
Efficient CO2 fixation under ambient pressure using poly(ionic liquid)-based heterogeneous catalysts†
Tanmoy
Biswas
and
Venkataramanan
Mahalingam
 *
*
Department of Chemical Sciences and Center for Advanced Functional Materials (CAFM), Indian Institute of Science Education and Research (IISER) Mohanpur, Kolkata, West Bengal 741252, India. E-mail: mvenkataramanan@yahoo.com; Fax: +91-33-25873020; Tel: +919007603474
First published on 22nd October 2018
Abstract
We report the use of ionic liquid based polymers for the conversion of epoxides into cyclic carbonates under ambient pressure (1.02 atm). The polymeric catalysts were prepared via thiol–ene click chemistry using trithiocyanuric acid (TTCA) and divinyl imidazolium-based ionic liquids in the presence of AIBN as a radical initiator under thermal conditions. TTCA acts as a planar C3 symmetric thiol molecule and helps in CO2 absorption due to the presence of a triazine moiety. Additionally, TTCA helps to achieve the optimum separation of catalytically active sites i.e. imidazolium bromide moieties. The resulting poly(ionic liquid) together with KI as a co-catalyst has been found to be very efficient for the conversion of a wide number of epoxide substrates into their corresponding cyclic carbonates under CO2 filled balloon conditions. The catalyst was able to retain its activity up to 6 cycles.
Global warming and climate change are the major problems in today's society. Excessive deforestation and fossil fuel burning result in a huge amount of carbon dioxide (CO2) accumulation in the atmosphere. Minimization of the excess amount of CO2 in the atmosphere is very much essential to control the drastic climate change.1 In fact, technological development has reached an extent where the separation of CO2 from the atmosphere has become possible.2 Recently, there have been immense research efforts to utilize the excess CO2 and convert it into chemicals and fuels.3 Moreover, CO2 can be efficiently utilised as a one-carbon (C1) source. One of the ways to utilize this excess CO2 is the conversion of epoxides to cyclic carbonates as they are very useful materials as highly polar aprotic solvents, monomers of polycarbonates, solvents for lithium ion batteries, core moieties of some drug molecules, etc.4 Industrial methods for the preparation of cyclic carbonates involve the use of highly corrosive phosgene as the C1 source. Moreover, hazardous hydrogen chloride is produced as one of the by-products.5 Replacement of phosgene with CO2 is also very advantageous due to its wide availability. There are many reports on the conversion of epoxides into cyclic carbonates using CO2. For example, salen–metal complexes, metalloporphyrins, metal organic frameworks, immobilized catalysts, modified molecular sieves, ion-exchange resins, quaternary ammonium salts, quaternary phosphonium salts, nitrogen-rich polymers, natural products, metal–crown ether complexes, g-C3N4 and zeolites were used as catalysts for conversion of epoxides into cyclic carbonates.6 However, one of the major problems associated with the epoxide to cyclic carbonate conversion is the requirement of high CO2 pressure.
One of the main difficulties in the activation of CO2 is its high thermodynamic stability and kinetic inertness.7 Consequently, it is necessary to develop efficient methods to perform the task under relatively less drastic conditions. In fact, there are few reports in the literature about the cyclic carbonate formation under atmospheric pressure.8 Thus, it is quite challenging to prepare cyclic carbonates from epoxides and CO2 under transition metal-free, solvent-free and ambient conditions and is also highly appreciated from the green chemistry viewpoint.
To achieve the goal of epoxide to cyclic carbonate conversion, our idea is to use ionic liquids (ILs) as they are highly efficient in absorbing CO2.9 Moreover, functionalised IL compounds are proven to be more efficient for epoxide to cyclic carbonate conversion as they provide additional stability to the intermediates produced during the reaction.10 However, the solubility of ILs in the reaction mixture is an issue during catalyst recovery after the completion of the reaction. To overcome this problem, heterogeneous polymeric IL-based materials were used as catalysts. However, the majority of previously reported poly(ionic liquid)-based materials were used under high CO2 pressure, which was the main disadvantage associated with them.11 Consequently, it is essential to introduce more reactivity by a proper choice of monomer during the synthesis. In fact, it is well established in the literature that two dimensional (2D) materials are highly efficient as heterogeneous catalysts due to their high surface area which results in excellent reactivity.12 For example, Gao and co-workers reported an IL-attached 2D covalent organic framework (COF) which efficiently reduces CO2 in the presence of silanes at 1 atmosphere pressure.13 Similarly, Dyson et. al. reported polyimidazolium based polymeric IL materials, which were able to convert epoxides under 1 atmospheric CO2 pressure.14 Additionally, Chen and co-workers also reported an IL-attached aluminium salen polymer which was efficient in bringing about cyclic carbonate conversion under 1 MPa pressure.15 In fact, there are many ILs composed of imidazolium salts having halide counter anions. The nucleophilicity of these halides plays an important role in the conversion as it initiates the reaction by opening up the epoxide ring.16 It has already been well established that quaternary ammonium iodide is able to provide excellent reactivity for the conversion but the major problem associated with it is its homogeneous nature. Our motivation was to use a cheap and heterogeneous iodide source i.e. KI for this purpose. For example, Werner and his group have used KI as a co-catalyst along with an amino alcohol as a homogeneous catalyst for epoxide to cyclic carbonate conversion.17a Similarly, Chen and co-workers used sugarcane bagasse as a heterogeneous catalyst and KI as a co-catalyst for the conversion of a wide number of substrates.17b Additionally, Liu and co-workers utilized KI as a co-catalyst in the presence of zinc-containing highly porous silica i.e. Zn-SBA-15.17c However, the above reports used high CO2 pressure for the epoxide to cyclic carbonate conversion. This motivated us to develop a suitable catalyst by combining KI and polymeric ILs. We expect that the catalytic efficiency of polymeric ILs would be enhanced in the presence of KI as a co-catalyst. We hope that this mixture would bring about the epoxide conversion under ambient CO2 pressure. To achieve this goal, our idea was to develop a thiol–ene click chemistry route for the polymerization of ILs. To enhance the reactivity, di-cationic ILs were chosen instead of the monocationic ones. Our strategy is to use a planar C3 symmetric triazine-containing thiol molecule like trithiocyanuric acid (TTCA). In fact, to our knowledge this is the first report where polymeric ILs are prepared via thiol–ene click chemistry using TTCA as the thiol source. Moreover, triazine-containing materials are reported to be effective for CO2 absorption and conversion.18 We expect that the planarity of TTCA may provide additional planarity to the target catalyst which is expected to enhance the reactivity (please see Fig. 1 and S1(a) in the ESI† for reaction schemes).
