
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
PbZrTiO3 ferroelectric oxide as an electron extraction material for stable halide perovskite solar cells†
Amador
Pérez-Tomas
 a,
Haibing
Xie
a,
Zaiwei
Wang
c,
Hui-Seon
Kim
c,
Ian
Shirley
a,
Silver-Hamill
Turren-Cruz
b,
Anna
Morales-Melgares
a,
Benedicte
Saliba
a,
David
Tanenbaum
a,
Michael
Saliba
b,
Shaik Mohammed
Zakeeruddin
b,
Michael
Gratzel
b,
Anders
Hagfeldt
*c and
Monica
Lira-Cantu
*ac
a,
Haibing
Xie
a,
Zaiwei
Wang
c,
Hui-Seon
Kim
c,
Ian
Shirley
a,
Silver-Hamill
Turren-Cruz
b,
Anna
Morales-Melgares
a,
Benedicte
Saliba
a,
David
Tanenbaum
a,
Michael
Saliba
b,
Shaik Mohammed
Zakeeruddin
b,
Michael
Gratzel
b,
Anders
Hagfeldt
*c and
Monica
Lira-Cantu
*ac
aCatalan Institute of Nanoscience and Nanotechnology (ICN2), CSIC and The Barcelona Institute of Science and Technology (BIST), Campus UAB, Bellaterra, E-08193 Barcelona, Spain. E-mail: monica.lira@icn2.cat
bLaboratory of Photonics and Interfaces, Institute of Chemical Sciences and Engineering, Ecole Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland
cLaboratory of Photonics and Interfaces, Institute of Chemical Sciences and Engineering, Ecole Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland
First published on 5th December 2018
Abstract
State-of-the-art halide perovskite solar cells employ semiconductor oxides as electron transport materials. Defects in these oxides, such as oxygen vacancies (Ovac), act as recombination centres and, in air and UV light, reduce the stability of the solar cell. Under the same conditions, the PbZrTiO3 ferroelectric oxide employs Ovac for the creation of defect-dipoles responsible for photo-carrier separation and current transport, evading device degradation. We report the application of PbZrTiO3 as the electron extraction material in triple cation halide perovskite solar cells. The application of a bias voltage (poling) up to 2 V, under UV light, is a critical step to induce charge transport in the ferroelectric oxide. Champion cells result in power conversion efficiencies of ∼11% after poling. Stability analysis, carried out at 1-sun AM 1.5 G, including UV light in air for unencapsulated devices, shows negligible degradation for hours. Our experiments indicate the effect of ferroelectricity, however alternative conducting mechanisms affected by the accumulation of charges or the migration of ions (or the combination of them) cannot be ruled out. Our results demonstrate, for the first time, the application of a ferroelectric oxide as an electron extraction material in efficient and stable PSCs. These findings are also a step forward in the development of next generation ferroelectric oxide-based electronic and optoelectronic devices.
Halide perovskite solar cells (PSCs) have emerged as a competitive photovoltaic technology with power conversion efficiencies (PCEs) surpassing the 22.7% mark.1 One of the main bottlenecks of the technology is their long-term stability.2 Understanding the different degradation mechanisms of the constituent materials, as well as interface instabilities, is of crucial importance for commercialization.3 Semiconductor oxides (SO) constitute a fundamental part of highly efficient photovoltaic technologies4–11 such as PSCs.11,12 Oxides are applied as dense thin films or as mesoporous oxide layers, working as hole or as electron transport materials (HTM or ETM, respectively) in normal or inverted PSC configurations.10,11,13–15 Electron transport semiconductor oxides, like TiO2, are characterized by an oxygen vacancy (Ovac)-mediated conductivity16 caused by a deviation in stoichiometry, the presence of impurities, or both.17 In oxygen-containing atmospheres, and especially under UV light, holes generated at the nonstoichiometric oxide surface react with the oxygen adsorbed at Ovac increasing charge recombination and degradation of the solar cell.17,18 Different methods have been employed to passivate or eliminate these Ovac. For example, the application of organic interfacial modifiers with anchoring groups specifically selected to bond with oxides,3,19 the application of doped oxides19,20 or the application of less reactive SnO2 which results in less hygroscopicity, fewer Ovac at its surface, and less UV-damage.17,18 Another possibility is the application of a coating of secondary oxides, like ZrO2 or Al2O3, applied to suppress surface defects, avoid interfacial recombination, and enhance device stability.21–23 More recently, Seok, et al., reported a highly stable PSCs by the application of an UV-light resistant oxide, the La-doped BaSnO3, applied as electron transport layer.24 A less-explored option is the application of complex oxides with singular properties, such as ferroelectric, magnetic or ferroic oxides. Given their wide bandgap values and insulating properties, their application as electron transport materials (ETM) in emerging solar cells seems unlikely.25 For example, ferroelectric oxides (FE-oxides) display spontaneous electric polarization where the application of an electric field (poling) reorients domains within individual grains. Owing to this characteristic, different sources of conduction can be observed, such as polarization switching, breakdown and leakage.26,27 Conduction through tunnel junctions28,29 as in TiO2/Al2O3 electrodes,21 or domain walls30,31 are other forms of current transport usually observed in nanometres thick films. Moreover, their large dielectric constant implies the presence of a low exciton binding energy which translates in low electron/hole recombination and more defect-tolerant charge carrier transport.32 Other ferroelectric materials have been applied in PSCs. For example ferroelectric polymers, such as PVDF, has been employed as an additive during the fabrication processes improving the perovskite morphology and the final device efficiency.33–41 It has also been applied as an insulating tunnelling layer between the halide perovskite and the electron transport layer,42 increasing carrier transfer.43 Nevertheless, the low photoactivity that FE oxides show under UV light makes them very attractive to enhance perovskite solar cell stability. In FE-oxides, UV light can photoinduce the enhancement of the remnant polarization, as recently reported for PbZrTiO3 (PZT) films.44 The formation of defect dipoles, responsible for charge separation and current transport, depends on the presence of defect charges (Ovac). Thus, instead of acting as photoreactive sites as in SnO2 or TiO2, Ovac in PZT are removed after poling under UV light increasing its transport properties,44 suggesting a different working mechanism which can benefit the stability of solar cells.
