
A garnet structure-based all-solid-state Li battery without interface modification: resolving incompatibility issues on positive electrodes†
Chih-Long
Tsai
 *abc,
Qianli
Ma
abc,
Christian
Dellen
ab,
Sandra
Lobe
ab,
Frank
Vondahlen
ab,
Anna
Windmüller
ab,
Daniel
Grüner
ab,
Hao
Zheng
a,
Sven
Uhlenbruck
abc,
Martin
Finsterbusch
abc,
Frank
Tietz
abc,
Dina
Fattakhova-Rohlfing
abc,
Hans Peter
Buchkremer
a and
Olivier
Guillon
abc
*abc,
Qianli
Ma
abc,
Christian
Dellen
ab,
Sandra
Lobe
ab,
Frank
Vondahlen
ab,
Anna
Windmüller
ab,
Daniel
Grüner
ab,
Hao
Zheng
a,
Sven
Uhlenbruck
abc,
Martin
Finsterbusch
abc,
Frank
Tietz
abc,
Dina
Fattakhova-Rohlfing
abc,
Hans Peter
Buchkremer
a and
Olivier
Guillon
abc
aForschungszentrum Jülich GmbH, Institute of Energy and Climate Research (IEK), 52425 Jülich, Germany. E-mail: c.tsai@fz-juelich.de
bJülich Aachen Research Alliance: JARA-Energy, 52425 Jülich, Germany
cHelmholtz Institute Münster: Ionics in Energy Storage (IEK-12), 52425 Jülich, Germany
First published on 8th November 2018
Abstract
The development of high-performance Li7La3Zr2O12 (LLZO)-based all-solid-state lithium batteries (SSLB) is usually hampered by highly resistive interfaces due to the need for sintering at elevated temperatures to form ionic diffusion paths through the grains. Many strategies have been proposed to solve the problem but the achievements have been limited. Herein, a new design principle is introduced, based on co-sintering crystalline LCO and Ta-substituted LLZO instead of using the more reactive Li–Co–O precursors and Al-substituted LLZO, which allows the fabrication of high specific areal density and low cell area resistance without the interface modification of LLZO-based SSLB. Detailed studies using micro-Raman and EDS mapping revealed that the well-sintered interfaces are free from detrimental secondary phases. To demonstrate that a true bulk-type SSLB can be constructed by this straightforward strategy, the material loading for a composite positive electrode was increased to about 10 times that in previous reports, which resulted in a high areal capacity of 1.63 mA h cm−2 (i.e. 110 mA h g−1) when discharged with a current density of 50 μA cm−2. It also allows one to discharge the fabricated SSLB at a very high current density of 500 μA cm−2 at 50 °C due to the minimized cell areal resistance. The new fabrication strategy for the LLZO-based SSLB paves the way for achieving SSLB with high safety and energy density.
Introduction
Li-ion batteries (LIBs) have been one of the most popular energy storage devices, since their first commercialization in 1991 by Sony, because of their extraordinary performance, high energy density, and long cycle life. Their applications nowadays are not only for small portable electronic devices but also for large-scale energy storage such as for electric vehicles and stationary energy storage. However, safety from using LIBs has been one of the greatest concerns due to the use of flammable organic liquid electrolytes. Researchers have, therefore, been pursuing the replacement of organic liquid electrolytes with nonflammable inorganic ones in LIBs along with the use of metallic Li as the anode to increase their energy densities.One of the keys to developing an all-solid-state Li battery (SSLB) is having a suitable solid state ionic conductor as the electrolyte. From a material selection point of view, a good solid electrolyte material should include the following properties:
(i) High ionic conductivity, ∼10−3 S cm−1, at room temperature.
(ii) Low electronic conductivity, <10−8 S cm−1, to prevent self-discharge.
(iii) Wide electrochemical window.
(iv) Good chemical stability over operating temperatures and towards electrodes.
(v) Low toxicity and cost.
In the event of using rigid materials such as the oxide class of ionic conductors for SSLB development, more requirements need to be fulfilled, as follows, for the solid electrolyte due to the necessity of elevated sintering temperatures to form low resistive ionic diffusion paths during the fabrication of SSLBs.
(vi) Matching of thermal expansion coefficients (CTE) with used electrode active materials.
(vii) Chemically stable with electrode active materials up to/nearby their sintering temperatures.
(viii) Sintering temperature matches to that of electrode active materials.
Among the inorganic solid-state Li-ion conductors, ionic conductors based on garnet structured Li7La3Zr2O12 (LLZO) are unique because of their fast Li-ion conduction (∼1 mS cm−1 at RT), low electronic conduction, wide electrochemical window and chemical and electrochemical stability towards metallic Li.1–6 Therefore, the electrochemical community has shown keen interest in using this material as a solid electrolyte and metallic Li as the anode in SSLB development for boosting their intrinsic safety as well as their gravimetric and volumetric energy densities.
For the selection of positive active electrode material, one should first consider the specific energy density, especially when SSLBs need to compete with the current LIBs, apart from the requirements that are mentioned above for sintering processes. The material itself also prefers to have good Li-ion and electronic conductivities so that micro-sized particles can allow the electrochemical performance of an SSLB, instead of nano-sized particles, which is difficult to maintain at elevated sintering temperatures. When Li-ion conduction of the selected electrode is low, a composite electrode that is composed of active electrode and electrolyte materials can be used for providing Li-ion conductive paths into the composite electrode. In the case of a low electronic conductive electrode, an extra phase needs to be considered to offer the electronic conductive paths. However, it has to be borne in mind that the more inactive material in the electrode, the lower the energy density of a battery is. Additionally, more troublesome issues can be expected from material compatibility and processing.
The compatibilities between LLZO and different positive active electrode materials have been reported by different groups.7–10 Positive electrode materials including LiMn2O4, LiFePO4, LiCoMnO4, LiFe0.5Mn1.5O4, LiNi0.5Mn1.5O4, LiNi1/3Co1/3Mn1/3O2 and LiCoO2 (LCO) have been examined for their compatibilities with LLZO at elevated temperatures. Among them, most of the materials reacted with LLZO at temperatures lower than 600 °C, while LiNi1/3Co1/3Mn1/3O2 is stable up to 800 °C and LCO develops a second phase that is only observable by Raman spectroscopy after 10 hours of dwell time at 900 °C.8 Uhlenbruck et al. subsequently reported that the rapid reaction between Ta-substituted LLZO (LLZ:Ta) and LCO only takes place at a temperature of 1085 °C by using differential scanning calorimetry measurements.11 Also, LCO has similar CTE to LLZO (1.3 × 10−5 K−1 for LCO and 1.5 × 10−5 K−1 for LLZO),12,13 which allows a fast sintering process for an LCO/LLZO composite electrode without crack formations and delamination from the LLZO solid electrolyte. It is also beneficial to use LCO as a positive active electrode material because it offers the highest total conductivity, with predominantly electronic and partially ionic conductivity of ∼4 S cm−1 for Li0.47CoO2 and ∼0.1 S cm−1 for Li0.97CoO2 at RT, as compared to the other positive active electrode materials, once Li deficiency is created by the charging process.14 A second phase for providing electronic conductive paths may able to be avoided by using LCO as the positive active electrode material. It is worth mentioning that the use of carbon material in a composite electrode, such as carbon black in conventional LIBs to increase their electronic conductivity within the electrodes, is not applicable for SSLB fabrication when using oxide class positive active electrode materials and LLZO. This is because a low oxygen partial pressure environment created by the oxidation of carbon to form carbon dioxide at elevated temperature could reduce positive active electrode materials such as LCO into cobalt oxide, which is not more electrochemically active at 4 V vs. Li/Li+.
Two different approaches for using garnet-structured LLZO solid electrolytes in SSLB fabrications have been reported. One of them involves using thin film technologies such as pulsed laser deposition,15 sputter deposition16 and sol–gel deposition17,18 to deposit active electrode materials onto dense LLZO pellets to form SSLBs. The results demonstrated electrochemical reactions from the deposited active materials but the peak current densities of the CV scans were very low, due to their low loadings and the utilization of active electrode materials, which make these approaches not practical for high energy density SSLB fabrications, Table 1.
