
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSecondary amine selective Petasis (SASP) bioconjugation†
Yonnette E.
Sim
 a,
Ogonna
Nwajiobi
b,
Sriram
Mahesh
b,
Ryan D.
Cohen
ac,
Mikhail Y.
Reibarkh
c and
Monika
Raj
*b
a,
Ogonna
Nwajiobi
b,
Sriram
Mahesh
b,
Ryan D.
Cohen
ac,
Mikhail Y.
Reibarkh
c and
Monika
Raj
*b
aDepartment of Chemistry and Biochemistry, Seton Hall University, South Orange, New Jersey 07079, USA
bDepartment of Chemistry and Biochemistry, Auburn University, Auburn, Alabama 36830, United States. E-mail: mzr0068@auburn.edu
cDepartment of Process Research and Development, Merck & Co., Inc., Rahway, New Jersey 07065, USA
First published on 7th November 2019
Abstract
Selective modification of proteins enables synthesis of antibody-drug conjugates, cellular drug delivery and construction of new materials. Many groups have developed methods for selective N-terminal modification without affecting the side chain of lysine by judicious pH control. This is due to lower basicity of the N-terminus relative to lysine side chains. But none of the methods are capable of selective modification of secondary amines or N-terminal proline, which has similar basicity as lysine. Here, we report a secondary amine selective Petasis (SASP) reaction for selective bioconjugation at N-terminal proline. We exploited the ability of secondary amines to form highly electrophilic iminium ions with aldehydes, which rapidly reacted with nucleophilic organoboronates, resulting in robust labeling of N-terminal proline under biocompatible conditions. This is the first time the Petasis reaction has been utilized for selective modification of secondary amines on completely unprotected peptides and proteins under physiological conditions. Peptide screening results showed that the reaction is highly selective for N-terminal proline. There are no other chemical methods reported in literature that are selective for N-terminal proline in both peptides and proteins. This is a multicomponent reaction leading to the synthesis of doubly functionalized bioconjugates in one step that can be difficult to achieve using other methods. The key advantage of the SASP reaction includes its high chemoselective and stereoselective (>99% de) nature, and it affords dual labeled proteins in one pot. The broad utility of this bioconjugation is highlighted for a variety of peptides and proteins, including aldolase and creatine kinase.
Introduction
The site-selective synthetic modification of proteins enables the construction of biomolecular hybrids for a multitude of bioanalytical,1 therapeutic, chemical biology2 and bioengineering applications.3–5 The synthesis of these constructs requires chemoselective and regioselective bioconjugation reactions that proceed under ambient, aqueous conditions at a kinetically favorable rate. Common methods for selective protein modification include alkylation of introduced cysteine residues,6 targeting artificial amino acids with distinct reactivity,7–9 native chemical ligation,10,11 and enzymatic labeling techniques.12,13There have been several reported methods for specific labeling of particular amino acids at the N-terminus, such as cysteine,10 tryptophan,14 serine,15,16 and threonine.15,16 Some groups developed methods for targeting the N-terminus of proteins, while avoiding lysine modification by controlling the reaction pH.17–19 Our group utilized a transamination approach and aldol chemistry for selective modification of protein N-termini.20 Recently, Francis and co-workers developed a robust method for the N-terminal modification of proteins.21 But none of these methods are capable of labeling secondary amines or N-terminal proline in a selective manner in both peptides and proteins.
Here, we report a multicomponent secondary amine selective Petasis (SASP) bioconjugation method for selective labeling of N-terminal proline of peptides and proteins (Fig. 1). Previously, a method for modification at N-terminal proline was attempted through oxidative coupling with an amino phenol;22 however, it leads to the modification of any amino acid at the N-terminus of peptides. The chemoselectivity was not observed for proline over other amino acids in peptides. The oxidative approach also leads to the modification of cysteine; thus, cysteine protection was required for selective labeling.22 To the best of our knowledge, there has been no chemical method reported for selective labeling of secondary amines, such as proline, in peptides and proteins under physiological conditions. A major chemical challenge that accompanies the development of a reaction that selectively modifies proline (pKa = 10.5) is the presence of competing, more reactive primary amino acids (pKa = 9.5), and the equally reactive side chain of lysine (pKa = 10.6).22 The secondary amine selective Petasis (SASP) reaction accomplishes this goal by exploiting the ability of secondary amines to form more electrophilic reactive iminium ions (1A, Fig. 2) towards nucleophilic organoboronates as compared to imines (1B, Fig. 2) obtained from primary amines.23,24 This makes the secondary amine selective Petasis (SASP) bioconjugation quite different from other methodologies attempted in literature. Peptide substrates were first screened to identify the site of modification. The results showed high selectivity for N-terminal proline over any other amino acids. We did not observe modification of any other amino acids. The reaction was then applied to protein substrates, showing similarly high conversions when an N-terminal proline residue was present. This multicomponent reaction dually labeled the peptides and proteins in one pot under physiological conditions with various cargoes. Moreover, the SASP reaction generated a chiral centre at the site of conjugation with high stereoselectivity. This was most likely due to the tendency of proline to induce asymmetric induction through the iminium ion mechanism.25 Unique selectivity, operational simplicity, no catalyst requirement, fast reaction kinetics, ability to generate a stable C–C bond, and physiological conditions are the major strengths of the SASP bioconjugation.
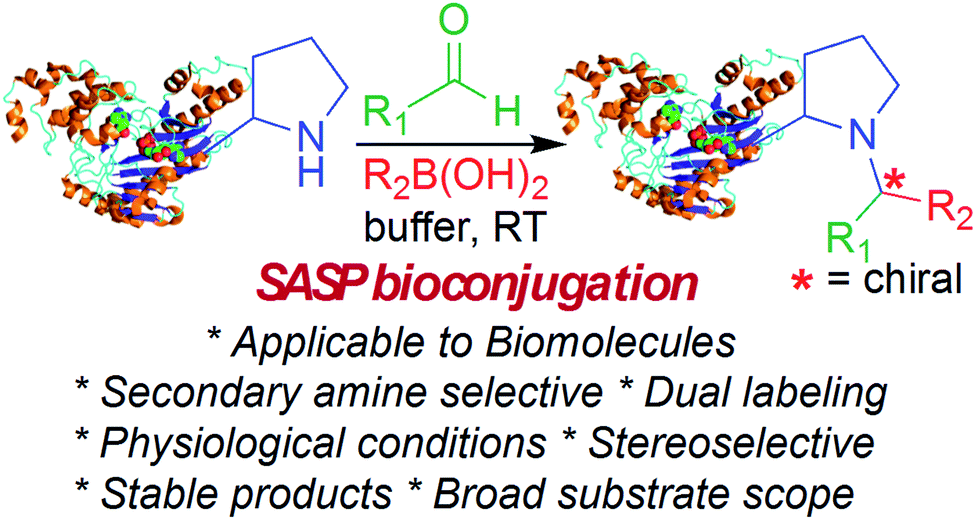 | ||
