
Alkali–oxygen treatment prior to the mechanical pulping of hardwood enhances enzymatic hydrolysis and carbohydrate recovery through selective lignin modification
Jie
Wu
,
Richard
Chandra
 * and
Jack
Saddler
* and
Jack
Saddler
Forest Products Biotechnology/Bioenergy Group, Department of Wood Science, Faculty of Forestry, University of British Columbia, 2424 Main Mall, Vancouver BC, V6T 1Z4, Canada. E-mail: richardchandra77@gmail.com
First published on 30th October 2018
Abstract
Aspen chips were impregnated with sodium carbonate (70 °C, overnight) and oxygen (110 °C, 2 hours) prior to pre-steaming (190 °C, 15 min) and mechanical refining. This pretreatment protocol resulted in the removal of 44% of the lignin while retaining 55% of the hemicellulose (which was enriched with carboxylic acid groups (78 mmol kg−1)) in the water-insoluble fraction. Lignin removal/modification improved fiber swelling and increased cellulose accessibility, resulting in a biomass substrate that could be readily hydrolyzed (80% cellulose hydrolysis, 100% xylan hydrolysis) at an enzyme loading of 20 mg g−1 cellulose. To further enhance the recovery of the hemicellulose component and the susceptibility of the water-insoluble fraction to enzymatic hydrolysis, a pre-hydrolysis (170 °C, 1 hour) step was added prior to alkali–oxygen impregnation. This resulted in a total recovery of 72% of the original hemicellulose from both the solid and liquid fractions. Alternatively, lowering the steaming temperature to 130 °C resulted in the preservation of 72% of the hemicellulose in the water insoluble fraction. Although less lignin was removed at the lower, 130 °C temperature, the resulting lignin contained a greater amount of acid groups (107 mmol of carboxylic acid groups per kg). Substrates containing lignin which were enriched in acid groups showed increased swelling and a decrease in the tendency of the lignin to bind enzymes through hydrophobic interactions. This substantially reduced the negative effects of lignin on enzymatic hydrolysis.
Introduction
Pretreatment, followed by enzymatic and microbial conversion, is one of the main processes that has been investigated for the potential development of a biomass-based biorefinery. The main roles of pretreatment during the enzyme-mediated conversion of biomass to renewable fuels and chemicals is to both maximize the accessibility of the cellulose to cellulase enzymes and recover as much of the hemicellulose in a useable form as possible.1,2 To maximize the value obtained from the biomass feedstock, pretreatments are also expected to recover the cellulose and hemicellulose while isolating a lignin component that has potential utility for downstream value-added chemical applications.3At an industrial scale, one of the most effective ways to pretreat and fractionate biomass is the pre-hydrolysis kraft pulping process (PHK). In this process the majority of the hemicellulose (>60%) is separated from the woody biomass followed by a subsequent kraft pulping process that removes most of the lignin.4 This provides a nearly pure cellulosic substrate (dissolving pulp) that is used for higher value applications such as the production of cellulose esters and ethers. However, the current price of industrial chemical pulps such as fully bleached kraft pulp or a PHK dissolving pulp that contain <1% lignin, is currently >$1000(US) per ton.5 Therefore, the high cost of making these types of substrates precludes their use for biochemical conversion. Unlike kraft pulping, where lignin removal results in a yield of approximately 50% of the original biomass, mechanical pulping typically employs a “mechanical refining” step to separate wood chips into fibers at yields close to 95% of the original biomass. Mechanical pulps are traditionally used to make newsprint. However, the demand for newsprint has dwindled significantly, decreasing by greater than 50% over the past decade.6 Thus there is considerable interest in the possible “re-purposing” of existing mechanical refining/pulping infrastructure as the “front-end” of an enzyme/microbial based biorefinery process.7–11
Some of the advantages of using a mechanical pulping approach include, size reduction from chips to pulp, the virtual complete recovery of the cellulose, hemicellulose and lignin components in the solid pulp substrate, onsite waste-water treatment, etc., as well making use of all of the utilities, permitting, etc., that were needed to establish the industrial site in the first place. However, as has been shown in other studies,12–14 a key challenge with utilizing a mechanical pulping approach to pretreating woody substrates is that the substrates continue to be quite recalcitrant to enzymatic hydrolysis. One of the major issues is that the lignin remains associated with the mechanically refined substrates, significantly hindering the swelling and limiting the accessibility of the enzymes to the cellulose.12–14 As well as inhibiting swelling, lignin has also been shown to bind enzymes through hydrophobic interactions.15–18 Consequently, most mechanical pulping based pretreatments that have been investigated to date have employed some type of chemical treatment to either remove lignin and/or modify hemicellulose prior to the mechanical pulping process.7,8,10,11
Similar to studies with mechanical pulping, lignin has been shown to be an impediment to the hydrolysis of steam-pretreated substrates. However, although removing the lignin from steam-pretreated substrates has been shown to improve enzymatic hydrolysis,19–21 in many cases, the treatments involved chemical loadings of >50% and/or resulted in a compromised recovery of the cellulose itself.20,22 Thus, more recent research has looked at the use of milder chemical treatments to modify lignin rather than targeting complete lignin removal.10,11,13,23 For example, integrating sulfonation and oxidation to fortify sulfonic and carboxylic acid groups on the substrate lignin has been shown to improve subsequent enzymatic hydrolysis without the need for complete lignin removal.13,23,24 This work has shown that the incorporation of acid groups into the lignin macromolecule using oxidation or sulfonation increased the hydrophilicity of lignin,25,26 consequently enhancing cellulose accessibility by facilitating fiber swelling27 and by reducing non-productive binding of enzymes to lignin.13,28 The negative charges on lignin have also been implicated in decreasing non-productive enzyme binding to lignin by electrostatic repulsion since the majority of enzyme components exhibit a net negative charge at the pH (4–5) where hydrolysis is being performed.13,25,28
Pretreatments that employ sulfonation and mild oxidation should also be easily implemented since they are already utilized at a commercial scale by the pulp and paper industry.29 Previous work has shown that sulfonation of refiner mechanical pulps and steam pretreated softwood and hardwood biomass improved hydrolysis yields without removing lignin.13,14,24 However, the improvements in enzymatic hydrolysis were still impeded by the physical presence of the lignin in the substrate.13,14,24 In subsequent work the application of either oxygen or sulfite treatment to wood chips prior to steam pretreatment was shown to improve enzymatic hydrolysis yields and also resulted in a high retention of carbohydrates.13,23
One of the goals of the work reported here was to determine whether a similar approach could also be used to modify the lignin in aspen (hardwood) biomass prior to the application of mechanical pulping. As described below, alkali and oxygen were integrated into a pre-steaming step (110–130 °C) that is normally employed during the production of thermo-mechanical pulps with the hope that it would increase the acid groups on the resulting mechanical pulp substrate. In this way we could retain most of the carbohydrate components in the pulp associated “water-insoluble fraction” while improving the cellulose accessibility and ease of enzymatic hydrolysis. It was apparent that the use of a mild (130 °C) oxygen/steaming treatment was effective at both improving enzymatic hydrolysis while retaining/recovering most of the hemicellulose in the water insoluble substrate as part of a “one-pot” approach.