| Reference | Cathode composition | Operational temp. (°C) | Active material loading (mg cm−2) | Operational current density (μA cm−2) | Specific areal capacity (mA h cm−2) | No. of cycles |
|---|---|---|---|---|---|---|
| a Assuming 100% dense LCO thin film. b Only provided CV scan. c Calculated from provided C-rate. d Calculated from (specific areal capacity)/(specific capacity). | ||||||
| Kato et al., 2014 (ref. 15) | LiCoO2 PLD thin film | RT | 0.08a | 1 | 0.0064 | 25 |
| Kotobuki et al., 2011 (ref. 16) | LiCoO2 sputtered thin film | RT | 1.52a | N/A | N/A | 20b |
| Kotobuki et al., 2013 (ref. 17) | LiCoO2 sol–gel thin film | RT | 1 | N/A | N/A | 30b |
| Ren et al., 2017 (ref. 18) | LiCoO2 infiltrated sol–gel | 80 | 2.9 | 6.4 | 0.051 | 10 |
| Han et al., 2018 (ref. 24) | LiCoO2 + Li2CO3 + Li2.3C0.7B0.3O3 | 100 | 1 | 5.75 | 0.106 | 40 |
| 25 | 0.094 | 100 | ||||
| Kato et al., 2016 (ref. 27) | LiNi1/3Co1/3Mn1/3O2 + Li–Al–Ti–P–O glass ceramic | 100 | 3.7d | 50 | 0.50c | 90 |
| Ohta et al., 2013 (ref. 19) | LiCoO2 + Li3BO3 | 25 | 2.35d | 10 | 0.20c | 5 |
| Liu et al., 2016 (ref. 26) | LiCoO2 + Li3BO3 + In2SnO5 | RT | 3.0 | 5 | 0.056 | 6 |
| 80 | 3.0 | 0.279 | 1 | |||
| RT | 0.89 | 0.122 | 1 | |||
| RT | 5.89 | 0.058 | 1 | |||
| Liu et al., 2017 (ref. 21) | LiCoO2 + Li3BO3 + In2(1−x)Sn2xO3 | RT | 1.9d | 5 | 0.190c | 5 |
| Inada et al., 2016 (ref. 28) | TiNb2O7 | 60 | 0.4 | 2 | 0.068 | 1 |
| Liu et al., 2018 (ref. 23) | Li–Ti–O coated NMC + ITO + Li3BO3 | 80 | ∼1 | 5 | 0.123 | 5 |
| Koo et al., 2012 (ref. 25) | LiCoO2 thin film (LiPON electrolyte) | 25 | 2.02a | 46.5 | 0.106 | 100 |
| This work | LiCoO 2 + LLZO | 50 | 12.6 | 50 | 1.50 | 100 |
The other type of batteries is usually classified as “bulk-type” SSLBs due to the use of the composite positive electrode (CPE) or high yield deposition methods. The most commonly used CPE consists of LCO and Li3BO3 (LBO) or its derivatives, where LBO serves as a sintering additive that melts at 700 °C to cement LCO and LLZO together and provides Li-ion transport pathways due to its very low Li-ion conductivity, ∼2 × 10−6 S cm−1, at RT.9,19–24 It is suggested that the low melting point of LBO at 700 °C could effectively avoid the formation of a highly resistive interface during high-temperature sintering of SSLB. These batteries offer very good electrochemical performance up to 100 cycles, but less than 10 cycles were usually shown in the other reports, Table 1. Furthermore, the batteries were only able to demonstrate very low specific areal loadings, usually between 1 to 2 mg cm−2, which is in the same range as that for a thin film SSLB, and very low operational current densities, lower than 10 μA cm−2, Table 1. For example, Han et al. demonstrated their bulk-type batteries that were able to cycle at 100 °C and 25 °C for 100 cycles by using LCO, LLZO and Li2.3C0.7B0.3O3 as CPE.24 The batteries had a specific areal loading of ∼1 mg cm−2, equivalent to ∼0.1 mA h cm−2, and were cycled with a current density of 5.75 μA cm−2. The performance seems very promising but the specific areal capacity is similar to that of the recent thin film SSLB, which has a much higher operational current density of 46.5 μA cm−2, Table 1.25 Furthermore, the electrochemical performance of this type of SSLB is dramatically reduced by the low ionic conductivity of LBO and high tortuosity of Li-ion diffusion paths within the CPE when the specific areal capacity increases to a higher number, as shown by Liu et al.26
The other approach for preparing bulk-type SSLBs is using aerosol deposition.27,28 Kato et al. demonstrated their bulk-type SSLB by using aerosol deposition for their CPE fabrication, which contained LiNi1/3Co1/3Mn1/3O2 + Li–Al–Ti–P–O glass-ceramic on an LLZO pellet.27 The cell was able to deliver a high specific areal capacity of 0.5 mA h cm−2 with an operational current density of 50 μA cm−2 at 100 °C. This technology has the major advantage of no need for high-temperature sintering for SSLB fabrications, which avoids the interface reaction between the used active electrode material and solid electrolyte. Therefore, a low interface resistance of the constructed SSLB could be expected. Nevertheless, this method would require a robust substrate or solid electrolyte that can stand the high-pressure impact from ceramic particles and gas carrier during the deposition. How to reduce the dead weight of the necessary substrate or thick solid electrolyte to increase the overall battery energy density could be another challenge.
Herein, we demonstrate the fabrication of a bulk-type SSLB with very low cell area resistance that can be achieved by proper material selection and processing rather than complicated interface modifications. To make the fabricated bulk-type SSLBs different from those of similar thin film SSLBs, we increased the specific areal capacity to least 10 times that for thin film SSLBs so that the results can provide new insights into what could be expected and the challenges of making an LLZO based bulk-type SSLB. For the material selection, cubic phase LLZ:Ta, Li6.6La3Zr1.6Ta0.4O12, was selected as the solid electrolyte instead of Al-substituted LLZO. This is because the Al-substituent in LLZO has a preference for diffusing to LCO at high temperature, ∼700 °C, which degrades the cubic phase LLZO into a tetragonal phase to form a highly resistive interface due to the very low Li-ion conductivity of the tetragonal phase LLZO.9 For the positive active electrode material, a crystallized layered structure LCO was chosen because it has the highest chemical stability against LLZ:Ta,8,11 provides the highest electronic conductivity and partial Li-ion conductivity and has the closest CTE as LLZO as compared to the other positive active electrode materials.12–14 It is important to mention that the use of Li–Co–O precursors, such as those deposited by pulsed laser depositions, physical vapor depositions and sol–gel methods, is not favored due to their higher Gibbs free energy than crystalline LCO, which makes it react with LLZO at a much lower temperature (∼700 °C) than that for crystalline LCO at 1085 °C.9–11 A mass ratio of LLZ:Ta/LCO = 1 was chosen for the CPE fabrication, equivalent to a volume ratio of LLZ:Ta/LCO = 48.6/51.4. Even though only one-third of the volume ratio of LLZ:Ta in a CPE would be required for establishing Li-ion percolation paths, the use of higher volume ratio in this work shows that even with a larger interface area between LCO and LLZ:Ta, a high performance SSLB with low cell area resistance can still be obtained. It is expected that a lower volume ratio of LLZ:Ta/LCO = 1/2 or less, due to the partial Li-ion conductivity of LCO itself, could be used to increase the SSLB energy density. Even though many reports have provided different methods for reducing the interface resistance between LLZO and Li metal to relieve the Li dendrite problem,4,29–31 we often encounter the short circuit problem during SSLB tests, especially when the specific areal capacity is higher than ∼0.5 mA h cm−2. Therefore, on the negative electrode, indium metal was used to avoid Li dendrite formation during the electrochemical tests.
Based on the strategy, we have demonstrated that a bulk type SSLB with low cell area resistance can be constructed straightforwardly without complicated interface modification. The experimental results showed that crystalline LCO and LLZ:Ta maintained their phase stability with no noticeable chemical reaction in the CPE after the sintering at 1050 °C. The much higher specific areal capacity and operational current densities of the fabricate SSLB support our new design principle for obtaining high-performance LLZO-based SSLB. A discussion of how to further improve the electrochemical performance of LLZO-based SSLB is provided for the future development of high safety and energy density SSLBs.
Experimental methods
Preparation of LLZ:Ta discs
Al-contaminated, Ta-substituted LLZ (LLZ:Ta, i.e., Li6.6La3Zr1.6Ta0.4O12) powder was prepared via a four-step solid-state reaction. The starting materials, LiOH·H2O (Merck, 98%), La2O3 (Merck, 99.9%, pre-dried at 900 °C for 10 h), ZrO2 (Treibacher, 99.5%), and Ta2O5 (Inframat, 99.95%) were mixed in stoichiometric amounts, with 20 mol% LiOH·H2O in excess. An extra 2.5 mol% of α-Al2O3 (Inframat, 99.9%) was added to the powder as a sintering additive (also see ESI† for a more detailed discussion on adding extra α-Al2O3 as sintering additive). The prepared powder was dry milled and pressed into pellets for 20 h sessions of calcinations, once at 850 °C and twice at 1000 °C. Grinding and pressing were repeated between the calcination steps. The total amount of the prepared powder was 1000 g, so that the effect from the varying powder quality due to different batches on experimental results would be minimal.The final sintering step of LLZ:Ta was carried out at 1175 °C with 4 hours dwell time. Each pellet contains 7 g of calcined LLZ:Ta powder, fabricated by using a die with a diameter of 13 mm and uniaxial pressing by applying 150 MPa. After sintering, the pellets were sliced into thin discs with a thickness of ∼0.6 mm by using a low-speed diamond saw (Buehler, Switzerland) for later application. The detailed preparation process for LLZ:Ta discs can be found in other publications.3,4
SSLB fabrication
The SSLB fabrication was done by sintering CPE onto LLZ:Ta discs. The CPE was prepared using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mass ratio of LCO (MTI Corp., USA), 5.06 g cm−3, and LLZ:Ta, 5.35 g cm−3, powder. The powders were weighed and milled by using yttrium stabilized zirconia balls, and pure ethanol as a solvent for 24 hours to reduce the particle size. After milling, the particle size distribution was D(v, 0.5) = 1.03 μm and D(v, 0.9) = 1.58 μm, determined by Fraunhofer diffraction using a Horiba La-950V2 instrument (Horiba, Japan) (see ESI Fig. S1†). The slurry was further collected and dried at 60 °C. Then, the screen printing ink was prepared by using a three-roll mill (Exakt 50, Germany) to mix a slurry with a weight ratio of 6 wt% ethyl cellulose (Sigma-Aldrich) in terpineol (Sigma-Aldrich):8250 thinner (Dupont):solid loading of 3:2:5. Afterward, the ink was painted onto the LLZ:Ta discs by a brush and dried at 55 °C in air. The cells were sintered in a tube furnace (Nabertherm, Germany) with a heating rate of 2 K min−1 to 600 °C, followed by 10 K min−1 to 1050 °C and 30 minutes dwell time using an Al2O3 ceramic boat as a carrier in air. Then, free cooling was applied to the furnace for its temperature to drop to RT. The LCO loading on a cell was calculated by the difference in weight between the used LLZ:Ta disc and that for the cell after sintering. Typical cells used in this paper have CPE loadings between 24 and 32 mg cm−2 to give LCO loading between 12 and 16 mg cm−2.