| Fig. 1 SASP bioconjugation reaction. | ||
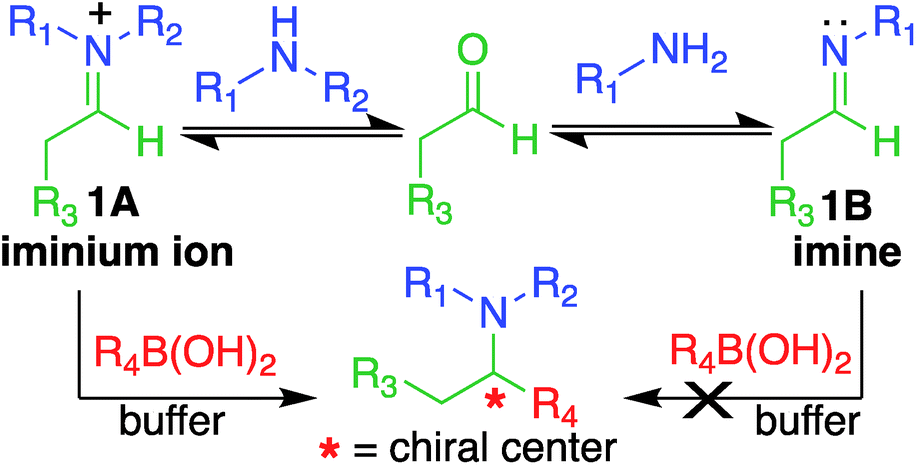 | ||
| Fig. 2 Proposed high reactivity and selectivity towards secondary amines compared to primary amines. | ||
Results and discussion
Design of Petasis reaction for selective modification of secondary amines
Our design of one-step, site-selective, and dual labeling of secondary amines/proline in completely unprotected peptides and proteins is derived from the well-known Petasis reaction, which forms stable C–N and C–C bonds at the site of conjugation between amine, aldehyde and organoboron reagent. Key features that caught our attention for choosing the Petasis reaction for developing a secondary amine selective-bioconjugation reaction are (i) the moderately high reactivity of the Petasis reaction for secondary amines in contrast to primary amines and ammonia; (ii) the exclusive preference of boronic acid to attack the carbon–nitrogen double bond, over any carbon–oxygen double bonds; (iii) the compatibility of Petasis reaction with water;26 (iv) stability of organoboron reagents towards air and moisture, (v) tolerance to a broad range of reactive functional groups; and (vi) the multicomponent nature of the Petasis reaction to provide a method for dual labeling of proteins in one-pot. In our design of the SASP reaction we were guided by a thorough kinetic study of the classical Petasis reaction indicating that formation of an imine/iminium ion intermediate is partially rate limiting.27 Taking note of the rapid rates at which amines condense with aldehydes and ketones, we anticipated that the rate of the iminium ion formation would be faster than the imine formation because of the high nucleophilicity of secondary amines (N = 17) as compared to primary amines (N = 12) in water24 (Fig. 2).Moreover, iminium ions obtained from secondary amines are more electrophilic as compared to imines toward organoboronates at physiological pH 7.3. This was most likely due to the low basicity of imines (pKa ∼ 9), which remained unprotonated under physiological conditions. These points suggested that the SASP reaction could be utilized for chemo- and site-selective labeling of N-terminal proline under physiological conditions. Thus this method was utilized for selective functionalization of biological molecules containing N-terminal proline that are native or readily introduced by standard site-directed mutagenesis.28
Evaluating the reactivity and selectivity of the SASP reaction on peptides
Initial experiments were designed to identify the optimal reaction conditions for labeling biological molecules containing N-terminal proline. The high chemoselectivity of the SASP reaction for N-terminal proline was determined by carrying out reactions with tripeptides![[X with combining low line]](https://www.rsc.org/images/entities/char_0058_0332.gif) AF, where X was substituted with 19 different canonical amino acids (pKa = 9.5) including amino acids with reactive side chains such as Lys, Thr, Ser, Asn, Gln, Glu, Asp, Tyr, Trp, Met and Cys (Fig. 3). These peptides were synthesized on solid support using standard Fmoc synthesis, cleaved from the resin, and purified by HPLC. After purification and lyophilization, peptides were exposed to salicylaldehyde SAL 2a and phenylboronic acid PBA 3a. The reactions were carried out in 25 mM phosphate buffer (pH 7.3)
AF, where X was substituted with 19 different canonical amino acids (pKa = 9.5) including amino acids with reactive side chains such as Lys, Thr, Ser, Asn, Gln, Glu, Asp, Tyr, Trp, Met and Cys (Fig. 3). These peptides were synthesized on solid support using standard Fmoc synthesis, cleaved from the resin, and purified by HPLC. After purification and lyophilization, peptides were exposed to salicylaldehyde SAL 2a and phenylboronic acid PBA 3a. The reactions were carried out in 25 mM phosphate buffer (pH 7.3)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMF (4:1) at room temperature, and reaction mixtures were analyzed by LC-MS (Fig. 3). DMF was used as a cosolvent due to the low solubility of PBA 3a in aqueous buffer. The results showed 70% conversion to Petasis product 4a with peptide PAF 1a containing proline at the N-terminus (Fig. 3). The formation of the Petasis product with peptides AF containing primary amino acids at the N-terminus was not detected, thereby showing that SASP exhibited high selectivity for secondary amines. A second observation of this screen was the fact that lysine and cysteine side chains were not modified under the reaction conditions.
:DMF (4:1) at room temperature, and reaction mixtures were analyzed by LC-MS (Fig. 3). DMF was used as a cosolvent due to the low solubility of PBA 3a in aqueous buffer. The results showed 70% conversion to Petasis product 4a with peptide PAF 1a containing proline at the N-terminus (Fig. 3). The formation of the Petasis product with peptides AF containing primary amino acids at the N-terminus was not detected, thereby showing that SASP exhibited high selectivity for secondary amines. A second observation of this screen was the fact that lysine and cysteine side chains were not modified under the reaction conditions.
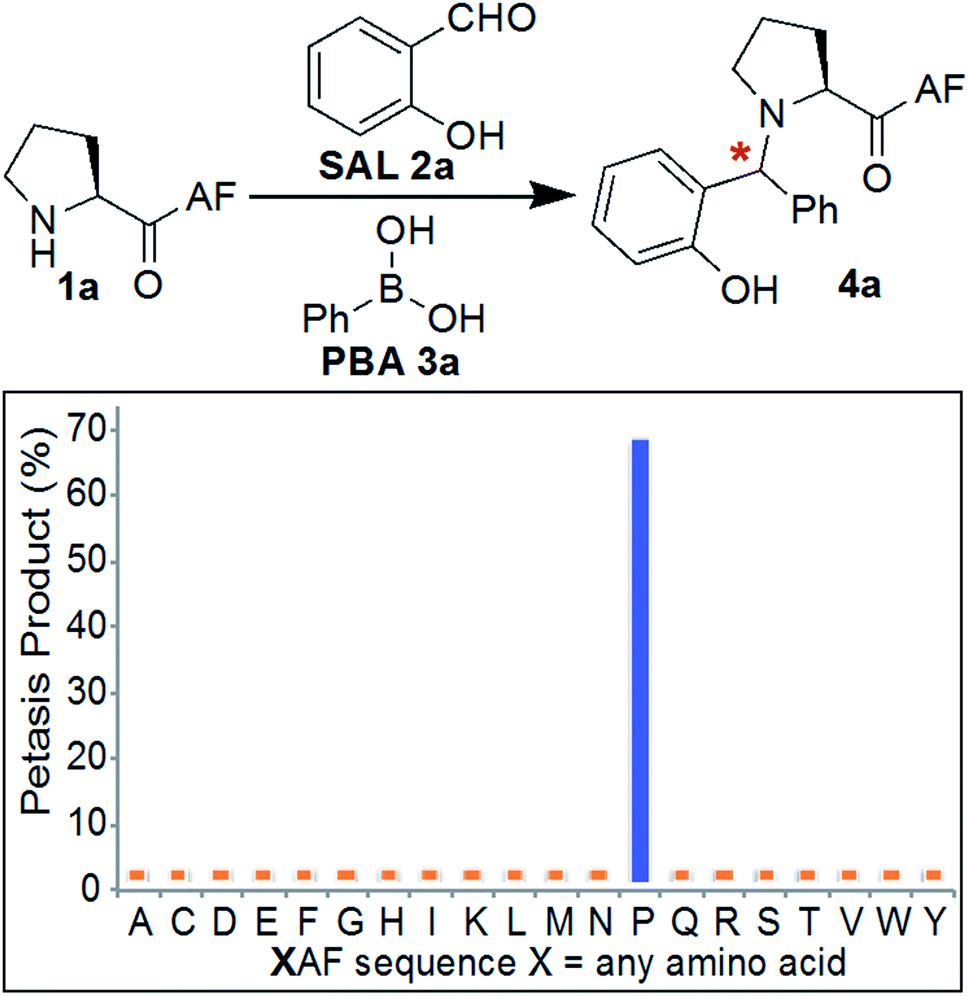 | ||
| Fig. 3 Chemoselectivity of SASP reaction for N-terminal proline. The reaction showed high selectivity for N-terminal proline as analyzed by LC/MS for a panel of XAF peptides, where X represents a variable amino acid. Reaction conditions: 2 mg XAF (12–17 mM, SAL 2a (3 equiv., 36–51 mM), PBA 3a (4 equiv., 48–68 mM) in buffer (pH 7.3):DMF (4:1) at room temperature for 4–24 h. | ||
Verification that SASP modifies only N-terminal proline
The chemoselectivity of SASP reaction towards N-terminal proline was further verified by competition reactions using a mixture of two peptides, PAF 1a andAF (X = Ala, Glu, Arg, Val, Asn, Asp, Ser, Trp, Tyr, Leu, Lys, and Met), in one pot (Fig. S1†). Different peptides were mixed together and exposed to SAL 2a and PBA 3a in 25 mM phosphate buffer (pH 7.3):DMF (4:1) solution. Among all combinations, the Petasis product 4a was observed from peptide PAF 1a only, while the other peptides XAF did not lead to any products under any tested conditions (Fig. S1†).
