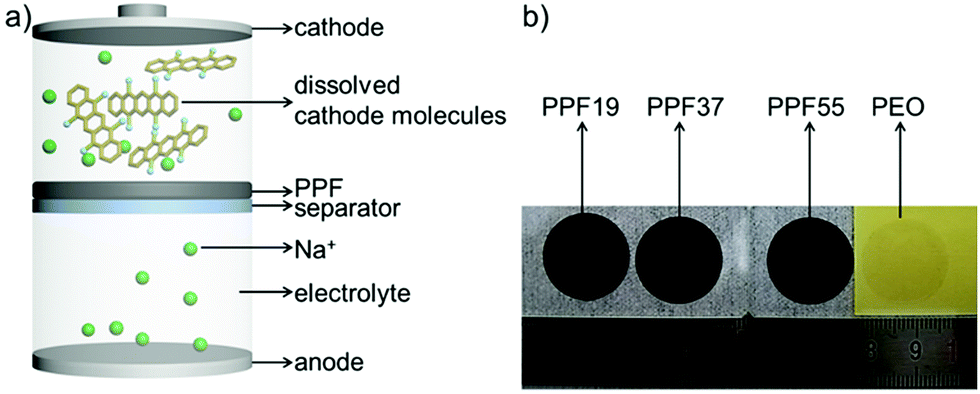 | ||
Fig. 1 (a) Schematic diagram of the free-standing protective film (PPF), which permits the passage of small Na-ions and prevents the passage of large active molecules. (b) Photo images of the free-standing protective films. PPF19, PPF37, PPF55 and PEO represent the films obtained by blending PEDOT:PSS and PEO in the weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9, 3:7, 5:5 and 0:10, respectively. :9, 3:7, 5:5 and 0:10, respectively. | ||
Results and discussion
Electrochemical performance of PT without protective films
PT is used as a proof-of-concept, which can deliver a theoretical capacity as high as 317 mA h g−1.46,47 However, the capacity decreased rapidly due to the dissolution of PT in electrolytes. The electrochemical performance of PT was evaluated by assembling PT electrodes in a half cell. 1 M NaPF6 in dimethyl ether (DME) was used as the electrolyte and sodium metal was used as the counter electrode. The PT electrode was prepared by mixing PT with Super P and poly(vinylidene fluoride) (PVDF) in the weight ratio of 8:1:1. The typical cyclic voltammogram (CV) curves of the PT electrode showed two main pairs of redox peaks located at 1.0 and 2.1 V during the first cathode scan (corresponding to 1.2 and 2.4 V during the first anode scan) at the scan rate of 0.5 mV s−1 (Fig. 2a). Every pair of peaks represented a two-electron transfer. The currents decreased gradually from the second cycle with a slight shift of the redox peaks (1.15 and 2.3 V for the reduction process, 1.25 and 2.5 V for the oxidization process). The shift of the redox peaks between the first and the subsequent cathode scans can probably be ascribed to the activation process and the polarization of the electrode materials. A higher applied potential was required for relaxing the strain/stress during interaction of the sodium ions.48 There were two distinct plateaus in the discharge voltage profiles, located at 2.3 and 1.1 V, which agreed well with the two reduction peaks on the CV curves (Fig. 2b). The charge processes also displayed two plateaus with very small hysteresis compared with the discharge process. Fig. 2c shows that at the rate of 50 mA g−1, the discharging capacity in the first cycle was as high as 285 mA h g−1, which is almost equal to the theoretical capacity of 317 mA h g−1. This indicated the high electrochemical performance of PT as a promising potential cathode for OSIBs. However, the capacity decreased rapidly to 62 mA h g−1 after 20 cycles and to 7 mA h g−1 after 50 cycles (Fig. 2b and c). The degradation can be ascribed to the dissolution of PT in the electrolytes.19 Moreover, the coulombic efficiency (CE) is only about 93% in the first cycle and still lower than 99% even after 35 cycles (Fig. 2c). The low CE can be ascribed to the diffusion of the dissolved PT molecules which might react with Na metal directly. The rapid performance degradation means that it makes no sense to investigate the high rate performance of PT (Fig. 2d and S1†). The decreased capacity is due to the dissolution rather than the high scan rate.
