
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stabilization of GaAs photoanodes by in situ deposition of nickel-borate surface catalysts as hole trapping sites†
Chaoran
Jiang
 ac,
Jiang
Wu
bd,
Savio J. A.
Moniz
a,
Daqian
Guo
b,
Mingchu
Tang
b,
Qi
Jiang
b,
Siming
Chen
b,
Huiyun
Liu
b,
Aiqin
Wang
*c,
Tao
Zhang
c and
Junwang
Tang
*a
ac,
Jiang
Wu
bd,
Savio J. A.
Moniz
a,
Daqian
Guo
b,
Mingchu
Tang
b,
Qi
Jiang
b,
Siming
Chen
b,
Huiyun
Liu
b,
Aiqin
Wang
*c,
Tao
Zhang
c and
Junwang
Tang
*a
aDepartment of Chemical Engineering, University College London, Torrington Place, London, WC1E 7JE, UK. E-mail: junwang.tang@ucl.ac.uk
bDepartment of Electronic & Electrical Engineering, Torrington Place, London, WC1E 7JE, UK
cState Key Laboratory of Catalysis, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, 116023, China. E-mail: aqwang@dicp.ac.cn
dInstitute of Fundamental and Frontier Science, University of Electronic Science and Technology of China, Chengdu 610054, P. R. China
First published on 21st January 2019
Abstract
Although semiconducting gallium arsenide (GaAs) possesses an ideal band gap for efficient solar-driven fuel synthesis, it is extremely unstable in aqueous media, undergoing facile photocorrosion and therefore is seldom used. We have addressed this stability problem to some extent using a strategy of introducing a Ni–B surface catalyst onto p/n junction GaAs by in situ photoassisted electrodeposition. A monolithic layer of Ni–B/Ga(As)Ox was generated during the Ni–B deposition process, resulting in a Ni–B/Ga(As)Ox/GaAs photoanode structure. Such a structure was optimized by varying the GaAs surface architecture, electrolyte pH value and Ni–B deposition time to achieve optimal photoelectrochemical performance, together with improved stability. The optimized photoanode Ni–B/Ga(As)Ox/shallow GaAs with 0.5 h Ni–B deposition time (∼900 nm thickness of the Ni–B/Ga(As)Ox layer) exhibited a very high photocurrent, leading to a nearly 22 hour stable photocurrent density of 20 mA cm−2, while bare GaAs exhibits 60% photocurrent loss after three hours under continuous one sun illumination (100 mW cm−2) in alkaline media (pH = 14). This remarkable performance in both photocurrent and stability directly addresses the current severe limitations in the application of GaAs photoanodes for solar fuel synthesis, and they may be applicable to other unstable photoelectrodes.
Introduction
The direct light-driven splitting of water is one of the most cost-effective methods to convert solar energy into a storable and transportable form of energy (H2).1 Great efforts have been made to seek suitable semiconductor materials which can be used as photoelectrodes in a photoelectrochemical cell (PEC); however the majority of the state of the art materials suffer from either low stability or poor efficiency (STH < 10%).2 The target of the solar to fuel field of research is to achieve at least 10% solar to hydrogen efficiency with affordable stability. Among the typical III–V materials, such as InGaP, GaAs, and InP, GaAs could be an ideal material for PEC water splitting due to its intrinsically small band-gap (1.42 eV), and appropriate band positions, promising a high theoretical solar energy conversion efficiency of 33%.3 Therefore there is substantial interest in using group III–V materials, in particular GaAs, as photoelectrodes for PEC water splitting. However the stability of III–V materials is one of the major challenges for their application in PEC cells due to the fast photocorrosion at the interface between III–V materials and electrolyte.4,5In a PEC water splitting system, oxygen evolution is the rate determining step and thus research on utilizing GaAs as a photoanode has attracted considerable attention.2 One of the first studies reporting GaAs photoanodes for solar fuel synthesis was by Zhou et al.6 The as-synthesized GaAs nanowire arrays exhibited a solar energy conversion efficiency of 8.1% without a co-catalyst. However, the hydrogen production was not affordable due to the electrode instability (50% current loss in one hour). Basically, the surface dissolves during the photoelectrochemical reaction and this fast surface etching results in H2 gas generation being limited to approximately one hour. This etching is caused by generated holes which are not used for the oxidation of water at the GaAs surface while being accumulated to oxidize the GaAs layer. This is partly because the consumption rate of holes for O2 generation on the GaAs surface is much lower than that of electrons on the counter electrode because four holes are required to produce an O2 molecule while only two electrons are necessary for the generation of a H2 molecule. Nocera et al. developed an earth abundant oxygen-evolving catalyst (cobalt phosphate, Co–Pi) to improve the hole utilization efficiency for the oxidation reaction of water.7 The devices consisting of a Co–Pi modified silicon-based photoanode for oxygen evolution and a NiMoZn catalyst for hydrogen evolution exhibited relatively good stability (10 h) for water splitting with a solar to hydrogen conversion efficiency of 4.7% for a wired configuration and 2.5% for a wireless configuration.8 Recently an efficiency of 12.3% was reported using a perovskite photovoltaic together with a bifunctional earth-abundant catalyst but authors underlined its poor stability.9
Various coating strategies have been explored to stabilize narrow gap semiconductors in a PEC cell. Shao-Horn et al. reported an n/p-GaAs (001) photocathode that operated in neural pH, stabilized by an epitaxial SrTiO3 surface layer to deliver a photocurrent of 3.1 mA cm−2 at 0.18 V with 24 h stability.10 Lewis et al. coated a GaAs electrode with conformal amorphous TiO2 as a protection layer alongside a thin layer of Ni metal as a co-catalyst. The prepared photoanodes exhibited a photocurrent of 15 mA cm−2 at −0.2 V (vs. SCE) in 0.1 M KOH with almost one day constant operation.11 The same group also fabricated a NiOx film on p+n-InP photoanodes as both a protection layer and a surface oxygen evolution catalyst via sputtering to achieve 48 hour solar driven water oxidation.12 In fact, both the protection layer and surface catalyst are significant for high and stable photocurrent. Lewis et al. also demonstrated that n-cadmium telluride photoanodes could be stabilized for water oxidation by using amorphous TiO2 films formed by atomic layer deposition.13
We previously reported that an earth-abundant catalyst, nickel borate (Ni–B), can be coupled onto ZnO photoanodes to dramatically enhance the holes' efficiency for the water oxidation reaction and also improve the ZnO stability.14 Herein, in order to fabricate a stable and efficient device for solar fuel synthesis, we have focused on the Ni–B surface catalyst as a hole trapping site on top of GaAs to suppress the photocorrosion reaction caused by accumulated holes on the GaAs surface and facilitate charge separation. The p–n GaAs junction was designed firstly to facilitate a hole transfer pathway while maintaining a good light absorption. In situ photoassisted electrodeposition of Ni–B onto the p–n GaAs photoanode was able to generate a monolithic layer of Ni–B/Ga(As)Ox on GaAs, which acts as a catalytic and passivation layer to protect GaAs against photocorrosion. Such a procedure resulted in a stable photocurrent and the surface treatment for the loading of water oxidation catalysts (Ni–B) on GaAs was also discussed in detail.