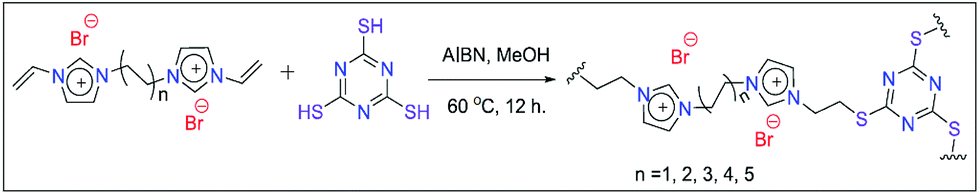 | ||
| Fig. 1 Scheme illustrating the synthetic protocol for the catalyst preparation via thiol–ene click chemistry. | ||
In this work, five dicationic ILs were used as starting materials and were prepared following the previous literature (see the Experimental section for details on IL synthesis).19 These materials were characterized by 1H and 13C NMR (Fig. S2 to S11, ESI†). The polymeric catalysts were prepared via thiol–ene click chemistry between 1,n-divinylimidazolium dibromides and TTCA in methanol, in the presence of AIBN as a radical initiator under thermal conditions (see the ESI† for details on polymer preparation).20 The polymeric catalysts were denoted as 1,n-IL-TTCA where “n” represents the number of carbon atoms present in the dibromoalkane moiety. The polymers were used as catalysts for the conversion of epoxides into cyclic carbonates in the presence of KI as a co-catalyst. This catalyst mixture was tested for a number of epoxides under CO2 filled balloon conditions.
All prepared polymers were characterized using solid-state 13C CP-MAS, TGA, PXRD, SEM, FTIR, TEM and CHN analyses. The presence of the imidazolium group, aliphatic carbon chain and triazine ring in all the samples was confirmed by 13C CP-MAS analysis. The peak corresponding to triazine appeared at 185 ppm which was also supported by the value obtained from ChemDraw. (Fig. S12 to S16, ESI†). Formation of the polymer was also supported by the TGA analysis under a N2 atmosphere. It was noted that all the polymeric materials are thermally stable up to 300 °C (Fig. S17, ESI†). Subsequently, 50% degradation was noted between 300 °C and 350 °C. The polymeric materials were found to be amorphous from the PXRD analysis (Fig. S18, ESI†). Surface morphology analysis using SEM reveals that the surface of all the polymers is rough in nature (Fig. S19, ESI†). The polymer formation was further verified using the FTIR analysis (see Fig. S20, ESI†). The absence of free S–H stretching at 2518 cm−1 and 2648 cm−1 suggests the complete conversion of thiol groups for the C–S bond formation. Additionally, the absence of the imine stretching peak (CN–H) near 2908 cm−1 (arising due to tautomerism) further confirms the polymer formation.21 Also, the above results confirm the absence of homo-polymerization of double bonds. We believe that the observed broadening of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching peak around 1645 cm−1 is due to the presence of some free double bonds owing to the use of excess monomers during the synthesis. For clarity, the FTIR spectra of the monomers are compared with those of 1,6-IL-TTCA and poly-1,6-IL and are shown in Fig. S20(b).† Moreover, the CHN analysis was performed for 1,6-IL-TTCA and the obtained percentages are 39.2%, 6.0% and 11.4% for C, H and N, respectively. Furthermore, the presence of sulfur and bromine was confirmed by elemental analysis during TEM measurement (see Fig. S21–S22 and Table S1†).
C stretching peak around 1645 cm−1 is due to the presence of some free double bonds owing to the use of excess monomers during the synthesis. For clarity, the FTIR spectra of the monomers are compared with those of 1,6-IL-TTCA and poly-1,6-IL and are shown in Fig. S20(b).† Moreover, the CHN analysis was performed for 1,6-IL-TTCA and the obtained percentages are 39.2%, 6.0% and 11.4% for C, H and N, respectively. Furthermore, the presence of sulfur and bromine was confirmed by elemental analysis during TEM measurement (see Fig. S21–S22 and Table S1†).
The above-mentioned polymer samples were utilized as catalysts for the conversion of epoxides into cyclic carbonates. All the reactions were performed under CO2 filled balloons. Epichlorohydrin was chosen as a model substrate to check the catalytic activity. We believe that the role of the triazine ring in the polymer is primarily to provide structural rigidity and secondarily to enhance CO2 absorption during the reaction. To check the reactivity for cyclic carbonate conversion, 50 mg of each polymer was used (Table 1, entry no. 7 to 11). It was observed that among other samples 1,6-IL-TTCA displays maximum conversion of epichlorohydrin into its corresponding cyclic carbonate (Table 1, entry no. 9). This may be due to the optimum separation of the active centres i.e. the imidazolium bromide moieties. We believe that the introduction of the long chain may introduce more hydrophobicity and lead to increased separation of active centres. This results in the lesser stabilization of the intermediates. When the amount of 1,6-IL-TTCA was further increased to 100 mg, the conversion increased up to 71% (Table 1, entry no. 13). To achieve complete conversion, salts like KI, KBr and KCl were used as co-catalysts. When KBr was used along with the polymer catalyst, 72% conversion was achieved, whereas 62% conversion was found in the case of KCl. It was also observed that individually the potassium halides are almost inactive for the conversion of epichlorohydrin (Table 1, entry no. 2, 4 & 5). Moreover, the 1,6-IL-TTCA and KI combination provided the complete conversion of epichlorohydrin (Table 1, entry no. 17) with a turnover number (TON) of 69.4. We believe that, during the