
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactivity control of a photocatalytic system by changing the light intensity†
Christoph
Kerzig
 * and
Oliver S.
Wenger
*
* and
Oliver S.
Wenger
*
Department of Chemistry, University of Basel, St. Johanns-Ring 19, 4056 Basel, Switzerland. E-mail: christoph.kerzig@unibas.ch; oliver.wenger@unibas.ch
First published on 30th October 2019
Abstract
We report a novel light-intensity dependent reactivity approach allowing us to selectively switch between triplet energy transfer and electron transfer reactions, or to regulate the redox potential available for challenging reductions. Simply by adjusting the light power density with an inexpensive lens while keeping all other parameters constant, we achieved control over one- and two-photon mechanisms, and successfully exploited our approach for lab-scale photoreactions using three substrate classes with excellent selectivities and good product yields. Specifically, our proof-of-concept study demonstrates that the irradiation intensity can be used to control (i) the available photoredox reactivity for reductive dehalogenations to selectively target either bromo- or chloro-substituted arenes, (ii) the photochemical cis–trans isomerization of olefins versus their photoreduction, and (iii) the competition between hydrogen atom abstraction and radical dimerization processes.
1 Introduction
The interplay of several photocatalyst properties such as molar absorption coefficients, excited-state energies, redox potentials, excited-state lifetimes and photostabilities determines the yield and selectivity of a given photo(redox) reaction. Therefore, it comes as no surprise that, under standardized conditions, every photoreaction has one ideal catalyst that needs to be identified. In direct consequence, a whole library of photoactive molecules with tailor-made properties has been prepared over the last few decades, by both organic1–4 and inorganic5–12 chemists.To overcome the need for several, frequently expensive catalysts with optimized properties, the concept of wavelength dependence and wavelength selectivity13 has been exploited to control the redox potential of single photocatalysts by regulating the irradiation light color.14,15 In a somewhat related approach, a photocatalytic allylation was combined with a light-color selective isomerization to control product formation.16 While these recent chromoselective methods15,16 are very elegant and clearly have an impact on photoredox catalysis, they both rely on irradiation equipment consisting of at least two high-power LEDs as light sources.
An attractive alternative, but yet underexplored strategy to gain control over photochemical reactivity is provided by regulating the light intensity while keeping all other parameters constant. The approach of intensity dependent one- (low intensity) and two-photon (high intensity) chemistry is rather well-established under laser flash photolysis conditions,14,17–22 which however necessitates expensive pulsed lasers that are inaccessible for most synthetic laboratories. Catalytic cycles comprising the consecutive absorption of two photons with an excited state as intermediate (i.e., without converting the excited state into a long-lived radical anion)23,24 were realized under continuous wave (cw) excitation conditions only very recently.25 The feasibility of this new kind of two-photon chemistry on the preparative scale with an inexpensive blue diode laser,26 whose acquisition costs are practically identical to those of comparable ready-to-use high-power LEDs, inspired the reactivity control approach presented herein. As we will show, the excitation of our recently developed water-soluble catalyst Irsppy27 (see Scheme 1 for its structure) with a single blue photon can initiate several dehalogenation and isomerization reactions, but when a second photon is absorbed within the lifetime of its excited triplet state (3Irsppy) using higher light intensities per irradiation area, highly aggressive hydrated electrons (eaq˙−) that react with the same substrates via a completely different pathway are produced, thereby allowing a fundamentally new reactivity control strategy and its application to selective photo(redox) applications.