Here we report the fabrication and lifetime analysis of PSCs applying a thin film of Pb[ZrxTi1−x]O3 (PZT) ferroelectric oxide as the ETM. The PZT was made as dense thin film and applied in planar configuration PSCs of the type: FTO/PZT/CsMAFA/spiro-OMeTAD/Au, where CsMAFA refers to the halide perovskite material with formula Cs5(MA0.17FA0.83)(95)Pb(I0.83Br0.17)3 and PZT refers to the nominal composition Pb[Zr0.6Ti0.4]O3. For comparison purposes, reference solar cells were made applying SnO2 as dense ETM as in FTO/SnO2/CsMAFA/spiro-OMeTAD/Au. We also explored the effect of the PZT layer on the triple cation perovskite under illumination with respect to solar cell stability. Analysis of the solar cells were made at 1-sun AM 1.5 G, including UV light, in air (no encapsulation), at 45 °C and 55% RH. The solar cells were poled up to 2 V in order to polarize the PZT electrode. Poling resulted in a slow but steady improvement of the photovoltaic properties of the PZT-based PSC. The improvement was observed during the first 90 minutes after which the device stabilized and then maintained its photovoltaic properties under continuous illumination in air for hours. Our results demonstrate, for the first time, the possibility of applying the PZT ferroelectric oxide as ETM in air and UV-stable PSCs.
Results and discussion
Growth of crystalline PZT films
In the context of emerging photovoltaics, ferroelectric oxide (FE-oxide) layers with perovskite arrangement are of particular interest since they are a class of materials with unique functionalities, strong metal-oxide bonds, and high heat of formation resulting in very stable compounds. A highly crystalline structure enables the ferroelectric properties of the PZT, resulting in performance dependent on grain boundaries and the ferroelectric domain wall configuration.44 Nevertheless, high temperatures, usually above 600 °C, are required in general for the desired ABO3 perovskite oxide phase formation24 which limits the extrinsic conductivity of the transparent conducting FTO substrate. We circumvent this problem by the application of a fast (10 min) sintering method for the fabrication of PZT thin films on transparent FTO.11 The synthesis of the PZT, Pb(ZrxTi1−x)O3 (x = 0.6) (for exact composition see ESI†), is made by solution processing and the solution is deposited on the FTO by spin coating. The minimum annealing temperature at which good ferroelectric properties are retained is found at 625 °C. The ferroelectric perovskite crystalline structure of the PZT deposited on FTO was confirmed by X-ray diffraction (XRD) and scanning electron microscopy (SEM) analysis, as shown in Fig. 1 respectively (see also ESI Fig. S1 and S2†). XRD theta-2theta (Fig. 1a) and cross-sectional X-TEM mapping (ESI Fig. S1†) shows that the PZT thin film belongs to the symmetry point group ∞ m with space group Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (221) cubic, where the lattice constant has been determined to be a = 4.077 Å (ESI Fig. S1†). Hysteresis loops demonstrating the ferroelectric character of the PZT film were carried out to all thin films. Fig. 3j and k shows the hysteresis loops of the PZT thin film, synthesized at 625 °C, before and after irradiation with UV light (Fig. 3j) and visible light (k). The analyses were made first in the dark and then after exposed to different light sources: (near-ultraviolet light-emitting diode (UVA-LED, 365 ± 10 nm/3.40 ± 0.12 eV) and white diode (White-LED, 400–700 nm/1.77–3.10 eV). For all the other PZT obtained at different temperatures see Fig. S2 (under ESI†). On the basis of controlled experiments examining the effect of the PZT thin film thickness, a series of different PZT thickness were fabricated (see Fig. 1c and S4–S8 in ESI†). The screening of the thickness was accomplished by diluting the PZT solution in acetic acid (c/co) as described in methods (ESI Fig. S4†). Analysis revealed that PZT films ranged from thick 100 nm films (corresponding to an undiluted solution c/co of 1/0) to thin <10 nm films (c/co of 1/10). Two dissimilar samples were then selected, a thick 100 nm and a thin ∼10 nm film (Fig. 1a and b). The final PZT surface morphology, analysed by AFM and SEM (Fig. 1e–h), seems to be poorly affected by the film thickness but also likely influenced by the well-crystallized FTO substrate which has a rutile crystal structure with a tetragonal unit cell (P42/mnm) matching the perovskite oxide structure. Nevertheless, careful analysis of the surface of a series of different PZT thin films made at different thicknesses showed a slight increase in surface roughness when increasing thin film thickness (ESI Fig. S5†). The surface morphology of the thicker films (e.g. 100 nm) are free of micro-cracks, pores or holes, whereas some pinholes can be present in thinner thin films.
m (221) cubic, where the lattice constant has been determined to be a = 4.077 Å (ESI Fig. S1†). Hysteresis loops demonstrating the ferroelectric character of the PZT film were carried out to all thin films. Fig. 3j and k shows the hysteresis loops of the PZT thin film, synthesized at 625 °C, before and after irradiation with UV light (Fig. 3j) and visible light (k). The analyses were made first in the dark and then after exposed to different light sources: (near-ultraviolet light-emitting diode (UVA-LED, 365 ± 10 nm/3.40 ± 0.12 eV) and white diode (White-LED, 400–700 nm/1.77–3.10 eV). For all the other PZT obtained at different temperatures see Fig. S2 (under ESI†). On the basis of controlled experiments examining the effect of the PZT thin film thickness, a series of different PZT thickness were fabricated (see Fig. 1c and S4–S8 in ESI†). The screening of the thickness was accomplished by diluting the PZT solution in acetic acid (c/co) as described in methods (ESI Fig. S4†). Analysis revealed that PZT films ranged from thick 100 nm films (corresponding to an undiluted solution c/co of 1/0) to thin <10 nm films (c/co of 1/10). Two dissimilar samples were then selected, a thick 100 nm and a thin ∼10 nm film (Fig. 1a and b). The final PZT surface morphology, analysed by AFM and SEM (Fig. 1e–h), seems to be poorly affected by the film thickness but also likely influenced by the well-crystallized FTO substrate which has a rutile crystal structure with a tetragonal unit cell (P42/mnm) matching the perovskite oxide structure. Nevertheless, careful analysis of the surface of a series of different PZT thin films made at different thicknesses showed a slight increase in surface roughness when increasing thin film thickness (ESI Fig. S5†). The surface morphology of the thicker films (e.g. 100 nm) are free of micro-cracks, pores or holes, whereas some pinholes can be present in thinner thin films.