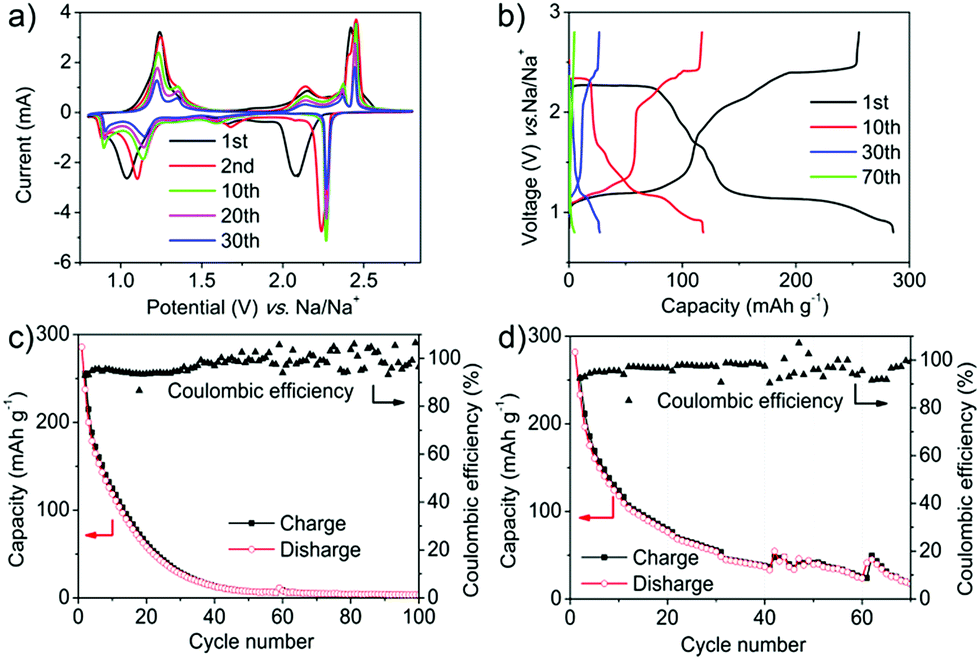 | ||
| Fig. 2 Electrochemical performances of PT electrodes without protective films. (a) Typical CV curves of PT electrodes at the scan rate of 0.5 mV s−1. (b) Voltage profiles at the rate of 50 mA g−1 (the 1st, 10th, 30th and 70th cycles as representatives). (c) Cycling performance at the rate of 50 mA g−1. (d) Rate cyclability (left to right: 50, 100, 200, 500, 1000, 2000 to 50 mA g−1, 10 cycles for each rate). | ||
Electrochemical performance of PT with protective films
The PT electrodes were then assembled with PPF films (Fig. 1a). Motivated by the success of PEDOT:PSS-cast separators for enhancing the cyclability of PT electrodes in our previous work,19 we first tried to get the free-standing films of PEDOT:PSS directly in view of its good electric and ionic conductivities.49 However, we found that it was difficult to achieve free-standing films of PEDOT:PSS. The films were very fragile and broke when peeled off from the substrates, which can probably be ascribed to the poor mechanical properties of PEDOT:PSS (complex-salt nature) films. Inspired by the good mechanical properties and moderate ionic conductivity of PEO which has been extensively studied as a solid-state electrolyte (SSE), a composite film was developed by blending PEDOT:PSS with PEO at different weight ratios. We found that when the content of PEDOT:PSS was higher than 50 wt%, the blending systems became fragile and would be broken. Fig. 1b shows the optical images of the obtained free-standing films by blending PEDOT:PSS and PEO in a weight ratio of 1:9, 3:7 and 5:5. The weight of PEDOT:PSS for every sample was kept constant for investigating the role of PEDOT:PSS for the protective films. The thicknesses of the films were hence in the range from 10 to 100 μm (Fig. S2†). The thickness was controllable. The different thicknesses of the samples are due to the constant weight of PEDOT:PSS in every sample, leading to the largest thickness of PPF19 (104 μm) and smallest thickness of PPF55 (11 μm). The obtained free-standing film of pure PEO is also presented for comparison. It is clear that the pure PEO is almost colorless. However, after adding 10 wt% of PEDOT:PSS, the color of the films changed to black (Fig. 1b).