Materials and methods
Biomass and chemicals
Aspen wood chips were obtained from a pulp mill in Western Canada. The chips were screened at ranges between 2.5 × 2.5 cm and 5.0 × 5.0 cm. The moisture content of the wood chips (6.22% ± 0.43%) was determined by measuring the loss of chips weight in the oven at 105 °C overnight. Sodium carbonate and sodium chlorite were purchased from Fisher Scientific and Acros organics respectively. Oxygen was received from Praxair Canada Inc. Direct orange 15 was received from Pylam Products. The dye was isolated using the method illustrated in Chandra et al.30 To fractionate the high molecular weight portion. Commercial enzymes Cellic CTec 3 (cellulase enzyme cocktail) and HTec (xylanase) used in the enzymatic hydrolysis were received from Novozymes (Novozymes, Bagsvaerd, Denmark).Alkaline-oxygen impregnated mechanical refining
Aspen chips were autoclaved before impregnation with 15% sodium carbonate (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 liquid:wood ratio) in the 70 °C water bath overnight. The purpose of autoclaving was to remove the inherent air from the chips, which facilitates the penetration of sodium carbonate into the chips.31 Aspen chips were then impregnated with oxygen in the Parr reactor at 100 psig and 110 °C for 2 hours prior to steam treatment at either 190 or 130 °C for 15 minutes in a stainless steel basket. After the steam treatment, the chips were mechanically refined by a commercially juicer (super angel juicer model 8500) and a lab scale PFI mill for 10000 revolutions according to TAPPI standard T-248 method.
:1 liquid:wood ratio) in the 70 °C water bath overnight. The purpose of autoclaving was to remove the inherent air from the chips, which facilitates the penetration of sodium carbonate into the chips.31 Aspen chips were then impregnated with oxygen in the Parr reactor at 100 psig and 110 °C for 2 hours prior to steam treatment at either 190 or 130 °C for 15 minutes in a stainless steel basket. After the steam treatment, the chips were mechanically refined by a commercially juicer (super angel juicer model 8500) and a lab scale PFI mill for 10000 revolutions according to TAPPI standard T-248 method.
Pre-hydrolysis treatment
Pre-hydrolysis treatment was carried out in a lab scale rotating digester (Aurora products Ltd. Savona, BC, Canada). In brief, 250 g (OD basis) of aspen chips were soaked with water in the cooking vessel at a liquid:wood ratio of 4:1. The cooking vessel was then moved into the digester. The temperature was heated to 170 °C within 1.5 hours and stayed at 170 °C for another hour. The vessel was then cooled down in the cold water before the pre-hydrolysis liquor was removed, filtered and undergo subsequent mechanical refining.
Enzymatic hydrolysis
The protein content of Cellic CTec 3 and HTec were 180 and 21 mg ml−1 respectively, measured by the ninhydrin assay described by Mok et al.32 During the enzymatic hydrolysis (in duplicate) at 10% solid loading, substrates were mixed with acetate buffer (50 mM, pH 5.0) and enzymes in the 50 ml glass septa bottles, and moved to a shaking incubator at 50 °C and speed of 180 rpm for 72 hours. Enzymatic hydrolysis of the pretreated substrates (in duplicate) at 2% solid loading was conducted in the 2 ml screw cap tubes (Eppendorf) with acetate buffer (50 mM, pH 5.0) and enzymes. The tubes were moved to a rotating incubator at 50 °C for 48 hours. In the case of the hydrolysis with bovine serum albumin (BSA), samples were impregnated with acetate buffer (50 mM, pH 5.0) containing 10 mg ml−1 of BSA at room temperature overnight prior to the addition of enzymes.Klason
Chemical composition of substrates was analyzed using TAPPI standard T-22 om-88 method. Briefly, 0.2 g of ground dry substrate was mixed with 3 ml of 72% H2SO4 for 2 hours prior to the addition of 112 ml deionized water and autoclave at 121 °C for 1 hour. The acid-insoluble lignin was filtered and weighed out using 30 ml fritted glass crucible. The acid-soluble lignin was analyzed by determining the absorbance at 205 nm. The sugars were measured by Dionex (Sunnyvale, CA) HPLC (ICS-3000).Acid group titration
Conductometric titration of acid groups was a modified version of the method developed by Katz et al.33 Briefly, 0.15 g (oven-dry basis) of pre-washed material was weighed out and mixed with 15 ml of 0.1 N HCl in the 15 ml falcon tube overnight. The sample was then washed by filtration with 250 ml nanopure water through a small Buchner funnel. After the filtration, the sample was transferred to a plastic beaker with the filter paper and re-suspended in 50 ml of 0.001 M NaCl solution containing 200 μL of 0.05 N HCl. The mixture was then titrated with 20 μL of 0.05 N NaOH. The conductivity of pulp suspension was measured by a conductivity meter and recorded after each addition to plot the conductometric titration curve. The total content of carboxylic acid groups was calculated using the following equation:| X = C × V/m |
Simons' stain
Direct Orange 15 (DO) dye was used to measure the accessibility of cellulose, according to previous studies by Chandra et al.30 Briefly, a set of 10 mg (oven-dry basis) of material were mixed with PBS buffer and increasing concentration of DO dye in 2 ml screw cap tubes overnight. The tubes were then moved to a shaking incubator at 60 °C and speed of 180 rpm overnight. The tubes were subsequently centrifuged and the absorbance of supernatant at 450 nm was measured by a spectrophotometer.Water retention value (WRV)
WRV was measured (in triplicate) using TAPPI Useful Method-256. In brief, approximately 0.5 g (oven-dry basis) of the pulp was soaked in 50 ml water overnight prior to filtration through 200-mesh screen in the WRV unit. The resulting pulp pad was then centrifuged at 900 g for 30 minutes at 25 °C. The subsequent sample was weighed and dried in the oven at 105 °C overnight. WRV was calculated by the equation:| WRV = (wet mass − dry mass)/(dry mass) |
Fiber length measurement
The length weighted fiber length was measured by the Fiber Quality Analyzer (FQA, LDA02, OpTest Equipment, Inc., Hawkesbury, ON, Canada) using ISO 16065 setting as measurement method. The pulp suspension was diluted prior to the measurement in order to make sure less than 40 fibers were measured per second.Viscosity
Viscosity of selected substrates was measured by a capillary viscometer (Cannon Ubbelohde Viscometer, Cannon Instrument Co., State College, PA) using TAPPI Standard Method T230 om-08. Prior to viscosity measurements, the lignin in the substrates was removed by sodium chlorite bleaching process according to Kumar et al.24 Briefly, 4 g sodium chlorite and 0.5 ml glacial acetic acid was added to 5 g of substrate at 15:1 liquor to wood ratio. The reaction was carried out in the fume hood at room temperature for 3 hours. The slurry after reaction was filtered and washed extensively in the Buchner funnel. The delignification process was repeated in order to delignify the substrates.