Next, peptide PAF 1a and SAL 2a were exposed to more water soluble p-methoxyphenyl boronic acid PMB 3b and reaction was carried out under physiological conditions in 25 mM phosphate buffer (pH 7.3):DMSO (4:1). The results showed progressively greater reactivity as 90% conversion to Petasis product 4b was achieved within 2 hours as determined by LC-MS. The product 4b of Petasis reaction between peptide PAF 1a, SAL 2a and PMB 3b was synthesized on larger scale and characterized using two-dimensional NMR and high resolution mass spectrometry HRMS (Fig. S2†). The greater reactivity of 3b is most likely due to the electron-donating group (EDG) at the para position that significantly increases nucleophilicity of the phenyl carbanion.29 Further, the high selectivity of the SASP reaction for proline in the presence of highly reactive PMB 3b was verified by carrying out the reaction with a mixture of two peptides PAF 1a and AF (where, X = Glu, Lys, Thr, Trp, Tyr, Leu, and His) and SAL 2a in one pot under physiological conditions (Fig. S3†). Again, the Petasis product 4b was observed from peptide PAF 1a only. As further validation of the reaction site specificity, no modification of lysine and cysteine was observed with peptides, PFKAFV and PFCAF, containing both N-terminal proline (pKa 10.5) and the reactive side chain of lysine (pKa = 10.6) and cysteine (Fig. S4†), respectively.
We also investigated SASP reactions of peptide PAF 1a and PMB 3b with water-soluble aldehydes such as 2-pyridinecarboxyaldehyde (2PCA) 2b and 3-hydroxy-2-pyridinecarbaldehyde (HPCA) 2d under physiological conditions. The results showed moderate conversion to Petasis products with both the aldehydes. On detailed analysis we found that this is due to the tendency of these aldehydes to generate an imidazolidinone product with the N-terminus as reported by Francis et al.21 To minimize the formation of the imidazolidinone product and to increase the yield of the Petasis product with aldehydes such as 2PCA 2b and HPCA 2d, we carried out optimization studies with HPCA 2d. The reagent ratios of aldehyde and boronic acid were varied. Peptide PAF 1a (15 mM) was reacted with aldehyde HPCA 2d (1.5–3 equiv., 22.6–45 mM) and PMB 3b (4–8 equiv., 60–120 mM) in 25 mM phosphate buffer (pH 7.3):DMSO (4:1) at room temperature for 4 h (Table S1†). It was demonstrated that conversion to Petasis product was highest using 1.5 equiv. of HPCA 2d and 8 equiv. of PMB 3b. We also observed the fast reaction rate under such conditions. The Petasis product 4c of peptide PAF 1a, 2PCA 2b and PMB 3b was then synthesized on a larger scale and characterized by 2D NMR and HRMS (Fig. S5†).
Next, we synthesized a water-soluble analogue of SAL by combining 4-formyl-3-hydroxybenzoicacid with propargylamine (Fig. S6†). The resulting SAL analogue 2a′ is highly soluble in water. We investigated the selectivity of aldehydes such as water-soluble SAL analogue 2a′, 2PCA 2b and HPCA 2d by carrying out reactions with various N-terminal tripeptides XAF (where, X = Glu, Thr, Ala, Gly, Phe, Tyr, Leu, Met, Pro and Gln) and PMB 3b under optimized conditions. The peptides XAF 1 (12–17 mM) were reacted with aldehydes 2a′, 2b or 2d (1.5 equiv., 18–25.5 mM) and PMB 3b (8 equiv., 96–136 mM) in 25 mM phosphate buffer (pH 7.3):DMSO (4:1) for 4 h at 37 °C. The reactions were quenched by dilution with cold water and by freezing at −80 °C followed by lyophilization. With aldehydes 2b and 2d, the proline terminal peptide reached the highest levels of conversion; although, modifications of other N-terminal peptides were also observed under the reaction conditions (Fig. 4). Despite efforts to optimize conditions for all N-termini for 2PCA and HPCA mediated Petasis reaction, proline still stood out as the most reactive species. As observed earlier with SAL 2a, the Petasis reaction with water-soluble SAL analogue 2a′ was also highly chemoselective for N-terminal proline. The formation of the Petasis product with peptides AF containing primary amino acids at the N-terminus was not detected with water-soluble SAL analogue 2a' (Fig. 4 and S7†).