reaction, some amount of cyclic carbonate was produced by the contribution of the polymer alone which slightly solubilises potassium halides. The extent of solubility of the potassium halides facilitates the anion exchange and the nucleophilicity and the good leaving group nature of the exchanged anions results in the difference in the conversion (Table 1, entry no. 15, 16 & 17).22 This hypothesis was further supported by the almost similar conversion in the case of KBr as a co-catalyst (Table 1, entry no. 15). Among the three halides, Cl− had the lowest nucleophilicity and the least efficient leaving group character. Thus the exchange of Cl− with Br−, from the polymer backbone, results in lower conversion compared to that obtained with the parent polymer alone (Table 1, entry no. 16 & 13). To find the optimum reaction conditions, the reaction time and temperature were also varied. In fact, a decrease in reaction time from 20 h and temperature from 105 °C resulted in a lower conversion (Table 1, entry no. 18 & 19). The results conclude that the 1,6-IL-TTCA and KI combination at 105 °C for 20 h are the optimum conditions. These conditions were used for the other epoxides (Table 1, entry no. 17).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Amount of catalyst | Conditions, temp. & time | Conversion (%) |
| a Conditions: substrate, epichlorohydrin 15 mmol; a CO2 filled balloon (1.02 atm, 99.5% pure, for balloon pressure measurement, see Fig. S23, ESI, & for the experimental set-up, see Fig. S24, ESI). Conversion was measured using 1H NMR spectroscopy. | ||||
| 1 | No cat. | — | 105 °C, 20 h | 0 |
| 2 | KI | 6.6 mol% | 105 °C, 20 h | 9 |
| 3 | KI | 1.7 mol% | 105 °C, 20 h | 0 |
| 4 | KBr | 6.6 mol% | 105 °C, 20 h | 0 |
| 5 | KCl | 6.6 mol% | 105 °C, 20 h | 0 |
| 6 | TTCA | 2.0 mol% | 105 °C, 20 h | 0 |
| 7 | 1,2-IL-TTCA | 50 mg | 105 °C, 20 h | 22 |
| 8 | 1,4-IL-TTCA | 50 mg | 105 °C, 20 h | 34 |
| 9 | 1,6-IL-TTCA | 50 mg | 105 °C, 20 h | 53 |
| 10 | 1,8-IL-TTCA | 50 mg | 105 °C, 20 h | 50 |
| 11 | 1,10-IL-TTCA | 50 mg | 105 °C, 20 h | 33 |
| 12 | 1,6 IL-TTCA + KI | 50 mg + 6.6 mol% | 105 °C, 20 h | 88 |
| 13 | 1,6-IL-TTCA | 100 mg | 105 °C, 20 h | 71 |
| 14 | Poly-1,6-IL | 100 mg | 105 °C, 20 h | 10 |
| 15 | 1,6-IL-TTCA + KBr | 100 mg + 6.6 mol% | 105 °C, 20 h | 72 |
| 16 | 1,6-IL-TTCA + KCl | 100 mg + 6.6 mol% | 105 °C, 20 h | 62 |
| 17 | 1,6-IL-TTCA + KI | 100 mg + 6.6 mol% | 105 °C, 20 h | 100 |
| 18 | 1,6-IL-TTCA + KI | 100 mg + 6.6 mol% | 105 °C, 10 h | 33 |
| 19 | 1,6-IL-TTCA + KI | 100 mg + 6.6 mol% | 85 °C, 20 h | 69 |
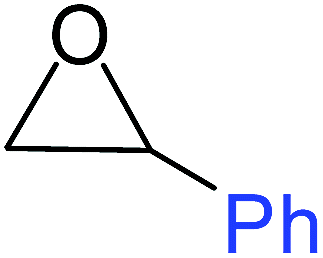
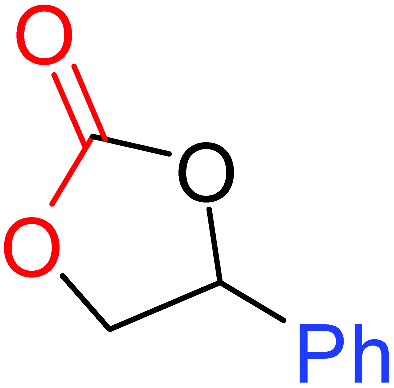
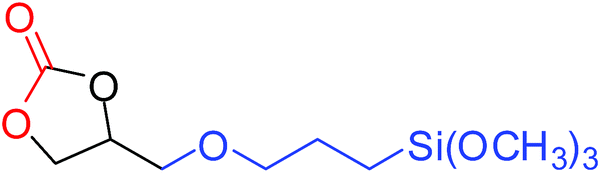
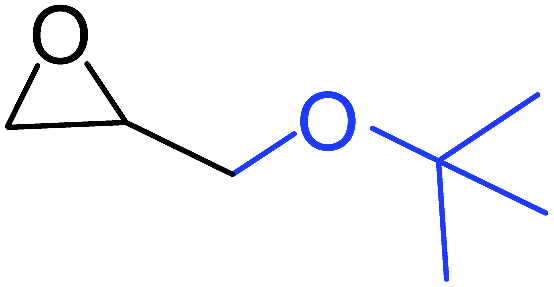
To understand the reactivity of our catalyst, different functionalized epoxides were tested for cyclic carbonate conversion under the optimized reaction conditions. For example, allyl glycidyl ether, phenyl glycidyl ether, isopropyl glycidyl ether, glycidyl methacrylate and styrene oxide showed complete conversion (Table 2, entry no. 2 to 6). The observed reactivity is likely due to the presence of electronegative heteroatoms (e.g. oxygen or chloride), which help to stabilize the electron-rich intermediates produced during the reaction. In the case of styrene oxide, the phenyl rings help to stabilize the intermediates. However, 87% conversion was found in the case of (3-glycidyloxypropyl)trimethoxysilane. This slight decrease in conversion may be due to the bulkiness associated with the trimethoxysilane group (Table 2, entry 7). This phenomenon was further observed with t-butylglycidyl ether where only 50% conversion was observed (Table 2, entry 8). However, 1,2-epoxy-9-decene displayed 11% conversion which may be due to the presence of a much less polar carbon chain in the epoxide molecule (Table 2, entry 9). To check the efficiency of the catalyst, the study was extended to diepoxides. Conversion of diepoxides to corresponding cyclic carbonates is interesting as they are useful in polyurethane preparation via the use of relatively less toxic chemicals.23 We used 1,2,7,8-diepoxyoctane which exhibits similar reactivity to 1,2-epoxy-9-decene. This may be due to the presence of a relatively non-polar carbon chain in the diepoxide. Similarly, poly(ethylene glycol)diglycidyl ether (average Mn = 500) also showed relatively lower conversion (27%). We presume that this may be due to the presence of a long carbon chain attached to the epoxide moiety which is chemically inert compared to glycidyl ethers, and high viscosity of these three epoxides also affects their conversion. However, the conversions increased up to 74% and 47% after the reaction time was increased from 20 h to 68 h (Table 2, entries 10 & 11). Although neopentyl glycol diglycidyl ether contains two electronegative oxygen atoms, we believe that the observed lower conversion is likely due to its higher viscosity and the bulkiness of the quaternary carbon atom (Table 2, entry 12). However, 75% conversion was observed when 1,4-butanediol diglycidyl ether was used as the substrate. This is likely due to the similarity of the substrate to simple glycidyl ethers which showed excellent reactivity (Table 2, entry 13).