Results and discussion
As described earlier, one of the goals of this study was to assess the potential of integrating an alkaline oxygen step into the initial steaming of wood chips prior to mechanical pulping/refining. Previous work had shown that the addition of an alkali–oxygen treatment at the milder temperatures of 110 and 135 °C prior to steam pretreatment/explosion at 210 °C resulted in enhanced hemicellulose recovery while improving the ease of hydrolysis of the cellulose component.23 Therefore, we first wanted to assess if a similar alkali–oxygen pretreatment could be successfully applied to aspen chips during steaming, prior to mechanical pulping.As earlier work had suggested that mechanical refining alone would do little to enhance enzymatic hydrolysis, we first wanted to assess how effective the combined action of pre-hydrolysis and mechanical refining might be as previous work had shown that hydrothermal auto-hydrolysis or dilute acid with subsequent mechanical refining could increase the ease of hydrolysis of woody biomass.34–36 However, in most cases, high enzyme/protein loadings were required to achieve hydrolysis yields that surpassed 70%.35 Similar to these previous results, applying a pre-hydrolysis step to the aspen wood chips prior to mechanical pulping enhanced the ease of hydrolysis of the substrate from 18 to 52% at a protein loading of 20 mg g−1 cellulose (Fig. 1). However, despite some improvement in hydrolysis resulting from the pre-hydrolysis treatment only 50% of the cellulose was hydrolyzed after 48 hours.
 | ||
| Fig. 1 Enzymatic hydrolysis of mechanical pulp produced using aspen wood chips with and without pre-hydrolysis (enzymatic hydrolysis performed at 2% solids (w/v) at an enzyme loading of 20 mg protein per g cellulose for 48 hours). | ||
As discussed and shown in earlier studies,13,14 the residual lignin remaining in the substrate after refining was likely restricting more effective hydrolysis. Thus, a range of pretreatment conditions were next assessed to see if we could modify or solubilize the lignin while retaining as much of the hemicellulose content with the insoluble cellulosic fraction as possible (Table 1). We first looked at utilizing alkaline oxygen treatment to modify lignin in the biomass improve the ease of hydrolysis of the carbohydrates contained in the mechanically refined substrates. Initially, the alkali–oxygen–steam treatment was applied at 190 °C to both the pre-hydrolyzed and original aspen chips prior to the mechanical refining. Previous work which had integrated alkaline oxygen into the steam pretreatment process showed that a temperature of 190 °C was effective for enhancing enzymatic hydrolysis while retaining much of the hemicellulose13,23 in the water insoluble fraction.
| Treatments | Cellulose (%) | Hemicellulose (%) | Lignin (%) | Solid yields (%) | Cellulose recovery (%) | Hemicellulose recovery (%) | Lignin removal (%) |
|---|---|---|---|---|---|---|---|
| a Explanation of treatments: 190 °C AO: alkali–oxygen impregnation prior to pre-steaming at 190 °C and mechanical refining; 190 °C Alkali: alkali oxygen impregnation prior to pre-steaming at 190 °C and mechanical refining; 190 °C Oxygen: oxygen impregnation prior to pre-steaming at 190 °C and mechanical refining; 190 °C Water: water impregnation prior to pre-steaming at 190 °C and mechanical refining; Pre-hydrolysis 190 °C AO: pre-hydrolysis with subsequent alkali–oxygen impregnation prior to pre-steaming at 190 °C and mechanical refining. b 50% of hemicellulose recovered from pre-hydrolysis liquor and the other 50% was stored in the solid fraction. c AO treatment recovered 74% of solid from pre-hydrolyzed chips. d 50% of hemicellulose recovered from pre-hydrolysis liquor and 22% of hemicellulose recovered from the solid fraction after alkali–oxygen treatment. | |||||||
| Aspen chips | 46 | 19 | 28 | N/A | N/A | N/A | N/A |
| Mechanical refining only | 46 | 19 | 28 | 100 | 100 | 100 | 0 |
| Pre-hydrolysis and mechanical refining | 56 | 11 | 26 | 78 | 100 | 100b | 18 |
| Pre-hydrolysis 190 °C AO | 70 | 7.3 | 30 | 58c | 84 | 72d | 39 |
| 190 °C AO | 66 | 15 | 23 | 71 | 98 | 56 | 42 |
| 190 °C Alkali | 57 | 13 | 24 | 76 | 90 | 53 | 35 |
| 190 °C Oxygen | 56 | 9.4 | 26 | 77 | 91 | 38 | 28 |
| 190 °C Water | 62 | 9.0 | 29 | 75 | 96 | 35 | 24 |
It was apparent (Table 1) that an alkaline pH was beneficial to retaining and recovering the hemicellulose component in the solid pulp after the pre-hydrolysis when combining alkali–oxygen and mechanical treatments. In contrast, performing the pre-hydrolysis prior to mechanical refining and/or alkaline oxygen treatment under acidic conditions, decreased the recovery of the hemicellulose in the insoluble fraction to less than 22% but, when combined with the hemicellulose in the soluble stream, a total hemicellulose recovery of 78% was achieved (Table 1). It was also evident that treatments performed in the absence of alkali, using either hot water or oxygen, resulted in lower hemicellulose recoveries in the water insoluble fraction (Table 1). Similar to reactions that are known to occur during pre-hydrolysis, the “190 °C Water” treatment likely solubilized the hemicellulose via “auto-hydrolysis” where the inherent acetyl groups on the hemicellulose component as an acid catalyst to hydrolyze and solubilize the hemicellulose. The use of oxygen in the absence of alkali has also been shown37 to result in the oxidation of a portion of the hemicellulose component to carboxylic acids. This, consequently acidifies the pretreatment resulting in the solubilization of hemicellulose and likely occurred with the “190 °C Oxygen” treatment. Although the addition of alkali to the pretreatment increased the recovery of hemicellulose in the solid fraction, it was evident that pre-steaming at 190 °C may have been excessively severe as the hemicellulose retention in the solid pulp fraction was just over 50% (Table 1). In earlier work, increased solubilization of hemicellulose at higher temperature oxidative treatments has been shown to result from radical initiated reactions that damage and solubilize hemicellulose.38