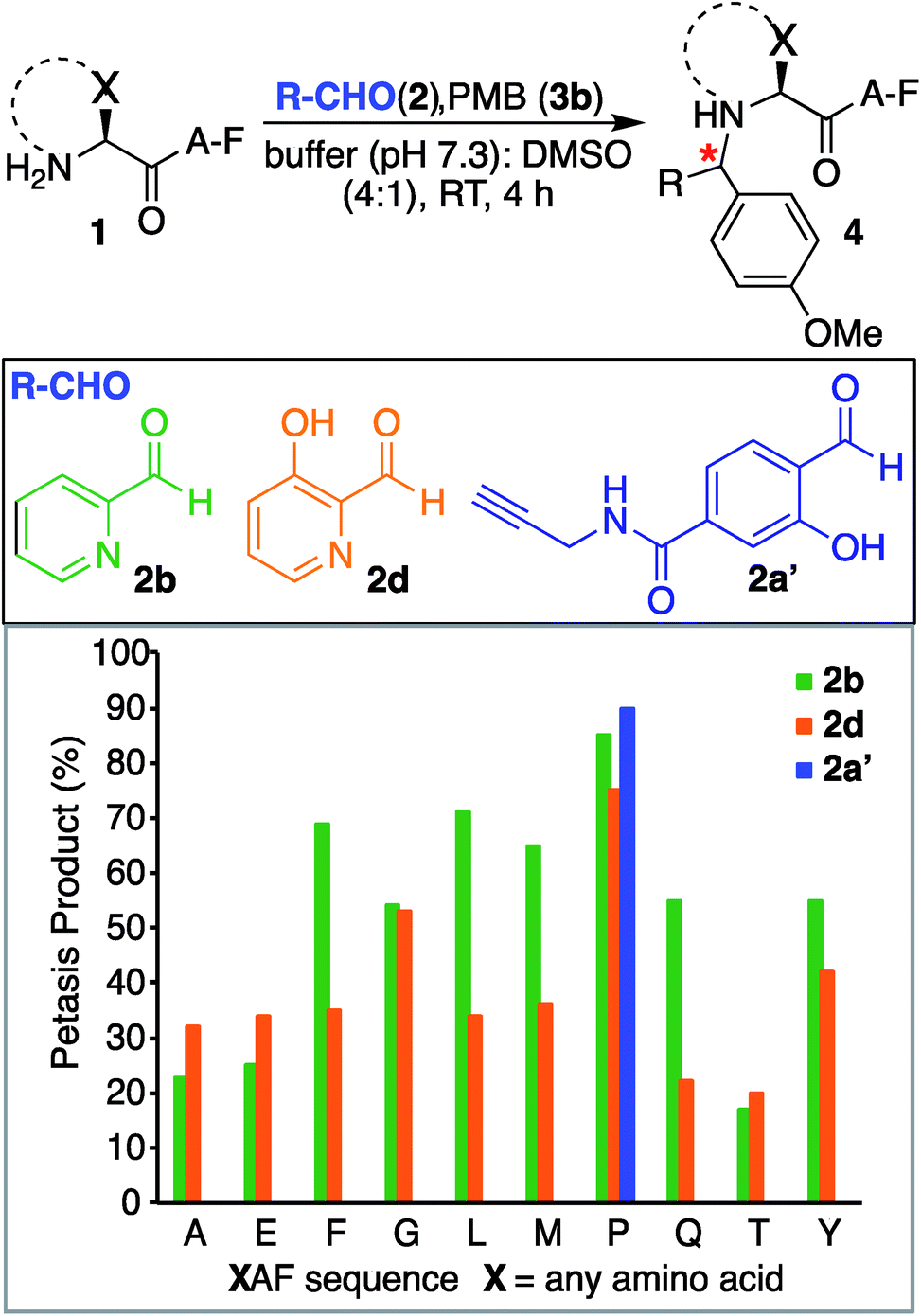 | ||
| Fig. 4 Peptide modification with Petasis reaction. Peptides with sequence XAF, where X represents a variable amino acid were screened for Petasis reaction with PMB 3b and a variety of aldehydes 2b, 2d and water-soluble SAL analogue 2a′. % conversion to Petasis products were determined by LC/MS. Reaction conditions: XAF (12–17 mM), RCHO 2 (1.5 equiv. 18–25.5 mM), PMB 3b (8 equiv. 96–136 mM) in 25 mM buffer (pH 7.3):DMSO (4:1) and 37 °C. The samples were incubated for 4 h. | ||
Stability studies of the SASP product.
Given the different reactivity of aldehydes with proline, we verified the stability of the product to a variety of conditions. The purified Petasis product 4b obtained from peptide PAF 1a, SAL 2a and PMB 3b was exposed to acidic and basic pH (pH 3.5–10.5) at room temperature. The peptides were analysed after regular intervals by LC-MS. Over the course of 24 h, the majority of the Petasis product 4b was stable at a pH range of 6.5 to 9.5; whereas at low pH 3.5 to 5.5, half of the Petasis product 4b was hydrolyzed to the starting peptide PAF 1a, as analyzed by LC-MS (Fig. S8†). We hypothesized that Petasis product 4b becomes protonated under acidic conditions leading to the hydrolysis of Petasis product by nucleophilic attack from the neighboring ortho-hydroxyl group to a newly generated chiral centre of the Petasis product 4b. Notably, pH had a huge effect on the degree of decomposition. Through substrate variation, it is likely that products of increased stability can be obtained for applications. To validate the process of hydrolysis, we analyzed the stability of Petasis product 4c obtained from the reaction of peptide PAF 1a with 2-PCA 2b (with no ortho-hydroxyl group) and PMB 3b using LC-MS (Fig. S9†). No degradation of the Petasis product 4c was observed under any of tested conditions demonstrating the hydrolytic stability of the product. The ability of the linkage to withstand these conditions makes this method applicable for the development of new materials for different biological applications.Effects of pH on the rate of the SASP reaction
The rate of the SASP reaction at different pH conditions ranging from 5.5 to 9.5 was explored on the model peptide PAF 1a with SAL 2a and PMB 3b. Although the rate is quite high at neutral pH 7.3 with 62% conversion to the Petasis product 4b in 15 min, the highest conversion (80% within 15 min) was observed at pH 9.5 (Fig. S10†). However, considering the biocompatibility of our method, physiological pH is the ideal condition for promoting this reaction with peptides and proteins. Next, we explored the reactivity of primary amines at lower pH, as it could be possible to protonate them and therefore add boronic acids. We screened N-terminal primary tripeptides XAF with SAL 2a/2PCA 2b and PMB 3b at pH 5.5. The results showed that SAL 2a is highly selective for N-terminal proline even at low pH 5.5. The results with 2PCA 2b showed high reactivity with N-terminal primary amines at low pH 5.5 as compared to physiological pH 7.3 (Fig. S11†). It could be because imines generated by primary amines can protonate faster at lower pH, and therefore are more reactive towards boronic acids.Stereoselective nature of SASP bioconjugation
Another unique feature of the SASP reaction is its ability to generate a new chiral centre at the site of conjugation with high stereoselectivity. This reaction could be of a great benefit in the derivatization of peptides and proteins involved in drug discovery since it has been reported that regioselectivity and stereoselectivity are crucial for assuming the active binding conformation.30The analysis of the SASP product 4b showed the formation of a newly generated chiral centre with high stereoselectivity (de >99%) and with the absolute configuration of (R) as determined by analysis of dipolar couplings obtained from ROESY NMR spectroscopy and by comparing the observed proton and carbon chemical shifts versus those calculated from density functional theory (Fig. 5a and S12†). In addition, the DP4+ method,31 which applies a Bayesian statistical treatment to the NMR data, predicted the R-configuration to be the correct stereoisomer with greater than 99% probability. The high stereoselectivity of the SASP bioconjugation was most likely directed by the chirality of N-terminal proline and by the boron which guides the nucleophilic attack of a phenylcarbanion on the iminium ion intermediate from the Re face since attack from the Si face is hindered, instead of the electronic effects of other substituents32,33 (Fig. 5b). To further validate the role of proline for obtaining high stereoselectivity with R absolute configuration, the SASP reaction was performed with peptide pAF containing D-proline under the optimized conditions. The Petasis product 4B was obtained from the reaction of peptide pAF with SAL 2a and PMB 3b and characterized by 2D NMR (Fig. S13†). The analysis of the Petasis product 4B showed formation of a newly generated chiral centre with (S) absolute configuration (de > 99%) which is opposite from the product obtained from peptide PAF with L-proline (Fig. S13†). This result showed that proline not only functions as a site of conjugation in the SASP reaction but also acts as a chiral asymmetric catalyst responsible for asymmetric induction in the Petasis product 4b, 4B and 4c (Fig. 5b and S5, S12 and S13†).