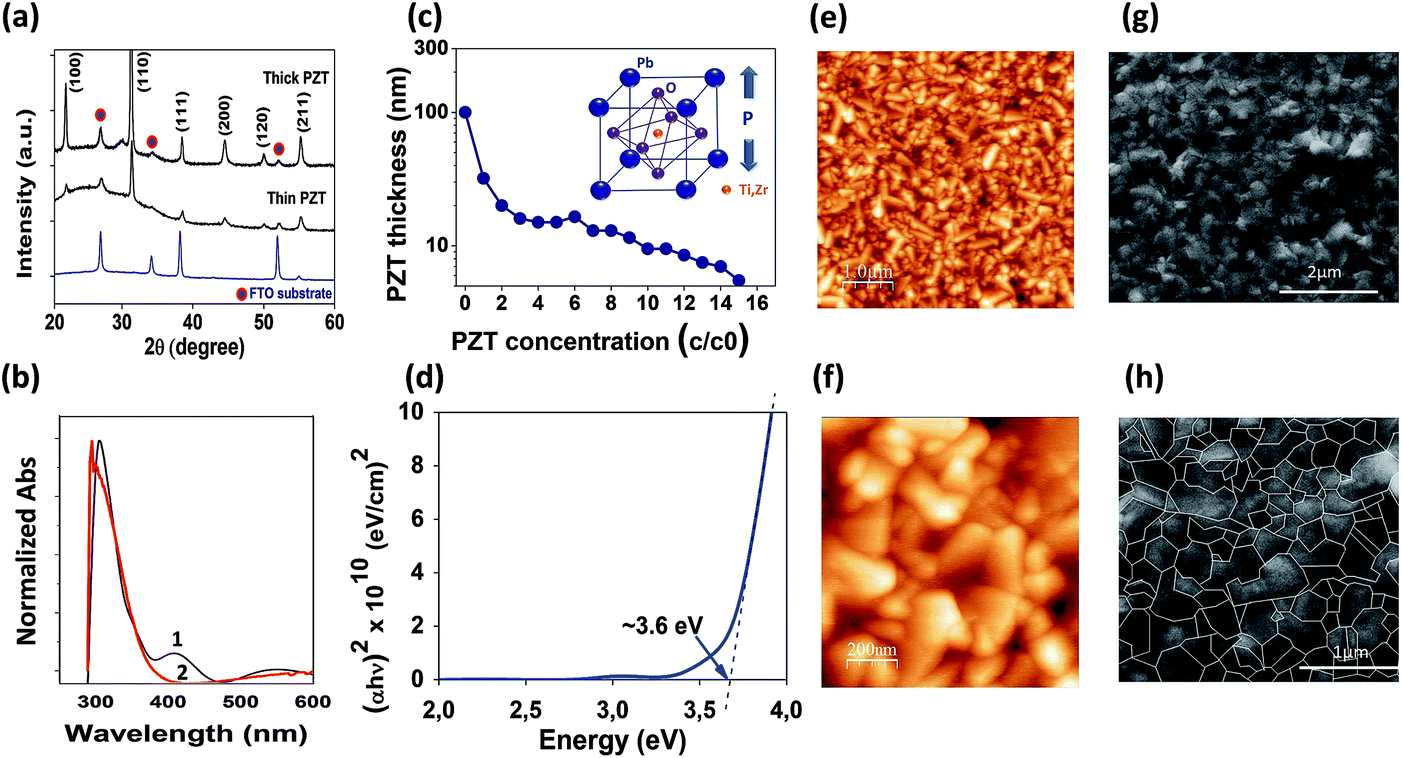 | ||
| Fig. 1 Synthesis of Pb[ZrxTi1−x]O3 (PZT) thin film on transparent FTO. (a) X-ray diffraction analysis of the PZT thin (∼10 nm) and thick (100 nm) films on FTO substrates, annealed at 625 °C. (b) UV vis analysis of the thick (1) and thin (2) PZT films; (c) representation of the crystal structure of the PZT, and the relation between the PZT thickness and the PZT concentration applied during thin film fabrication; (d) PZT band gap at 3.6 eV; (e–f) AFM surface analysis of the ∼10 nm thin PZT film; (g and h) surface SEM images (g) and grain boundaries (marked in white, (h)) of the ∼10 nm thin PZT film. | ||
Photovoltaic performance of the PZT-based PSCs
Here we fabricate complete PSCs consisting of the FTO/PZT thin films, followed by the CsMAFA perovskite of about 600 nm. Spiro-OMeTAD was used as the hole transport material and a thin layer (80 nm) of Au as the back metal electrode. For comparison purposes, reference solar cells were also fabricated as previously described45 applying the SnO2 as the ETM. The PZT-based PSC schematics are shown in Fig. 2a. In this configuration, the PZT takes the place of the electron transport material (ETM). The solar cells were analysed at 1-sun AM 1.5 G (including UV light) under ambient conditions without encapsulation. We collected statistical data on devices applying different PZT thickness (ESI Fig. S6 and S7†) where efficiency increases as the thickness of the PZT decreases, with a maximum observed at ∼10 nm thickness as shown in Fig. 2c. Below the ∼10 nm (c/co of 1/10), the photovoltaic performance decreases possibly due to the presence of pinholes or conduction via tunnelling (only observable in a-few nanometers thick films29,46,47). For comparison purposes, we selected two PZT films with highly dissimilar thickness: ∼10 nm (c/co of 1/10) and 100 nm (c/co of 1/1). In general, the majority of the as-prepared devices showed no photovoltaic response or exhibited poor PCEs with values around 0.2% and 4% for PSCs applying the 100 nm and 10 nm PZT films, respectively, as shown in Fig. 2c and d (see also Table 1, and ESI Table S1†). These results show an evident suppression of the electron injection from the PZT to the FTO electrode, especially for thick PZT films, highlighting the high dielectric and insulating nature of PZT. The solar cells were then subjected to poling up to 2 V under 1 sun AM 1.5 G. During poling, the photovoltaic properties of the PZT-based solar cells increased gradually until stabilization. This improvement was almost negligible for the thick 100 nm PZT based PSC (ESI Fig. S7†) and drastic for the thin ∼10 nm PZT-based PSC (Fig. 2c–f). The champion PSCs corresponds to solar cells with the ∼10 nm PZT thin film (Fig. 2c–f) which showed a dramatic and steady increase in PCE scaling from an initial 1.49% as-prepared, before poling (Fig. 2e), up to 11.16% efficiency after poling (Fig. 2f, Table 1). Fig. 2d depicts the histogram of the PCE for the PSCs using the champion cell of ∼10 nm thick PZT film, before and after poling, displaying a very reproducible pattern of efficiency increase for all devices. Besides the difference in thicknesses, the dissimilarity observed on photovoltaic response between the solar cells with the thin (∼10 nm) and the thick (100 nm) PZT film can be related to the surface roughness morphology and grain size of the PZT layer. The polarization-dependent conductivity (ferroelectric properties)48,49 is highly reliant on synthesis conditions, morphology, defect concentration and the orientation and size of the oxide grains.50 Especially the presence of different grain size indicates different intrinsic stress in the films due to the difference in grain boundaries, resulting in different dielectric properties.51 Thus, the different thickness, surface roughness morphology and grain size of the PZT films could benefit the photovoltaic response after poling for the PSC with the ∼10 nm thin PZT which shows the largest roughness (Ra ∼26 nm) in comparison to the 100 nm thick PZT (Ra ∼ 20 nm) (ESI Fig. S2†). | ||