As well as retaining approximately 50% of the hemicellulose in the water insoluble substrate, the 190 °C AO and 190 °C Alkali pretreatments were also quite effective in solubilizing the lignin (Table 1). This was consistent with previous studies39,40 which showed a higher selectivity for lignin removal when alkaline treatments were compared to acidic treatments. It has been shown that, under alkaline conditions, the de-protonation of phenolic groups by hydroxide ions leads to the formation of quinone methide structures, which results in the cleavage of α-aryl ether bonds.41 The β-aryl ether bonds are also cleaved by hydroxide ions and the lignin fragments are solubilized by the alkaline solution. In the work reported here, alkali treatment without the addition of oxygen removed up to 35% of the lignin from the aspen chips. The addition of oxygen to the alkaline treatment increased lignin removal to more than 40%, likely due to the synergistic action of alkali and oxygen oxidizing the lignin macromolecule.42 The higher lignin removal after 190 °C AO treatment was likely due to the increased solubility of the oxygen-modified lignin in an alkali solution.42 The addition of oxygen to the alkaline treatments (190 °C AO) also increased the recovery of the cellulose (Table 1), which is also indicative of the higher selectivity of the oxygen treatment.
Earlier work had shown that applying a pre-hydrolysis step to poplar chips enhanced the delignification when a subsequent acidic organosolv pretreatment was utilized.43 Thus, it was likely that the initial acid lignin fragmentation reactions occurring during pre-hydrolysis were extended during the subsequent organosolv pretreatment. However, in the work reported here it was apparent that the lignin remaining in the biomass after pre-hydrolysis was slightly less amenable to oxygen mediated lignin removal (Pre-hydrolysis-AO) when compared to the application of the 190 °C AO treatment (Table 1). It is worth noting that the pre-hydrolysis of the wood chips in the current work was performed at 170 °C for a residence time of 1 hour including a 57 minute pre-heating stage. This extensive treatment probably resulted in the acidic condensation of lignin, likely compromising its downstream reactivity with oxygen.44 It is also possible that the residual acetic acid that remained in the wood chips after the pre-hydrolysis stages could have partially neutralized the alkali added during the AO stage. The partial neutralization could have compromised the oxygen delignification as the pre-hydrolyzed chips did not undergo washing prior to the application of the AO stage.
It has been shown that the main effect of alkaline-oxygen (AO) treatment on substrate lignin is a stepwise oxidation, typically commencing on the phenolic groups42 and that alkaline-oxygen treatment prior to steaming partially removed lignin, while increasing the amount of acid groups on the substrate lignin. It was apparent that, in addition to solubilizing 40% of the lignin, the 190 °C AO treatment also left the residual substrate lignin in a highly oxidized state as it contained 78 mmol of carboxylic acid group per kg of substrate (Table 2). Although the pre-hydrolyzed lignin within the wood chips was less reactive towards the alkali–oxygen treatment, when compared to the single step AO treatment, the pre-hydrolysis AO treatment still contained 51 mmol carboxylic acid per kg substrate. Interestingly, the high level of sodium carbonate used for the alkali treatment in the absence of oxygen also resulted in the generation of a 67 mmol carboxylic acid groups per kg of substrate (Table 2). It has been suggested that alkali treatment results in the formation of acid groups within biomass due to the alkaline induced cleavage of ester bonds between lignin and hemicellulose. This has been observed to occur during the sodium carbonate (Alkaline) treatment.45,46 As mentioned earlier, the incorporation of acid groups into lignin can increase substrate swelling, consequently enhancing cellulose accessibility,27 as well as reducing the amount of non-productive binding of cellulases to lignin during enzymatic hydrolysis.25,28 Therefore, we next analyzed the increases in substrate accessibility and hydrophilicity using the Simon's stain and water retention value techniques.
| Treatments | Carboxylic acid groups (mmol kg−1) | Water retention value | DO dye adsorption (mg g−1 dry fiber) | LW fiber length (mm) | Viscosity (mPa s) |
|---|---|---|---|---|---|
| 190 °C AO | 78 | 3.1 ± 0.08 | 95 | 0.80 | 5.2 ± 0.1 |
| 190 °C Alkali | 67 | 2.8 ± 0.06 | 90 | 0.81 | 8.8 ± 0.1 |
| 190 °C Oxygen | 23 | 2.6 ± 0.02 | 88 | 0.66 | 6.7 ± 0.1 |
| 190 °C Water | 22 | 2.8 ± 0.04 | 77 | 0.60 | 8.6 ± 0.0 |
| Pre-hydrolysis 190 °C AO | 51 | 2.6 ± 0.13 | 74 | 0.62 | 6.4 ± 0.0 |
When substrate swelling was assessed by water retention value (WRV) measurement47,48 the 190 °C AO treated substrate also exhibited the highest WRV (Table 2). Both the WRV and the adsorption of the Direct Orange dye that constitutes the Simon' stain have been successfully used to predict the susceptibility of pretreated biomass to enzymatic hydrolysis.30,49 However, unlike the WRV which uses water to probe for substrate swelling, the Simon's stain utilizes the Direct Orange (DO) dye that has a high specificity for cellulose and a similar size to a typical cellulase enzyme.50–52 As well as having the highest accessibility, as indicated by the Simon's stain, it was also apparent that the 190 °C AO treatment affected the average viscosity/molecular weight of the cellulose. When oxygen was combined with alkali (190 °C AO), the viscosity decreased substantially compared to the alkaline treatment in the absence of oxygen (190 °C Alkali). As mentioned above, the alkaline oxygen treatment at 190 °C may have been excessively severe, resulting in the solubilization of hemicellulose and the non-specific radical induced molecular weight reduction of cellulose (Table 1).53 The non-specific nature of alkaline oxygen is one of the recognized limitations of oxygen delignification when applied at a commercial scale during pulp bleaching.54 However, although the 190 °C AO and 190 °C alkaline treatments decreased the molecular weight of the cellulose and/or compromised hemicellulose yield, these treatments resulted in substrates with the longest fibers. These results suggested that alkaline treatments primarily targeted the lignin in the middle lamella to provide cleaner fiber separation (Table 2). Overall, it appeared that both the 190 °C alkali and 190 °C AO treatments were the most effective as these treatments resulted in substrates that had the highest accessibility as measured by Simon's stain and WRV while retaining more than 50% of the hemicellulose in the solid fraction.