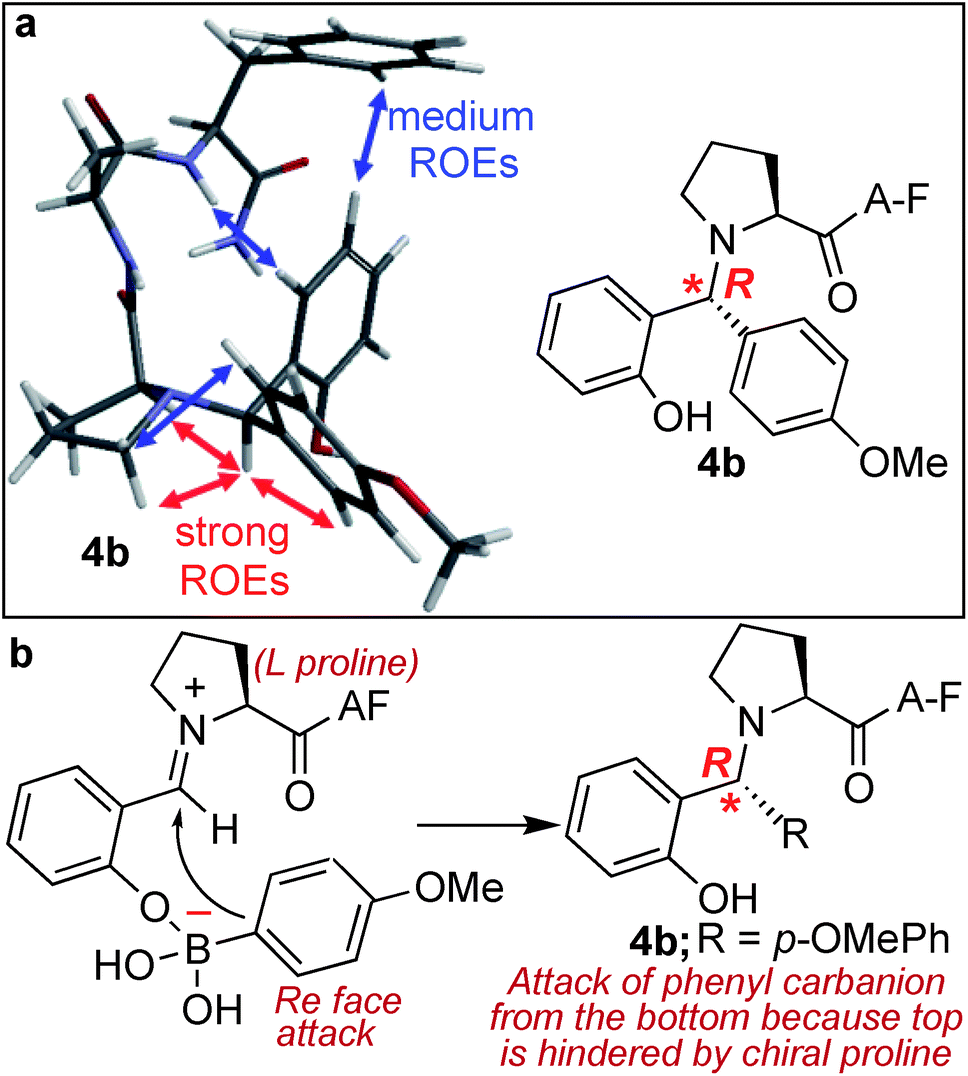 | ||
| Fig. 5 Chemo- and stereo-selective SASP bioconjuation under physiological conditions. (a) NMR ROEs of Petasis product 4b are consistent with R configuration from molecular mechanics Monte Carlo. (b) Plausible pathway for high stereoselectivity. | ||
Mechanistic studies of SASP reaction: role of aldehydes.
Aldehydes, such as salicylaldehyde SAL 2a, 2PCA 2b and HPCA 2d, were used for our initial studies with the view that the hydroxyl and pyridine groups at the ortho position relative to the aldehyde coordinates with PMB 3b by a Lewis acid–base mechanism (by dative bond), which brings the organoboronic acid in close proximity to the reaction centre (Fig. 6a). Also, this dative bond with organoboronic acid facilitates the release of a carbanion to react with an electrophilic iminium ion through a favorable five-membered cyclic transition state, thereby generating a stable C–C bond at the site of conjugation (Fig. 6a). To test this proposal, a set of other aldehydes such as glyoxylic acid 2c, 4-hydroxy benzaldehyde 2e and 2-nitro benzaldehyde 2f were investigated for their potential to modify the N-terminus of a model peptide PAF 1a using PMB 3b under optimized conditions (Fig. 6b, and S14†). Glyoxylic acid 2c showed progressively greater reactivity for the SASP reaction since it exhibits Lewis basic groups proximal to the reactive carbonyl carbon (Fig. 6b). Notably, Petasis products were not observed with 4-hydroxy benzaldehyde 2e, which contains a distal Lewis basic group, or from 2-nitro benzaldehyde 2f, which lacks Lewis basic group (Fig. 6b). The results showed that the presence of a Lewis base directly adjacent to the aldehyde was crucial for this reaction. Ultimately, SAL 2a, water-soluble SAL analogue 2a′, 2PCA 2b, glyoxylic acid 2c and HPCA 2d reagents emerged as the most effective and synthetically tractable aldehydes for further studies and modifications.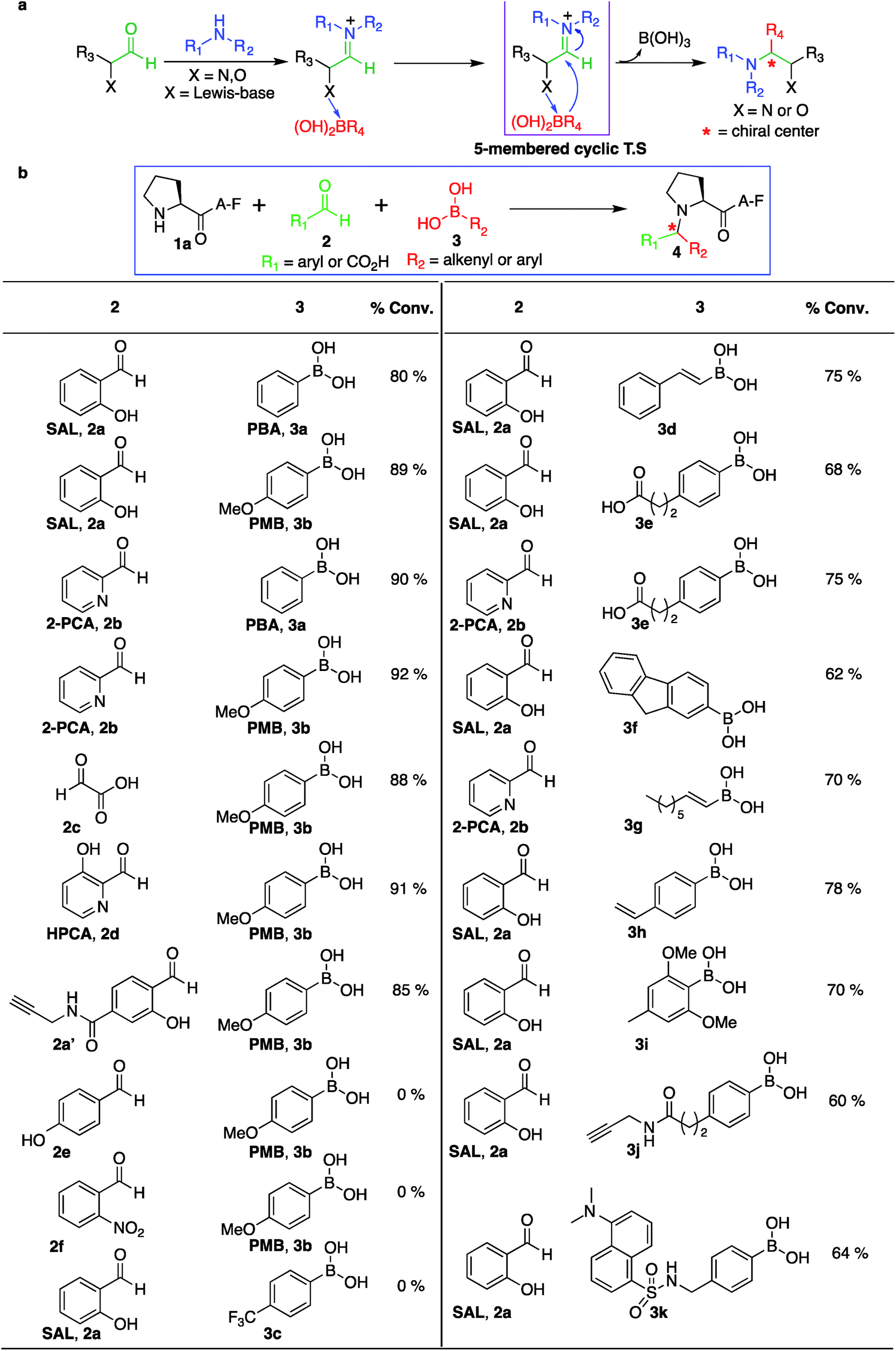 | ||
| Fig. 6 Selective dual labeling with diverse functional groups. (a) Significance of Lewis-base near to the aldehyde. (b) Aldehyde substrate scope and boronic acid scope with peptide PAF 1a. Reaction conditions. To a peptide PAF 1a (2 mg, 12 mM), in 0.5 mL solution of 25 mM phosphate buffer (pH 7.3):DMSO (4:1) was added SAL 2a or 2PCA 2b (1.5 equiv., 18 mM) and a variety of boronic acids 3 (8 equiv., 96 mM). The reactions were stirred at room temperature for 2–24 h. | ||
Role of boronic acids in SASP reaction