|
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Conversion (%) |
| a Conditions: catalyst 100 mg or 1.58 mol% (based on the imidazolium cation); substrate, 15 mmol; a CO2 filled balloon (1.02 atm, 99.5% pure); time, 20 h; and temp., 105 °C. b The reaction time was 68 h. Conversion was measured using 1H NMR spectroscopy (Fig. S25 to S37, ESI). | |||
| 1 |
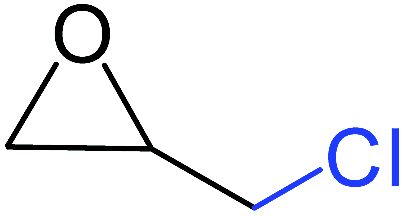
|
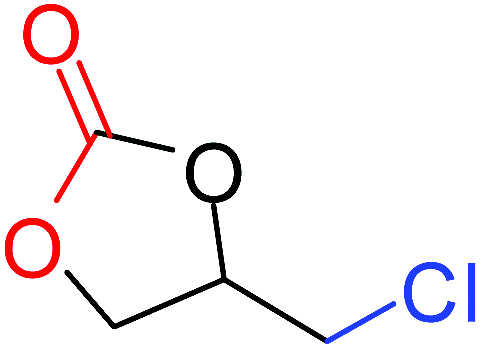
|
100 |
| 2 |
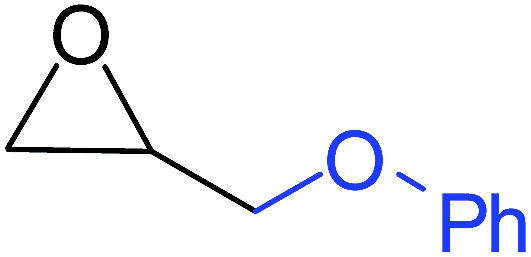
|
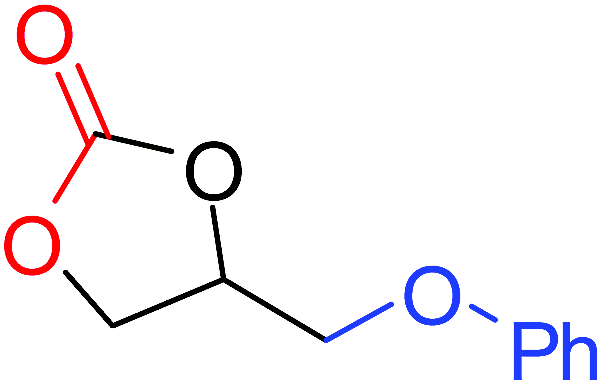
|
100 |
| 3 |
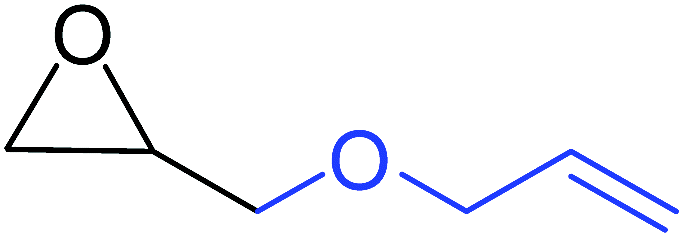
|
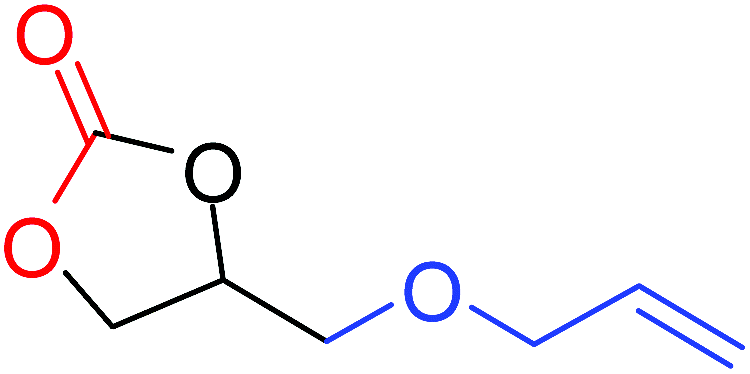
|
100 |
| 4 |
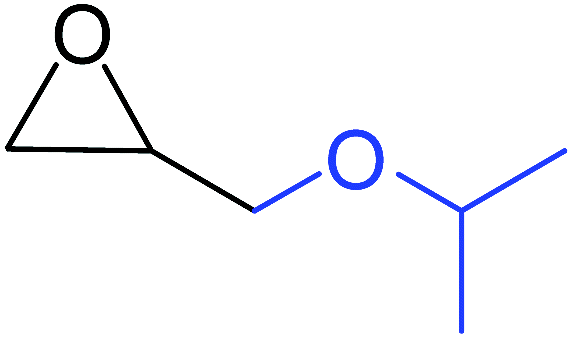
|
|
100 |
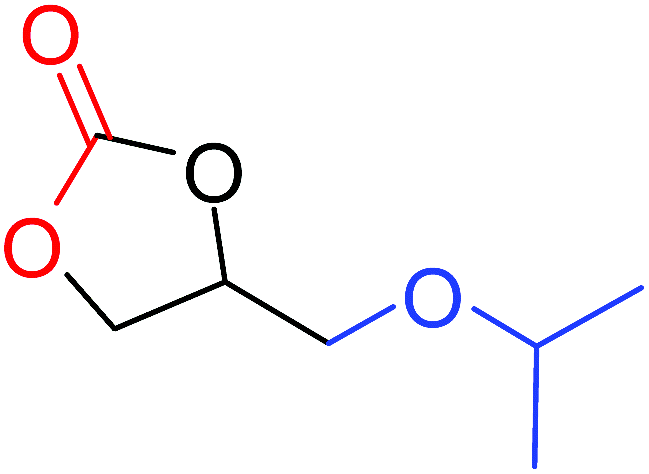
| 5 |
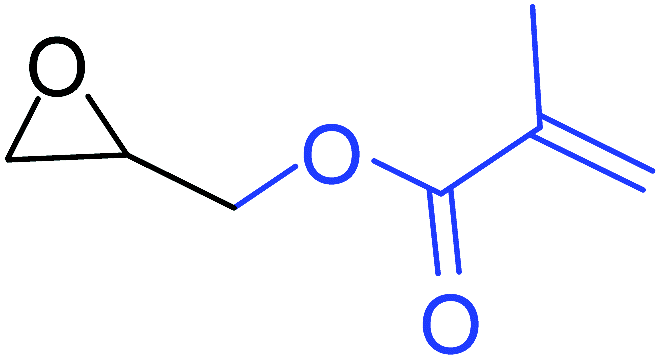
|
|
100 |
| 6 |
|
|
100 |
| 7 |

|
|
87 |
| 8 |
|
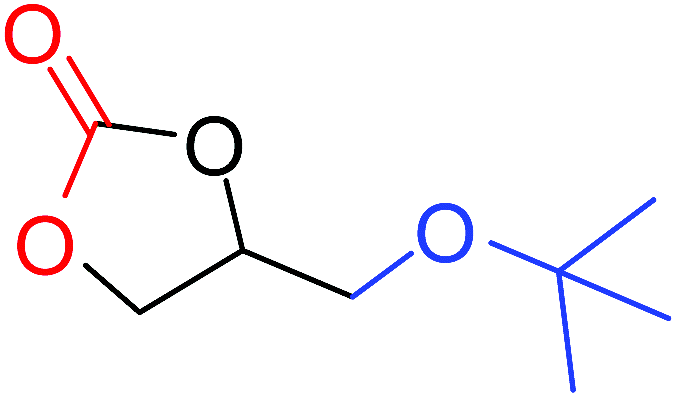
|
50 |
| 9 |
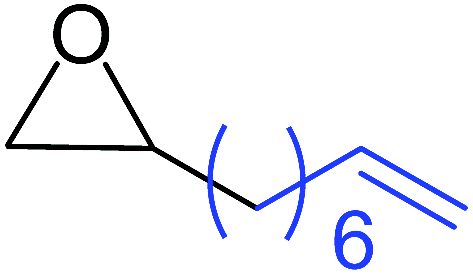
|
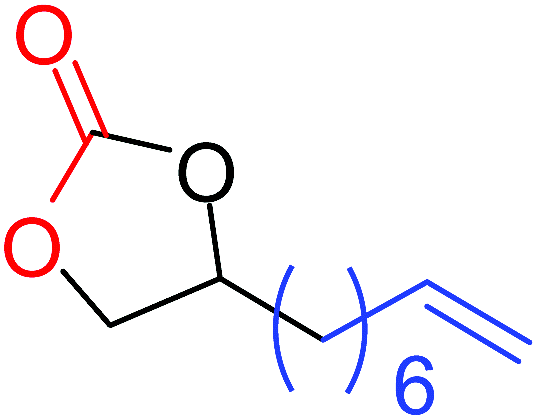
|
11 |
| 10 |

|

|
24 (74)b |
| 11 |
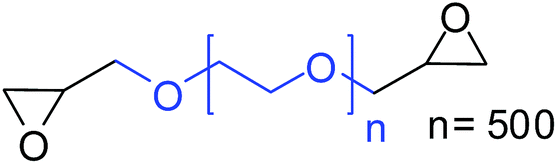
|
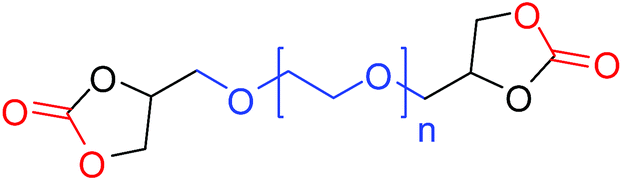
|
27 (47)b |
| 12 |
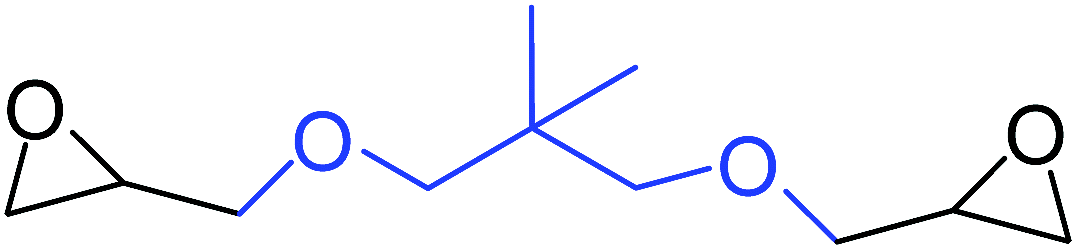
|
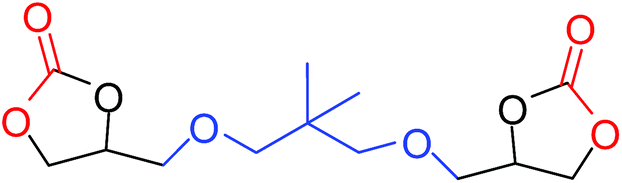
|
35 |
| 13 |
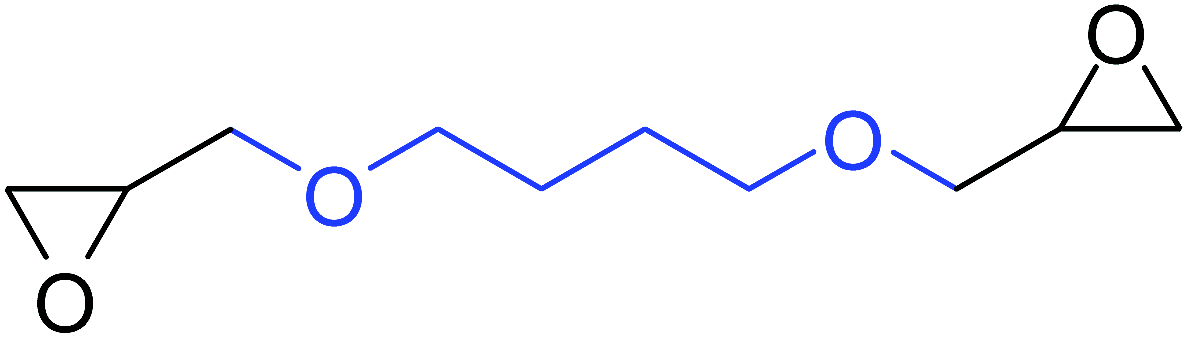
|
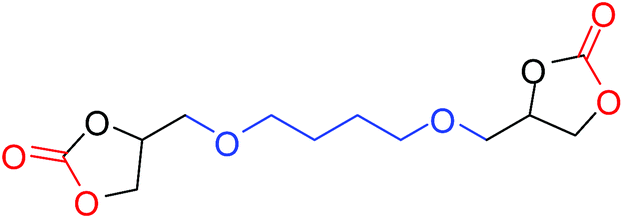
|