We firstly checked the pure PEO films as a protective layer for inhibiting the shuttle effect. Fig. S3a† shows the CV curves of the PT-based batteries assembled with pure PEO films, which were quite similar to the CV curves without protective films. The discharge voltage profiles also showed two major plateaus located at about 2.3 and 1.1 V, respectively (Fig. S3b†). The discharging capacity in the first cycle reached 283.5 mA h g−1 at the rate of 50 mA g−1, and then decreased to 212.1 mA h g−1 for the second cycle. Fig. 3c shows the cyclability of PT electrodes assembled with or without PPF films at the rate of 50 mA g−1. It is clear that the capacity became more stable after being assembled with PEO films, although the capacity still decreased rapidly to 48.8 mA h g−1 after 100 cycles. In addition, the CE was about 97% in the first cycle and increased to around 100% after 5 cycles (Fig. S3c†). The capacity decreased by 180 mA h g−1 in the first 10 cycles but only by 54 mA h g−1 for the next 90 cycles. The gradual degradation from the 10th to the 100th cycle and the high CE suggested the protective effect of PEO films, even though it was far from satisfactory. What's more, the batteries after being assembled with PEO films showed very poor rate performance, which were even worse than the batteries without any protective layers (Fig. 3d and S3d†). The poor rate performance can be ascribed to the relatively low ionic conductivity of PEO (normally lower than 10−6 S cm−1),50 indicating that the PEO films also blocked the transport of Na-ions.
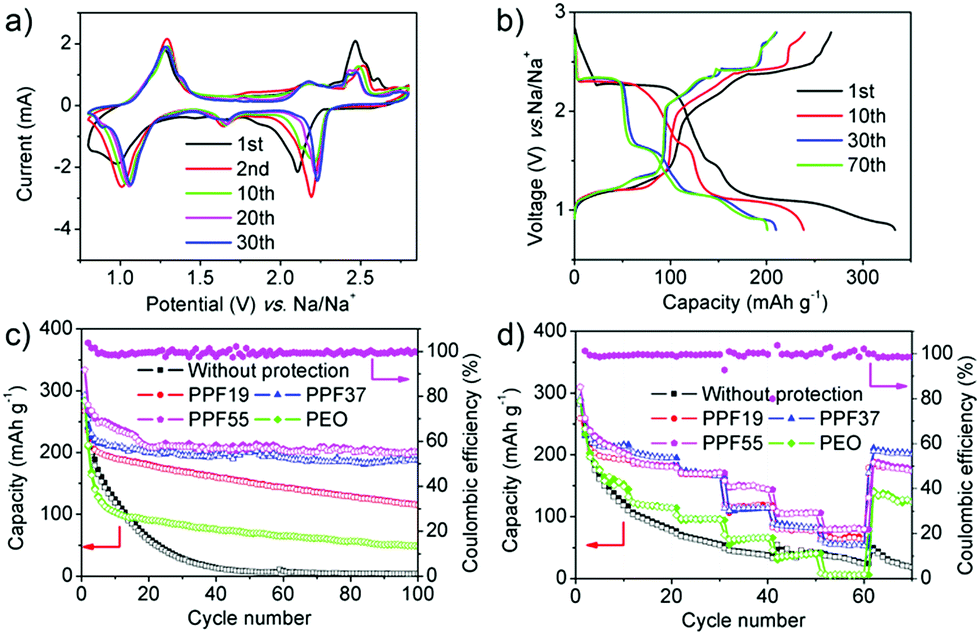 | ||
| Fig. 3 (a) Typical CV curves of PT electrodes assembled with PPF55 films at the scan rate of 0.5 mV s−1. (b) Voltage profiles of PT electrodes assembled with PPF55 films at the rate of 50 mA g−1 (the 1st, 10th, 30th and 70th cycles as representatives). (c) Cycling performance of PT electrodes assembled with or without protective films at the rate of 50 mA g−1. (d) Rate cyclability (left to right: 50, 100, 200, 500, 1000, 2000 to 50 mA g−1, 10 cycles for each rate). The coulombic efficiencies of the batteries assembled with PPF55 films are presented in (c) and (d) as an example. | ||
Based on our previous work as mentioned above,19 we then investigated the PPF19 films as a protective layer for inhibiting the shuttle effect. Fig. S4a† shows the CV curves of the PT-based batteries assembled with PPF19 films. It is obvious that the currents became much more stable than those of the batteries assembled with or without PEO films. Moreover, the capacity became very stable from the 10th cycle (190.1 mA h g−1) and remained at 115 mA h g−1 after 100 cycles (Fig. 3c, S4b and c†). The CE was around 100% from the first cycle (Fig. S4c†). All of this indicated that the PPF19 films indeed can suppress the shuttle effect of PT. It is noteworthy that the rate performance of the batteries assembled with PPF19 films was also improved (Fig. 3d and S4d†). This suggested that the ionic conductivity of PEDOT:PSS should be much better than that of PEO. Unfortunately, we can't measure the ionic conductivity of PEDOT:PSS, because PPF19 films are already electrically conductive even though only 10 wt% of