| Fig. 2 Photovoltaic performance of PSCs of the type: FTO/PZT/CsMAFA/spiro-OMeTAD/Au. (a) Schematic representation of the planar and normal configuration PSC. (b) SEM analysis of a PZT-based PSC. (c) PCE (%) observed at different PZT dilutions or concentrations (c/co). (d) Histograms of the ∼10 nm thin PZT-based PCSs before and after 90 min of poling. (e) and (f) I–V curves for the ∼10 nm thin PZT-based PSC before (e) and after (f) 90 min of poling in air. Solar cells were analysed at 1-sun AM 1.5 G, including UV light, in air (no encapsulation), at 45 °C and 55% RH. | ||
| Cell | Before poling | After poling | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Scan | J sc (mA cm−2) | V oc (V) | FF (%) | PCE (%) | Scan | J sc (mA cm−2) | V oc (V) | FF (%) | PCE (%) | |
| PZT | Forward | 2.40 | 0.99 | 27.0 | 0.54 | Forward | 18.26 | 0.91 | 43.8 | 7.31 |
| Reverse | 2.28 | 1.01 | 31.8 | 0.64 | Reverse | 18.51 | 0.91 | 65.8 | 11.16 | |
| SnO2 | Forward | 22.48 | 1.13 | 0.78 | 19.55 | Forward | 9.36I | 1.02 | 27.3 | 2.63 |
| Reverse | 22.84 | 1.12 | 0.76 | 19.03 | Reverse | 2.43 | 0.94 | 27.7 | 0.63 | |
Electrical poling under 1 sun AM 1.5 G
Poling, the process of applying an external electric field to a ferroelectric material, induces the reorientation of dipole moments within individual grains improving the mobility of charge carriers.52 Poling conditions carried out under 1 sun AM 1.5 G, were observed to be strongly affected by the PZT thin film thickness, the light harvester material (active layer), and the method at which the electric bias is applied. In our samples, progressive IV poling (with increasing initial voltage of 20–50 mV) were observed to result in enhanced photovoltaic performance of the FE-oxide based PSC if compared to the application of a single high voltage. Thus, careful optimization of all parameters was carried out (see ESI Fig. S2, S8–S11†). We first analysed the voltage limit for poling. We have previously observed high robust response of solar cells based on the P3HT:PCBM active layer even after the application of voltages as high as 40 V.53 In the case of the halide perovskite active layer, the ∼10 nm PZT-based PSCs barely sustain the application of 2 V, while the thick 100 nm PZT-based PSC requires stronger poling showing a voltage limit at around 4–5 V. Moreover, the scan rate should also be limited to low speeds, with optimum values below 2 mV s−1. Finally, we observed that the solar cell was unresponsive after several scans in the forward direction (i.e. biasing the solar cell from the minimum positive voltage value) during poling, thus poling was carried out under reverse mode only. In summary, the poling was carried out in air at under 1 sun AM 1.5 G (including UV light), through subsequent IV-curves up to maximum of +2 V. The scan direction was the reverse mode (i.e. biasing the solar cell from the maximum positive voltage value) and the scan rate was 2 mV s−1. IV curves were performed every 5 min until the photovoltaic parameters, in particular current density, Jsc, reached a maximum and stabilized (for details on poling optimization see ESI Fig. S7, S8 and S15†).Fig. 3a and b shows the effect of electrical poling on the photovoltaic response of the champion PZT-based PSC (red line) and the comparison with a reference device based on SnO2 (blue line). The PZT-based PSC increased its photovoltaic response from 1.49% up to 11.16% after poling. This response validates the effective activation of the photovoltaic parameters through poling for the PZT which evolve slowly until stabilization of the PCE after 90 minutes. Poling the reference solar cell (made with SnO2) results in a clear degradation of the photovoltaic response after poling (Fig. 3b). Although this observed degradation of the SnO2-based PSC is expected (due to the severe testing environment and conditions) it permits to demonstrate the dissimilar working mechanism between both the PZT and the SnO2 ETMs. The poling effect is observed to be highly reproducible for a series of analysed samples as shown by the error bars depicted in Fig. 3c–f (also in Fig. S7†). These results demonstrate the crucial importance of poling under light irradiation, for the successful transformation of the insulating PZT into an electron transport material in our solar cells.