As mentioned earlier, a major goal of this work was to enhance the cellulose accessibility of mechanically pretreated aspen biomass while recovering as much of the hemicellulose as possible, either through pre-hydrolysis or by retaining hemicellulose in the solid pulp substrate component. However, as shown previously, the retention of hemicellulose in lignocellulosic substrates can compromise the ease of cellulose hydrolysis55,56 unless enzyme cocktails are supplemented with hemicellulase activities.57,58 Therefore, considering the high xylan content (ranging from 11 to 14%) of the substrates pretreated under alkaline conditions, the enzyme cocktails were supplemented with xylanases to assess the potential benefits of this enzyme addition.
In order to achieve high hydrolysis yields at the solids loading of 10% (w/v), the total protein loading was used at 20 mg/g cellulose (15 mg cellulase and 5 mg xylanase). It was apparent that the hydrolysis yields reflected the incorporation of acid groups in the substrates (Fig. 2, Table 2) as previous work had shown that an increase in acid groups improved the swelling and hydrophilicity of the substrate as well as decreasing the non-productive binding of the enzyme to the lignin.27,28 It was also evident that the xylan component of the substrates treated under alkaline conditions (190 °C AO, 190 °C alkali) were more prone to hydrolysis (Fig. 2). Although the enzyme preparations were loaded to the substrate based on cellulose content, the hydrolysis of the alkaline pretreated substrates that contained less cellulose and a higher xylan content resulted in almost complete hydrolysis of the xylan, despite the use of a lower enzyme/protein loading (Table 1, Fig. 2). This was likely due to the deacetylation of the xylan as mild alkaline treatments have been shown to facilitate the removal of acetyl groups on the hemicellulose from hardwood59,60 and agriculture biomass.7,61 The removal of acetyl groups has also been shown to result in an increase in subsequent xylan and cellulose hydrolysis yields by liberating the reaction sites on xylan from xylanases.62
 | ||
| Fig. 2 Cellulose and xylan hydrolysis of substrates obtained when initial aspen biomass and pre-hydrolyzed aspen biomass were pretreated at 190 °C with alkaline-oxygen (AO) and separate alkali, oxygen, or water prior to mechanical pulping. (Hydrolysis was conducted at 10% solid using enzyme loading of 15 mg cellulase and 5 mg xylase per g cellulose for 72 hours.) | ||
As a result of the lower hemicellulose content and decreased response of the lignin to the alkaline-oxygen treatment the Prehydrolysis-190 °C AO treatment resulted in lower hydrolysis yields for both the xylan and cellulose components. This was despite its higher cellulose content and the use higher enzyme loadings (Table 1). It was likely that the alkaline oxygen treatment applied to the substrate prior to mechanical refining increased the selectivity of lignin modification through the incorporation of carboxylic acid groups, consequently facilitating subsequent enzymatic hydrolysis. However, compared to the Prehydrolysis-190 °C AO treatment, which resulted in the recovery of 78% of the original hemicellulose (Table 1), the 190 °C AO treatment recovered just over 50% of the hemicellulose in the solid substrate. As shown previously,7,63 the dissolution of hemicelluloses has been shown to be associated with their degradation/and or formation of fermentation inhibitors such as furans and acetic acid. As well as decreasing fermentation inhibitors, as discussed in the Introduction, one of the main advantages of utilizing a mechanical pulping approach is the high retention of carbohydrates in the solid “pulp” substrate which is compromised when a chemical treatment dissolves the hemicellulose prior to mechanical treatment. Therefore, we hypothesized that the use of a lower temperature during pre-steaming would increase the retention of the hemicelluloses in the water insoluble substrate, while still allowing for a modification of the lignin to facilitate the enzymatic hydrolysis of the retained carbohydrates during subsequent enzymatic hydrolysis. To determine if the use of less severe conditions would help we next assessed alkali–oxygen treatment with a steaming temperature of 130 °C, which is closer to the pre-steaming conditions used in a commercialized mechanical pulping process.64
At the lower 130 °C temperature, although hemicellulose recovery increased from 55 to 73% (130 °C AO, Table 3) the lignin removal decreased from 42 to 25%. However, the carboxylic acid groups on the 130 °C AO substrate increased from 100 to 107 mmol kg−1, indicating that the alkali–oxygen reaction with lignin had shifted from lignin removal towards lignin modification (Table 3). The 130 °C AO substrate also showed a slightly higher accessibility to the Direct Orange dye as well as a decrease in the viscosity of cellulose. This suggested that the cellulose underwent some oxidative damage even at the lower 130 °C steaming temperature (Table 3). Despite the higher amount of carboxylic acid groups on the 130 °C treated substrate, they were less susceptible to enzymatic hydrolysis (Fig. 3, Table 3), probably due to the lower amount of lignin removed at the 130 °C temperature.