conductive PEDOT:PSS was added. However, we believed that it was safe to draw the conclusion that the improved electrochemical performance of PPF films compared with pure PEO can be ascribed to the higher electric and ionic conductivities, because: (1) as mentioned in our previous work,19 PSS shows strong ion exchange properties of Na-ions and has been applied in water softening; and (2) PSS analogues51 that also have sulfonate functional groups have been reported as solid-state electrolytes, showing much higher ionic conductivity than PEO.
Inspired by these intriguing findings, we further increased the ratio of PEDOT:PSS to 30% and 50%. As mentioned above, further increase in the content of PEDOT:PSS will lead to fragile films. Remarkably, the currents in the CV curves after being assembled with PPF37 films became much more stable and the shapes of curves were still retained with the three kinds of cells mentioned above (Fig. S5a†). The first discharge process delivered a capacity of 293 mA h g−1 at the rate of 50 mA g−1 (Fig. 3c and S5b†). Although the capacity also decreased in the first 10 cycles to 211.2 mA h g−1, the capacity tended to be stable afterwards. The capacity decreased to 199 mA h g−1 after 20 cycles and stabilized at ∼190 mA h g−1 even after 100 cycles (Fig. 3c). The CE remained at about 100% from the first cycle (Fig. S5c†). All of this showed that the PPF37 films can efficiently inhibit the shuttle effect of PT molecules, leading to high coulombic efficiency and cyclability. The batteries assembled with PPF37 films also exhibited excellent rate performance (Fig. 3d and S5d†). Further increase in the content of PEDOT:PSS (PPF55) led to a slight enhancement of electrochemical performance at a low rate (50 mA g−1), as shown in Fig. 3a–c. The similar performance (close to the theoretical capacity) between the cells with PPF37 and PPF55 showed that the capacity became “saturated” when the content of PEDOT:PSS increased to 30%, indicating that the shuttle effect was already suppressed. The first discharge process in PPF55-based cells delivered a capacity of 333 mA h g−1 at the rate of 50 mA g−1. The capacity is a little higher than the theoretical capacity, which is probably due to the contribution of Super P and PEDOT:PSS (Fig. S6†).52 The capacity remained at 201.4 mA h g−1 even after 100 cycles. This performance is much better than that in our previous work (∼73 mA h g−1) by using a PEDOT:PSS-separator as the protective layer, which might break during the assembly of batteries and only alleviated the shuttle effect of PT to a certain extent.19 It's also noteworthy that the content of the active materials is higher in this work (80%) than in our previous paper (50 or 70%), without using any special binders or complex methods but showed better encapsulation of the materials. More attractively, the batteries assembled with PPF55 films displayed much enhanced rate performance (Fig. 3d and S7†). In view of the similar low-rate performance of the cells assembled with PPF55 films to those assembled with PPF37, the enhanced rate performance of PPF55 films can probably be ascribed to the smaller thickness, which benefited the ionic transport (Fig. S2†). In addition, although the PPF55 (11 μm) and PPF37 (42 μm) films were much thinner than PPF19 (104 μm), the cycle stability of PPF55- and PPF37-based cells was much higher than that of PPF19. This indicated the high quality of PPF55 (dense but with high ionic conductivity) and the importance of PEDOT:PSS for suppressing the shuttle effect. Besides, the thickness of PPF55 (11 μm) is also much smaller than that of PEO (33 μm); however, PPF55-based cells showed much better protection of PT (higher capacity) and higher rate capability than PEO-based cells, which reconfirmed that PEDOT:PSS should possess higher electric and ionic conductivities.
Experimental
Materials and characterization
5,7,12,14-Pentacenetetrone (PT), carbon black (Super P), poly(vinylidene fluoride) (PVDF) and 1-methyl-2-pyrrolidinone (NMP) were purchased from Alfa Aesar. Polyethylene oxide (PEO) and conductive poly(3,4-ethylenedioxythiophene)-poly(styrene sulfonate) (PEDOT:PSS, conductivity: 1 S cm−1) were purchased from Sigma Aldrich. All of the reagents and solvents were used as purchased without any purification or treatment. Scanning electron microscopy (SEM) images were acquired using a JSM-7600F scanning electron microscope. The CV experiments were conducted with a BioLogic VSP potentiostat. The CV curves were scanned in the potential range from 0.8 to 2.8 V at a scan rate of 0.5 mV s−1.Preparation of PEDOT:PSS/PEO free-standing films (PPF55 for example)
PEO (58.5 mg) was added into 20 ml deionized water in a beaker and stirred for 2 h at 50 °C. When the PEO was dissolved completely, it was cooled down to room temperature. PEDOT:PSS (58.5 mg) was then added into the beaker and stirred for another 2 h. The mixture was poured into a Petri dish (diameter: 65 mm) and kept still for 5 h on a shockproof table at r.t. It was directly placed in the oven to remove water at 50 °C and then further dried in vacuum for another 6 h. The film (PPF55) was then stripped off from the Petri dish, leading to a freestanding film with thickness about 11 μm. PPF films with different content ratios were obtained similarly with different weight ratios of PEDOT:PSS and PEO, giving freestanding films with thickness in the range of 10–100 μm. PEO films (same weight of PEO with PPF37) were obtained similarly without adding PEDOT:PSS.Electrochemical characterization
PT electrodes were obtained by mixing PT with a conductive additive and binder at a ratio of 8:1:1, and then coating the slurry on a Cu foil using a doctor-blade, leading to a mass loading for every sample of about 1.7 mg cm−2. The electrodes were dried at 90 °C under vacuum overnight. The electrochemical performance was investigated by using 2032 coin cells. The batteries were assembled in an Ar-filled glove box (water < 1 ppm, oxygen < 1 ppm). Sodium was used as a counter electrode and the two electrodes were separated by a glass fiber separator (Whatman, GF/B, pore size: ∼1 μm) with or without protective films (PEO or PPF). 1 M NaPF6 in dimethyl ether (DME) was used as the electrolyte. Galvanostatic experiments were conducted in the potential range of 0.8–2.8 V (vs. Na/Na+) using a LANHE-CT2001A test system (Wuhan, China) at room temperature.