The third reagent involved in the multi-component SASP reaction is organoboronic acid. Based on the initial SASP reaction and the proposed mechanism, organoboronic acid acts as a nucleophile. The high nucleophilicity of the organoboronic acid significantly accelerates the rate of the SASP reaction. With this reactivity behavior in mind, various aryl- and alkenyl-boronic acids (3a–i) were screened for their potential to modify a model peptide PAF 1a with SAL 2a or 2PCA 2b under physiological conditions (Fig. 6b and S14†). The results showed that arylboronic acid with electron donating group EDGs such as para-methoxyboronic acid (PMB) 3b exhibited progressively greater reactivity (90% conversion in 2 h) as compared to arylboronic acid containing electron withdrawing groups (EWGs) such as 4-trifluoromethylphenylboronic acid 3c (0% conversion in 24 h) (Fig. 6b). Interestingly, we observed the formation of a Petasis product with straight chain octenylboronic acids under the reaction conditions. To confirm the structure of the Petasis product with octenylboronic acid, we carried out NMR on the isolated product obtained from the reaction of morpholine with octylenylboronic acid and SAL 2a/2PCA 2b (Fig. S15†). The substrate scope was further explored with boronic acid derivatives containing an alkyne group 3j and a dye, dansyl methylphenylboronic acid, 3k which can then be utilized for click bioconjugation and cell imaging studies, respectively. These derivatives of boronic acids (3j and 3k) were synthesized from commercially available 4-(2-carboxyethyl)benzeneboronic acid 3e and 4-(aminomethyl)phenylboronic acid (for synthesis, see Fig. S16†)SASP reaction on biologically active peptides
The broad substrate scope of the SASP bioconjugation was determined by carrying out reactions with bioactive peptides with varying amino acid composition such as angiotensin I (PDRVYIHPFHL), a peptide hormone that causes vasoconstriction and a subsequent increase in blood pressure, bradykinin (PPGFSPFR), an inflammatory mediator, and hypertrehalosemic neuropeptide (PVNFSPGWGT)29 by using SAL 2a and PMB 3b under optimized conditions. The amino acid composition of the peptide exhibited minimal impact on the degree or selectivity of the modification, as observed by good to excellent yields for singly modified peptides at N-terminal proline in all cases (Fig. 7 and S17†). The SASP reaction was carried out on a peptide PICSLYQLENYCN derived from chain A of insulin with water-soluble SAL derivatives (Fig. 7). The results showed single modification to Petasis conjugate at N-terminal proline with >80% conversion. The reaction with a corresponding peptide ICSLYQLENYCN containing primary amino acid at the N-terminus did not lead to the formation of any Petasis product.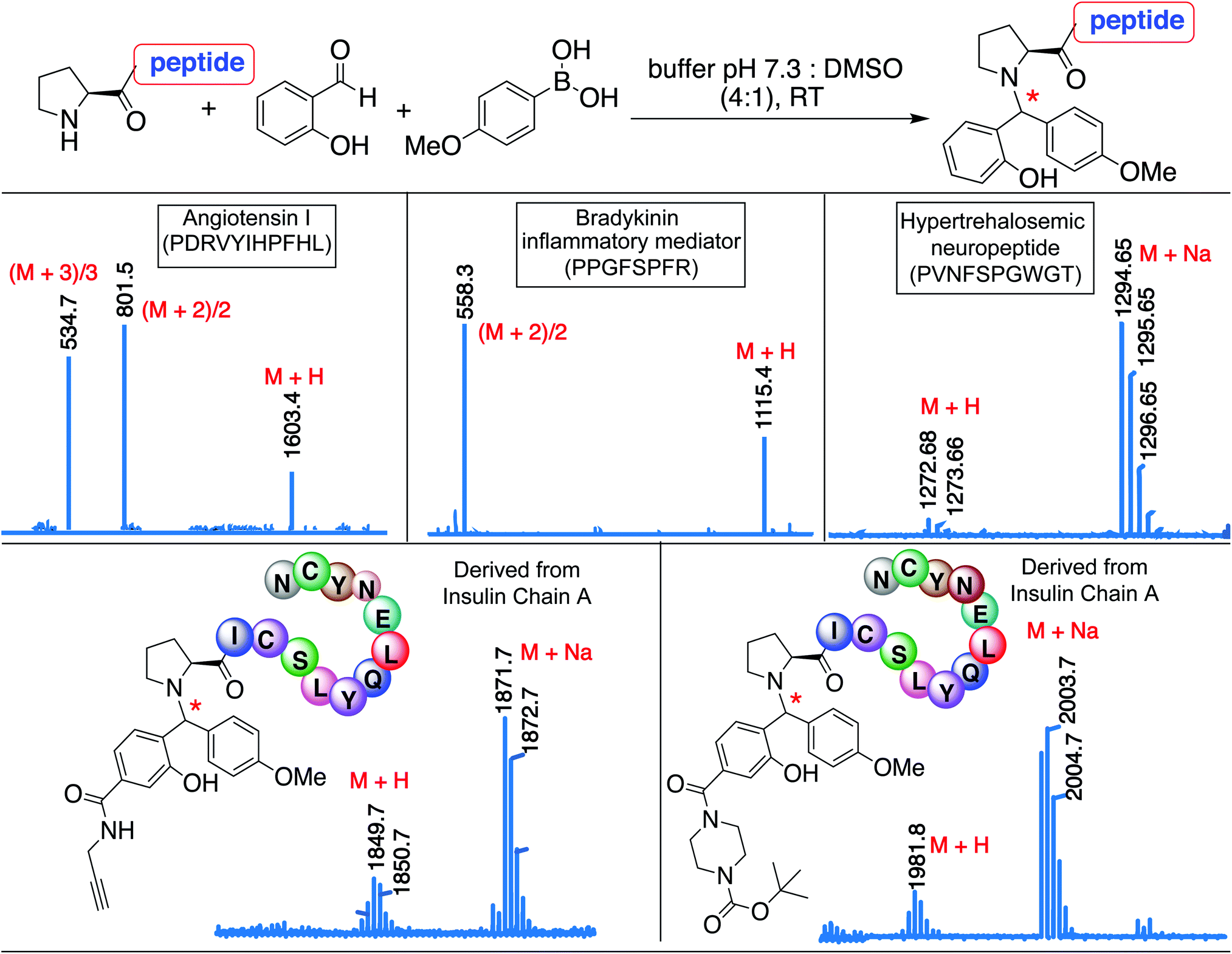 | ||
| Fig. 7 Secondary amine selective Petasis reaction of bioactive peptides as analyzed by ESI-MS for angiotensin I, bradykinin, and hypertrehalosemic neuropeptide reactions with SAL 2a and PMB 3b. ESI-MS for insulin chain A derived peptides with water soluble SAL derivatives. Reaction conditions. 2 mM solution of the bioactive peptide was reacted with SAL 2a (1.5 equiv., 3 mM) and PMB 3b (8 equiv., 16 mM) in 0.5 mL 25 mM phosphate buffer pH 7.3:DMSO (4:1) and reactions were stirred at room temperature for 15 h. | ||