75 |
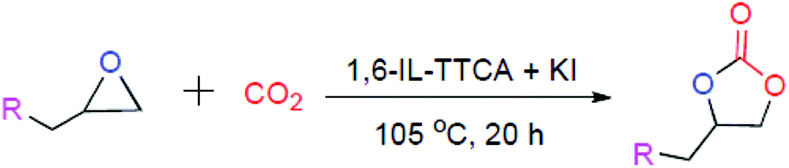
To get more insight, the proposed mechanism for the cyclic carbonate conversion using this catalyst combination is shown in Fig. 2. First, the epoxide molecules are expected to approach the polymer likely due to the possibility of hydrogen bonding and ion–dipole interaction. This favours a facile counter anion attack on the epoxide which produces the halo-alkoxide anion. Subsequently, this anion attacks the CO2 molecule resulting in the catalytic incorporation of CO2 into the epoxide molecule. The final stage of the cycle involves the intramolecular ring closure resulting in the production of the cyclic carbonate molecules. In addition, we strongly presume that the incorporation of the triazine helps to absorb CO2 and separates the imidazolium centres which results in the excellent reactivity. Although, there is a chance that such reactivity may arise due to the homopolymer of 1,6-IL. To examine the fact, poly-1,6-IL was synthesized and used as a catalyst [see Fig. S1(b), ESI†]. Here, only 10% conversion was observed (Table 1, entry no. 14). This result indicates that the reactivity arises due to the thiol–ene polymeric catalyst. Moreover, another reason for the reactivity is the optimum separation of the active centres, i.e. imidazolium groups.