:1 mass ratio of LCO (MTI Corp., USA), 5.06 g cm−3, and LLZ:Ta, 5.35 g cm−3, powder. The powders were weighed and milled by using yttrium stabilized zirconia balls, and pure ethanol as a solvent for 24 hours to reduce the particle size. After milling, the particle size distribution was D(v, 0.5) = 1.03 μm and D(v, 0.9) = 1.58 μm, determined by Fraunhofer diffraction using a Horiba La-950V2 instrument (Horiba, Japan) (see ESI Fig. S1†). The slurry was further collected and dried at 60 °C. Then, the screen printing ink was prepared by using a three-roll mill (Exakt 50, Germany) to mix a slurry with a weight ratio of 6 wt% ethyl cellulose (Sigma-Aldrich) in terpineol (Sigma-Aldrich):8250 thinner (Dupont):solid loading of 3:2:5. Afterward, the ink was painted onto the LLZ:Ta discs by a brush and dried at 55 °C in air. The cells were sintered in a tube furnace (Nabertherm, Germany) with a heating rate of 2 K min−1 to 600 °C, followed by 10 K min−1 to 1050 °C and 30 minutes dwell time using an Al2O3 ceramic boat as a carrier in air. Then, free cooling was applied to the furnace for its temperature to drop to RT. The LCO loading on a cell was calculated by the difference in weight between the used LLZ:Ta disc and that for the cell after sintering. Typical cells used in this paper have CPE loadings between 24 and 32 mg cm−2 to give LCO loading between 12 and 16 mg cm−2.
After the sintering process, samples were polished on the LLZ:Ta side to remove possible impurities and thin the solid electrolyte down to ∼300 μm by using SiC sandpaper. Plasma etching (Gambetti, Italy) was applied for cleaning the LLZ:Ta surface before Au thin film was sputtered on top of the CPE to serve as a current collector and on LLZ:Ta to help indium adhesion using a desktop sputter coater (Cressington 108 auto coater, UK). Then, the batteries were assembled using indium foil as the anode, and heated up to 200 °C to increase the bonding between the LLZ:Ta and indium anode on a hot plate before they were put into Swagelok cells. The plasma etching, thin film Au depositions and cell assembly were all done in high purity Ar-filled glove-boxes.
Sample characterizations
The samples were characterized for their phase purity using X-ray diffraction (XRD). For this purpose, a Bruker D 4 Endeavor device using Cu Kα radiation equipped with a 1D detector LYNXEY and a DIFFRACplus BASIC package released in 2009 were used. For microstructural investigations, samples were embedded in EpoFix epoxy (Struers, Germany) and mirror-polished. Images of the microstructure were taken by using a scanning electron microscope (SEM, Zeiss Merlin and Zeiss Supra 50 VP) combined with energy dispersive X-ray spectroscopy (EDS, Oxford Instruments X-max 80).Micro-Raman spectroscopy was carried out with a Renishaw inVia Raman microscope using a solid-state 532 nm excitation laser and 1800 lines per mm grating. To avoid laser-induced phase changes or damage to the samples, investigations of safe laser power were carried out on the materials before the measurements. The final laser power was set to 0.25 mW. The high-resolution Raman mapping was carried out by applying the live track function (an autofocus software solution) within the WiRE 5.1 software. The spectra were collected for an area of 55 μm × 42 μm by using a 100× objective. A step size of (x, y) = (0.3 μm, 1.3 μm) and a 5 second spectral acquisition time were chosen for maximal resolution and minimal time per spectrum. The total number of acquired spectra for the mapping was 6072. The high-resolution mapping was processed and analyzed within the WiRE 5.1 software, by using a statistical noise filter and a statistical component analysis algorithm.
Electrochemical performances were recorded at 50 °C by using a BioLogic VMP-300 multipotentiostat combined with a climate chamber (Vötsch Industrietechnik VT 4002EMC, Germany). The electrochemical impedance spectroscopies were measured with frequency varied from 7 MHz to 10 mHz with an electrical field perturbation of 10 mV. For each measurement, a constant-current–constant-voltage (CC–CV) process was used for charging the battery. The battery was charged to 3.65 V vs. Li–In with a constant current density of 50 μA cm−2 and then the battery was held at a constant voltage of 3.65 V vs. Li–In until the current dropped to 5 μA cm−2. The battery was set to OCV for 5 minutes before EIS measurements were started.
For the C-rate tests, CC-CV was used for the charging process. The discharged measurements with current densities of 50, 100, 200, 300 and 500 μA cm−2 were done in sequence with the same cell. For the cyclic voltammetry (CV) measurement, the battery was cycled between 2.4 and 3.6 V vs. Li–In with a scan rate of 20 μV s−1. The long-term galvanostatic cycle was performed by using CC–CV for the charging process and a constant current density of 50 μA cm−2 for the discharging process until the voltage dropped to 2.4 V vs. Li–In.
Results and discussion
The success of sintering the CPE on a LLZ:Ta pellet without noticeable chemical reaction between LCO and LLZ:Ta is the first key step to constructing a LLZO based SSLB. Herein, we use an electrolyte supported SSLB to demonstrate our new SSLB design principle that is shown in Fig. 1 along with its cross-section image from scanning electron microscopy (SEM). The constructed SSLB has a CPE about 50 μm thick and for the solid electrolyte, the thickness is about 300 μm. The chemical information of the CPE after sintering on a LLZ:Ta pellet at 1050 °C was first examined by X-ray diffraction (XRD) (see ESI Fig. S3†). No discernable impurity phase was detectable by XRD after the sintering process at 1050 °C, which is in agreement with results obtained by Uhlenbruck et al.11 Although XRD is not sensitive to low amounts of impurity phases, it shows that the LCO and LLZ:Ta remain in their original phases in the CPE after the high-temperature sintering process at 1050 °C.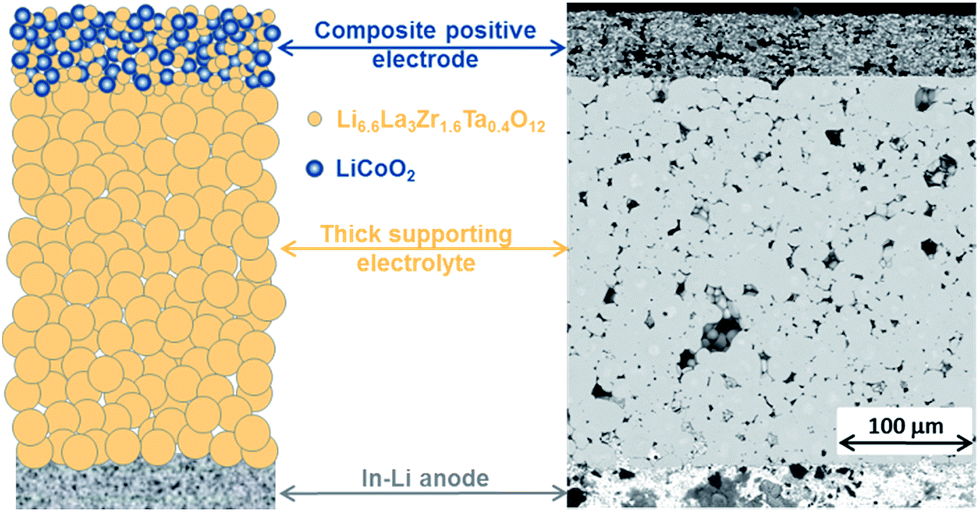 | ||
| Fig. 1 A sketch of the fabricated electrolyte supported SSLB and its SEM cross-sectional image. | ||
To compensate for the low sensitivity of XRD, high-resolution micro-Raman spectroscopy and mapping were applied for impurity phase identification of the sintered CPE as well as the used pristine materials, Fig. 2. The spectra from pristine LCO and LLZ:Ta are in agreement with previous reports that could be assigned to different Raman bands (see ESI Fig. S4† and for a detailed discussion of the spectra).32–39 A built-in method “empty modeling” in the WiRE 5.1 software was used to analyze the 6072 collected spectra during the Raman mapping. The method is a form of multivariate curve resolution-alternating least squares method that allows component information to be rotated into physically meaningful components. The results show that the collected 6072 spectra can be modeled by three spectra, namely spectra for LCO, LLZ:Ta and used epoxy, whereas a fourth model would return a spectrum that is a reverse-type of LCO, i.e. turn peaks in the spectrum into valleys.