Experimental section
1 p/n junction GaAs growth
The GaAs photoanodes were grown by using a Veeco Gen 930 molecular beam epitaxy system. Before loading the substrate into the epitaxy system, the GaAs substrate was thermally degassed to remove contaminants. The GaAs photoelectrodes were homoepitaxially grown on an n-type GaAs substrate (650 μm thickness). A thin layer of native oxide was thermally removed by holding the substrate at high temperature (600 °C) under the protection of arsenic flux for eight minutes. The GaAs growth temperature was 580 °C. An n-type GaAs buffer layer of 200 nm was first grown on the substrate. After the growth of the buffer layer, a p-type GaAs layer of 2000 nm was grown. The doping density was 2 × 1018 cm−3. The growth rate was ∼1 monolayer per second, controlled by adjusting the Ga source temperature. The temperature variation was very small within ± 0.1 °C controlled by using a PID controller, thus enabling a high reproducibility of the GaAs photoelectrode synthesis. Doping of the GaAs layers to make a p or n type semiconductor was achieved in situ during epitaxy growth, where Si and Be were used as n-type and p-type dopants, respectively. The growth was monitored with a reflection high-energy electron diffraction system, which includes a high energy electron gun, phosphor screen, and CCD camera.2 Pre-treatment of the GaAs surface
The as-prepared p/n junction GaAs samples were either etched or polished to promote attachment with surface catalysts Ni–B. For etched GaAs samples, a thin Ni layer (2 nm) was deposited on the sample surface by evaporation followed by thermal annealing at 500 °C to form surface Ni nanodots. The sample was then chemically etched by using the Ni nanodots as a mask. To further increase the surface roughness, the samples were polished using 1 micron grinding papers with a mechanical polisher. After polishing, the samples were washed with a solution mixture of H3PO4/H2O2/HCl in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 ratio and dried for use.
:1:1 ratio and dried for use.
3 Preparation of GaAs photoanodes
Ohmic contacts to the GaAs photoanodes were formed by evaporation of Ni/AuGe/Ni/Au metals on the back of the substrate and rapid thermal annealing at a preferred temperature of 400 °C. The GaAs electrodes were then wired by soldering a Cu wire to the alloyed metal contact. The samples were encapsulated by using epoxy (Sigma, resin:hardener = 1:1). The Ni–B catalyst was deposited onto the GaAs samples by photoassisted deposition in a three-electrode system with GaAs samples as working electrodes, Ag/AgCl as a reference electrode, and a Pt mesh as a counter electrode. Before loading Ni–B on the pre-treated GaAs surface, it was immersed into methanol solution for 60 s, followed by a 1:1 volume ratio of HCl and deionized (DI) water for another 60 s, and then rinsed with a large amount of DI water and dried under the stream of argon to make a clean surface. The photoassisted electrodeposition of Ni–B on GaAs was carried out by applying a constant bias of 0.5 V (vs. Ag/AgCl) in a solution of 0.1 M potassium borate containing 1 mM Ni (NO3)2 with varying deposition times under AM 1.5 G light illumination (100 mW cm−2).
4 Materials characterization
SEM images were obtained by using a Jeol JSM-7401F scanning electron microscope with an EDS detector. High-resolution XPS was performed by using a Thermo Scientific K-alpha photoelectron spectrometer with monochromatic AlKα radiation; peak positions were calibrated to carbon (284.5 eV) and plotted with CasaXPS software. Atomic force microscopy (AFM) images of the surface morphology were obtained using a Veeco Nanoscope atomic force microscope. The X-ray diffraction (XRD) patterns were obtained by using a Jordan Valley Bede D1 system.5 Photoelectrochemical measurements
The photoelectrochemical measurements were carried out in a gas-tight cell equipped with a quartz window. The prepared GaAs samples were used as the working electrodes. A Pt mesh and Ag/AgCl (3 M KCl) were used as the counter electrode and reference electrode, respectively. An electrolyte containing 0.1 M KOH (pH = 14) in deionized water, which was purged with argon gas prior to use for 20 min to remove dissolved oxygen, was used. The light source was a 150 W xenon lamp (Newport) equipped with a standard AM 1.5 G filter. The light intensity was calibrated to 100 mW cm−2 using a silicon photodiode, together with a hand held optical meter (Newport, 1918-R). A potentiostat (Ivium technology) was used to apply the bias at a scan rate of 20 mV s−1 between −1.5 and 1.0 V (vs. Ag/AgCl). The electrochemical impedance spectroscopy (EIS) data were obtained through the same electrochemical measurement system under AM 1.5-light illumination at an applied bias of −0.6 V (vs. Ag/AgCl) in 0.1 M potassium hydroxide solution with a frequency range of 100000 and 1 Hz of a sinusoidal perturbation with 10 mV amplitude.
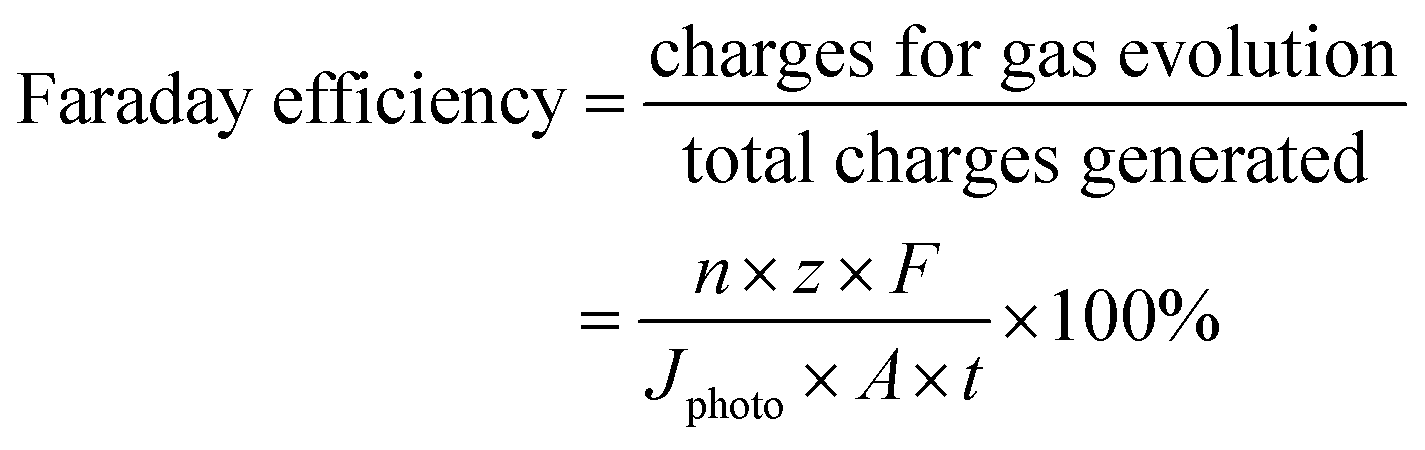 | (1) |
485 C mol−1; z = 4 is the number of electrons involved from the oxygen evolution reaction and z = 2 is the number of electrons involved from the hydrogen evolution reaction; Jphoto is the photocurrent density (A cm−2) generated during the measurement time (t) (seconds); A is the illumination area of the photoelectrode (cm2).