| Treatment | Cellulose recovery (%) | Hemicellulose recovery (%) | Lignin removal (%) | Carboxylic acid groups (mmol kg−1) | Water retention value | DO dye adsorption (mg g−1 dry fiber) | LW fiber length (mm) | Viscosity (mPa s) | Cellulose hydrolysisa (%) | Xylan hydrolysisa (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| a Enzymatic hydrolysis was conducted at 10% solid at enzyme loading of 15 mg cellulase and 5 mg xylase per g cellulose for 72 hours. | ||||||||||
| 130 °C AO | 96 | 73 | 25 | 107 | 3.1 ± 0.15 | 95 | 0.84 | 6.7 ± 0.1 | 75 ± 2.2 | 100 ± 7.1 |
| 130 °C Alkaline | 95 | 71 | 21 | 100 | 3.0 ± 0.08 | 81 | 0.85 | 8.0 ± 0.1 | 69 ± 0.5 | 97 ± 3.9 |
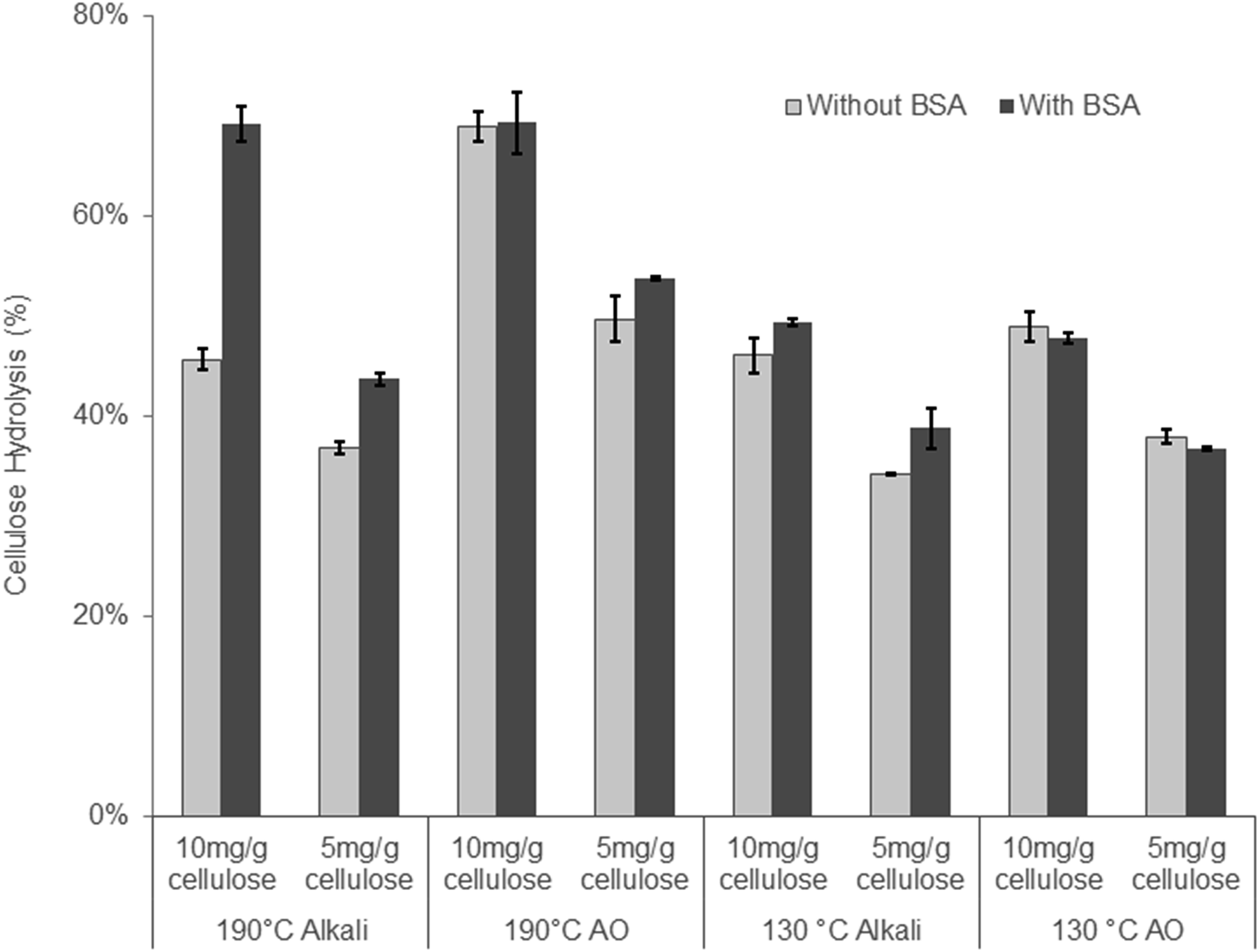 | ||
| Fig. 3 Enzymatic hydrolysis of AO and alkali treated substrates with and without the addition of BSA. (Hydrolysis was conducted at 2% solid using enzyme loading of 8 mg cellulose/2 mg xylanase and 4 mg cellulase/1 mg xylanase per gram of cellulose for 48 hours.) | ||
When the 130 °C Alkali and the 130 °C AO treated substrates were compared, the addition of oxygen only resulted in a slight improvement (∼7%) in cellulose hydrolysis (Table 3). As this result was unexpected, we reduced the enzyme loading to ensure that the higher 20 mg g−1 protein loading had not “masked” any differences between the two substrates. As well as potential “masking” of substrate differences at the higher enzyme loading, non-productive binding between enzymes and lignin through hydrophobic interactions has been shown to have negative impacts on enzymatic hydrolysis,26,65 especially at low enzyme loadings.24 The addition of bovine serum albumin (BSA) to enzymatic hydrolysis has long been known to bind to lignin through hydrophobic interactions, thereby acting as a lignin “blocking agent” to increase enzymatic hydrolysis yields.66 Recent studies have also shown that the increase in enzymatic hydrolysis of a given substrate that has been pre-incubated with BSA is an indication of the hydrophobic nature of lignin and its potential to non-productively bind cellulase enzymes through hydrophobic interactions.40,67 Therefore, the enzymatic hydrolysis of the substrates treated under alkaline conditions at 190 and 130 °C was performed at enzyme loadings of 5 and 10 mg g−1 cellulose on substrates both with and without pre-incubation with BSA. When comparing the 190 °C AO and 190 °C Alkali substrates, it was apparent that reducing the enzyme loading to 10 mg g−1 cellulose exposed differences between these substrates as the hydrolysis yield of the 190 °C AO was 26% higher than the 190 °C alkali. However, when hydrolyzing the substrates pre-incubated with BSA, the substrates reached similar hydrolysis yields, indicating the hydrophobic nature of the lignin component of the 190 °C alkali substrate. Therefore, despite containing a high amount of acid groups, likely through the cleavage of lignin-carbohydrate complexes, the lignin in the 190 °C alkaline treatment also appeared to be quite hydrophobic. As mentioned earlier, under alkaline conditions the phenolic groups in lignin are deprotonated to form quinone methides (QM). In the absence of a strong nucleophile such as HS−, the quinone methides undergo alkaline condensation reactions that increase lignin hydrophobicity.41 This likely occurred with the 190 °C alkaline substrate. Typically, when oxygen is added to the alkaline reaction, it removes an electron from the phenolate anion and subsequently reacts with the lignin to incorporate acid groups, consequently increasing the hydrophilicity of lignin. The increased lignin hydrophilicity in the alkali–oxygen reaction likely explains the negligible effect of hydrolysis after adding BSA to the 190 °C AO substrate.