 | ||
| Fig. 3 Effect of poling in air and UV light. Evolution of the IV curves during poling for PSCs made with (a) PZT and (b) SnO2. (c–f) Evolution of the photovoltaic parameters of the solar cells with time under ambient atmosphere and UV light (no encapsulation) for the SnO2 – based PSC (blue) and the PZT-based PSC (red). IPCE (g) and normalized IPCE (h) for the champion solar cell; (i) comparison between Jsc obtained from IV curves and from the IPCE spectra at different poling times. Poling times: (1) 0.1 h, (2) 1.0 h, (3) 2.5 h, (4) 5.0 h. Solar cells were analysed at 1-sun AM 1.5 G, including UV light, in air (no encapsulation), at 45 °C and 55% RH. Hysteresis loops for the PZT before and after illumination with UV light (j) and visible light (k) and conductance measurements of a PZT thin film in the dark and under 1 sun AM 1.5 G continuous illumination, including UV light, and under air atmosphere (l). | ||
To analyse the evolution of the transport properties of the PZT-based PSC under UV light and poling, we carried out experiments of IPCE (or EQE) against poling time. We then compared the Jsc obtained from the IPCE and the one obtained from the IV curves at specific times. The poling experiments were carried out at 0.1 h, 1.0 h, 2.5 h and 5 h following the same poling procedure. After each poling period the sample was taken out of the sun simulator and immediately analysed by IPCE. The first observation is that, due to the insulating properties of the PZT, very low charge transport is initially observed (Fig. 3g-1) by increasing the poling time from 0.1 h to 5 h, the conversion of photons to electrons increases from 4% to 60% (Fig. 3g-1 and 4). The transformation of the PZT with time and the improvement of IPCE (and solar cell efficiency) can also be explained by the shape of the IPCE spectra. Fig. 3h shows the normalized IPCE spectra shown in Fig. 3g. The initial spectra obtained after 0.1 h of poling (Fig. 3h-1) revealed a very weak absorption of the PZT oxide at wavelengths below 340 nm, corresponding with a very poor semiconductor. At this time, no absorption of the halide perovskite CsMAFA (between 400–800 nm) is observed since no charge transport takes place from the PZT to the halide perovskite. As the poling under UV light continues, this peak at 340 nm, red shifts to 380 nm (from 3.64 to 3.26 eV), a response observed during the reduction of the band gap in oxides. This red shift is attributed to oxide doping observed in classical oxides like TiO2 (ref. 54 and 55) but also observed in other oxides like PZT.56 It has also been attributed to the continuous and steady removal of Ovac under UV light exposure or inert atmospheres for several binary oxides.6,7 The increase of the poling time, from 0.1 h to 1.0 h and 2.5 h, results in the gradual improvement of the IPCE absorption between 400 nm and 800 nm, which is the region that corresponds to the absorption of the halide perovskite CsMAFA material (Fig. 3h-1 to 4), an indication of charge transport throughout the PSC. We should also emphasize that the shape of the final IPCE obtained after 5 h poling (Fig. 3g-4 and h-4) corresponds exactly with the shape of the IPCE of a PSC applying the classical SnO2 oxide as ETL as shown in ESI Fig. S9†.57
Finally, we compared the response observed for the current density (Jsc) obtained from IV curves and IPCE after poling (Fig. 3i). In both cases, the Jsc shows a steadily increase until reaching similar values around ∼14 mA cm−2 after the poling process. We observed however, that for intermediate poling times between (1) 0.1 h, (2) 1.0 h and (3) 2.5 h, the Jsc obtained from IPCE is always lower than the Jsc obtained from the IV curves (Fig. 3i). The main difference between both characterization methods is that IV curves are carried out under full illumination under the sun simulator, right after poling. In the case of the IPCE characterization, illumination and poling are eliminated in order to situate the sample under the IPCE set up for analysis. In this short period of time, the removal of UV light and poling in PZT samples brings the material back to its original state and electrostatic repulsion between polarization charges and defect charges (Ovac) immediate occurs, depolarizing the PZT film. Since remnant polarization is defined as the polarization remaining in the material after the poling voltage is eliminated or reduced to zero, we attributed the increase in Jsc (from IPCE) to the enhancement of the remnant polarization when increasing poling time, in perfect agreement with the mechanism reported for PZT by Cao, et al.44
Thus, we infer here that the as-prepared PZT-based PSC with polycrystalline PZT thin films (as shown in Fig. 1 and ESI Fig. S6†) have domains which are random. When the solar cell is poled under UV light, part of the defect-dipoles located at Ovac, are separated and the charges are driven to opposite interfaces of the film. This effect is enhanced by UV light irradiation which permits the excitation of the remaining defect-dipole charges that are expelled to opposite interfaces by the poling field as reported for PZT.44 The process takes place slowly throughout the first minutes of the analysis. With time, the PZT behaves as an electron transport material in the solar cell and delivers the best photovoltaic performance. The effect is reversible if the bias voltage and UV light is eliminated but can become permanent if the poling time increases (as observed in Fig. 3i), in agreement to the enhancement of the remnant polarization with poling as described for PZT.44 The transformation of the PZT from an insulator into a conductor is proved by the hysteresis (ferroelectric) loops analysed to the PZT before and after light irradiation (Fig. 3j and k). After analysis under white-LED the hysteresis loop shows a clear ferroelectric response. Under UV-LED irradiation the PZT samples presented significant hysteresis suppression in the hysteresis loops as observed in Fig. 3j. This effect, known as polarization suppression58 (or ferroelectric fatigue59), is a well-known effect in ferroelectric materials60 and it is directly related to the conductivity of the perovskites: good ferroelectric properties can only be observed on materials with low conductivity, improving conductivity will inevitably increase the leakage current and suppress ferroelectricity.60 These results are also supported by conductivity measurements carried out to the PZT thin film in the dark and after light irradiation (Fig. 3g). The PZT samples analysed in the dark show low conductivity which increases once the sample is irradiated with light. The effect is reversible once the PZT thin film is stored in the dark again.
Given that the defect-dipoles are reliant on the multicrystalline nature of the PZT (density of grain boundaries and domain walls), we infer that the different response of the PSC after poling is attributed to the different PZT thickness (and surface roughness) as shown in Fig. 1 (and Fig. S6 ESI†). We also observed that the PZT-based PSCs showed large hysteresis after poling and that the hysteresis index (HI) strongly depends on the poling time (Fig. 2 and ESI Fig. S14†). Since hysteresis is related to factors like applied conditioning voltage, device configuration or UV light, among many others61 we attribute the observed hysteresis to the effect of testing conditions (ambient air or the presence of UV light) but especially to the large poling voltage applied. Especially, the hysteresis observed from ion migration in halide perovskites can be strongly affected by the poling procedure. Identifying a specific conduction mechanism in a device comprising a ferroelectric material can be rather complex, especially in a dynamic system under the continuous effect of poling and UV light. Nevertheless, since several possible conduction mechanisms can be possible (for example bulk effects including ohmic conduction, charge carrier hopping, space charge limited conduction, interface effects, tunnelling, conduction through domain walls and the conduction mechanisms related to defects like oxygen vacancies in oxides58–60), there is a possibility that poling and UV light irradiation can also be causing the accumulation of charges or the migration of ions (or the combination of all of them) in the active, transport layer or at interfaces in our devices.