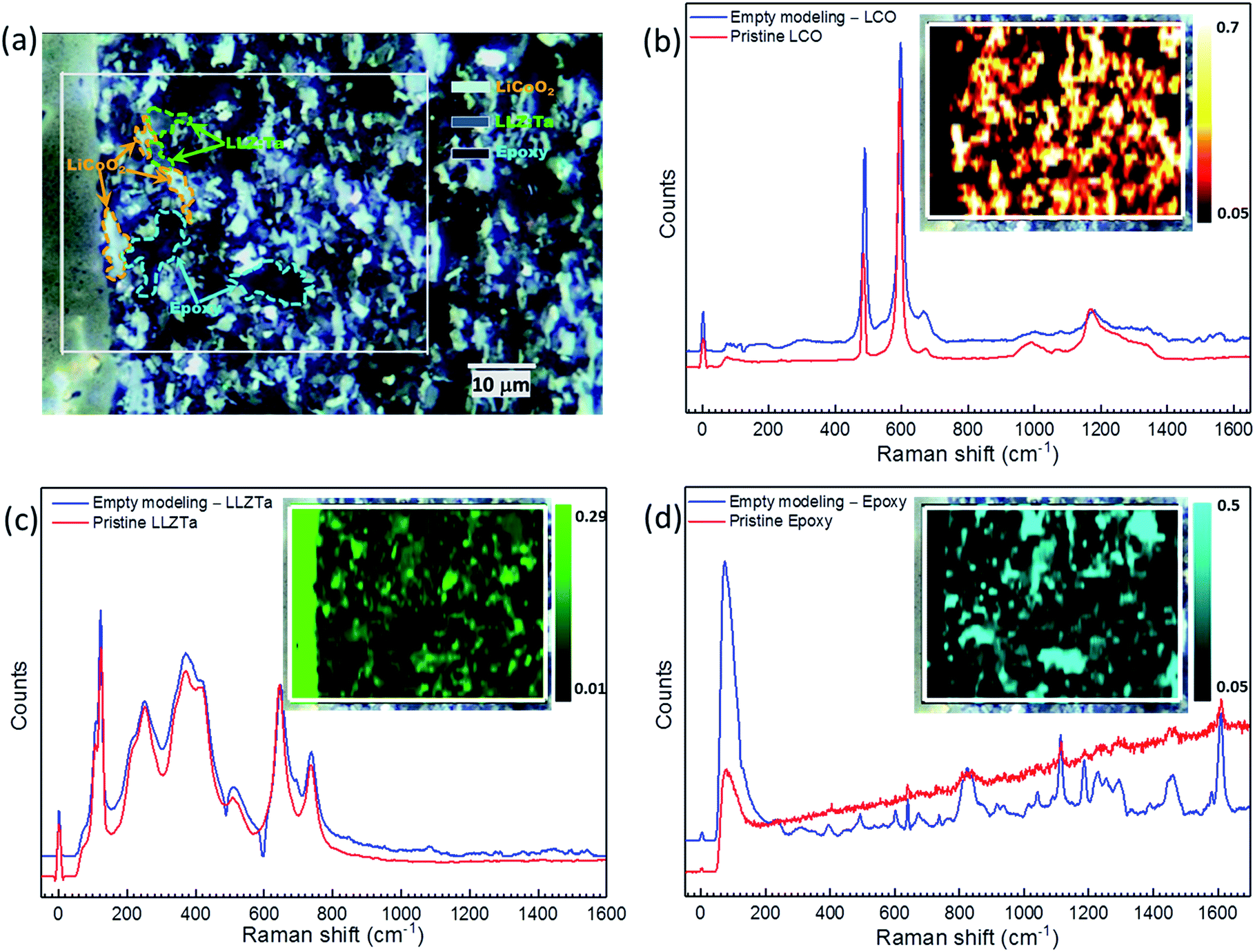 | ||
| Fig. 2 High-resolution micro-Raman mapping of a SSLB cross-section. (a) Optical image of the SSLB cross-section and its mapping area. The Raman mappings and the spectra for (b) LCO, (c) LLZ:Ta and (d) epoxy. | ||
Detailed studies of sintered CPE using micro-Raman spectroscopy and high-resolution mapping revealed that the formation of impurity phases at LCO and LLZ:Ta interfaces can be excluded. The high-resolution micro-Raman mapping clearly shows that both LCO and LLZ:Ta phases are maintained after the sintering process at 1050 °C. They were randomly distributed across the CPE while the epoxy was detected in the area of pores. Ren et al. reported that LCO and LLZ:Ta reacted at 900 °C to form LaCoO3, which gives a Raman band at 685 cm−1, and Kim et al. used nano-beam electron diffraction to show that PLD deposited Li–Co–O on LLZO pellet reacted to form La2CoO4 at 700 °C.8,10 Despite the PLD deposited Li–Co–O being more reactive to LLZO than crystalized LCO, the formation of La2CoO4 requires low oxygen partial pressure due to the low oxidation state of Co2+.40–42 The formation of La2CoO4 is unlikely since our sintering process was carried out at high temperature and under an oxidative environment. For LaCoO3, Ren's referenced literature suggested that the band at 675 cm−1 was assigned to the second-order Raman scattering of LaCoO3 but not the band at 685 cm−1.8,43 Also, Hara et al. observed laser irradiation decomposition from LCO to Co3O4 that gave a Raman band at 694 cm−1 during Raman measurement.32 In consideration of the strong peak intensity and position, it is rather difficult to agree that the band at 685 cm−1 in Ren's report originated from either LaCoO3 or Co3O4 due to the large 10 wavenumber difference. Therefore, LaCoO3 and Co-substituted LLZ:Ta, designed as Li6.3Co0.1La3Zr1.6Ta0.4O12, were synthesized for micro-Raman spectroscopy analysis (see ESI† for detailed Experimental methods and discussion). The measured spectrum for LaCoO3 is in agreement with Ishikawa et al. that the Raman bands could be assigned to 162 cm−1 (Eg, La vibration), 231 cm−1 (A1g, rotation around c), 478 cm−1 (Eg, bending), 547 cm−1 (Eg, quadrupole) and 645 cm−1 (A2g, breathing) at RT for which Raman bands at 162 cm−1 and 654 cm−1 are pronounced with low accumulative time of the spectrum (see ESI Fig. S5†).44 For Co-substituted LLZ:Ta, the Raman spectrum from a single crystal showed an identical pattern to that for LLZ:Ta, except for an extra peak at 693 cm−1 (see ESI Fig. S6†). A further study of the Co-substituted LLZ:Ta found that the relative intensity of the peak at 693 cm−1 varied from the excitation wavelength used in the Raman spectra that indicates that the peak at 693 cm−1 is due to photoluminescence from the substituted Co within LLZ:Ta instead of a Raman band for Co-substituted LLZ:Ta (see ESI Fig. S7† and for detailed discussion).45 Since a Raman band at 162 cm−1 was not observed in the inspected CPE from the empty modeling spectra or the individual and average spectrum from the collected 6072 spectra, Fig. 2 (also see ESI Fig. S4(d) and S8†), it indicates that the formation of LaCoO3 can be excluded in the sintered CPE. Nevertheless, a weak intensity band assigned by curve fitting at 689 cm−1 was observed in LLZ:Ta grains, indicating that a low concentration of Co was diffused into LLZ:Ta grains during the sintering process (see ESI Fig. S8†). It is important to note that the observation of no reaction byproduct of LCO and LLZ:Ta by XRD and high resolution micro-Raman spectroscopy does not mean that there is totally no interfacial reaction between LCO and LLZ:Ta, especially at such a high sintering temperature of 1050 °C. Yet, it is possible to minimize any reaction between LCO and LLZ:Ta to as low as possible by optimizing the processing conditions that lead to a well-functioning SSLB with a low cell area resistance.