The trends observed with the substrates treated at 130 °C were similar but less pronounced. This was probably a result of the decreased amount of lignin condensation occurring as the temperature was decreased from 190 to 130 °C. However, reducing the temperature of the AO pretreatment from 190 to 130 °C increased the hemicellulose recovery in the water insoluble component (Tables 1 and 3) while the cellulose in this fraction was readily hydrolyzed, resulting in a yield of 70% at enzyme loading of 20 mg g−1 cellulose.
Conclusions
A mechanical refining pretreatment of aspen chips could result in the retention of the vast majority of the cellulose and hemicellulose components in the water insoluble fraction. However, the lignin component significantly impeded enzymatic hydrolysis, likely through a combination of restricting swelling and physically blocking access to the cellulose and non-productive binding of enzymes to lignin. It was apparent that the lignin contained in the aspen biomass treated with alkali in the absence of oxygen had a higher tendency to non-productively bind cellulases during enzymatic hydrolysis. Rather than using a pre-hydrolysis step to isolate hemicellulose in a separate fraction prior to the alkaline-oxygen treatment, lowering the pretreatment temperature resulted in lignin modification while recovering more than 70% of hemicellulose in association with the cellulose fraction in the water insoluble fraction. When a commercial cellulase mixture was supplemented with xylanases, this substrate could be readily hydrolyzed, recovering most of the hemicellulose and cellulose as monomeric sugars. Following this “one-pot” approach it was possible to enhance enzyme-mediated hydrolysis of the cellulose and hemicellulose components of mechanically refined biomass substrates without the need for extensive lignin removal.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors like to acknowledge the financial support from the Natural Science and Erngineering Council of Canada (NSERC) and the Korea Institute of Science and Technology (KIST). The authors would also like to thank Novozymes (Davis, CA) for the provision of enzymes for this study.Notes and references
- N. Mosier, C. Wyman, B. Dale, R. Elander, Y. Y. Lee and M. Ladisch, Bioresour. Technol., 2005, 96, 673–686 CrossRef CAS PubMed.
- R. P. Chandra, R. Bura, W. E. Mabee, A. Berlin, X. Pan and J. N. Saddler, Adv. Biochem. Eng./Biotechnol., 2007, 108, 67–93 CrossRef CAS PubMed.
- P. Alvira, E. Tomás-Pejó, M. Ballesteros and M. J. Negro, Bioresour. Technol., 2010, 101, 4851–4861 CrossRef CAS PubMed.
- H. Li, A. Saeed, M. S. Jahan, Y. Ni and A. Van Heiningen, J. Wood Chem. Technol., 2010, 30, 48–60 CrossRef CAS.
- Current lumber, pulp and panel prices | Natural Resources Canada, https://www.nrcan.gc.ca/forests/industry/current-prices/13309, accessed 3 July 2018.
- Unece and Fao, Geneva Timber For. Study, 2006, vol. 65 Search PubMed.
- X. Chen, J. Shekiro, R. Elander and M. Tucker, Ind. Eng. Chem. Res., 2012, 51, 70–76 CrossRef CAS.
- X. Chen, W. Wang, P. Ciesielski, O. Trass, S. Park, L. Tao and M. P. Tucker, ACS Sustainable Chem. Eng., 2016, 4, 324–333 CrossRef CAS.
- B. W. Koo, T. H. Treasure, H. Jameel, R. B. Phillips, H. M. Chang and S. Park, Appl. Biochem. Biotechnol., 2011, 165, 832–844 CrossRef CAS PubMed.
- G. S. Wang, X. J. Pan, J. Y. Zhu, R. Gleisner and D. Rockwood, Biotechnol. Prog., 2009, 25, 1086–1093 CrossRef CAS PubMed.
- J. Y. Zhu, X. J. Pan, G. S. Wang and R. Gleisner, Bioresour. Technol., 2009, 100, 2411–2418 CrossRef CAS PubMed.
- A. Boussaid and J. N. Saddler, Enzyme Microb. Technol., 1999, 24, 138–143 CrossRef CAS.
- R. P. Chandra, Q. L. Chu, J. Hu, N. Zhong, M. Lin, J. S. Lee and J. Saddler, Bioresour. Technol., 2016, 199, 135–141 CrossRef CAS PubMed.
- C. A. Mooney, S. D. Mansfield, M. G. Touhy and J. N. Saddler, Bioresour. Technol., 1998, 64, 113–119 CrossRef CAS.
- A. Berlin, N. Girkes, A. Kurabi, M. Tu, D. Kilburn and J. Saddler, Appl. Biochem. Biotechnol., 2005, 121, 163–170 CrossRef PubMed.
- A. Berlin, M. Balakshin, N. Gilkes, J. Kadla, V. Maximenko, S. Kubo and J. Saddler, J. Biotechnol., 2006, 125, 198–209 CrossRef CAS PubMed.
- L. Kumar, R. Chandra and J. Saddler, Biotechnol. Bioeng., 2011, 108, 2300–2311 CrossRef CAS PubMed.
- M. Tu, R. P. Chandra and J. N. Saddler, Biotechnol. Prog., 2007, 23, 398–406 CrossRef CAS PubMed.
- C. Cara, E. Ruiz, I. Ballesteros, M. J. Negro and E. Castro, Process Biochem., 2006, 41, 423–429 CrossRef CAS.
- X. Pan, X. Zhang, D. J. Gregg and J. N. Saddler, Appl. Biochem. Biotechnol., Part A, 2004, 115, 1103–1114 CrossRef.
- B. Yang, A. Boussaid, S. D. Mansfield, D. J. Gregg and J. N. Saddler, Biotechnol. Bioeng., 2002, 77, 678–684 CrossRef CAS PubMed.
- M. Hamaguchi, M. Cardoso and E. Vakkilainen, Energies, 2012, 5, 2288–2309 CrossRef CAS.
- Q. Chu, R. P. Chandra, C. S. Kim and J. N. Saddler, ACS Sustainable Chem. Eng., 2017, 5, 4011–4017 CrossRef CAS.
- L. Kumar, V. Arantes, R. Chandra and J. Saddler, Bioresour. Technol., 2012, 103, 201–208 CrossRef CAS PubMed.
- H. Lou, J. Y. Zhu, T. Q. Lan, H. Lai and X. Qiu, ChemSusChem, 2013, 6, 919–927 CrossRef CAS PubMed.