Stability analysis in air and UV light
We further studied the stability of PSCs under continuous 1-sun AM 1.5 G light irradiation (Fig. 4). After poling, the PSCs were analysed by consecutive IV curves taken each few minutes in reverse mode with a scan rate of 2 mV s−1. The I–V scan starting voltage was established at +1.6 V and the scan final voltage was −0.1 V. Degradation of halide PSCs under ambient conditions (without encapsulation) is well-documented for devices applying electron transport oxides like TiO2 or SnO2.3,11,14 Thus, long-term stability studies are usually carried out under encapsulation conditions or inert atmospheres.24 In our stability tests, we want to demonstrate the effective response of the PZT thin film under severe conditions: 1 sun continuous light irradiation (including UV light), in air, at 45 °C and 55% RH and without encapsulation. Fig. 4 shows the results of the initial 10 h of the stability analysis carried out after poling. Strikingly, our PZT-based PSC shows a remarkable stability under air and UV light, while the SnO2-based PSC degrades in minutes. Although one can argue that the degradation of the reference device, the SnO2-based PSC, is most likely to be intensified due to the application of bias voltage (poling), the fact that our PZT-based PSCs sustain and improve its photovoltaic properties demonstrates a different working mechanism and an improved stability in air and UV light.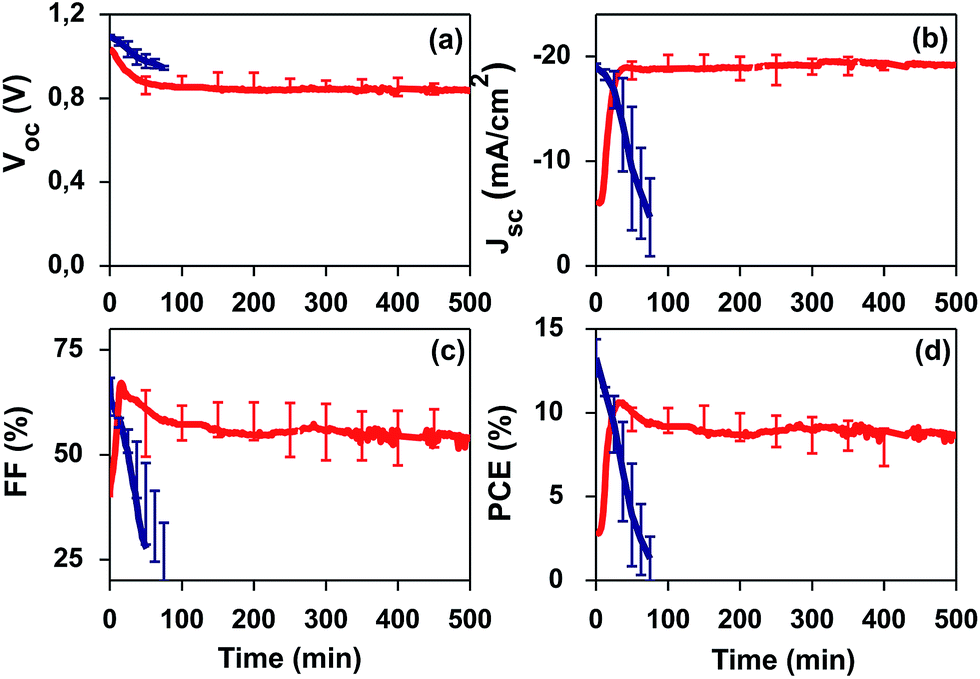 | ||
| Fig. 4 Stability of PSCs in air and UV light (unencapsulated devices) after poling. Stability analysis of a PZT-based PSC (red) and a reference PSC applying SnO2 (blue). Analyses were made at 1 sun AM 1.5 G continuous illumination, including UV light, and under air atmosphere (55% RH, 45 °C) without encapsulation. | ||
Conclusions
We introduced the ferroelectric oxide Pb(ZrxTi1−x)O3 (x = 0.6), PZT, as the electron extraction layer in triple cation, Cs5(MA0.17FA0.83)(95)Pb(I0.83Br0.17)3 (CsMAFA) halide perovskite solar cells. Poling the solar cells under UV light in air, triggered the transport properties of the PZT resulting in the enhancement of the photovoltaic properties of the devices with time. This improvement is directly influenced by the thickness of the PZT with the best response observed for PZT films of ∼10 nm. The champion PZT-based solar cell resulted in 11% PCE after poling under standard 1-sun AM 1.5 G sunlight (including UV light, in air and without encapsulation). The solar cells were stable in air and UV light for several hours demonstrating the stability of the CsMAFA perovskite. We also demonstrated a different working mechanism of the PZT in comparison with a classical oxides like SnO2 or TiO2, establishing for the first time the possibility to apply (and transform) an insulating ferroelectric oxide to work as electron extraction layer in PSCs. Our experiments indicate the effect of ferroelectricity, however alternative conducting mechanisms affected by the accumulation of charges or the migration of ions (or the combination of them) cannot be ruled out. We expect our findings to have a major impact in a new generation of ETMs for PSCs that can also affect the long-term stability of emerging photovoltaics, but also to open up new possible functionalities of ferroelectric oxides in a wide range of novel electronic and optoelectronic applications.Author contributions
M. L.-C. wrote the manuscript with feedback from the co-authors. M. L.-C. and A. H. supervised the study. M. L.-C., H. X. and A. P conceived and designed the experiments. H. X., Z. W., H.-S. K. and S.-H. T.-C. fabricated and characterized the devices. H. X. carried out the conductivity measurements. A. P. and I. S. fabricated and characterized the PZT substrates and developed and optimized the poling method. A. M. performed the SEM and AFM analysis. B. S. carried out the UV-vis and catalysis analyses. D. T., M. S., S. M. Z and M. G. coordinated the work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors would like to thank Prof. Gustau Catalán for the hysteresis loops made at his laboratory. To Dr Belen Ballesteros for the X-TEM images and analysis. M. G. and S. M. Z. thank the European Union's Horizon 2020 programme, through a FET Open research and innovation action under grant agreement No. 687008 and the King Abdulaziz City for Science and Technology (KACST) for the financial support. H.-S. K. is grateful for the postdoctoral fellowship grant (NRF-2016R1A6A3A03012393). M. S. acknowledges support from the co-funded Marie Skłodowska Curie fellowship, H2020 grant agreement no. 665667. To the Spanish MINECO through the Severo Ochoa Centers of Excellence Program under Grant SEV-2013-0295 and for the postdoctoral contract for H. X.; for the grant ENE2016-79282-C5-2-R and the OrgEnergy Excelence Network CTQ2016-81911-REDT. To the Agència de Gestió d'Ajuts Universitaris i de Recerca for the support to the consolidated Catalonia research group 2017 SGR-329 and the Xarxa de Referència en Materials Avançats per a l'Energia (Xarmae). To the Agencia Estatal de Investigación (AEI) and Fondo Europeo de Desarrollo Regional (FEDER) for the contract ENE2015-74275-JIN to A. P. To the CERCA Programme/Generalitat de Catalunya and to the European COST Action StableNextSol project MP1307. To the Fullbright fellowship program for the support to I. S.References
- W. S. Yang, B.-W. Park, E. H. Jung, N. J. Jeon, Y. C. Kim, D. U. Lee, S. S. Shin, J. Seo, E. K. Kim, J. H. Noh and S. I. Seok, Science, 2017, 356, 1376–1379 CrossRef CAS PubMed.