The solar to hydrogen conversion efficiency (η) was estimated by using the following equation:
 | (2) |
Results and discussion
p/n junction GaAs samples were fabricated by using commercial n-type GaAs (001) as the substrate for the epitaxial growth of 2000 nm p-type GaAs. The GaAs surface was treated before loading of Ni–B in order to increase the roughness of the GaAs surface for better attachment of the Ni–B catalyst, which would improve the stability of the photoanode and catalyse the water oxidation reaction. The surface of the GaAs samples was either textured by etching or polished with a mechanical polisher, annotated as textured (etched), shallow (moderately polished) or deep GaAs (deep polished) photoanodes based on the surface roughness while the untreated was named flat photoanode. Fig. 1 shows the AFM images of the surface treated GaAs before loading of Ni–B. The root mean square (RMS) roughness of the as-grown GaAs (flat GaAs) photoanode is about 0.25 nm. The RMS roughness of textured GaAs changes to 1.94 nm and the average peak to valley height is ∼10 nm. The RMS roughness increases after surface treatment with the mechanical polisher, resulting in shallow and deep dents on the surface. The measured RMS roughness is 8 nm and 39.2 nm for the shallow and deep treated GaAs surfaces, respectively and the average peak to valley height is increased by an order of five from the shallow to the deep treated GaAs photoanode.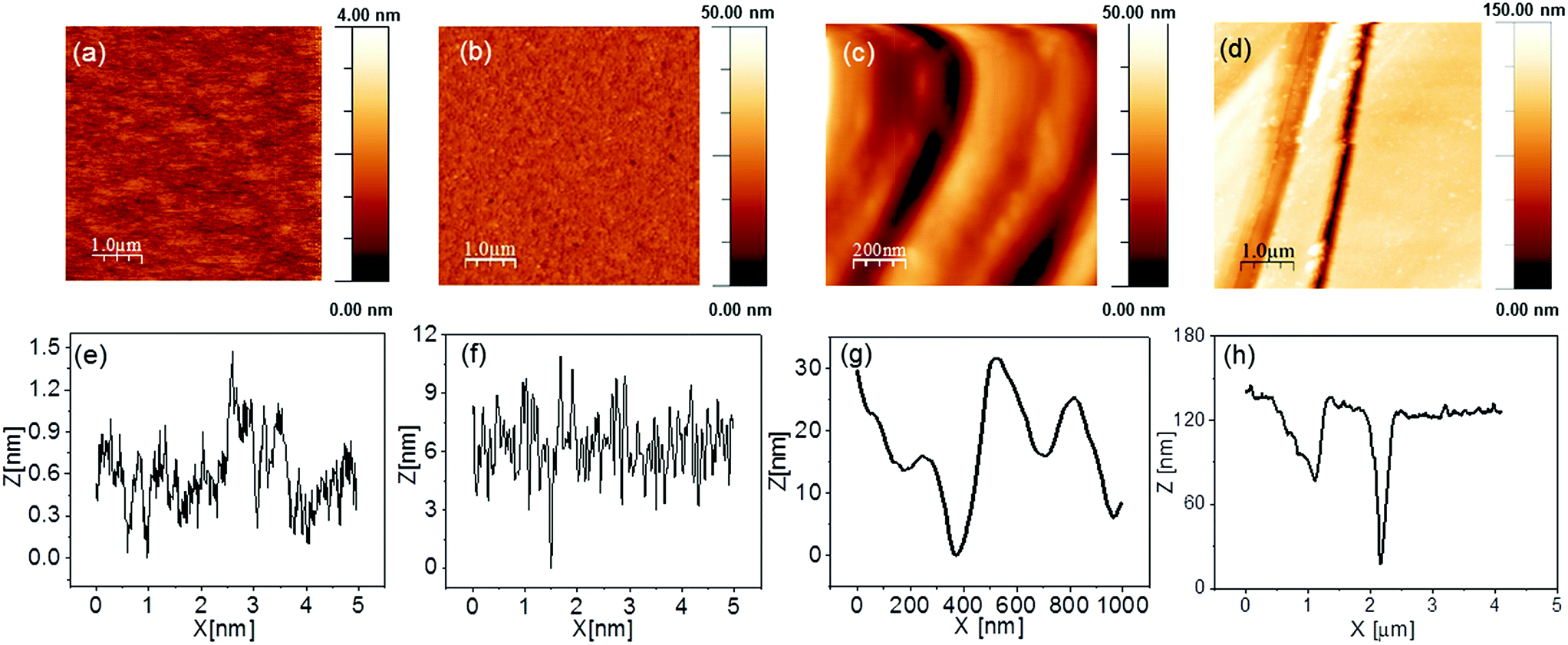 | ||
| Fig. 1 AFM images and the corresponding width–height plot of various GaAs films; (a and e) flat GaAs; (b and f) textured GaAs; (c and g) shallow GaAs; (d and h) deep GaAs. | ||
Fig. S1† shows the AFM image and the corresponding width–height plot of Ni–B/Ga(As)Ox/textured GaAs photoanodes with 0.5 h Ni–B photoassisted electrodeposition. After Ni–B loading, the RMS roughness of the textured GaAs surface increases from 1.94 nm to 30 nm and the peak to valley height of the textured GaAs surface increases from 10 nm to 15 nm, which indicates the change of the GaAs surface roughness after Ni–B modification.
X-ray photoelectron spectroscopy measurements (XPS) were carried out to monitor the valence change of the components in the fabricated photoanodes. The XPS spectra of 0.5 h photoassisted electrodeposited Ni–B onto shallow GaAs electrodes are shown in Fig. 2. Three Ga-based 3d peaks are observed. The peak at 19.1 eV corresponds to the GaAs substrate. The gallium oxides (GaOx) are composed of a mixture of Ga2O (Ga1+) and Ga2O3 (Ga3+) at a binding energy of 19.7 eV and 20.7 eV, respectively. The O 2s signal originates from gallium oxide formation during the photoassisted electrodeposition process. These values well match with the literature.15 The As 3d spectrum also confirms the presence of the As–Ga substrate (41.2 eV). The arsenic oxides (AsOx) are composed of a mixture of As2O3 (44.1 eV), As3+ native oxide (45.0 eV) and As2O5 (45.9 eV).16 Two Ni 2p peaks are observed at 855.2 and 872.8 eV, which likely correspond to Ni2+ or Ni3+. As the binding energies of these two states suffer from a high degree of overlap, it is difficult to distinguish the exact nature of Ni with confidence.17 For boron, the expected singlet peak is found at 191.1 eV, indicative of a B3+ environment and in agreement with the literature.14,18 Also, the surface residual potassium species from the potassium borate electrolyte are confirmed by XPS of K 2p spectra (Fig. S2†). Such samples are denoted as Ni–B/Ga(As)Ox/shallow GaAs. Moreover, the X-ray diffraction patterns of Ni–B/Ga(As)Ox/shallow GaAs reveal a highly crystallized GaAs substrate with (001) orientation and the amorphous nature of Ni–B (Fig. S3†).19–21 Further increasing photoassisted electrodeposition time to 1 h results in the disappearance of GaAs peaks from both Ga 3d and As 3d spectra, but the Ni 2p and B 1s spectra remain the characteristic of the Ni–B catalyst (Fig. S4†). This observation indicates that the thickness of the Ni–B/Ga(As)Ox layer could be controlled during the photoassisted electrodeposition of the Ni–B catalyst.