Conclusions
In summary, inspired by the success of PEDOT:PSS films in alleviating the shuttle effect to a certain extent in our previous work and the good mechanical properties and moderate ionic conductivity of PEO which has been extensively studied as an SSE, we developed a free-standing protective film with blended PEDOT:PSS and PEO for suppressing the shuttle effect of active materials and enhancing the cyclability. We found that the single component of PEDOT:PSS or PEO is not sufficient to protect the active materials. The free-standing protective films with blended PEDOT:PSS and PEO in certain ratios can significantly enhance the capacity (close to theoretical capacity), coulombic efficiency (from 93% to ∼100%) and cyclability (201.4 mA h g−1 after 100 cycles). This performance is much better than that of our previous work (∼73 mA h g−1) by using a PEDOT:PSS-separator as the protective layer, which may be broken during the assembly of batteries. The content of the active materials is also higher in this work (80%) than in our previous paper (50 or 70%), without using any special binders or complex methods but showed better encapsulation of the materials. The ultrathin PPF55 (11 μm) showed the best improvements, indicating the high quality of PPF55 (dense but with high ionic conductivity) for suppressing the shuttle effect. There is no special requirement for the active materials. Hence, this strategy should be applicable to many electrode materials. Our work presents a facile method for inhibiting the shuttle effect and thereby enhancing the capacity, coulombic efficiency and cyclability of organic sodium-ion batteries. What's more, the utilization of SSE materials and free-standing films would pave the way to simplify the processing technology by combining the protective layer with the separator through further optimizing the materials and mixing ratio.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National 1000-Talents Program, the National Natural Science Foundation of China (51773071), Wuhan Municipal Science and Technology Bureau (2017010201010141) and the Fundamental Research Funds for the Central Universities (HUST: 2017KFYXJJ023, 2017KFXKJC002, 2018KFYXKJC018).Notes and references
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- M. Tang, H. Li, E. Wang and C. Wang, Chin. Chem. Lett., 2018, 29, 232–244 CrossRef CAS.
- Q. Zhao, Y. Lu and J. Chen, Adv. Energy Mater., 2017, 7, 1601792 CrossRef.
- Y. Park, D.-S. Shin, S. H. Woo, N. S. Choi, K. H. Shin, S. M. Oh, K. T. Lee and S. Y. Hong, Adv. Mater., 2012, 24, 3562–3567 CrossRef CAS PubMed.
- E. Edison, S. Sreejith, C. T. Lim and S. Madhavi, Sustainable Energy Fuels, 2018, 2, 2567–2582 RSC.
- S. Wu, W. Wang, M. Li, L. Cao, F. Lyu, M. Yang, Z. Wang, Y. Shi, B. Nan, S. Yu, Z. Sun, Y. Liu and Z. Lu, Nat. Commun., 2016, 7, 13318 CrossRef CAS PubMed.
- X. Zhao, W. Cai, Y. Yang, X. Song, Z. Neale, H.-E. Wang, J. Sui and G. Cao, Nano Energy, 2018, 47, 224–234 CrossRef CAS.
- C. Zhao, Y. Cai, K. Yin, H. Li, D. Shen, N. Qin, Z. Lu, C. Liu and H.-E. Wang, Chem. Eng. J., 2018, 350, 201–208 CrossRef CAS.
- N. Yabuuchi, K. Kubota, M. Dahbi and S. Komaba, Chem. Rev., 2014, 114, 11636–11682 CrossRef CAS PubMed.
- S. Wang, L. Wang, Z. Zhu, Z. Hu, Q. Zhao and J. Chen, Angew. Chem., Int. Ed., 2014, 53, 5892–5896 CrossRef CAS PubMed.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947–958 CrossRef CAS.
- S.-W. Kim, D.-H. Seo, X. Ma, G. Ceder and K. Kang, Adv. Energy Mater., 2012, 2, 710–721 CrossRef CAS.
- B. Häupler, A. Wild and U. S. Schubert, Adv. Energy Mater., 2015, 5, 1402034 CrossRef.