- G. Grancini, C. Roldán-Carmona, I. Zimmermann, E. Mosconi, X. Lee, D. Martineau, S. Narbey, F. Oswald, F. De Angelis, M. Graetzel and M. K. Nazeeruddin, Nat. Commun., 2017, 8, 15684 CrossRef CAS PubMed.
- M. Lira-Cantú, Nat. Energy, 2017, 2, 17115 CrossRef.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- J. Xia, N. Masaki, M. Lira-Cantu, Y. Kim, K. Jiang and S. Yanagida, J. Am. Chem. Soc., 2008, 130, 1258–1263 CrossRef CAS PubMed.
- M. Lira-Cantu and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2006, 90, 2076–2086 CrossRef CAS.
- M. Lira-Cantu, K. Norrman, J. W. Andreasen and F. C. Krebs, Chem. Mater., 2006, 18, 5684–5690 CrossRef CAS.
- G. Teran-Escobar, J. Pampel, J. M. Caicedo and M. Lira-Cantu, Energy Environ. Sci., 2013, 6, 3088–3098 RSC.
- J. Zhang, B. Xu, L. Yang, A. Mingorance, C. Ruan, Y. Hua, L. Wang, N. Vlachopoulos, M. Lira-Cantú, G. Boschloo, A. Hagfeldt, L. Sun and E. M. J. Johansson, Adv. Energy Mater., 2017, 7, 1602736 CrossRef.
- A. Perez-Tomas, A. Mingorance, Y. Reyna and M. Lira-Cantu, in The Future of Semiconductor Oxides in Next Generation Solar Cells, ed. M.Lira-Cantu, Elsevier Singapore, 2017, p. 566 Search PubMed.
- M. Lira-Cantu, The future of semiconductor oxides in next generation solar cells, Elsevier Singapore, 2017 Search PubMed.
- M. Saliba, T. Matsui, J. Y. Seo, K. Domanski, J. P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- A. Mingorance, H. Xie, H.-S. Kim, Z. Wang, M. Balsells, A. Morales-Melgares, N. Domingo, N. Kazuteru, W. Tress, J. Fraxedas, N. Vlachopoulos, A. Hagfeldt and M. Lira-Cantu, Adv. Mater. Interfaces, 2018, 0, 1800367 CrossRef.
- Y. Reyna, A. Perez-Tomas, A. Mingorance and M. Lira-Cantu, in Molecular Devices for Solar Energy Conversion and Storage, ed. H.Tian, G.Boschloo and A.Hagfeldt, Springer-Verlag Berlin, Berlin, 2018, pp. 477–531 Search PubMed.
- A. Hagfeldt and M. Lira-Cantu, Appl. Surf. Sci., 2018 Search PubMed , In Press.
- M. K. Nowotny, T. Bak and J. Nowotny, J. Phys. Chem. B, 2006, 110, 16270–16282 CrossRef CAS PubMed.
- A. Raccanelli and A. Maddalena, J. Am. Ceram. Soc., 1976, 59, 425–430 CrossRef CAS.
- J. W. Erickson and S. Semancik, Surf. Sci., 1987, 187, L658–L668 CrossRef CAS.
- Y. Hou, X. Du, S. Scheiner, D. P. McMeekin, Z. Wang, N. Li, M. S. Killian, H. Chen, M. Richter, I. Levchuk, N. Schrenker, E. Spiecker, T. Stubhan, N. A. Luechinger, A. Hirsch, P. Schmuki, H.-P. Steinrück, R. H. Fink, M. Halik, H. J. Snaith and C. J. Brabec, Science, 2017, 358, 1192–1197 CrossRef CAS PubMed.
- W. Chen, Y. Wu, Y. Yue, J. Liu, W. Zhang, X. Yang, H. Chen, E. Bi, I. Ashraful, M. Grätzel and L. Han, Science, 2015, 350, 944–948 CrossRef CAS PubMed.
- Y. H. Lee, J. Luo, M.-K. Son, P. Gao, K. T. Cho, J. Seo, S. M. Zakeeruddin, M. Grätzel and M. K. Nazeeruddin, Adv. Mater., 2016, 28, 3966–3972 CrossRef CAS PubMed.
- E. Palomares, J. N. Clifford, S. A. Haque, T. Lutz and J. R. Durrant, Chem. Commun., 2002, 1464–1465 RSC.
- A. Mei, X. Li, L. Liu, Z. Ku, T. Liu, Y. Rong, M. Xu, M. Hu, J. Chen, Y. Yang, M. Grätzel and H. Han, Science, 2014, 345, 295–298 CrossRef CAS PubMed.
- S. S. Shin, E. J. Yeom, W. S. Yang, S. Hur, M. G. Kim, J. Im, J. Seo, J. H. Noh and S. I. Seok, Science, 2017, 356, 167–171 CrossRef CAS PubMed.
- A. Pérez-Tomás, M. Lira-Cantú and G. Catalan, Adv. Mater., 2016, 28, 9644–9647 CrossRef PubMed.
- L. Pintilie, in Ferroelectrics - Physical Effects, ed. M.Lallart, InTech, Rijeka, 2011, p. ch. 05 Search PubMed.
- I. Stolichnov and A. Tagantsev, Ferroelectrics, 1999, 225, 147–154 CrossRef.
- Z. Wen, C. Li, D. Wu, A. Li and N. Ming, Nat. Mater., 2013, 12, 617–621 CrossRef CAS PubMed.
- E. Y. Tsymbal and A. Gruverman, Nat. Mater., 2013, 12, 602–604 CrossRef CAS PubMed.
- T. Sluka, A. K. Tagantsev, P. Bednyakov and N. Setter, Nat. Commun., 2013, 4, 1808 CrossRef PubMed.