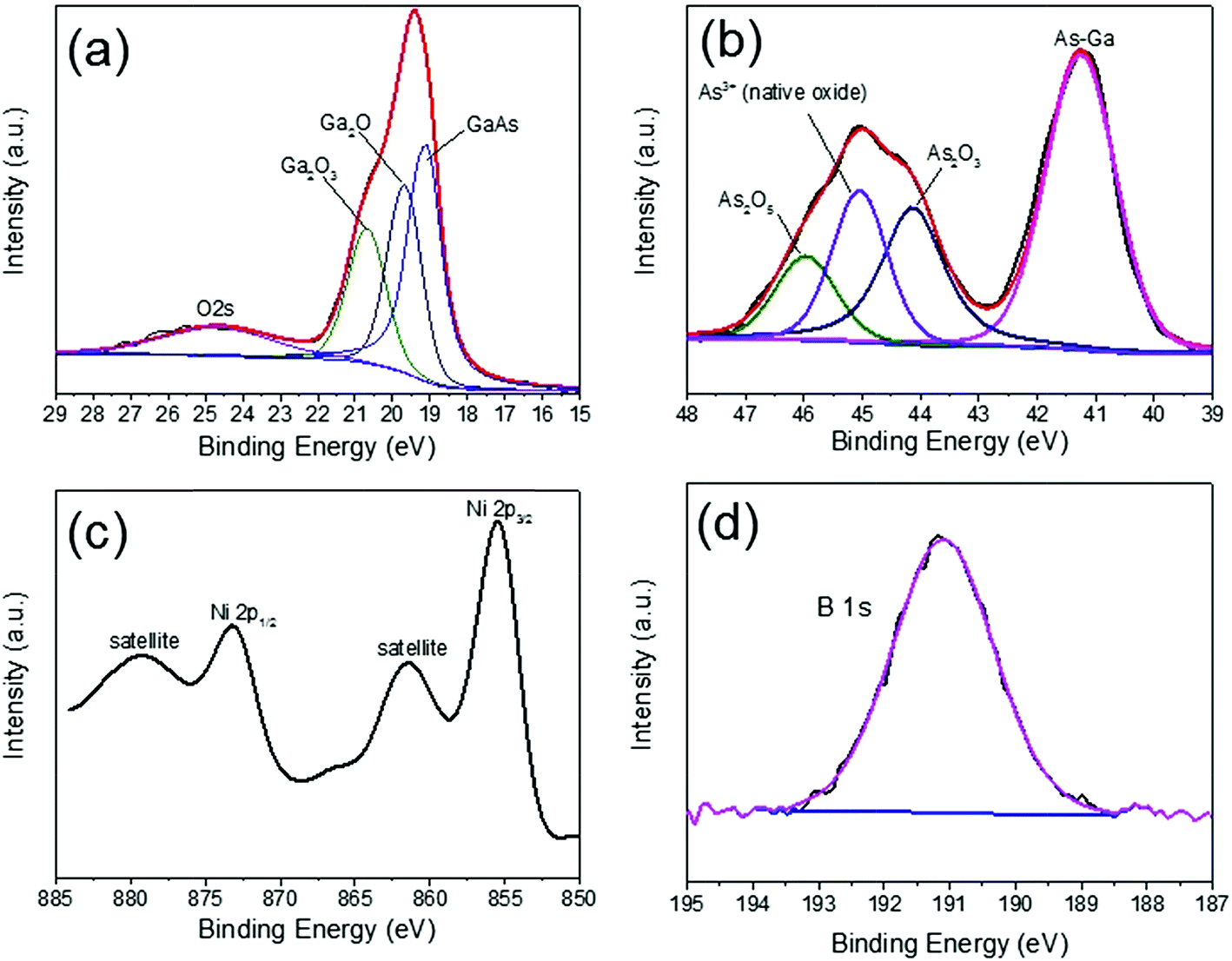 | ||
| Fig. 2 XPS spectra of Ga 3d, As 3d, Ni 2p and B1s of the Ni–B/Ga(As)Ox/shallow GaAs photoanode with the 0.5 h photoassisted electrodeposited Ni–B catalyst. | ||
The SEM image of a shallow GaAs surface is shown in Fig. S5a,† which indicates an uncorroded GaAs electrode before the photoelectrochemical reaction. Fig. 3a and b show the cross-sectional and top-down morphology of shallow GaAs decorated with a Ni–B catalyst with 0.5 h deposition time, respectively. The cross-sectional SEM image in Fig. 3a displays a layer of Ni–B/Ga(As)Ox with a thickness of ca. 900 nm on top of the GaAs surface. The monolithic Ni–B/Ga(As)Ox layered structure might have resulted from Ni–B loading and Ga(As)Ox passivation layer formation during the photoassisted simultaneous electrodeposition. The line analysis using SEM-EDS (the inset in Fig. 3a) throughout the Ni–B/GaAs layer indicates the co-existence of Ni, Ga, As and O in the top 900 nm while more Ni and O exist in the top 550 nm maybe in the form of NiOx. B is only observed on the surface (Fig. 3d), suggesting that Ni–B is only formed on the surface. The top-down SEM image (Fig. 3b) and the corresponding elemental mapping images (Fig. 3c–g) further provide the evidence of Ni–B/Ga(As)Ox layer formation on top of GaAs and furthermore, and evenly distributed Ni element is observed. This observation illustrates the in situ formation of the Ga(As)Ox layer from loading the Ni–B catalyst on the GaAs substrate, which is consistent with XPS results.