- Y. Ding and G. Yu, Chem, 2017, 3, 917–919 CAS.
- Z. Song and H. Zhou, Energy Environ. Sci., 2013, 6, 2280–2301 RSC.
- Y. Liang, Z. Chen, Y. Jing, Y. Rong, A. Facchetti and Y. Yao, J. Am. Chem. Soc., 2015, 137, 4956–4959 CrossRef CAS PubMed.
- X. Dong, Z. Guo, Z. Guo, Y. Wang and Y. Xia, Joule, 2018, 2, 902–913 CrossRef CAS.
- C. Wang, H. Dong, L. Jiang and W. Hu, Chem. Soc. Rev., 2018, 47, 422–500 RSC.
- C. Wang, C. Jiang, Y. Xu, L. Liang, M. Zhou, J. Jiang, S. Singh, H. Zhao, A. Schober and Y. Lei, Adv. Mater., 2016, 28, 9182–9187 CrossRef CAS PubMed.
- B. Xiao, J. Liu, Q. Sun, B. Wang, M. N. Banis, D. Zhao, Z. Wang, R. Li, X. Cui, T.-K. Sham and X. Sun, Adv. Sci., 2015, 2, 1500022 CrossRef PubMed.
- T. Yokoji, H. Matsubara and M. Satoh, J. Mater. Chem. A, 2014, 2, 19347–19354 RSC.
- C. Luo, J. Wang, X. Fan, Y. Zhu, F. Han, L. Suo and C. Wang, Nano Energy, 2015, 13, 537–545 CrossRef CAS.
- Y. Yang, G. Yu, J. J. Cha, H. Wu, M. Vosgueritchian, Y. Yao, Z. Bao and Y. Cui, ACS Nano, 2011, 5, 9187–9193 CrossRef CAS PubMed.
- J.-Q. Huang, T.-Z. Zhuang, Q. Zhang, H.-J. Peng, C.-M. Chen and F. Wei, ACS Nano, 2015, 9, 3002–3011 CrossRef CAS PubMed.
- J.-Q. Huang, Q. Zhang, H.-J. Peng, X.-Y. Liu, W.-Z. Qian and F. Wei, Energy Environ. Sci., 2014, 7, 347–353 RSC.
- S. Bai, X. Liu, K. Zhu, S. Wu and H. Zhou, Nat. Energy, 2016, 1, 16094 CrossRef CAS.
- C. Jiang, M. Tang, S. Zhu, J. Zhang, Y. Wu, Y. Chen, C. Xia, C. Wang and W. Hu, Angew. Chem., Int. Ed., 2018 DOI:10.1002/anie.201809907.
- Y. Meng, G. Lin, H. Ding, H. Liao and C. Wang, J. Mater. Chem. A, 2018, 6, 17186–17191 RSC.
- H. Liao, H. Wang, H. Ding, X. Meng, H. Xu, B. Wang, X. Ai and C. Wang, J. Mater. Chem. A, 2016, 4, 7416–7421 RSC.
- C.-H. Chang, S.-H. Chung and A. Manthiram, Sustainable Energy Fuels, 2017, 1, 444–449 RSC.
- C. Wang, Y. Xu, Y. Fang, M. Zhou, L. Liang, S. Singh, H. Zhao, A. Schober and Y. Lei, J. Am. Chem. Soc., 2015, 137, 3124–3130 CrossRef CAS PubMed.
- C. Wang, Y. Fang, Y. Xu, L. Liang, M. Zhou, H. Zhao and Y. Lei, Adv. Funct. Mater., 2016, 26, 1777–1786 CrossRef CAS.
- H.-G. Wang, S. Yuan, D.-L. Ma, X.-L. Huang, F.-L. Meng and X.-B. Zhang, Adv. Energy Mater., 2014, 4, 1301651 CrossRef.
- Y. Jing, Y. Liang, S. Gheytani and Y. Yao, Nano Energy, 2017, 37, 46–52 CrossRef CAS.