Protein scope for SASP reaction
The SASP reaction with N-terminal amino group was first tested on native proteins such as myoglobin and α-lactalbumin, which contain several lysine residues (pKa = 10.6) and lack proline at their respective N-termini. The native proteins were reacted with water-soluble 2PCA 2b and reactive PMB 3b under optimized reaction conditions. We do not see any modification on these proteins as analyzed by MS and gels (Fig. 8a). The protein reactions were explored with higher amount of 2PCA 2b (5 equiv.) and PMB 3b (50 equiv.) but any modification on these proteins were not observed. This could be because they have less reactive primary amino acids at the N-terminus.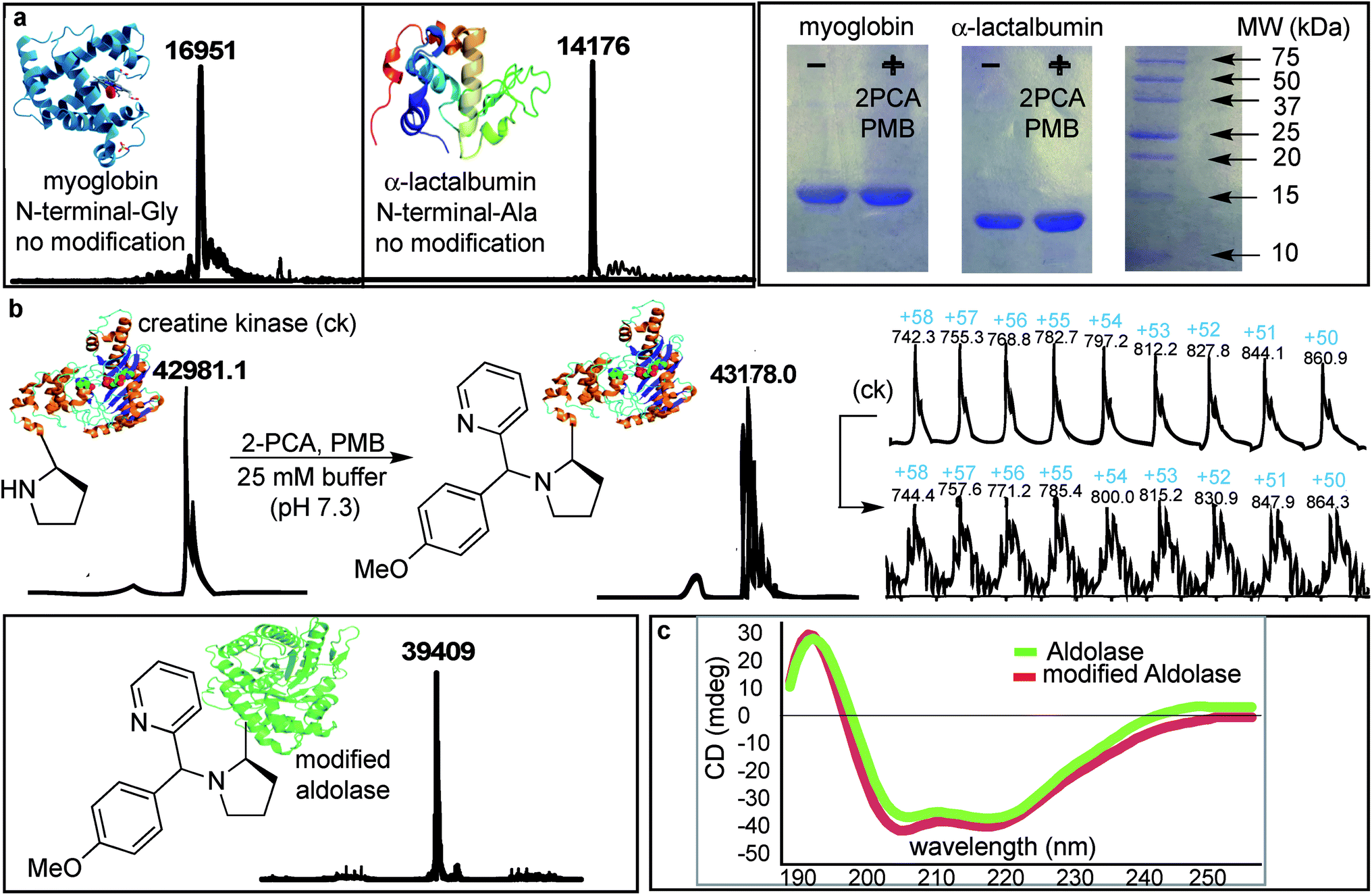 | ||
| Fig. 8 Chemo- and site-selective dual labeling of proteins with 2-PCA 2b and PMB 3b. (a) No modification was observed for myoglobin and α-lactalbumin lacking N-terminal proline. SDS PAGE images of proteins before and after SASP reaction. (b) Proteins, creatine kinase and aldolase with N-terminal proline underwent modification to SASP product, as characterized by ESI-MS. (c) CD spectra of aldolase before and after SASP reaction showed no change in the secondary structure of a protein. Reaction conditions: A protein (100 μM) was dissolved in 400 μL of 25 mM phosphate buffer pH 7.3. 2PCA (0.5 mM) and PMB (5 mM) was added in the solution. The reaction was agitated at room temperature for 16 h. | ||
Next, N-terminal proline containing native proteins such as aldolase and creatine kinase were evaluated for the reaction with 2PCA 2b and PMB 3b. The protein reactions were carried out with a slight excess of 2-PCA 2b (5 equiv.) and PMB 3b (50 equiv.) in 25 mM phosphate buffer (pH 7.3) at room temperature for 16 h (Fig. 8b).
Analysis by MS demonstrated that creatine kinase [m = 43178 (+197 Da)] and aldolase [m = 39409 (+197 Da)] were modified by the formation of the Petasis product. We did not observe any second modification indicating that reaction is highly selective for N-terminal-proline. Interestingly, the Petasis reaction with 2PCA 2b worked with a primary amino acid at the N-terminus of peptides, but in proteins, the Petasis product was observed with more reactive N-terminal proline containing proteins. Peptide screening in Fig. 4 showed that N-terminal amino acids can undergo Petasis reaction with 2PCA 2b, but the proline terminal peptide has highest conversion. Despite efforts to optimize conditions for all N-termini, proline still stood out as the most reactive species. When applied to protein substrates, the reaction showed a stronger requirement for the N-terminal proline therefore we did not observe modification of myoglobin and α-lactalbumin.
After purification, incubation of N-terminally modified creatine kinase from a pH range of 3.5 to 9.5 showed full product stability over a 24 h period as detected by MS (Fig. S18†).