 | ||
| Fig. 3 Ni–B/Ga(As)Ox/shallow GaAs electrode: (a) cross-sectional SEM image and the line analysis throughout the cross section (inset). (b) Top-down SEM images. (c–g) SEM-DES micrograph and maps of distribution of elements on the electrode surface for gallium (Ga), oxygen (O), arsenide (As), nickel (Ni), and boron (B). | ||
The PEC performance of the as-prepared GaAs based photoanodes was investigated in a three-electrode configuration. Initially, we investigated the effect of Ni–B surface catalysts on PEC water oxidation performance. Fig. 4 shows the photocurrent density as a function of applied voltage under one sun illumination (100 mW cm−2) and dark current for both bare shallow GaAs and Ni–B/Ga(As)Ox/GaAs photoanodes in 0.1 M potassium hydroxide electrolyte (pH = 14). In comparison with bare shallow GaAs, which has a dark current onset potential of 0.5 V (vs. Ag/AgCl), the dark current onset is cathodically shifted to 0.25 V (vs. Ag/AgCl) for the Ni–B/Ga(As)Ox/shallow GaAs photoanode (Fig. 4b). This significant shift in onset potential for dark current (by 0.25 V) after Ni–B loading indicates the catalytic effect and improved water oxidation kinetics by Ni–B. However, no significant photocurrent onset potential shift is observed between bare shallow GaAs and Ni–B/Ga(As)Ox/shallow GaAs photoanodes (Fig. 4a). The flat band position of Ni–B/Ga(As)Ox/shallow GaAs was determined by Mott–Schottky measurements to be −1.6 V vs. Ag/AgCl (Fig. S6† at pH = 14), consistent with previously reported GaAs.22 The conduction band is believed to approximate to −1.6 V vs. Ag/AgCl. Based on the band gap of ca. 1.4 eV, the valence band of GaAs is ca. −0.2 V relative to Ag/AgCl. Taking into account that the potential for water oxidation, E (O2/H2O), is about 0.19 V (vs. Ag/AgCl) at pH = 14,2,11 the onset potential should be about −1.2 V (vs. Ag/AgCl), which is almost consistent with our experimental observation. The saturated photocurrent is observed when the bias is above −0.6 V (vs. Ag/AgCl), which is due to the increased water oxidation and simultaneous reduction of photocorrosion by the Ni–B catalyst. The overall photocurrent density also increases by loading the Ni–B catalyst onto GaAs, resulting in photocurrent density increasing from 10 to 20 mA cm−2 at −0.6 V (vs. Ag/AgCl). Furthermore, the oxygen evolution on both the GaAs and Ni–B/Ga(As)Ox/GaAs photoanodes was analyzed at a photocurrent of 20 mA cm−2, as shown in Fig. 4c. One can see that there is no oxygen gas produced at all on the bare GaAs photoanode. However, linear oxygen evolution is observed with time on the Ni–B/Ga(As)Ox/GaAs photoanode, proving that the enhanced photocurrent is due to water oxidation by the Ni–B catalyst. Therefore, we attribute such an enhancement in photocurrent to the catalytic effect of the Ni–B catalyst while loading on GaAs. In addition, the Ni–B catalysts could mitigate hole–electron recombination by acting as a hole-trapping site under sufficient bias applied (above −0.6 V vs. Ag/AgCl (pH = 14)) to increase charge separation efficiency as proved previously.14
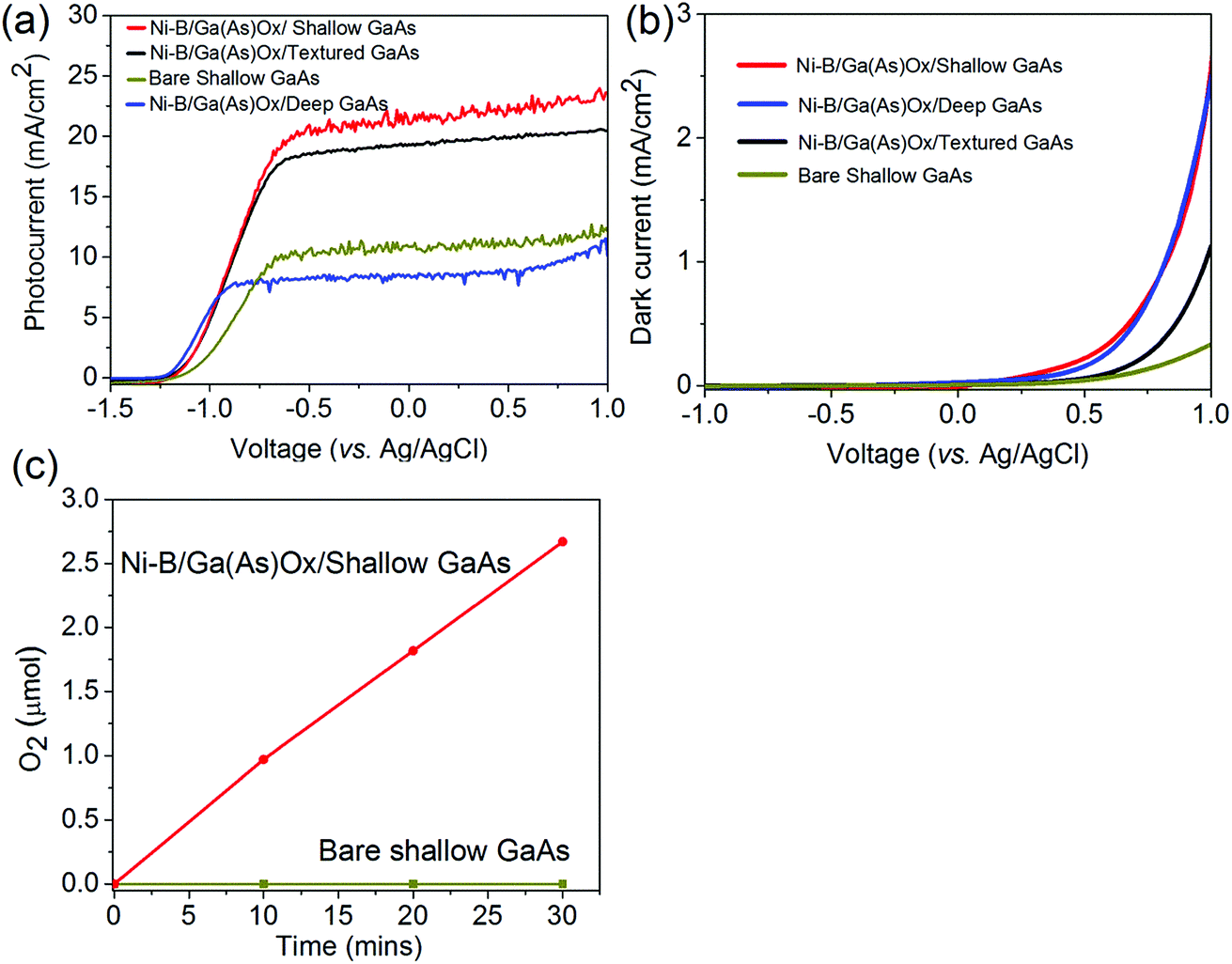 | ||
| Fig. 4 Photocurrent (a) and dark current (b) of Ni–B/Ga(As)Ox/GaAs photoelectrodes with 0.5 h Ni–B photoassisted electrodeposition and various GaAs surface architectures by using bare shallow GaAs as a control sample. All photoelectrodes are evaluated in 0.1 M potassium hydroxide electrolyte under one sun illumination (100 mW cm−2). (c) Time profile of O2 generation during the photoelectrochemical water splitting reaction in a gas-tight three electrode cell at a constant applied potential of −0.6 vs. Ag/AgCl using Ni–B/Ga(As)Ox/shallow GaAs and bare shallow GaAs photoanodes (counter electrode: Pt mesh; reference electrode: Ag/AgCl; electrolyte: 0.1 M potassium hydroxide; light source: AM 1.5 light irradiation,100 mW cm−2). | ||
Furthermore, we optimised the Ni–B/Ga(As)Ox/GaAs electrodes for PEC water oxidation performance based on three factors including: (1) GaAs surface architecture; (2) electrolyte pH value; (3) Ni–B surface catalyst deposition time.