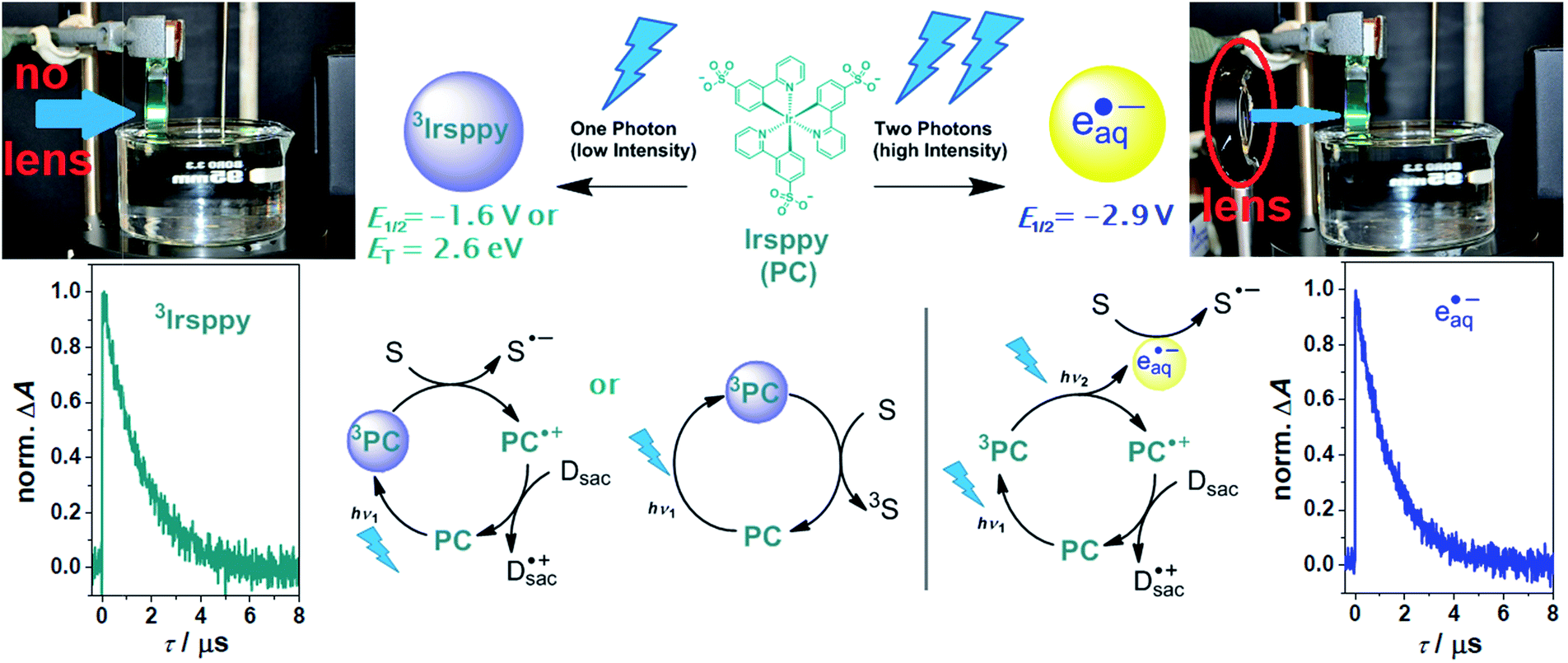 | ||
| Scheme 1 Reactivity control of our photocatalytic system with a continuous wave (cw) laser (447 nm, 1 W) using the natural laser beam for one-photon excitation (left) or the collimated beam for consecutive two-photon excitation (right). Upper row, structure of the photocatalyst (PC) Irsppy, reactive intermediates produced upon blue one- (left) or two-photon (right) excitation, their reactivities in terms of redox potentials available for reductions (E1/2vs. NHE) or triplet energy (ET), as well as photographs illustrating the experimental conditions. Lower row, one- and two-photon mechanisms for substrate (S) activation, and kinetic data showing the unquenched lifetimes of both reactive species (3Irsppy and eaq˙−, respectively). For further explanations, see text and ESI.† | ||
2 Results and discussion
With a triplet excited state energy of 2.65 eV and an oxidation potential of 1.01 V vs. NHE,253Irsppy accessible by one blue photon can either act as sensitizer for challenging triplet–triplet energy transfer reactions28 or as photoredox catalyst for many one-electron substrate reductions (E1/2(Irsppy+/3Irsppy) = −1.64 V vs. NHE, see also left part of Scheme 1).25,27 As we reported recently, the remarkable photostability of Irsppy combined with its favorable excited-state properties, permits an efficient two-photon ionization producing the superreductant eaq˙− with its standard potential of −2.9 V vs. NHE,29 even under cw excitation conditions (right part of Scheme 1). Interestingly, the redox energy available for reductions is almost doubled when going from 3Irsppy to eaq˙−, whereas the unquenched lifetimes of both species are practically identical in our system (see kinetic data presented in Scheme 1 and Section 1.3 of the ESI for details†). During our recent investigations on the two-photon ionization of Irsppy25 using the chloroacetate assay22,30,31 to probe eaq˙− formation, we did not observe any eaq˙− production with the natural beam of the 447 nm cw laser operating at 1 W optical output (in Scheme 1, the beam of that laser is visualized by the catalyst emission), but the biphotonic ionization operates efficiently when the diode laser beam is collimated to a spot smaller than 1 mm2 using a lens. In this study, we put to good use a lens as inexpensive key element to switch between one-photon (3Irsppy) and two-photon (eaq˙−) induced lab-scale reactions under otherwise very similar conditions. All photoreactions were carried out in 4 mL cuvettes with septum caps (pathlength, 10 mm) employing triethanolamine (TEOA) as electron donor for catalyst regeneration and hydrogen atom donor allowing the formation of well-defined reaction products.25,32 A detailed description of our experimental setup is given in Section 3.1 of the ESI.