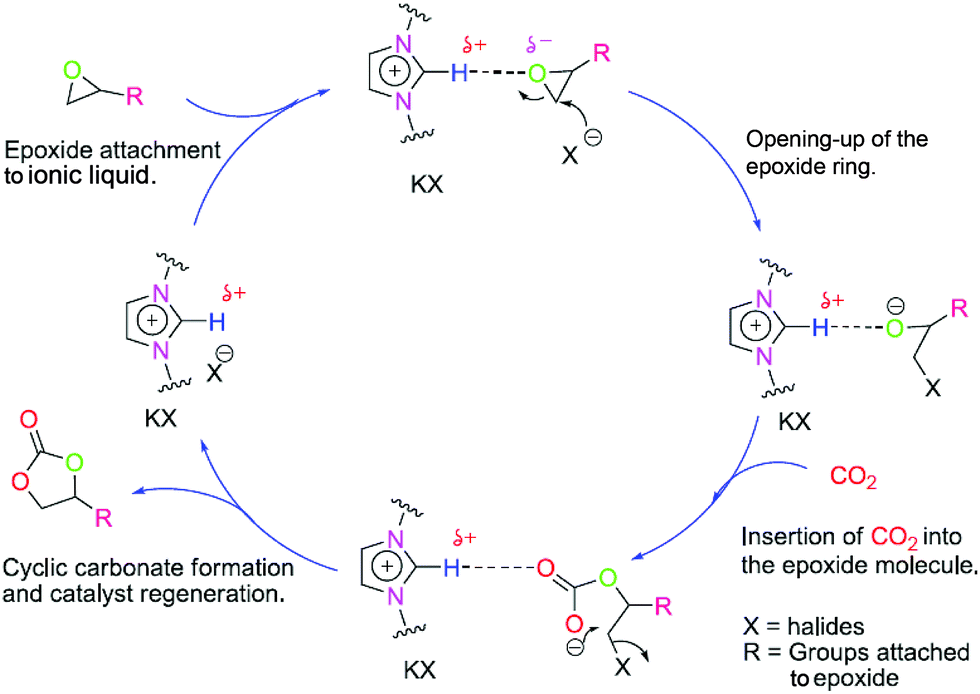 | ||
| Fig. 2 Proposed mechanism for the 1,6-IL-TTCA/KI catalyzed epoxide to cyclic carbonate conversion. | ||
To check the stability and heterogeneity of our catalyst recyclability experiments were performed in the presence and absence of the co-catalyst (see Fig. 3 and S38 in ESI†). A sharp decrease in the conversion was observed after the second cycle. Interestingly, further addition of 1.7 mol% KI resulted in complete conversion. This is because from the third cycle the catalysts reacted again synergistically resulting in complete conversion. A similar phenomenon occurred in the 5th cycle also. It is expected that with an increase in the number of catalytic cycles, the efficiency of the catalyst would decrease. We strongly believe that the retention of reactivity is obtained due to the anion exchange occurring during the reaction. To confirm our hypothesis, the polymer catalyst after 6 cycles was collected, washed with water for the complete removal of all the inorganic salts and subjected to EDAX analysis (please see the Experimental section, ESI†, for the washing protocol) The iodide incorporation in the polymer as a counter anion was found to be 0.62% which supports the anion exchange (see Table S2 and Fig. S39 in the ESI†). To further understand the stability of the catalyst, the material obtained after the 6th cycle was characterized again using TGA, SEM and PXRD analysis. The TGA profile reveals that the stability of the catalyst is preserved even after the 6th cycle (Fig. S40, ESI†). The SEM analysis showed that the catalyst retains its morphology after six cycles (Fig. S41, ESI†). From the PXRD analysis of the sample, it was noted that the catalyst was amorphous like the parent one but there is a slight 2° shift to a lower 2θ value. We presume that this is likely due to the increase in interlayer spacing. The observed decrease in intensity might be indicative of the destacking of the layers (Fig. S42, ESI†).
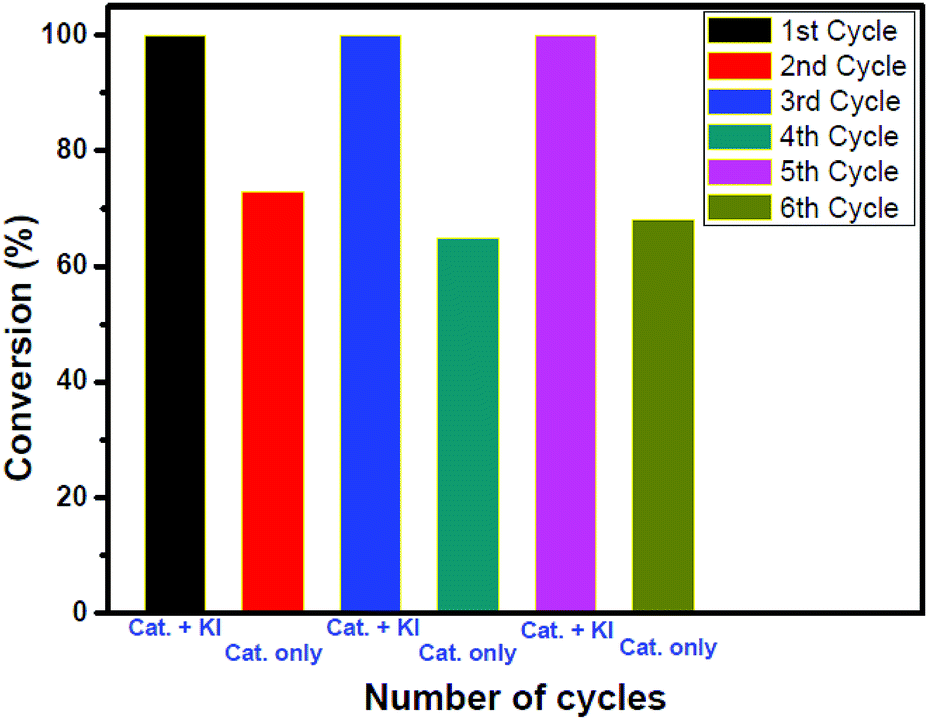 | ||
| Fig. 3 Bar diagram showing the recyclability of the catalyst. Conditions: substrate, epichlorohydrin 15 mmol; in the third and fifth cycles 1.7 mol% KI was added; a CO2 filled balloon (1.02 atm, 99.5% pure); time, 20 h; and temp., 105 °C. Conversion was measured using 1H NMR spectroscopy. | ||
Conclusions
In conclusion, we report for the first time the synthesis of a polymeric ionic liquid (IL) via thiol–ene click chemistry using trithiocyanuric acid, a dicationic IL. The resulting poly(ionic liquid) together with KI as a co-catalyst has been found to be very efficient for the conversion of epoxides into their corresponding cyclic carbonates under ambient CO2 pressure (i.e. under balloon conditions). The catalyst combination was very effective for the conversion of several epoxides to their corresponding cyclic carbonates. The recyclability study suggests that the heterogeneous catalyst was able to retain its activity up to 6 cycles. The developed catalyst utilized a cheap iodide source to improve the reactivity and provided good conversion for a wide range of substrates. Overall a bypass method was developed for the costly alkyl iodide reagent. We believe that further modification of the ionic liquid moiety or the use of different thiol precursors may provide a superior catalyst in future.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
M. V. thanks the Department of Science and Technology (DST) for the project “EMR/2014/000204” and the Indian Institute of Science Education and Research (IISER) Kolkata for funding. T. B. thanks the UGC for a fellowship.Notes and references
- (a) F. Schierbaum, Carbon Dioxide as Chemical Feedstock, ed. M.Aresta, Wiley-VCH, 2010, vol. 62 Search PubMed; (b) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510 CrossRef CAS PubMed.