1 Effect of GaAs surface architecture
The surface architecture and roughness of the GaAs substrate are believed to affect the loading profile of the Ni–B catalyst, which was then studied by using textured, shallow, and deep GaAs samples. The current–voltage curves in Fig. 4a show the photocurrent of the as-treated GaAs photoanodes with Ni–B loading, which is strongly dependent on surface roughness. The deep GaAs surface likely has a thicker Ni–B/Ga(As)Ox layer than the textured and shallow GaAs surface while keeping other deposition parameters constant due to the larger surface area generated. The thickness of the Ni–B/Ga(As)Ox monolithic layer deposited on shallow and deep GaAs surfaces is measured to be 900 nm (Fig. 3a) and 1200 nm (Fig. S7†), respectively. The thick Ni–B/Ga(As)Ox layer on the GaAs photoanode surface results in not only light loss due to Ni–B absorption but also a high possibility of charge carrier recombination in the bulk due to the long diffusion length of photo-generated holes to the reaction sites.23 The deep GaAs surface treated with the mechanical polisher may also have larger surface recombination centers. Consequently, in comparison with Ni–B/Ga(As)Ox/deep GaAs with 0.5 h Ni–B loading by photoelectrochemistry, which exhibits a photocurrent of 8 mA cm−2 at −0.6 V (vs. Ag/AgCl), the photocurrent of Ni–B/Ga(As)Ox/shallow GaAs and Ni–B/Ga(As)Ox/textured GaAs photoanodes increases to 20 mA cm−2 and 18 mA cm−2 at −0.6 V (vs. Ag/AgCl) under one sun illumination, respectively. The noisy photocurrent as shown is due to the bubbles generated on the electrodes as well as the vigorous stirring during the photocurrent measurements. Because of the high photocurrents achieved on both shallow and textured samples, they were used for the subsequent studies.2 Effect of the electrolyte pH value
Next, the effect of electrolyte pH on the PEC activity of Ni–B/Ga(As)Ox/textured GaAs photoanodes was investigated. As shown in Fig. S8,† the photocurrent onset potential shifts to a more negative potential (0.059 V per pH unit) with the pH value increasing. For the Ni–B/Ga(As)Ox/textured GaAs photoanodes evaluated in 0.1 M potassium borate (pH = 9.2), photoinduced holes accumulate rapidly on the surface due to efficient electron–hole separation when applying a bias higher than −0.4 V (vs. Ag/AgCl), whereas there are not enough OH− groups in the electrolyte to be oxidized by accumulated holes, and thus the surface electron–hole recombination occurs, resulting in a large current drop at high applied bias. This also proves the high efficiency of the Ni–B catalyst for water oxidation in part. When increasing the concentration of OH− ions in the electrolyte (pH = 10.2), no current drop is observed and upon further increasing the pH value to 14, the highest photocurrent of 18 mA cm−2 was obtained. Therefore, the electrolyte was fixed as 0.1 M KOH solution (pH = 14) for further PEC measurements.3 Effect of Ni–B surface catalyst deposition time
The loading amount of Ni–B by photoelectrochemistry depends on not only the surface architecture of GaAs electrodes, but also the loading time. Since the Ni–B loading amount on the GaAs surface dominates the stability and activity of the photoanode, various Ni–B/Ga(As)Ox/GaAs photoanodes were prepared by photoelectrodeposition with the electrolyte, applied potential and light intensity fixed, but varying deposition times from 0 h to 1 h for further study. Fig. 5a and the inset show the photocurrent and dark current of the Ni–B/Ga(As)Ox/shallow GaAs photoanodes, respectively. The Ni–B/Ga(As)Ox/shallow GaAs photoanode with 1 h Ni–B loading time exhibits the lowest onset potential under dark conditions, which is cathodically shifted by about 0.3 V compared to bare shallow GaAs and Ni–B/Ga(As)Ox/shallow GaAs photoanodes with 0.5 h Ni–B loading time. This observation proves the excellent catalytic effect of Ni–B with a long deposition time. However, this photoanode does not exhibit the highest photocurrent under illumination. According to the Nyquist plots (Fig. 5b), the electrodes exhibit semicircles with different radii. The fittings for the semicircles indicate that bare shallow GaAs has an interfacial charge transfer resistance (Rct) of 15 kΩ under light irradiation. The Rct of the Ni–B/Ga(As)Ox/shallow GaAs photoanode with 0.5 h Ni–B loading time is significantly reduced by a factor of 10 (1.5 kΩ) compared to bare shallow GaAs. Hence the charge transfer should be enhanced by the Ni–B surface catalyst, which may result in enhanced hole transfer to surface Ni states. However, while increasing the photo-assisted deposition time to 1 h, a dense Ga(As)Ox layer is formatted together with Ni–B deposition on the GaAs surface and thus results in increased resistance (4 kΩ). Meanwhile, dense Ga(As)Ox may cause poor light absorption. Therefore, it is crucial to control the Ni–B photo electrodeposition time. The Ni–B/Ga(As)Ox/shallow GaAs photoanode with 0.5 h Ni–B deposition time exhibits a saturated photocurrent of 20 mV cm−2 at −0.6 V (vs. Ag/AgCl), which is two-fold higher than that of bare shallow GaAs (10 mA cm−2 at −0.6 V (vs. Ag/AgCl)). We also investigated the current–voltage characteristics of textured GaAs with varying Ni–B photo-assisted electrodeposition time. The photocurrent (Fig. S9a†) and dark current (Fig. S9b†) of Ni–B/Ga(As)Ox/textured GaAs exhibit a current–voltage characteristic analogous to Ni–B/Ga(As)Ox/shallow GaAs photoanodes. Ni–B/Ga(As)Ox/textured GaAs with 1 h Ni–B loading time still shows the lowest onset potential (0.2 V vs. Ag/AgCl) under dark conditions and Ni–B/Ga(As)Ox/textured GaAs with 0.5 h Ni–B loading time exhibits the highest photocurrent (18 mA cm−2 at −0.6 V vs. Ag/AgCl). The photocurrent of GaAs photoanodes increases dramatically after Ni–B loading because the Ni–B complex functions as a hole trapping site for in situ charge separation, thus reducing surface recombination and improving the water oxidation kinetics.24,25 During the photoelectrochemical process, the electron–hole pairs generated by the light-absorbing semiconductor are separated by an internal electrical field. Photogenerated electrons move towards the Pt counter electrode through the external circuit under external bias (for H2 evolution) and photogenerated holes migrate to the Ni–B surface and oxidize Ni from 2+ to 3+, and then O2 evolution occurs by withdrawal of electrons from water, accompanied by a change in the Ni oxidation state back to 2+.14 | ||
| Fig. 5 (a) Photocurrent and dark current (inset) of the Ni–B/Ga(As)Ox/shallow GaAs photoanode with varying deposition time; (b) impedance analysis (Nyquist plots) of bare shallow GaAs and Ni–B/Ga(As)Ox/shallow GaAs electrodes; all photoanodes were evaluated in 0.1 M potassium hydroxide (pH = 14) under one sun illumination (100 mW cm−2). | ||
Altogether, the Ni–B loading amount, which can be controlled by the GaAs surface texture and deposition time by photoassisted electrodeposition, has a significant effect on the PEC performance of Ni–B/Ga(As)Ox/GaAs photoanodes. Therefore, the best performing photoanode with the highest photocurrent is Ni–B/Ga(As)Ox/shallow GaAs with 0.5 h photoelectrochemical Ni–B catalyst deposition, which exhibits a saturated photocurrent of 20 mA cm−2 at a potential of −0.6 V (vs. Ag/AgCl) in 0.1 M KOH electrolyte under one sun illumination, in comparison to 6, 18, and 8 mA cm−2 at −0.6 V (vs. Ag/AgCl) on the flat, textured and deep GaAs photoanodes, respectively.