†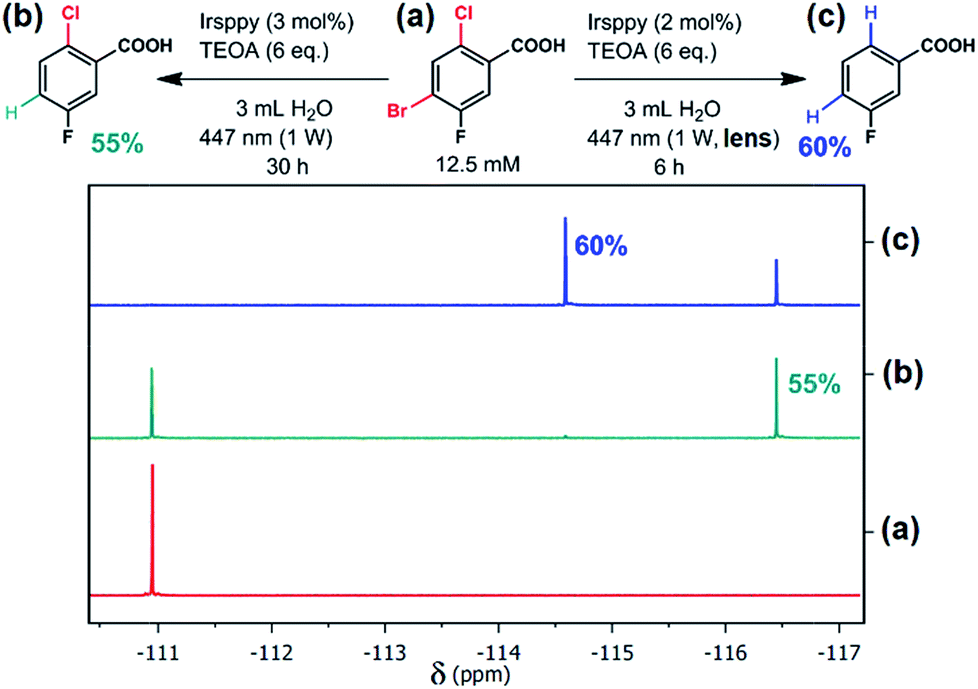 | ||
| Fig. 1 Selective debromination and subsequent dechlorination carried out with 4-bromo-2-chloro-5-fluorobenzoic acid (a) using the new blue-light driven photocatalytic system in Ar-saturated solutions ((b), one-photon conditions; (c), two-photon conditions) with the reaction parameters given at the equations. Quantitative 19F NMR measurements are displayed before (a) and after (b and c) irradiation. For further details, see text and Section 3.3 of the ESI.† | ||
2.1 Redox-potential control for reductive dehalogenations
Thermodynamically, the reductive activation of C–Cl bonds is much more challenging than that of C–Br bonds,33–35 and we speculated that 3Irsppy can merely debrominate, whereas eaq˙− will certainly be able to dechlorinate29 as well. As first substrate to test our reactivity control approach, we selected 4-bromo-2-chloro-5-fluorobenzoic acid (which is present as carboxylate under our experimental conditions, see Chapter 3 of the ESI for details† and Fig. 1a for the structure). Efficient activation of the C–F bond of that substrate is unlikely to occur,29 but the fluoro substituent allows us to exploit 19F NMR spectroscopy36 for selective and sensitive detection of the reaction outcome.First irradiation experiments (3 h of 447 nm illumination) using our catalytic system (Irsppy and TEOA in alkaline water) without the lens, i.e., under one-photon conditions, gave the debromination product 2-chloro-5-fluorobenzoic acid in very low yields (4%) as observed by quantitative 19F NMR spectroscopy (for details, see Section 3.3 of the ESI†). Given that the first mechanistic step, the oxidative quenching of 3Irsppy by 4-bromo-2-chloro-5-fluorobenzoate, is very slow (∼3 × 105 M−1 s−1, Section 2.2 of the ESI†), a significant increase in catalyst loading and reaction time was necessary to observe acceptable yields of the debromination product (55%, Fig. 1b). Importantly, only traces of 3-fluorobenzoic acid (<1%) could be detected resulting in a debromination selectivity as high as 62 (Fig. 1b and Section 3.3 of the ESI†).