The ionic transport number of a LLZ:Ta pellet that was co-sintered with CPE remained the same as for a pristine pellet. After removing the sintered CPE by sandpaper, a DC open circuit potential method with a voltage of 4 V to simulate charged LCO was applied to the used LLZ:Ta to examine its ionic transport number by using sputtered Au thin films as electrodes.46 The recorded initial current density was as high as 235 μA cm−2 and stabilized to ∼0.036 μA cm−2, which represents the electronic leakage through the LLZ:Ta pellet, Fig. 3. When the bias was switched off, a reverse current density of 37.35 μA cm−2 due to the backflow of accumulated Li-ions was measured. Therefore, the ionic transport number for LLZ:Ta was calculated to be ∼1 after sintering with CPE at 1050 °C, which indicated that the self-discharge of the fabricated SSLB is negligible.
 | ||
| Fig. 3 DC open circuit potential measurement of LLZ:Ta which was sintered with a composite positive electrode at 1050 °C. The ionic transport number is calculated to be ∼1. | ||
The cyclic voltammetry (CV) scan of the prepared SSLB using indium as the anode is shown in Fig. 4. The inset in Fig. 4 is the first galvanostatic charge–discharge of the SSLB using a constant current density of 20 μA cm−2 before it was subjected to the CV scan. The charge–discharge curves show typical LCO behavior, where the first charging specific areal capacity was 2.01 mA h cm−2 (i.e. 140 mA h g−1), while that for discharging was 1.62 mA h cm−2 (i.e. 113 mA h g−1). The irreversible capacity in the first cycle is often seen in different SSLBs. Since In was used as the anode in the tested SSLB, it is possible that some of the Li atoms were trapped in the Li–In alloy and were not able to re-intercalate into LCO. Nevertheless, this is often explained by side reactions between solid electrolyte and electrode or the large polarization due to the loss of Li-ion conduction paths to LCO, which is caused by the volume change in LCO during the charging-discharging process, when using the sulfide class of solid electrolytes.47–50 When using an oxide class of solid electrolytes, the irreversible capacity of the first cycle is often explained by the decomposition of organolithium compounds when using LBO as the sintering additive;9,19,24 the LCO/LLZO interface irreversibly decomposed at a voltage > 3 V vs. Li/Li+ when Li–Co–O was sintered directly on LLZO.9 Since the SSLBs in this study were sintered at 1050 °C in air and operated at 50 °C, the decomposition of organolithium and side reactions between LCO and LLZO can be ruled out due to the extremely high sintering temperature in an oxidative environment. Only the loss of Li-ion conduction paths and the irreversible formation of new interfaces could be the reasons for the observed irreversible capacity. Here, the loss of conduction paths is more conceivable as compared to the other reason, which will be shown in the cell's microstructure analysis.
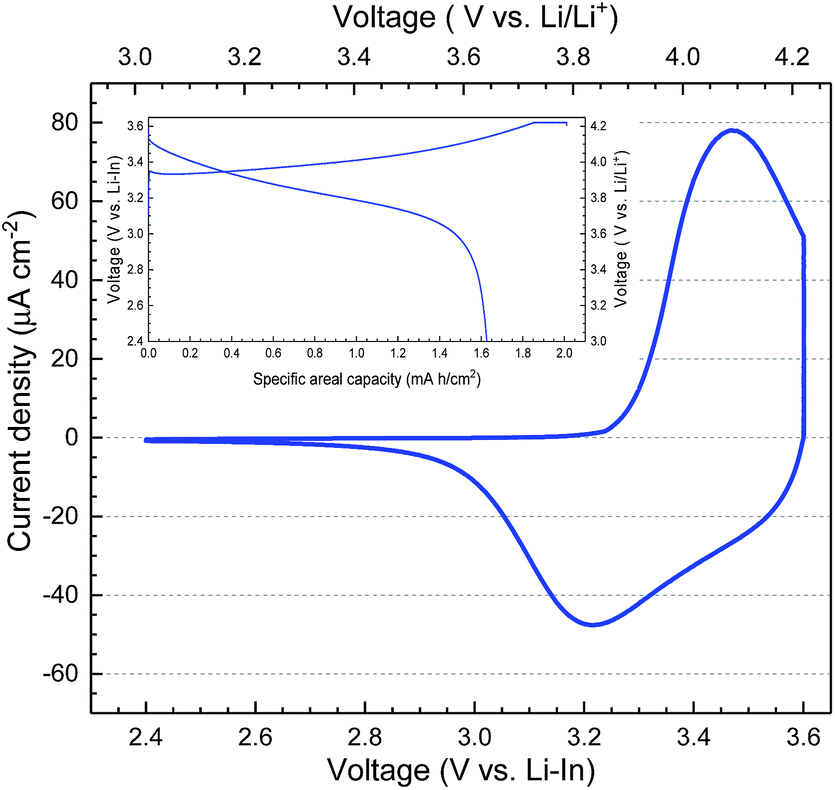 | ||
| Fig. 4 The cyclic voltammetry scan between 2.4 and 3.6 V vs. Li–In of the prepared SSLB. The inset is the first cycle charge–discharge performance using a constant current density of 20 μA cm−2 of a SSLB before it was subjected for the CV scan. | ||
The CV scan in Fig. 4 shows only a redox reaction from LCO, which provides evidence for the successful sintering of SSLB at 1050 °C without impurity phase formation using our strategy. The broad peaks in the CV scan are associated with the first-order phase transition peak of LCO at 3.47 V vs. Li–In (4.09 V vs. Li/Li+) for the positive scan and that at 3.20 V vs. Li–In (3.82 V vs. Li/Li+) for the negative scan. Two peaks are separated by 0.27 V, indicating that the total resistance across the cell is still relatively high when compared to thin film SSLB51 but similar to that of conventional LIB.52 This is the first report that shows the proper LCO redox peaks by using LLZO as the solid electrolyte in the SSLB. It is a result of having the right selection of compatible materials to prevent the formation of highly resistive interfaces to lag Li-ion transport within the fabricated SSLB, which gives a relatively low cell area resistance when compared to the other reported cell concepts.16,26,53,54
The low cell area resistance allows the fabricated cell to charge–discharge in much higher current densities than the other reported cell concepts.9,19–23,28,53,55Fig. 5 shows the SSLB discharge profile with different discharge current densities. It is worth noting that the discharge measurements with current densities of 50, 100, 200, 300 and 500 μA cm−2 were done in sequence with the same cell. Therefore, the capacity degradation due to the cycling of the cell was not taken into account for the capacity calculation. When discharged with a constant current density of 50 μA cm−2, the specific areal capacity of the SSLB was 1.63 mA h cm−2 (i.e. 110 mA h g−1) with a first discharge voltage point (FDVP) at 3.59 V vs. Li–In. With the increase in the discharge current density up to 500 μA cm−2, the FDVP decreased dramatically to 2.90 V vs. Li–In and gave a specific areal capacity of only 0.11 mA h cm−2. The fast decrease of FDVP along with the steep increases of the slopes of the discharge curves are both indicators of suffering from high ionic transport resistance across the tested SSLB when the discharging current density is high.
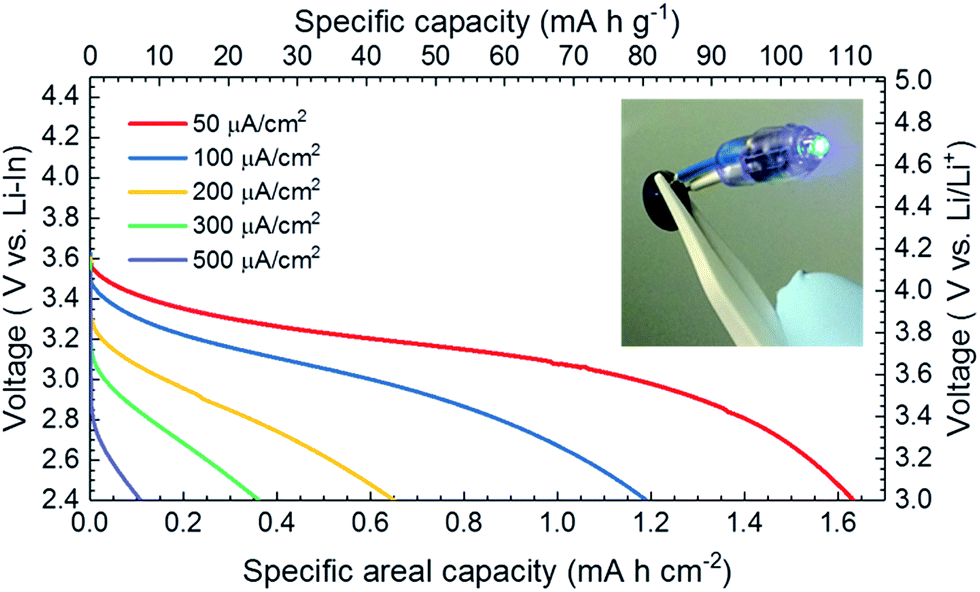 | ||
| Fig. 5 SSLB discharge profile with different discharge current densities. The discharge profile was obtained in sequence from low to high current density of the cell. Therefore, the capacity fading due to the cycling of the cell was not taken into account for the capacity calculation. Inset is the SSLB with a black colored CPE in the front that lights up an LED. | ||
Long-term galvanostatic cycling showed that the fabricated cell undergoes high capacity degradation due to the gradual increase in cell polarization, Fig. 6. The coulombic efficiency of the first cycle was comparable to that of the other tested SSLBs, with only 81.5% and a discharge specific areal capacity of 1.48 mA h cm−2 (i.e.117 mA h g−1) and a FDVP at 3.59 V vs. Li–In. The coulombic efficiency increased rapidly from 81.5% in the first cycle to 96.4% in the second cycle and stabilized at ∼99% after 15 cycles. After 100 cycles, the discharged specific areal capacity was decreased to 0.45 mA h cm−2 (i.e. 36 mA h g−1) with a FDVP at 3.36 V vs. Li–In, Fig. 6(b). The cell capacity degradation was quite high and was faster at the beginning cycles than that at later ones (see ESI Fig. S9†). The cell area resistance can be calculated by dividing the voltage difference between the open circuit voltage, assuming the cell reaches equilibrium voltage during the charging process at 3.65 V vs. Li–In, and the FDVP by discharging current density. The calculated area resistance of the tested SSLB was increased from 1138 Ω cm2 at the first cycle to 5804 Ω cm2 at the 100th cycle, Fig. 6(b), which is in agreement with the electrochemical impedance spectroscopy (EIS) results from before and after the cycling of the SSLB (see ESI, Fig. S10†). Although the EIS data are preliminary, it is generally accepted that the intercept of the EIS data and x-axis at high frequency represents the area resistance from the solid electrolyte, ∼35 Ω cm2, which remained the same after cycling. Therefore, the degradation of the SSLB capacity can be explained by the increase in polarization from the electrodes that made its discharging voltage curve steeper to reach down to the cutoff voltage before the cell was completely discharged.