The stability of a photoelectrode under prolonged illumination in aqueous solution is one of the major criteria for its commercial application, which is a critical issue for GaAs-based electrodes as discussed in the Introduction.26 Therefore, the stability of both the bare shallow GaAs and Ni–B/Ga(As)Ox/shallow GaAs photoanodes was monitored in 0.1 M potassium hydroxide solution (pH = 14) at a constant applied potential of −0.6 V (vs. Ag/AgCl) under one sun illumination, see Fig. 6a. The photocurrent of the bare shallow GaAs photoanode decays very sharply during the first three hours of irradiation, resulting in 60% photocurrent loss caused by the rapid etching of the GaAs surface by surface accumulated holes. After that it shows a relatively stable photocurrent of ca. 6 mA cm−2 because of a dense oxide protection layer formed (Fig. S10b†). In contrast, the optimized Ni–B/Ga(As)Ox/shallow GaAs photoanode with 0.5 h Ni–B deposition time shows excellent stability (zero photocurrent loss within 22 h) when measured in 0.1 M KOH electrolytes with continuous argon purge and electrolyte replacement every 12 hours. It has been reported that glass corrodes in an alkaline solution and the produced contaminants, such as lead and silica, can influence the electrochemical activity on Pt electrodes.27 This effect is evidenced by the gradual decrease of photocurrent during the first 12 hours, which recovers after the replacement of a fresh electrolyte. To minimize such contamination at the solid–liquid interface, the electrolyte was replaced with a fresh one after 12 hours. The photoelectrochemical stability of Ni–B/Ga(As)Ox/shallow GaAs is comparable with that of GaAs photoanodes protected by atomic-layer deposited amorphous TiO2 in combination with a thin layer of Ni metal surface catalysts, exhibiting a saturated photocurrent of 15 mA cm−2 with 24 h durability under identical experimental conditions.11 The morphology of the obtained GaAs based electrodes after 6 hours of PEC reaction was revealed by the SEM images as shown in Fig. S10.† Typically, a 4 μm thick Ga(As)Ox passivation layer is formed on the bare shallow GaAs surface due to the photooxidation of GaAs (Fig. S10b†). As expected, after 6 hours of PEC reaction, Ni–B/Ga(As)Ox/shallow GaAs with 0.5 h Ni–B deposition time has the same Ni–B/Ga(As)Ox layer thickness of ca. 900 nm as the freshly prepared Ni–B/Ga(As)Ox/shallow GaAs (Fig. 3a and S10a†). In addition, as shown in Fig. S11,† the Ni 2p spectra of Ni–B/Ga(As)Ox/shallow GaAs after PEC measurements reveal no obvious changes in chemical states, which is also in good agreement with the previously reported enhanced stability of Ni–B protected ZnO and BiVO4 photoelectrodes.14,18 The faradaic efficiency of O2 and H2 generation by employing the Ni–B/Ga(As)Ox/shallow GaAs photoanode together with a Pt counter electrode is about 36% and 75%, respectively (details are given in ESI, Fig. S12†). In contrast, the faradaic efficiency of O2 production on the bare shallow GaAs photoanode is almost zero during the identical measurement (Fig. 4c), proving that the Ni–B surface cocatalyst acts as a hole trapping site on the GaAs surface to promote the water oxidation reaction and prevent the photocorrosion of the photoanode in part although it is not ideal at present, which we believe is due to voids formed in the top Ni–B/Ga(As)Ox layer as shown in Fig. 3b. However such a facile strategy has been successfully demonstrated and the synthesis of a more dense top layer free of voids is underway.
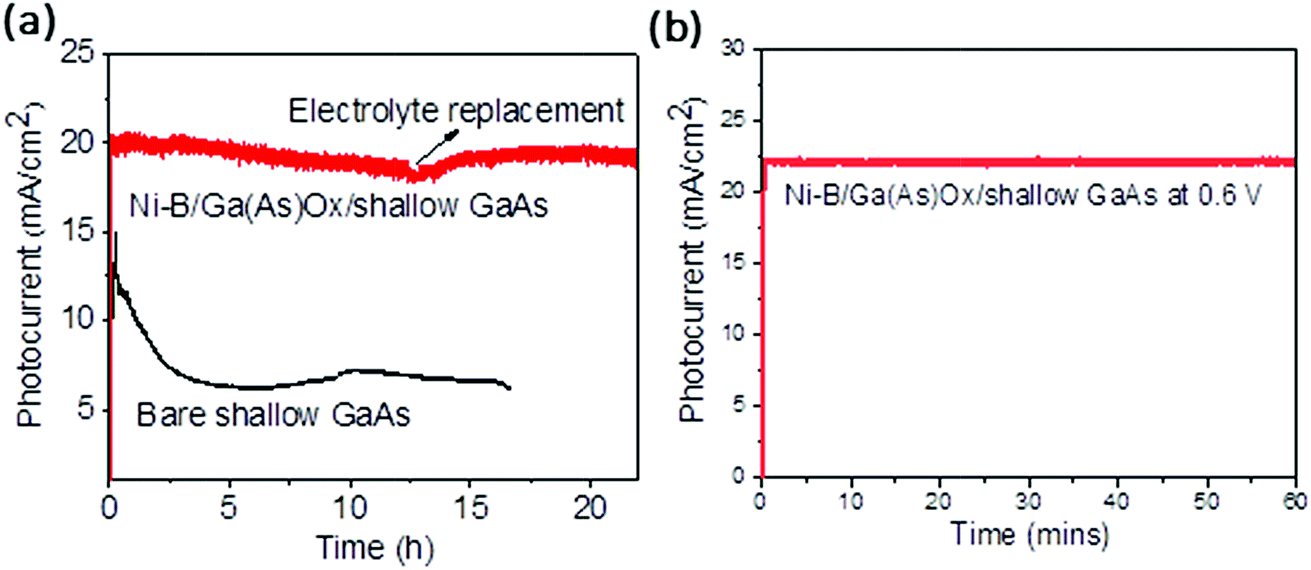 | ||
| Fig. 6 Current–time plot of Ni–B/Ga(As)Ox/shallow GaAs with continuous argon purge and electrolyte replacement every 12 hours in 0.1 M potassium hydroxide electrolyte (pH = 14): (a) measured in a three-electrode system with a constant applied potential of −0.6 V (vs. Ag/AgCl) by using bare shallow GaAs as a control sample and (b) a two-electrode system when the bias is 0.6 V. | ||
The high activity and stability of the Ni–B/Ga(As)Ox/shallow GaAs photoanode are attributed to (i) the surface protection layer of Ga(As)Ox, which is deposited in situ on the GaAs surface during photoassisted electrodeposition of Ni–B and more importantly (ii) the efficient catalytic effect of Ni–B for water oxidation. Without Ni–B modification, all holes generated in GaAs are consumed for GaAs oxidation, which is the typical photocorrosion process of GaAs, whereas surface Ni–B catalysts can efficiently trap the photogenerated holes and speed up the water oxidation reaction, and thus suppress the GaAs self-oxidation reaction. The deposited porous Ni–B/Ga(As) layer allows the direct water attack of the substrate, which is the reason why the faradaic efficiency of O2 production is smaller than that of H2. In addition, conducting PEC measurements in potassium hydroxide (KOH) electrolyte not only yields higher photocurrents but also has little effect on the photoelectrode stability. The energy conversion efficiency, i.e., conversion efficiency from incident light energy to stored H2 chemical energy, was calculated to be ca 15% based on the observed photocurrent and H2 Faraday efficiency. Furthermore, the photocurrent was measured in a two-electrode system for Ni–B/Ga(As)Ox/shallow GaAs (Fig. 6b), indicating its photostability again. These results demonstrate a promising strategy, comprised of the in situ formation of a Ni–B/Ga(As)Ox protection layer on the GaAs substrate, which ultimately leads to a stable and efficient photoanode for solar energy conversion.