Simply by putting the lens between cw light source and cuvette (top right photograph in Scheme 1), and irradiating our catalytic system in the presence of the substrate 4-bromo-2-chloro-5-fluorobenzoic acid under similar conditions as in Fig. 1b, we observed complete consumption of the starting material and 3-fluorobenzoic acid as main product after six hours (Fig. 1c). The employed two-photon conditions that permit efficient eaq˙− production evidently initiate not only debromination but also dechlorination. Compared to the one-photon reaction (Fig. 1a), the reduction of the reaction time to 20% with an even higher yield of the desired two-fold dehalogenated product (Fig. 1c) seems surprising at first glance. However, the diffusion controlled one-electron reduction kinetics of both bromo- and chloro-substituted benzoate derivatives29 allowing fast reductive lab-scale dehalogenations provide an explanation for these experimental findings.
The results of this section clearly demonstrate that increasing the thermodynamic reactivity of a catalytic system while maintaining a long lifetime, i.e., a high kinetic reactivity, of the initiating species (longer than 1 microsecond, see Scheme 1) is very beneficial for photoredox applications: first, we introduced the light intensity as selectivity parameter to reductive photoredox activations; second, our approach relying on eaq˙− as superreductant ensures relatively short reaction times for challenging and consecutive photoreactions, which otherwise frequently require more than one day of intense photoirradiation.15,35,37
2.2 From triplet–triplet energy transfer (TTET) to electron transfer (ET) chemistry
Having observed intensity dependent redox-potential regulation for photoreductions, we now turn to the question whether a completely different substrate activation mechanism can operate under one- and two-photon conditions. A one-electron reduction is the only conceivable pathway for eaq˙− initiated reactions, whereas 3Irsppy with its high triplet energy (2.65 eV, see above) could additionally activate substrates through a triplet–triplet energy transfer (TTET) event, which is quite promising given the ongoing rapid development of triplet-sensitized photocatalytic reactions.28,38,39Cinnamates, the salts and esters of cinnamic acids, have a (photo)isomerizable double bond with the trans isomer being more stable. Recent photochemical investigations on both the triplet-sensitized trans–cis isomerization28,40–42 and the hydrated electron induced hydrogenation of double bonds43,44 prompted us to apply our reactivity control approach to this substance class.
We started our studies with unsubstituted trans-cinnamic acid, which was present in its carboxylate form under our experimental conditions (see ESI Section 3.4 for details†). This substrate has a triplet energy of about 2.4 eV,45i.e., more than 0.2 eV lower than that of 3Irsppy, thermodynamically allowing a TTET and subsequent double bond isomerization. Using Irsppy as photocatalyst, photoinduced electron transfer can be excluded for thermodynamic reasons since the cinnamate radical anion formation requires a significantly higher (∼0.8 V)46 excited-state oxidation potential than 3Irsppy can provide. We indeed observed fast quenching of 3Irsppy by the trans-cinnamate ion. A Stern–Volmer analysis gave a rate constant of 1.20 × 109 M−1 s−1 (see ESI, Fig. S2†), which is in line with diffusion-controlled quenching taking coulombic interactions into account,47,48 and we detected the corresponding cis isomer in preparative photolysis experiments employing 1H NMR product analysis (one-photon conditions, 79% yield, details are given in Section 3.4 of the ESI†). Collimating the cw laser output with a lens to achieve two-photon conditions for eaq˙− generation, a novel product was observed in the 1H NMR spectrum, whose signals are in agreement with those of β-phenylpropionate.46 However, a reliable quantification could not be carried out due to spectral overlap of the desired product peaks with trans/cis-cinnamate (aromatic region of the spectrum) and TEOA-derived (aliphatic region) signals.