- (a) G. Holmes and D. W. Keith, Philos. Trans. R. Soc., A, 2012, 370, 4380–4403 CrossRef CAS PubMed; (b) M. Mahmoudkhani, K. R. Heidel, J. C. Ferreira, D. W. Keith and R. S. Cherry, Energy Procedia, 2009, 1, 1535–1542 CrossRef CAS.
- (a) C. Wu, Z. Zhang, Q. Zhu, H. Han, Y. Yang and B. Han, Green Chem., 2015, 17, 1467–1472 RSC; (b) X. Sun, Q. Zhu, X. Kang, H. Liu, Q. Qian, Z. Zhang and B. Han, Angew. Chem., Int. Ed., 2016, 55, 6771–6775 CrossRef CAS PubMed.
- (a) J.-Q. Wang, J. Sun, W.-G. Cheng, C.-Y. Shi, K. Dong, X.-P. Zhang and S.-J. Zhang, Catal. Sci. Technol., 2012, 2, 600–605 RSC; (b) J. H. Clements, Ind. Eng. Chem. Res., 2003, 42, 663–674 CrossRef CAS; (c) J. Bayardon, J. Holz, B. Schäffner, V. Andrushko, S. Verevkin, A. Preetz and A. Börner, Angew. Chem., Int. Ed., 2007, 46, 5971–5974 CrossRef CAS PubMed.
- A.-A. G. Shaikh and S. Sivaram, Chem. Rev., 1996, 96, 951–976 CrossRef CAS PubMed.
- (a) Q.-W. Song, Z.-H. Zhou and L.-N. He, Green Chem., 2017, 19, 3707–3728 RSC; (b) M. Cokoja, M. E. Wilhelm, M. H. Anthofer, W. A. Herrmann and F. E. Kühn, ChemSusChem, 2015, 8, 2436–2454 CrossRef CAS PubMed; (c) T. Ema, Y. Miyazaki, J. Shimonishi, C. Maeda and J. Hasegawa, J. Am. Chem. Soc., 2014, 136, 15270–15279 CrossRef CAS PubMed; (d) J. W. Comerford, I. D. V. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC; (e) J. A. Castro-Osma, M. North, W. K. Offermans, W. Leitner and T. E. Mìller, ChemSusChem, 2016, 9, 791–794 CrossRef CAS PubMed; (f) C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef; (g) L. M. Rodriguez, J. O. Garmilla and A. W. Kleij, ChemSusChem, 2016, 9, 1–8 CrossRef; (h) Q. Su, J. Sun, J. Wang, Z. Yang, W. Cheng and S. Zhang, Catal. Sci. Technol., 2014, 4, 1556–1562 RSC; (i) Q. Su, X. Yao, W. Cheng and S. Zhang, Green Chem., 2017, 19, 2957–2965 RSC; (j) T. Biswas and V. Mahalingam, New J. Chem., 2017, 41, 14839–14842 RSC; (k) S. Bhunia, R. A. Molla, V. Kumari, S. M. Islam and A. Bhaumik, Chem. Commun., 2015, 51, 15732–15735 RSC; (l) S. K. Das, S. Chatterjee, S. Bhunia, A. Mondal, P. Mitra, V. Kumari, A. Pradhan and A. Bhaumik, Dalton Trans., 2017, 46, 13783–13792 RSC; (m) J. A. Castro-Osma, J. Martínez, F. D. L. Cruz-Martínez, M. P. Caballero, J. Fernández-Baeza, J. Rodríguez-López, A. Otero, A. Lara-Sánchez and J. Tejeda, Catal. Sci. Technol., 2018, 8, 1981–1987 RSC; (n) F. D. L. Cruz-Martínez, J. Martínez, M. A. Gaona, J. Fernández-Baeza, L. F. Sánchez-Barba, A. M. Rodríguez, J. A. Castro-Osma, A. Otero and A. Lara-Sánchez, ACS Sustainable Chem. Eng., 2018, 6, 5322–5332 CrossRef; (o) M. Liu, X. Lu, L. Shi, F. Wang and J. Sun, ChemSusChem, 2017, 10, 1110–1119 CrossRef CAS PubMed; (p) M. Liu, J. Lan, L. Liang, J. Sun and M. Arai, J. Catal., 2017, 347, 138–147 CrossRef CAS.
- M. Peters, B. Kçhler, W. Kuckshinrichs, W. Leitner, P. Markewitz and T. E. Müller, ChemSusChem, 2011, 4, 1216–1240 CrossRef CAS PubMed.
- (a) X. Liu, S. Zhang, Q.-W. Song, X.-F. Liu, R. Maa and L.-N. He, Green Chem., 2016, 18, 2871–2876 RSC; (b) R. Babu, A. C. Kathalikkattil, R. Roshan, J. Tharun, D.-W. Kimb and D.-W. Park, Green Chem., 2016, 18, 232–242 RSC; (c) L. Wang, G. Zhang, K. Kodama and T. Hirose, Green Chem., 2016, 18, 1229–1233 RSC; (d) L. Wang, K. Kodama and T. Hirose, Catal. Sci. Technol., 2016, 6, 3872–3877 RSC; (e) R. Ma, L.-N. He and Y.-B. Zhou, Green Chem., 2016, 18, 226–231 RSC; (f) Y. Xie, T.-T. Wang, X.-H. Liu, K. Zou and W.-Q. Deng, Nat. Commun., 2013, 4, 1960–1967 CrossRef PubMed.
- (a) M. Vafaeezadeh, J. Aboudi and M. M. Hashemi, RSC Adv., 2015, 5, 58005–58009 RSC; (b) M. Ramdin, T. W. D. Loos and T. J. H. Vlugt, Ind. Eng. Chem. Res., 2012, 51, 8149–8177 CrossRef CAS; (c) L. Zhang, J. Chen, J. X. LV, S. F. Wang and Y. Cui, Asian J. Chem., 2013, 25, 2355–2358 CAS; (d) M. Wang, L. Zhang, L. Gao, K. Pi, J. Zhang and C. Zheng, Energy Fuels, 2013, 27, 461–466 CrossRef CAS.