- Z. Song, Y. Qian, M. L. Gordin, D. Tang, T. Xu, M. Otani, H. Zhan, H. Zhou and D. Wang, Angew. Chem., Int. Ed., 2015, 54, 13947–13951 CrossRef CAS PubMed.
- H. Li, M. Tang, Y. Wu, Y. Chen, S. Zhu, B. Wang, C. Jiang, E. Wang and C. Wang, J. Phys. Chem. Lett., 2018, 9, 3205–3211 CrossRef CAS PubMed.
- Z. Song, Y. Qian, T. Zhang, M. Otani and H. Zhou, Adv. Sci., 2015, 2, 1500124 CrossRef PubMed.
- M. Tang, S. Zhu, Z. Liu, C. Jiang, Y. Wu, H. Li, B. Wang, E. Wang, J. Ma and C. Wang, Chem, 2018, 4, 2600–2614 CAS.
- M.-H. Yoon, H. Yan, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2005, 127, 10388–10395 CrossRef CAS PubMed.
- Z. Xue, D. He and X. Xie, J. Mater. Chem. A, 2015, 3, 19218–19253 RSC.
- C. Jiang, H. Li and C. Wang, Sci. Bull., 2017, 62, 1473–1490 CrossRef CAS.
- S. Patra, A. B. Puthirath, T. V. Vineesh, S. Narayanaru, B. Soman, S. Suriyakumar, A. M. Stephan and T. N. Narayanan, Sustainable Energy Fuels, 2018, 2, 1870–1877 RSC.
- J. Rivnay, S. Inal, B. A. Collins, M. Sessolo, E. Stavrinidou, X. Strakosas, C. Tassone, D. M. Delongchamp and G. G. Malliaras, Nat. Commun., 2016, 7, 11287 CrossRef PubMed.
- C. M. Proctor, J. Rivnay and G. G. Malliaras, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 1433–1436 CrossRef CAS.
- S. Fabiano, N. Sani, J. Kawahara, L. Kergoat, J. Nissa, I. Engquist, X. Crispin and M. Berggren, Sci. Adv., 2017, 3, e1700345 CrossRef PubMed.
- M. Yao, H. Senoh, T. Sakai and T. Kiyobayashi, Int. J. Electrochem. Sci., 2011, 6, 2905–2911 CAS.
- M. Yao, S.-I. Yamazaki, H. Senoh, T. Sakai and T. Kiyobayashi, Mater. Sci. Eng., B, 2012, 177, 483–487 CrossRef CAS.
- Y. Wang, Y. Ding, L. Pan, Y. Shi, Z. Yue, Y. Shi and G. Yu, Nano Lett., 2016, 16, 3329–3334 CrossRef CAS PubMed.
- L. Yan, X. Gao, J. P. Thomas, J. Ngai, H. Altounian, K. T. Leung, Y. Meng and Y. Li, Sustainable Energy Fuels, 2018, 2, 1574–1581 RSC.
- E. Quartarone and P. Mustarelli, Chem. Soc. Rev., 2011, 40, 2525–2540 RSC.
- R. Bouchet, S. Maria, R. Meziane, A. Aboulaich, L. Lienafa, J.-P. Bonnet, T. N. T. Phan, D. Bertin, D. Gigmes, D. Devaux, R. Denoyel and M. Armand, Nat. Mater., 2013, 12, 452–457 CrossRef CAS PubMed.
- C.-M. Wu, P.-I. Pan, Y.-W. Cheng, C.-P. Liu, C.-C. Chang, M. Avdeev and S.-k. Lin, J. Power Sources, 2017, 340, 14–21 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SEM images of cross-section of the free-standing protective films; detailed electrochemical performances of PT electrodes assembled with PEO, PPF19, PPF37 and PPF55; electrochemical performances of the protective film, showing the low capacity contribution of the protective films in the cells. See DOI: 10.1039/c8se00477c |
| This journal is © The Royal Society of Chemistry 2019 |