- J. Guyonnet, I. Gaponenko, S. Gariglio and P. Paruch, Adv. Mater., 2011, 23, 5377–5382 CrossRef CAS PubMed.
- S. K. Wallace, K. Svane, W. P. Huhn, T. Zhu, D. B. Mitzi, V. Blum and A. Walsh, Sustainable Energy Fuels, 2017, 1339–1350 RSC.
- Y. Yuan, T. J. Reece, P. Sharma, S. Poddar, S. Ducharme, A. Gruverman, Y. Yang and J. Huang, Nat. Mater., 2011, 10, 296 CrossRef CAS PubMed.
- B. Yang, Y. Yuan, P. Sharma, S. Poddar, R. Korlacki, S. Ducharme, A. Gruverman, R. Saraf and J. Huang, Adv. Mater., 2012, 24, 1455–1460 CrossRef CAS PubMed.
- S. Y. Yang, J. Seidel, S. J. Byrnes, P. Shafer, C. H. Yang, M. D. Rossell, P. Yu, Y. H. Chu, J. F. Scott, J. W. Ager Iii, L. W. Martin and R. Ramesh, Nat. Nanotechnol., 2010, 5, 143 CrossRef CAS PubMed.
- J. Huang, J. Yu, Z. Guan and Y. Jiang, Appl. Phys. Lett., 2010, 97, 143301 CrossRef.
- K. S. Nalwa, J. A. Carr, R. C. Mahadevapuram, H. K. Kodali, S. Bose, Y. Chen, J. W. Petrich, B. Ganapathysubramanian and S. Chaudhary, Energy Environ. Sci., 2012, 5, 7042–7049 RSC.
- K.-S. Shin, T. Y. Kim, G. C. Yoon, M. K. Gupta, S. K. Kim, W. Seung, H. Kim, S. Kim, S. Kim and S.-W. Kim, Adv. Mater., 2014, 26, 5619–5625 CrossRef CAS PubMed.
- S. Zhang, Y. Lu, B. Lin, Y. Zhu, K. Zhang, N.-Y. Yuan, J.-N. Ding and B. Fang, Sol. Energy Mater. Sol. Cells, 2017, 170, 178–186 CrossRef CAS.
- A.-N. Cho and N.-G. Park, ChemSusChem, 2017, 10, 3687–3704 CrossRef CAS PubMed.
- C. Sun, Y. Guo, B. Fang, J. Yang, B. Qin, H. Duan, Y. Chen, H. Li and H. Liu, J. Phys. Chem. C, 2016, 120, 12980–12988 CrossRef CAS.
- Q. Wang, Q. Dong, T. Li, A. Gruverman and J. Huang, Adv. Mater., 2016, 28, 6734–6739 CrossRef CAS PubMed.
- K. Yang, Z.-Y. Deng and H.-J. Feng, Appl. Phys. Lett., 2017, 111, 143902 CrossRef.
- D. Cao, C. Wang, F. Zheng, L. Fang, W. Dong and M. Shen, J. Mater. Chem., 2012, 22, 12592–12598 RSC.
- M. Saliba, T. Matsui, K. Domanski, J. Y. Seo, A. Ummadisingu, S. M. Zakeeruddin, J. P. Correa-Baena, W. R. Tress, A. Abate, A. Hagfeldt and M. Grätzel, Science, 2016, 354, 206–209 CrossRef CAS PubMed.
- V. Garcia and M. Bibes, Nat. Commun., 2014, 5, 4289 CrossRef CAS PubMed.
- E. Y. Tsymbal and H. Kohlstedt, Science, 2006, 313, 181–183 CrossRef CAS PubMed.
- S. E. Bernacki, MRS Proceedings, 2011, 243 Search PubMed.
- D.-F. Pan, G.-F. Bi, G.-Y. Chen, H. Zhang, J.-M. Liu, G.-H. Wang and J.-G. Wan, Sci. Rep., 2016, 6, 22948 CrossRef CAS PubMed.
- M. V. Kamenshchikov, A. V. Solnyshkin, A. A. Bogomolov and I. P. Pronin, Phys. Solid State, 2011, 53, 2080 CrossRef CAS.
- L. Lian and N. R. Sottos, J. Appl. Phys., 2000, 87, 3941–3949 CrossRef CAS.
- S. Y. Yang, J. Seidel, S. J. Byrnes, P. Shafer, C. H. Yang, M. D. Rossell, P. Yu, Y. H. Chu, J. F. Scott, J. W. Ager, L. W. Martin and R. Ramesh, Nat. Nanotechnol., 2010, 5, 143–147 CrossRef CAS PubMed.
- A. Pérez-Tomás, A. Lima, Q. Bilion, I. Shirley, G. Catalan and M. Lira-Cantú, Adv. Funct. Mater., 2018, 28, 1707099 CrossRef.
- N. Serpone, J. Phys. Chem. B, 2006, 110, 24287–24293 CrossRef CAS PubMed.
- M. M. Khan, S. A. Ansari, D. Pradhan, M. O. Ansari, D. H. Han, J. Lee and M. H. Cho, J. Mater. Chem. A, 2014, 2, 637–644 RSC.
- S. Samanta, M. Muralidhar, V. Sankaranarayanan, K. Sethupathi, M. S. Ramachandra Rao and M. Murakami, J. Mater. Sci., 2017, 52, 13012–13022 CrossRef CAS.
- M. Saliba, T. Matsui, J.-Y. Seo, K. Domanski, J.-P. Correa-Baena, M. K. Nazeeruddin, S. M. Zakeeruddin, W. Tress, A. Abate, A. Hagfeldtd and M. Gratzel, Energy Environ. Sci., 2016, 9, 1989–1997 RSC.
- W. L. Warren, D. Dimos, B. A. Tuttle, G. E. Pike, R. W. Schwartz, P. J. Clews and D. C. McIntyre, J. Appl. Phys., 1995, 77, 6695–6702 CrossRef CAS.
- Y. A. Genenko, J. Glaum, M. J. Hoffmann and K. Albe, J. Mater. Sci. Eng. B, 2015, 192, 52–82 CrossRef CAS.
- Z. Fan, K. Sun and J. Wang, J. Mater. Chem. A, 2015, 3, 18809–18828 RSC.
- B. Chen, M. Yang, S. Priya and K. Zhu, J. Phys. Chem. Lett., 2016, 7, 905–917 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8se00451j |
| This journal is © The Royal Society of Chemistry 2019 |