- (a) V. B. Saptal and B. M. Bhanage, ChemSusChem, 2017, 10, 1145–1151 CrossRef CAS PubMed; (b) Y. Toda, Y. Komiyama, A. Kikuchi and H. Suga, ACS Catal., 2016, 6, 6906–6910 CrossRef CAS; (c) J.-Q. Wang, J. Sun, W.-G. Cheng, K. Dong, X.-P. Zhang and S.-J. Zhang, Phys. Chem. Chem. Phys., 2012, 14, 11021–11026 RSC; (d) J. Sun, S. Zhang, W. Cheng and J. Ren, Tetrahedron Lett., 2008, 49, 3588–3591 CrossRef CAS.
- (a) S. Soll, P. Zhang, Q. Zhao, Y. Wangb and J. Yuan, Polym. Chem., 2013, 4, 5048–5051 RSC; (b) W. Zhang, Q. Wang, H. Wu, P. Wu and M. He, Green Chem., 2014, 16, 4767–4774 RSC; (c) J. Li, D. Jia, Z. Guo, Y. Liu, Y. Lyu, Y. Zhou and J. Wang, Green Chem., 2017, 19, 2675–2686 RSC; (d) A. Dani, E. Groppo, C. Barolo, J. G. Vitillo and S. Bordiga, J. Mater. Chem. A, 2015, 3, 8508–8518 RSC; (e) W. Wang, C. Li, L. Yan, Y. Wang, M. Jiang and Y. Ding, ACS Catal., 2016, 6, 6091–6100 CrossRef CAS; (f) M. Buaki-Sogó, A. Vivian, L. A. Bivona, H. García, M. Gruttadauria and C. Aprile, Catal. Sci. Technol., 2016, 6, 8418–8427 RSC; (g) M. Buaki-Sogo, H. Garcia and C. Aprile, Catal. Sci. Technol., 2015, 5, 1222–1230 RSC; (h) D. Xing, B. Lu, H. Wang, J. Zhao and Q. Cai, New J. Chem., 2017, 41, 387–392 RSC; (i) S. G. Esfahani, H. Song, E. Păunescu, F. D. Bobbink, H. Liu, Z. Fei, G. Laurenczy, M. Bagherzadeh, N. Yana and P. J. Dyson, Green Chem., 2013, 15, 1584–1589 RSC; (j) W.-L. Dai, B. Jin, S.-L. Luo, S.-F. Yin, X.-B. Luo and C.-T. Au, J. CO2 Util., 2013, 3, 7–13 CrossRef; (k) Y. Xie, Z. Zhang, T. Jiang, J. He, B. Han, T. Wu and K. Ding, Angew. Chem., Int. Ed., 2007, 46, 7255–7258 CrossRef CAS PubMed; (l) W. Wang, C. Li, L. Yan, Y. Wang, M. Jiang and Y. Ding, ACS Catal., 2016, 6, 6091–6100 CrossRef CAS; (m) C. Li, W. Wang, L. Yan, Y. Wang, M. Jiang and Y. Ding, J. Mater. Chem. A, 2016, 4, 16017–16027 RSC; (n) W. Wang, Y. Wang, C. Li, L. Yan, M. Jiang and Y. Ding, ACS Sustainable Chem. Eng., 2017, 5, 4523–4528 CrossRef CAS; (o) M. Liu, L. Liang, X. Li, X. Gaoa and J. Sun, Green Chem., 2016, 18, 2851–2863 RSC.
- D. Deng, K. S. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11, 218–230 CrossRef CAS PubMed.
- B. Dong, L. Wang, S. Zhao, R. Ge, X. Song, Y. Wanga and Y. Gao, Chem. Commun., 2016, 52, 7082–7085 RSC.
- W. Zhong, F. D. Bobbink, Z. Fei and P. J. Dyson, ChemSusChem, 2017, 10, 2728–2735 CrossRef CAS PubMed.
- Y. Chen, R. Luo, Q. Xu, J. Jiang, X. Zhou and H. Ji, ChemSusChem, 2017, 10, 2534–2541 CrossRef CAS PubMed.
- V. Calo', A. Nacci, A. Monopoli and A. Fanizzi, Org. Lett., 2002, 4, 2561–2563 CrossRef.
- (a) T. Werner and N. Tenhumberg, J. CO2 Util., 2014, 7, 39–45 CrossRef CAS; (b) W. Chen, L.-X. Zhong, X.-W. Peng, R.-C. Sun and F.-C. Lu, ACS Sustainable Chem. Eng., 2015, 3, 147–152 CrossRef CAS; (c) M. Liu, K. Gao, L. Liang, J. Sun, L. Sheng and M. Arai, Catal. Sci. Technol., 2016, 6, 6406–6416 RSC.
- (a) P. Puthiaraj, S.-M. Cho, Y.-R. Lee and W.-S. Ahn, J. Mater. Chem. A, 2015, 3, 6792–6797 RSC; (b) R. Gomes, P. Bhanja and A. Bhaumik, Chem. Commun., 2015, 51, 10050–10053 RSC; (c) P. Katekomol, J. Roeser, M. Bojdys, J. Weber and A. Thomas, Chem. Mater., 2013, 25, 1542–1548 CrossRef CAS; (d) J. Roeser, K. Kailasam and A. Thomas, ChemSusChem, 2012, 5, 1793–1799 CrossRef CAS PubMed.
- (a) J. Yuan and M. Antonietti, Macromolecules, 2011, 44, 744–750 CrossRef CAS; (b) M. Taghavikish, S. Subianto, N. K. Dutta and N. R. Choudhury, ACS Appl. Mater. Interfaces, 2015, 7, 17298–17306 CrossRef CAS PubMed.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- D. Koa, J. S. Lee, H. A. Patel, M. H. Jakobsen, Y. Hwang, C. T. Yavuz, H. C. B. Hansen and H. R. Andersen, J. Hazard. Mater., 2017, 332, 140–148 CrossRef PubMed.
- A. K. S. Labban and Y. Marcus, J. Solution Chem., 1991, 20, 221–232 CrossRef CAS.
- (a) M. S. Kathalewar, P. B. Joshi, A. S. Sabnis and V. C. Malshe, RSC Adv., 2013, 3, 4110–4129 RSC; (b) O. Figovsky, L. Shapovalov, A. Leykin, O. Birukova and R. Potashnikova, Chem. Chem. Technol., 2013, 7, 79–87 CAS; (c) X. Sheng, G. Ren, Y. Qin, X. Chen, X. Wang and F. Wang, Green Chem., 2015, 17, 373–379 RSC; (d) S. Verma, M. Nazish, R. I. Kureshy and N.-U. H. Khan, Sustainable Energy Fuels, 2018, 2, 1312–1322 RSC; (e) P. Patel, B. Parmar, R. I. Kureshy, N.-U. H. Khan and E. Suresh, ChemCatChem, 2018, 10, 2401–2408 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, PXRD, FT-IR, TGA, 13C CP MAS, SEM, EDAX, TEM and NMR, the image of the reaction set-up, and balloon pressure measurement. See DOI: 10.1039/c8se00401c |
| This journal is © The Royal Society of Chemistry 2019 |