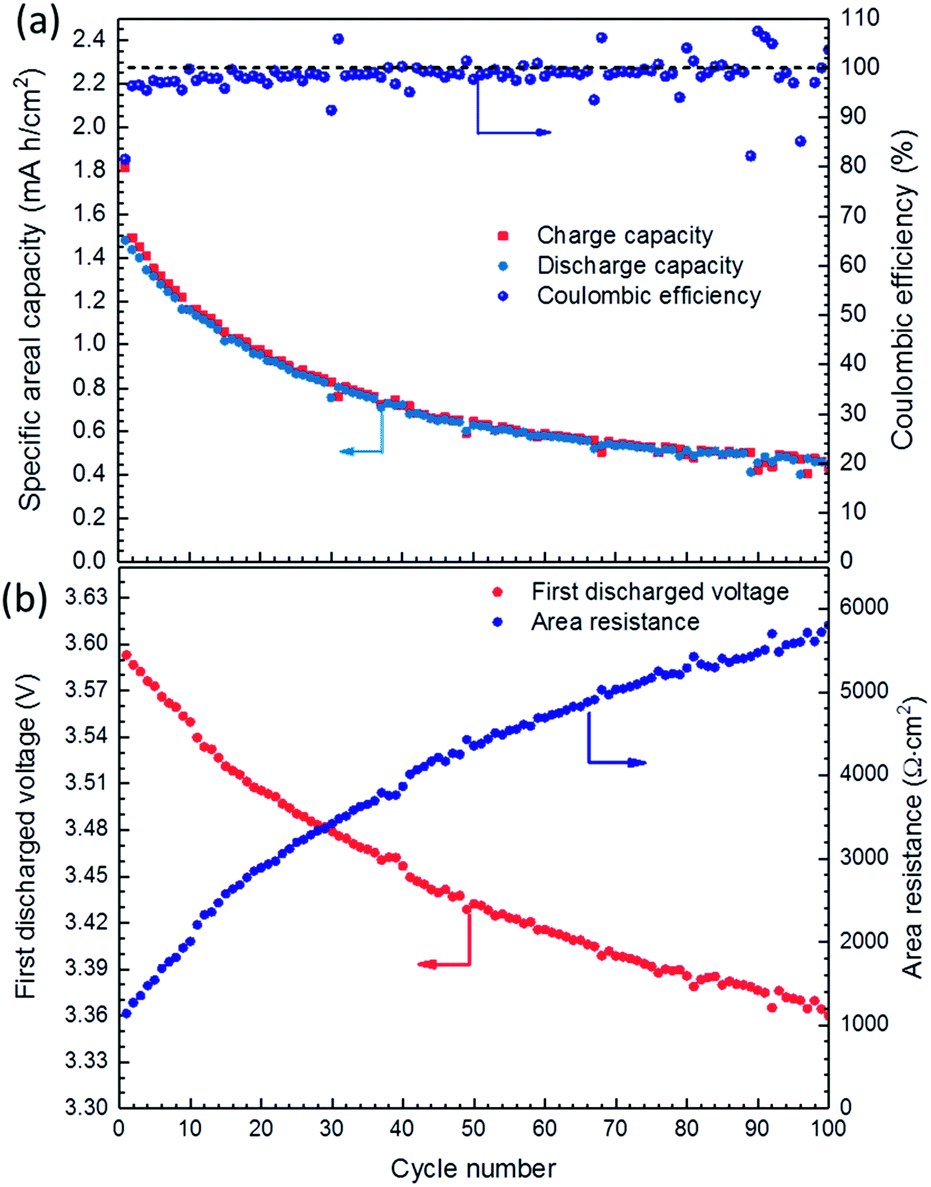 | ||
| Fig. 6 (a) Long-term charge–discharge cycling of SSLB; (b) the first discharge voltage points for the cycles and calculated area resistance of the SSLB. | ||
The microstructure of a sintered CPE, along with its energy-dispersive X-ray spectroscopy (EDS) mapping before any electrochemical test, is shown in Fig. 7. It can be seen that the contact necks between particles were well established by the sintering process that offered ionic and electronic conductive paths through the CPE. However, the relative density of the sintered CPE was not really high under the applied circumstances. Sub-micrometer pores are clearly visible between the grains while some macropores up to a couple tens of micrometers also exist in the sintered CPE. From the EDS mapping, Zr, La, Ta and Co elements are well confined within their own phases after the sintering process. Although the color mapping shows that oxygen (yellow) is enriched at the interfaces between LCO and LLZ:Ta, it could be a result of the overlapping of Zr (red) and Co (green) or different oxygen ion densities between LCO and LLZ:Ta. Therefore, a monochrome mapping was used to show the defined edges of the interfaces, which means that elemental inter-diffusion was able to be minimized under our SSLB fabrication strategy, Fig. 7(b)–(e). In particular, the monochrome EDS mappings showed that La and Co have rather clear boundaries, Fig. 7(c) and (e). If LCO and LLZ:Ta reacted to form LaCoO3, as suggested by Ren et al.,8 an internal diffusion of La and Co should be observed. The clean edges from EDS mappings for La and Co between the LCO and LLZ:Ta particles exclude any severe reactions between the two materials.
 | ||
| Fig. 7 (a) SEM and EDS mapping of the sintered composite positive electrode. Individual monochrome EDS mapping of (b) Zr, (c) La, (d) Ta and (e) Co. | ||
The microstructure of SSLB after 100 cycles suggests that the micro-cracks in the CPE from the repetitive electrode expansion and contraction is the origin of the capacity degradation in the garnet structure-based SSLB. Fig. 8 shows the microstructure from SEM images of SSLB after 100 cycles with some selected areas that are amplified for better scrutiny. The microstructure is similar to the one used for EDS mapping that contained sub-micrometer pores and macropores within the CPE. On the bottom part of the SEM image, it can be clearly seen that the sintering necks between the LLZ:Ta solid electrolyte and CPE were well established by the sintering process. When compared to the as-sintered cell, three differences were observed in microstructure of the cycled CPE.
 | ||
| Fig. 8 SEM cross-section images of a SSLB that underwent 100 galvanostatic charge–discharge cycles at 50 °C. | ||
First, the trans-granular fracture was found in most of the LCO grains. LCO grains next to macropores and the outer part of the CPE (to the current collector side) had more severe cracks than those on the inner side and compact areas. Secondly, most of the LLZ:Ta grains with trans-granular fracture were next to macropores and the outer part of CPE while those in compact areas and the inner side of the CPE were mostly crack free. Thirdly, the contacts between LCO and LLZ:Ta were damaged for those grains next to the macropores and the outer part of the CPE, while those in compact areas and the inner part of the CPE remained in good contact. The observation of both inter-granular and trans-granular fractures of the CPE grains could explain the high capacity degradation during the cycling. The trans-granular fracture of LCO grains can be understood from their volume change during galvanostatic charge–discharge, even though the change was less than 4%.56 The inter-granular fractures could be a result of non-uniform electrochemical reaction next to the free space, which causes an uneven volume change in LCO grains during cycling. Such an uneven volume change could build up stresses that are high enough to break up the CPE grains to release the stress into free space. The other point to note is that the contacts between LCO and LLZ:Ta in the compact areas and inner part of the CPE maintained good bonding. Since the galvanostatic cycling of this cell spanned about 4000 hours with voltage maintained higher than 3 V vs. Li/Li+, one would expect inter-granular fracture to be everywhere if irreversible LCO/LLZO interface decomposition occurred at a voltage > 3 V vs. Li/Li+.9 Therefore, the irreversible decomposition of the interface could also be ruled out, which makes the fracture of the ceramic grains from the repetitive electrode expansion and contraction the key factor behind the capacity degradation in garnet structure-based SSLB.