- H. Palonen, F. Tjerneld, G. Zacchi and M. Tenkanen, J. Biotechnol., 2004, 107, 65–72 CrossRef CAS PubMed.
- L. F. del Rio, R. P. Chandra and J. N. Saddler, Biotechnol. Bioeng., 2011, 108, 1549–1558 CrossRef CAS PubMed.
- S. Nakagame, R. P. Chandra and J. N. Saddler, Biotechnol. Bioeng., 2010, 105, 871–879 CAS.
- G. A. Smook, Handbook for Pulp and Paper Technologists, 1989 Search PubMed.
- R. P. Chandra and J. N. Saddler, Ind. Biotechnol., 2012, 8, 230–237 CrossRef CAS.
- S. Malkov, P. Tikka, V. Kuzmin and V. Baltakhinov, Nord. Pulp Pap. Res. J., 2002, 17, 420–426 CrossRef CAS.
- Y. K. Mok, V. Arantes and J. N. Saddler, Appl. Biochem. Biotechnol., 2015, 176, 1564–1580 CrossRef CAS PubMed.
- S. Katz, R. P. Beatson and A. M. Scallan, Sven. Papperstidn., 1984, 87, 48–53 Search PubMed.
- S. M. Kim, B. S. Dien and V. Singh, Biotechnol. Biofuels, 2016, 9, 97 CrossRef PubMed.
- W. Weiqi, W. Shubin and L. Liguo, Bioresour. Technol., 2013, 128, 725–730 CrossRef PubMed.
- H. Inoue, S. Yano, T. Endo, T. Sakaki and S. Sawayama, Biotechnol. Biofuels, 2008, 1, 2 CrossRef PubMed.
- R. Biswas, H. Uellendahl and B. K. Ahring, BioEnergy Res., 2015, 8, 1101–1116 CrossRef CAS.
- R. Yang, L. Lucia, A. J. Ragauskas and H. Jameel, J. Wood Chem. Technol., 2003, 23, 13–29 CrossRef CAS.
- R. Chandra and J. Saddler, J-FOR, 2013, 3, 6–14 Search PubMed.
- G. Siqueira, V. Arantes, J. N. Saddler, A. Ferraz and A. M. F. Milagres, Biotechnol. Biofuels, 2017, 10, 1–12 CrossRef PubMed.
- D. Fengel and G. Wegener, Wood: chemistry, ultrastructure, reactions, 1989 Search PubMed.
- F. Asgari and D. S. Argyropoulos, Can. J. Chem., 1998, 76, 1606–1615 CAS.
- I. A. Panagiotopoulos, R. P. Chandra and J. N. Saddler, Bioresour. Technol., 2013, 130, 570–577 CrossRef CAS PubMed.
- J. Li, G. Henriksson and G. Gellerstedt, Bioresour. Technol., 2007, 98, 3061–3068 CrossRef CAS PubMed.
- B. V. Kokta and A. Ahmed, Young, 1998, 191–211 Search PubMed.
- E. Sjöström, T. Janson, P. Haglund and B. Enström, J. Polym. Sci., Part C: Polym. Symp., 1965, 11, 221–241 CrossRef.
- M. Bendzalova, A. Pekarovicova, B. V Kokta and R. Chen, Cellul. Chem. Technol., 1996, 30, 19–32 CAS.
- Y. Ogiwara and K. Arai, Text. Res. J., 1968, 38, 885–891 CrossRef CAS.
- X. L. Luo, J. Y. Zhu, R. Gleisner and H. Y. Zhan, Cellulose, 2011, 18, 1055–1062 CrossRef CAS.
- X. Yu and R. H. Atalla, Powder Technol., 1998, 98, 135–138 CrossRef CAS.
- H. E. Grethlein, Bio/Technology, 1985, 3, 155–160 CrossRef CAS.
- J. E. Stone, A. M. Scallan, E. Donefer and E. Ahlgren, in Advances in Chemistry Series, 1969, vol. 95, pp. 219–241 Search PubMed.
- C. W. Dence, Pulp Bleach. Princ. Pract, 1996 Search PubMed.
- C. J. Biermann, Handbook of Pulping and Papermaking, 1996 Search PubMed.
- K. Öhgren, R. Bura, J. Saddler and G. Zacchi, Bioresour. Technol., 2007, 98, 2503–2510 CrossRef PubMed.
- S. I. Mussatto, M. Fernandes, A. M. F. Milagres and I. C. Roberto, Enzyme Microb. Technol., 2008, 43, 124–129 CrossRef CAS.
- J. Hu, V. Arantes and J. N. Saddler, Biotechnol. Biofuels, 2011, 4, 36 CrossRef CAS PubMed.
- J. Hu, R. Chandra, V. Arantes, K. Gourlay, J. Susan van Dyk and J. N. Saddler, Bioresour. Technol., 2015, 186, 149–153 CrossRef CAS PubMed.
- D. H. Cho, S. J. Shin, Y. Bae, C. Park and Y. H. Kim, Bioresour. Technol., 2010, 101, 4947–4951 CrossRef CAS PubMed.
- R. C. d. A. Castro, B. G. Fonseca, H. T. L. dos Santos, I. S. Ferreira, S. I. Mussatto and I. C. Roberto, Ind. Crops Prod., 2017, 106, 65–73 CrossRef CAS.
- G. Chaudhary, L. K. Singh and S. Ghosh, Bioresour. Technol., 2012, 124, 111–118 CrossRef CAS PubMed.
- D. Mitchell, K. Grohmann, M. Himmel, B. Dale and H. Schroeder, J. Wood Chem. Technol., 1990, 10, 111–121 CrossRef CAS.
- L. J. Jönsson and C. Martín, Bioresour. Technol., 2016, 103–112 CrossRef PubMed.
- P. F. Vena, Thermomechanical pulping (TMP), chemithermomechanical pulping (CTMP) and biothermomechanical pulping (BTMP) of bugweed (Solanum mauritianum) and Pinus patula, PhD dissertation, 2005.
- J. L. Rahikainen, R. Martin-Sampedro, H. Heikkinen, S. Rovio, K. Marjamaa, T. Tamminen, O. J. Rojas and K. Kruus, Bioresour. Technol., 2013, 133, 270–278 CrossRef CAS PubMed.
- B. Yang and C. E. Wyman, Biotechnol. Bioeng., 2006, 94, 611–617 CrossRef CAS PubMed.
- J. A. Rollin, Z. Zhu, N. Sathitsuksanoh and Y. H. P. Zhang, Biotechnol. Bioeng., 2011, 108, 22–30 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2019 |