Study of protein structure and function after the modification
To evaluate the effect of the N-terminal modification on the secondary and tertiary structure of aldolase, we compared the circular dichroism (CD) of the native and the SASP modified aldolase. The result showed no change in the secondary structure of the modified aldolase (Fig. 8c). Currently, we are using this dual-labeling strategy to introduce analogues of boronic acid and SAL, which act as IR probes or are detectable by 19F NMR spectroscopy.34Conclusions
In this study, we present SASP reactions for selective modification of N-terminal proline containing peptides and native-proteins. Proline can be introduced readily in N-terminal positions using site-directed mutagenesis.28 The SASP method reported here offer two distinct advantages over other N-terminal labeling methods. First, it is highly selective for N-terminal proline and does not lead to modification of any other amino acids. Second, it is a multicomponent reaction and dually labels peptides and proteins in a single step in one pot under physiological conditions, without the use of any catalysts. This reaction features excellent site specificity and promising scale-up potential.Moreover, the SASP reaction leads to the generation of a new chiral centre at the site of conjugation with high stereoselectivity (de >99%). This is due to the ability of proline to carry out asymmetric synthesis. The results on a wide variety of peptides and proteins strongly indicate that the SASP modifies N-terminal proline residues (pKa = 10.5) even in the presence of free cysteine and lysine residues which is particularly challenging due to the strong nucleophilicity of the thiol group and equivalent basicity of the γ−chain of lysine (pKa = 10.6) at relevant pH values.
Furthermore, the secondary amine selective nature of this reaction suggests that it could be very useful for selective tagging/enriching proteins with monomethyl lysine posttranslational modifications PTMs for determining their role in regulating various cellular signaling processes.35 This work is currently underway in our laboratory. Considering the easy setup, high selectivity for secondary amines, dual labeling nature and broad substrate scope with a variety of aldehydes and organoboronic acids, we anticipate that this method will become a highly useful tool in many scientific studies.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by NSF CAREER Award #1752654 granted to M.R.Notes and references
- S. Zalipsky, Bioconjugate Chem., 1995, 6, 150–165 CrossRef CAS.
- J. Rao, A. Dragulescu-Andrasi and H. Yao, Curr. Opin. Biotechnol., 2007, 18, 17–25 CrossRef CAS.
- L. S. Witus, C. Netirojjanakul, K. S. Palla, E. M. Muehl, C.-H. Weng, A. T. Iavarone and M. B. Francis, J. Am. Chem. Soc., 2013, 135, 17223–17229 CrossRef CAS.
- S. C. Alley, N. M. Okeley and P. D. Senter, Curr. Opin. Chem. Biol., 2010, 14, 529–537 CrossRef CAS.
- L. S. Witus and M. B. Francis, Acc. Chem. Res., 2011, 44, 774–783 CrossRef CAS PubMed.
- S. Szobota, P. Gorostiza, F. Del Bene, C. Wyart, D. L. Fortin, K. D. Kolstad, O. Tulyatham, M. Volgraf, R. Numano, H. L. Aaron, E. K. Scott, R. H. Kramer, J. Flannery, H. Baier, D. Trauner and E. Y. Isacoff, Neuron, 2007, 54, 535–545 CrossRef CAS.
- V. W. Cornish, K. M. Hahn and P. G. Schultz, J. Am. Chem. Soc., 1996, 118, 8150–8151 CrossRef CAS.
- A. Deiters, T. A. Cropp, M. Mukherji, J. W. Chin, J. C. Anderson and P. G. Schultz, J. Am. Chem. Soc., 2003, 125, 11782–11783 CrossRef CAS.
- J. C. M. van Hest, K. L. Kiick and D. A. Tirrell, J. Am. Chem. Soc., 2000, 122, 1282–1288 CrossRef CAS.
- P. E. Dawson, T. Muir, I. Clark-Lewis and S. Kent, Science, 1994, 266, 776–779 CrossRef CAS PubMed.
- T. W. Muir, Annu. Rev. Biochem., 2003, 72, 249–289 CrossRef CAS.
- P. Wu, W. Shui, B. L. Carlson, N. Hu, D. Rabuka, J. Lee and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3000–3005 CrossRef CAS.
- C. Uttamapinant, K. A. White, H. Baruah, S. Thompson, M. Fernández-Suárez, S. Puthenveetil and A. Y. Ting, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10914–10919 CrossRef CAS.
- X. Li, L. Zhang, S. E. Hall and J. P. Tam, Tetrahedron Lett., 2000, 41, 4069–4073 CrossRef CAS.
- K. F. Geoghegan and J. G. Stroh, Bioconjugate Chem., 1992, 3, 138–146 CrossRef CAS.
- Y. Zhang, C. Xu, H. Y. Lam, C. L. Lee and X. Li, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6657–6662 CrossRef CAS.
- D. P. Baker, E. Y. Lin, K. Lin, M. Pellegrini, R. C. Petter, L. L. Chen, R. M. Arduini, M. Brickelmaier, D. Wen, D. M. Hess, L. Chen, D. Grant, A. Whitty, A. Gill, D. J. Lindner and R. B. Pepinsky, Bioconjugate Chem., 2006, 17, 179–188 CrossRef CAS.
- R. Singudas, S. R. Adusumalli, P. N. Joshi and V. Rai, Chem. Commun., 2015, 51, 473–476 RSC.
- A. O. Chan, C. M. Ho, H. C. Chong, Y. C. Leung, J. S. Huang, M. K. Wong and C. M. Che, J. Am. Chem. Soc., 2012, 134, 2589–2598 CrossRef CAS.
- T. S. Howard, R. D. Cohen, O. Nwajiobi, Z. P. Muneeswaran, Y. E. Sim, N. N. Lahankar, J. T.-H. Yeh and M. Raj, Org. Lett., 2018, 20, 5344–5347 CrossRef CAS.
- J. I. MacDonald, H. K. Munch, T. Moore and M. B. Francis, Nat. Chem. Biol., 2015, 11, 326–331 CrossRef CAS.
- A. C. Obermeyer, J. B. Jarman and M. B. Francis, J. Am. Chem. Soc., 2014, 136, 9572–9579 CrossRef CAS.
- N. A. Petasis and I. Akritopoulou, Tetrahedron Lett., 1993, 34, 583–586 CrossRef CAS.
- F. Brotzel, Y. C. Chu and H. Mayr, J. Org. Chem., 2007, 72, 3679–3688 CrossRef CAS PubMed.
- M. Raj and V. K. Singh, Chem. Commun., 2009, 6687–6703 RSC.
- N. R. Candeias, L. F. Veiros, C. A. M. Afonso and P. M. P. Gois, Eur. J. Org. Chem., 2009, 1859–1863 CrossRef CAS.
- N. Schlienger, M. R. Bryce and T. K. Hansen, Tetrahedron, 2000, 56, 10023–10030 CrossRef CAS.
- P. H. Hirel, M. J. Schmitter, P. Dessen, G. Fayat and S. Blanquet, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 8247–8251 CrossRef CAS.
- F. Berrée, A. Debache, Y. Marsac and B. Carboni, Tetrahedron Lett., 2001, 42, 3591–3594 CrossRef.
- L. A. Nguyen, H. He and C. Pham-Huy, Int. J. Biomed. Sci., 2006, 2, 85–100 CAS.
- N. Grimblat, M. M. Zanardi and A. M. Sarotti, J. Org. Chem., 2015, 80, 12526–12534 CrossRef CAS.
- T. J. Southwood, M. C. Curry and C. A. Hutton, Tetrahedron, 2006, 62, 236–242 CrossRef CAS.
- N. A. Petasis, A. Goodman and I. A. Zavialov, Tetrahedron, 1997, 53, 16463–16470 CrossRef CAS.
- M. A. Danielson and J. J. Falke, Annu. Rev. Biophys. Biomol. Struct., 1996, 25, 163–195 CrossRef CAS.
- L. E. West, S. Roy, K. Lachmi-Weiner, R. Hayashi, X. Shi, E. Appella, T. G. Kutateladze and O. Gozani, J. Biol. Chem., 2010, 285, 37725–37732 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental methods, spectroscopic data and supplementary figures (PDF). See DOI: 10.1039/c9sc04697f |
| This journal is © The Royal Society of Chemistry 2020 |