To date, many approaches have been tried in order to obtain SSLBs based on oxide-class solid electrolytes because of their higher safety and easier handling as compared to the sulfide class. However, the progress is rather slow, mainly due to the high cell resistance that results from the high-temperature sintering process that is necessary when the wrong combination of materials is used. Presently, the chosen crystalline LCO and LLZ:Ta for CPE fabrication and LLZ:Ta as the solid electrolyte allows the sintering temperature to go up to 1050 °C to form low resistive ionic diffusion paths while their similar CTEs allow a fast sintering process with a minimized time at high temperature to avoid elemental inter-diffusion. High-resolution micro-Raman and EDS mapping reveal the LCO and LLZ:Ta phases are well maintained after the sintering process. Furthermore, the presented SSLB in Fig. 6 has an area resistance of ∼1150 Ω cm2 at 50 °C, with a CPE loading of 25.2 mg cm−2 (i.e. 12.6 mg cm−2 of LCO) that has about 10 times higher active electrode material loading but more than 10 times lower area resistance as compared to the cells using LBO or PVDF as the bonding agent.21,23,26,53 It, therefore, allows the cell to charge–discharge at much higher current densities than the other designs of cell concepts. The outstanding electrochemical performances with much higher areal specific capacity and operational current densities of the constructed SSLBs than the other cell concepts support our SSLB fabrication strategy to be a successful one, where the fabrication process can be straightforward without interface modification.
There are still many more things to be improved for better SSLB performances. For example, the relative density of the CPE should be as high as possible for better energy density, while the cell resistance will also be lower due to the lower tortuosity of ionic and electronic diffusion paths. Even though a model from Bucci et al. suggests that a fracture of the CPE could be prevented if an active electrode material's cycling expansion is lower than 7.5% when using LLZO as a solid electrolyte, trans- and inter-granular fractures were found in the SSLB after 100 cycles.57 This could be a result of the non-optimized microstructure of the CPE where the macropores should be avoided to reduce microstructure cracking and improve the SSLB cycling performance. It is also preferable to increase the electronic conductivity within the CPE. Since LCO only provides a low total conductivity up to 4 S cm−1 when charged up to Li0.47CoO2 at RT,14 a second phase that provides higher electronic conductivity could provide electrons to access reaction sites faster than going through LCO grains. This will allow higher C-rate performances of the SSLB. However, the finding of this electronic conductive phase may not be easy because it needs to be chemically stable to both LCO and LLZ:Ta and inert to oxygen up to 1050 °C.
From an energy density and practical point of view, the LLZ:Ta solid electrolyte should be as thin as possible, but thick enough to prevent material breakdown due to the high voltage across the cell. The presented SSLBs have LLZ:Ta solid electrolytes of ∼300 μm in thickness. Although an electrolyte supported SSLB is easier to fabricate for research purposes, it is not practical for application due to the high dead weight of the SSLB. It has been reported that it is difficult but possible to deposit LLZ:Ta as a thin film with a Li-ion conductivity as high as that for bulk ceramic.58 However, a deposition of LLZO thin film generally requires high-temperature treatment to crystallize the Li–La–Zr–O precursors into garnet-structured LLZO. This high-temperature heat treatment usually causes a chemical reaction between the active electrode material and Li–La–Zr–O precursors to form phases other than the garnet-structured LLZO as well as the formation of a short circuit due to the elemental inter-diffusion of the transition metal from the active electrode material to a solid electrolyte.59 Therefore, a new strategy for depositing a thin solid electrolyte to fabricate a positive electrode supported SSLB should be developed, or a thick film (<20 μm in thickness) deposited by tape casting or screen printing should be considered.60
Finally, the Li dendrite issue on SSLBs is still a challenge when metallic Li is used as the anode. Although there have been many publications regarding the Li dendrite problem, the experiments were usually done by using symmetric cells that only show a capacity lower than 1 mA h cm−2, which can be delivered as a short circuit free cell at RT.4,29–31 This is a much lower capacity than what it is needed for practical use, which is expected to be >3 mA h cm−2 to compete with the current LIBs. Increasing the Li utilization without dendrite formation for ceramic type SSLBs, i.e. without any use of liquid electrolyte to avoid fire hazards, is still another challenge.
Conclusions
A bulk-type SSLB based on a garnet-structured solid electrolyte was successfully prepared by the right choice of material and processing. In order to prevent chemical reactions that form highly resistive interfaces between the solid electrolyte and electrode material, crystalline LCO and Ta-substituted LLZO were chosen for SSLB fabrication due to the lower Gibbs free energy of crystalline LCO as compared to Li–Co–O and the higher stability of the cubic phase LLZ:Ta as compared to Al-substituted LLZO. The higher stability between crystalline LCO and LLZ:Ta, which allowed the developed SSLB to sinter at 1050 °C to form low resistive ionic diffusion paths without noticeable chemical reaction, was confirmed by micro-Raman and EDS mapping and EIS. Their similar CTEs allow for fast heating and cooling during the sintering process, shortening the sintering time at an elevated temperature so that elemental inter-diffusion, as well as the possibility of crack formation, within the CPE can be avoided. Much higher electrode material loading of the CPE, between 24 to 30 mg cm−2, makes the presented SSLB a true “bulk-type” SSLB. The resulting SSLBs have much lower area resistance than the other reported cell concepts that allow the cell to be charged and discharged with much higher current densities. However, the micro-cracking of the microstructure due to the volume change of LCO particles degrades the SSLB performance during the cycling. These fractures may be avoided by densifying the CPE to a macropore-free one. Furthermore, optimization of the SSLB for practical use by increasing the electronic conductivity of the CPE, using a thinner solid electrolyte and avoiding Li dendrite formation should be thoroughly investigated.Author contributions
C. L. T. conceived of and planned the experiments, wrote and edited the manuscript. C. L. T., Q. M., C. D., S. L, F. V., A. W., D. G., H. Z., S. U., F. T. conducted experiments, result discussions and interpretations. All authors have reviewed and approved of this article.Broader context
All-solid-state lithium batteries (SSLB) based on the garnet-structured Li7La3Zr2O12 solid electrolyte have been regarded as next-generation energy storage devices due to their high intrinsic safety and high energy density. However, the necessity of the sintering process at elevated temperatures during battery fabrication to form ionic diffusion paths through ceramic materials within the battery usually results in the formation of second phases at interfaces that dramatically lag the ionic transports. Therefore, developing a compatible interface between the used materials to achieve high interfacial ionic transport is one key strategy for realizing high-performance SSLB. Herein, we present a low interface resistance SSLB that can be straightforwardly achieved by material selections and processing. To support the strategy, an LLZO based SSLB with very high specific areal capacity and operational current densities is demonstrated. The results also provide new insights into bulk-type solid-state electrode/electrolyte interfacial chemistry and the mechanism of battery performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by Helmholtz Gemeinschaft Deutscher Forschungszentren e. V. under grant “Speicher und Vernetzte Infrastrukturen” and Helmholtz Institute Münster (HI MS), and by Bundesministerium für Bildung und Forschung (Federal ministry of education and research), Germany, under project no. 13N9973, 03X4634C and 03SF0477A is gratefully acknowledged. The authors are responsible for the content.References
- R. Murugan, V. Thangadurai and W. Weppner, Angew. Chem., Int. Ed. Engl., 2007, 46, 7778–7781 CrossRef CAS PubMed.
- H. Buschmann, J. Dolle, S. Berendts, A. Kuhn, P. Bottke, M. Wilkening, P. Heitjans, A. Senyshyn, H. Ehrenberg, A. Lotnyk, V. Duppel, L. Kienle and J. Janek, Phys. Chem. Chem. Phys., 2011, 13, 19378–19392 RSC.
- C.-L. Tsai, E. Dashjav, E.-M. Hammer, M. Finsterbusch, F. Tietz, S. Uhlenbruck and H. P. Buchkremer, J. Electroceram., 2015, 35, 25–32 CrossRef CAS.
- C.-L. Tsai, V. Roddatis, C. V. Chandran, Q. Ma, S. Uhlenbruck, M. Bram, P. Heitjans and O. Guillon, ACS Appl. Mater. Interfaces, 2016, 8, 10617–10626 CrossRef CAS PubMed.
- L. Buannic, B. Orayech, J.-M. López Del Amo, J. Carrasco, N. A. Katcho, F. Aguesse, W. Manalastas, W. Zhang, J. Kilner and A. Llordés, Chem. Mater., 2017, 29, 1769–1778 CrossRef CAS.
- F. Yonemoto, A. Nishimura, M. Motoyama, N. Tsuchimine, S. Kobayashi and Y. Iriyama, J. Power Sources, 2017, 343, 207–215 CrossRef CAS.
- L. Miara, A. Windmuller, C.-L. Tsai, W. D. Richards, Q. Ma, S. Uhlenbruck, O. Guillon and G. Ceder, ACS Appl. Mater. Interfaces, 2016, 8, 26842–26850 CrossRef CAS PubMed.
- Y. Ren, T. Liu, Y. Shen, Y. Lin and C.-W. Nan, J Materiomics., 2016, 2, 256–264 CrossRef.
- K. Park, B.-C. Yu, J.-W. Jung, Y. Li, W. Zhou, H. Gao, S. Son and J. B. Goodenough, Chem. Mater., 2016, 28, 8051–8059 CrossRef CAS.
- K. H. Kim, Y. Iriyama, K. Yamamoto, S. Kumazaki, T. Asaka, K. Tanabe, C. A. J. Fisher, T. Hirayama, R. Murugan and Z. Ogumi, J. Power Sources, 2011, 196, 764–767 CrossRef CAS.
- S. Uhlenbruck, S. Lobe, C. Dellen, C.-L. Tsai, B. Gotzen, D. Sebold, M. Finsterbusch and O. Guillon, J. Electroceram., 2017, 38, 197–206 CrossRef CAS.