Conclusions
Overall, we have demonstrated a novel strategy to achieve a very high and more importantly stable photocurrent by using GaAs photoanodes using a reproducible and robust method. The as-prepared photoanodes were characterized by AFM, SEM with EDS mapping, and XPS, which suggested in situ formation of a Ni–B/Ga(As)Ox layer during Ni–B photo-assisted electrodeposition. This Ga(As)Ox acts as a passivation layer which not only prevents the GaAs substrate from photocorrosion in part but also does not block photogenerated hole transfer to the Ni–B surface catalyst. The Ni–B on the p–n GaAs substrate plays a key role in reducing the photocorrosion (photooxidation of GaAs) from continuous operation and enhancing the water oxidation kinetics of the surface catalyst. The optimized photoanode with the highest photocurrent is Ni–B/Ga(As)Ox/shallow GaAs with a 0.5 h photoassisted electrochemical loaded Ni–B catalyst, which exhibits a saturated photocurrent of 20 mA cm−2 under one sun illumination compared with 10 mA cm−2 of the bare shallow GaAs photoanode at an identical potential of −0.6 V (vs. Ag/AgCl) in 0.1 M KOH electrolyte. In addition, the photocurrent can be maintained for at least 22 hours without decay under continuous operation while bare GaAs exhibits 60% photocurrent loss after three hours. Therefore, the current system demonstrates strong potential for scale-up to produce a device capable of efficient H2 synthesis and the novel strategy may be applicable to other unstable group III–V based photoelectrodes. A further step to reduce the voids of the top catalyst layer is under way.Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. J. acknowledges financial support from the China Scholarship Council (CSC File No. 201308060090). S. J. A. M. and J. T. acknowledge funding from EPSRC (EP/N009533/1), Leverhulme Trust (Grant No: RPG-2017-122) and Newton Advanced Fellowship grant (NA150418).References
- J. Turner, Nat. Mater., 2008, 7, 770–771 CrossRef CAS PubMed
.
- C. Jiang, S. J. Moniz, A. Wang, T. Zhang and J. Tang, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC
- J. R. Bolton, S. J. Strickler and J. S. Connolly, Nature, 1985, 316, 495–500 CrossRef CAS
- T. G. Deutsch, C. A. Koval and J. A. Turner, J. Phys. Chem. B, 2006, 110, 25297–25307 CrossRef CAS PubMed
- O. Khaselev and J. A. Turner, Science, 1998, 280, 425–427 CrossRef CAS PubMed
- S. Hu, C.-Y. Chi, K. T. Fountaine, M. Yao, H. A. Atwater, P. D. Dapkus, N. S. Lewis and C. Zhou, Energy Environ. Sci., 2013, 6, 1879–1890 RSC
- M. W. Kanan and D. G. Nocera, Science, 2008, 321, 1072–1075 CrossRef CAS PubMed
- S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645–648 CrossRef CAS PubMed
- J. Luo, J.-H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N.-G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed
- L. Kornblum, D. Fenning, J. Faucher, J. Hwang, A. Boni, M. Han, M. Morales-Acosta, Y. Zhu, E. Altman, M. Lee, C. H. Ahn, F. J. Walker and Y. Shao-Horn, Energy Environ. Sci., 2017, 10, 377–382 RSC
- S. Hu, M. R. Shaner, J. A. Beardslee, M. Lichterman, B. S. Brunschwig and N. S. Lewis, Science, 2014, 344, 1005–1009 CrossRef CAS PubMed
- K. Sun, Y. Kuang, E. Verlage, B. S. Brunschwig, C. W. Tu and N. S. Lewis, Adv. Energy Mater., 2015, 5, 1402276 CrossRef
- M. F. Lichterman, A. I. Carim, M. T. McDowell, S. Hu, H. B. Gray, B. S. Brunschwig and N. S. Lewis, Energy Environ. Sci., 2014, 7, 3334–3337 RSC
- C. Jiang, S. J. a. Moniz, M. Khraisheh and J. Tang, Chemistry, 2014, 20, 12954–12961 CrossRef CAS PubMed
- A. J. Henegar, A. J. Cook, P. Dang and T. Gougousi, ACS Appl. Mater. Interfaces, 2016, 8, 1667–1675 CrossRef CAS PubMed
- H. Kuhr, W. Ranke and J. Finster, Surf. Sci., 1986, 178, 171–178 CrossRef CAS
- M. J. Kenney, M. Gong, Y. Li, J. Z. Wu, J. Feng, M. Lanza and H. Dai, Science, 2013, 342, 836–840 CrossRef CAS PubMed
- S. K. Choi, W. Choi and H. Park, Phys. Chem. Chem. Phys., 2013, 15, 6499–6507 RSC
- T. Jin, P. Diao, D. Xu and Q. Wu, Electrochim. Acta, 2013, 114, 271–277 CrossRef CAS
- M. Dincă, Y. Surendranath and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10337–10341 CrossRef PubMed
- E. Janik, P. Dluzewski, S. Kret, A. Presz, H. Kirmse, W. Neumann, W. Zaleszczyk, L. T. Baczewski, A. Pietruczik, E. Dynowska, J. Sadowski, W. Caliebe, G. Karczewski and T. Wojtowicz, Catalytic growth of ZnTe nanowires by molecular beam epitaxy: structural studies, Nanotechnology, 2007, 18, 475606 CrossRef
- P. Allongue and S. Blonkowski, Corrosion of III-V compounds; a comparative study of GaAs and InP: Part I. Electrochemical characterization based on Tafel plot measurements, J. Electroanal. Chem. Interfacial Electrochem., 1991, 316, 57–77 CrossRef CAS
- S. J. a. Moniz, J. Zhu and J. Tang, Adv. Energy Mater., 2014, 4, 1301590 CrossRef
- M. Gao, W. Sheng, Z. Zhuang, Q. Fang, S. Gu, J. Jiang and Y. Yan, J. Am. Chem. Soc., 2014, 136, 7077–7084 CrossRef CAS PubMed
- D. K. Bediako, Y. Surendranath and D. G. Nocera, J. Am. Chem. Soc., 2013, 135, 3662–3674 CrossRef CAS PubMed
- D. Bae, B. Seger, P. C. K. Vesborg, O. Hansen and I. Chorkendorff, Chem. Soc. Rev., 2017, 46, 1933–1954 RSC
- K. J. J. Mayrhofer, A. S. Crampton, G. K. H. Wiberg and M. Arenz, J. Electrochem. Soc., 2008, 155, 78–81 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8se00265g |
| This journal is © The Royal Society of Chemistry 2019 |