- A. A. Hubaud, D. J. Schroeder, B. J. Ingram, J. S. Okasinski and J. T. Vaughey, J. Alloys Compd., 2015, 644, 804–807 CrossRef CAS.
- E. J. Cheng, N. J. Taylor, J. Wolfenstine and J. Sakamoto, Journal of Asian Ceramic Societies, 2018, 5, 113–117 CrossRef.
- J. Molenda, A. StokLosa and T. Bak, Solid State Ionics, 1989, 36, 53–58 CrossRef CAS.
- T. Kato, T. Hamanaka, K. Yamamoto, T. Hirayama, F. Sagane, M. Motoyama and Y. Iriyama, J. Power Sources, 2014, 260, 292–298 CrossRef CAS.
- M. Kotobuki, K. Kanamura, Y. Sato and T. Yoshida, J. Power Sources, 2011, 196, 7750–7754 CrossRef CAS.
- M. Kotobuki and K. Kanamura, Ceram. Int., 2013, 39, 6481–6487 CrossRef CAS.
- Y. Ren, Y. Shen, Y. Lin and C.-W. Nan, Ionics, 2017, 23, 2521–2527 CrossRef CAS.
- S. Ohta, S. Komagata, J. Seki, T. Saeki, S. Morishita and T. Asaoka, J. Power Sources, 2013, 238, 53–56 CrossRef CAS.
- S. Ohta, J. Seki, Y. Yagi, Y. Kihira, T. Tani and T. Asaoka, J. Power Sources, 2014, 265, 40–44 CrossRef CAS.
- T. Liu, Y. Zhang, R. Chen, S.-X. Zhao, Y. Lin, C.-W. Nan and Y. Shen, Electrochem. Commun., 2017, 79, 1–4 CrossRef CAS.
- L. Feng, L. Li, Y. Zhang, H. Peng and Y. Zou, Solid State Ionics, 2017, 310, 129–133 CrossRef CAS.
- T. Liu, Y. Zhang, X. Zhang, L. Wang, S.-X. Zhao, Y.-H. Lin, Y. Shen, J. Luo, L. Li and C.-W. Nan, J. Mater. Chem. A, 2018, 6, 4649–4657 RSC.
- F. Han, J. Yue, C. Chen, N. Zhao, X. Fan, Z. Ma, T. Gao, F. Wang, X. Guo and C. Wang, Joule, 2018, 2, 497–508 CrossRef CAS.
- M. Koo, K. I. Park, S. H. Lee, M. Suh, D. Y. Jeon, J. W. Choi, K. Kang and K. J. Lee, Nano Lett., 2012, 12, 4810–4816 CrossRef CAS PubMed.
- T. Liu, Y. Ren, Y. Shen, S.-X. Zhao, Y. Lin and C.-W. Nan, J. Power Sources, 2016, 324, 349–357 CrossRef CAS.
- T. Kato, S. Iwasaki, Y. Ishii, M. Motoyama, W. C. West, Y. Yamamoto and Y. Iriyama, J. Power Sources, 2016, 303, 65–72 CrossRef CAS.
- R. Inada, S. Yasuda, M. Tojo, K. Tsuritani, T. Tojo and Y. Sakurai, Front. Energy, 2016, 4, 28 Search PubMed.
- A. Sharafi, E. Kazyak, A. L. Davis, S. Yu, T. Thompson, D. J. Siegel, N. P. Dasgupta and J. Sakamoto, Chem. Mater., 2017, 29, 7961–7968 CrossRef CAS.
- K. K. Fu, B. Liu, Y. Zhu, S. Xu, Y. Yao, W. Luo, C. Wang, S. D. Lacey, J. Dai, Y. Chen, Y. Mo, E. Wachsman and L. Hu, Sci. Adv., 2017, 3, e1601659 CrossRef PubMed.
- C. Yang, L. Zhang, B. Liu, S. Xu, T. Hamann, D. McOwen, J. Dai, W. Luo, Y. Gong, E. D. Wachsman and L. Hu, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3770–3775 CrossRef CAS PubMed.
- K. Hara, T. Yano, K. Suzuki, M. Hirayama, T. Hayashi, R. Kanno and M. Hara, Anal. Sci., 2017, 33, 853–858 CrossRef CAS PubMed.
- T. Gross, L. Giebeler and C. Hess, Rev. Sci. Instrum., 2013, 84, 073109 CrossRef CAS PubMed.
- T. Gross and C. Hess, J. Power Sources, 2014, 256, 220–225 CrossRef CAS.
- S. K. Tripathy, M. Christy, N.-H. Park, E.-K. Suh, S. Anand and Y.-T. Yu, Mater. Lett., 2008, 62, 1006–1009 CrossRef CAS.
- J. Zhang, W. Gao, M. Dou, F. Wang, J. Liu, Z. Li and J. Ji, Analyst, 2015, 140, 1686–1692 RSC.
- Y. Lu, W. Zhan, Y. He, Y. Wang, X. Kong, Q. Kuang, Z. Xie and L. Zheng, ACS Appl. Mater. Interfaces, 2014, 6, 4186–4195 CrossRef CAS PubMed.
- F. Tietz, T. Wegener, M. T. Gerhards, M. Giarola and G. Mariotto, Solid State Ionics, 2013, 230, 77–82 CrossRef CAS.
- T. Thompson, J. Wolfenstine, J. L. Allen, M. Johannes, A. Huq, I. N. David and J. Sakamoto, J. Mater. Chem. A, 2014, 2, 13431–13436 RSC.
- P. Babkevich, D. Prabhakaran, C. D. Frost and A. T. Boothroyd, Phys. Rev. B, 2010, 82, 184425 CrossRef.
- R. Le Toquin, W. Paulus, A. Cousson, G. Dhalenne and A. Revcolevschi, Phys. B, 2004, 350, E269–E272 CrossRef CAS.
- M. A. Nowroozi, S. Ivlev, J. Rohrer and O. Clemens, J. Mater. Chem. A, 2018, 6, 4658–4669 RSC.
- J. Zhu, Y. Wang, X. Wang, X. Yang and L. Lu, J. Rare Earths, 2007, 25, 601–604 CrossRef.
- A. Ishikawa, J. Nohara and S. Sugai, Phys. Rev. Lett., 2004, 93, 136401 CrossRef CAS PubMed.
- D. Tuschel, Spectroscopy, 2016, 31, 14–23 CAS.
- R. A. Huggins, ionics, 2002, 8, 300–313 CrossRef CAS.
- Y. Seino, K. Takada, B.-C. Kim, L. Zhang, N. Ohta, H. Wada, M. Osada and T. Sasaki, Solid State Ionics, 2005, 176, 2389–2393 CrossRef CAS.
- K. Takada, A. Kajiyama, H. Sasaki, S. Kondo, M. Watanabe, M. Murayama and R. Kanno, Solid State Ionics, 2003, 158, 269–274 CrossRef CAS.
- K. Okada, N. Machida, M. Naito, T. Shigematsu, S. Ito, S. Fujiki, M. Nakano and Y. Aihara, Solid State Ionics, 2014, 255, 120–127 CrossRef CAS.
- S. Yubuchi, S. Teragawa, K. Aso, K. Tadanaga, A. Hayashi and M. Tatsumisago, J. Power Sources, 2015, 293, 941–945 CrossRef CAS.
- Y. Iriyama, T. Kako, C. Yada, T. Abe and Z. Ogumi, J. Power Sources, 2005, 146, 745–748 CrossRef CAS.
- I. Cho, J. Choi, K. Kim, M.-H. Ryou and Y. M. Lee, RSC Adv., 2015, 5, 95073–95078 RSC.
- J. van den Broek, S. Afyon and J. L. M. Rupp, Adv. Energy Mater., 2016, 6, 1600736 CrossRef.
- M. Huang, M. Shoji, Y. Shen, C.-W. Nan, H. Munakata and K. Kanamura, J. Power Sources, 2014, 261, 206–211 CrossRef CAS.
- J. van den Broek, J. L. M. Rupp and S. Afyon, J. Electroceram., 2017, 38, 182–188 CrossRef CAS.
- J. N. Reimers and J. R. Dahn, J. Electrochem. Soc., 1992, 139, 2091–2097 CrossRef CAS.
- G. Bucci, T. Swamy, Y.-M. Chiang and W. C. Carter, J. Mater. Chem. A, 2017, 5, 19422–19430 RSC.
- S. Lobe, C. Dellen, M. Finsterbusch, H. G. Gehrke, D. Sebold, C.-L. Tsai, S. Uhlenbruck and O. Guillon, J. Power Sources, 2016, 307, 684–689 CrossRef CAS.
- S. Lobe, A. Windmüller, C.-L. Tsai, F. Vondahlen, S. Uhlenbruck and O. Guillon, Ionics, 2018, 24, 2199–2208 CrossRef CAS.
- E. Yi, W. Wang, J. Kieffer and R. M. Laine, J. Power Sources, 2017, 352, 156–164 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional detail discussion of material synthesis and Raman spectroscopy investigations and other supplementary results. See DOI: 10.1039/c8se00436f |
| This journal is © The Royal Society of Chemistry 2019 |