To circumvent these analytical issues, we turned to trans-3-fluorocinnamate as substrate (Fig. 2). The addition of the fluoro substituent to our substrate should only slightly modify the pertinent energetics (as the unchanged TTET kinetics displayed in the lower left part of Fig. 2 indicate), but allows straightforward detection and quantification of both its cis isomer and the corresponding hydrogenated product via19F NMR spectroscopy.49 Our irradiation studies with trans-3-fluorocinnamate employing this procedure gave 75% of the cis isomer with the unmodified laser beam (and 25% remaining trans isomer), whereas a yield of 62% for the hydrogenation product was observed under two-photon conditions with otherwise unmodified experimental parameters (upper reaction equations in Fig. 2, NMR spectra are shown in Fig. S5 of the ESI†).
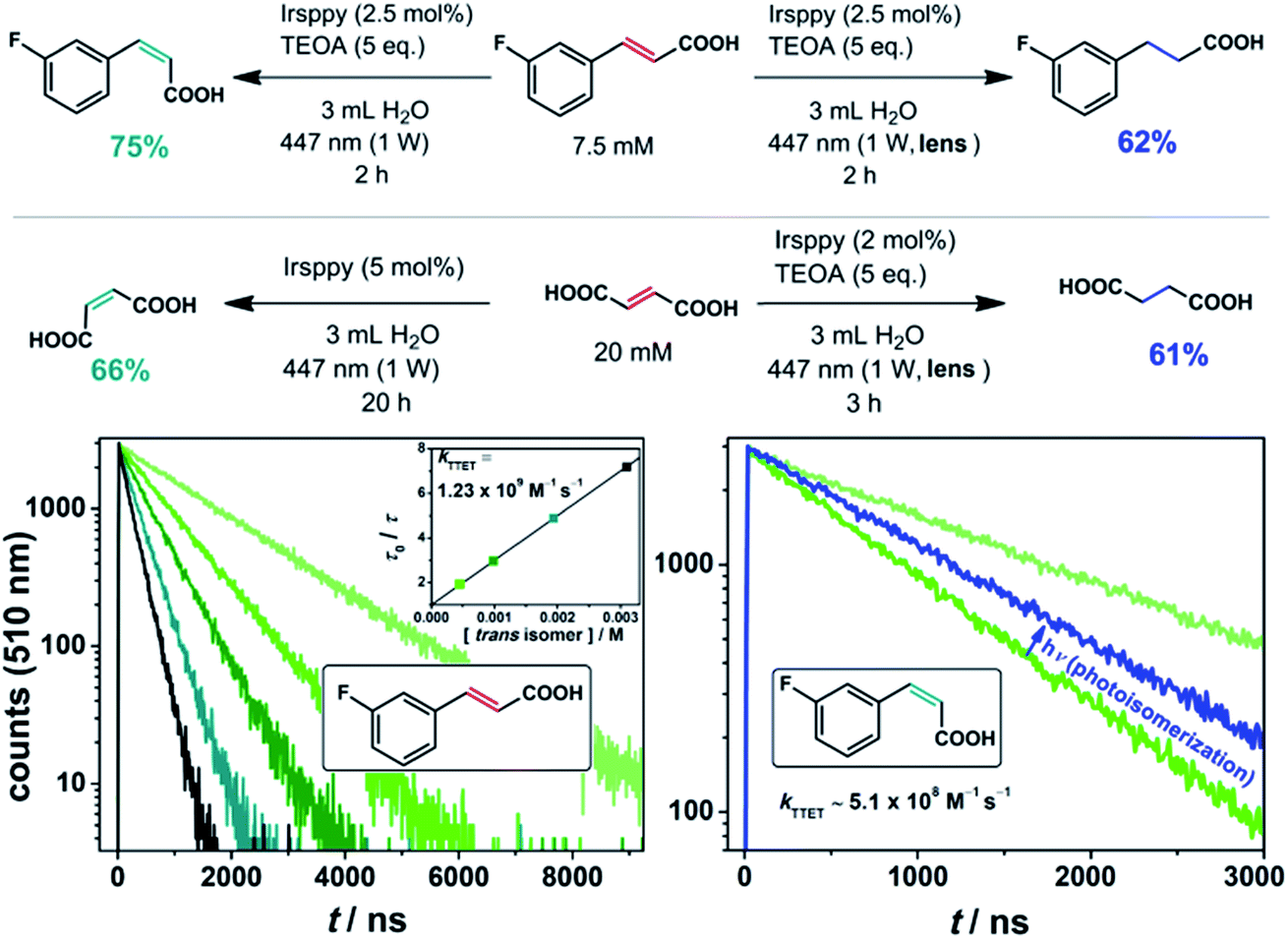 | ||
Fig. 2
trans–cis Isomerization (left, one-photon mechanism) and hydrogenation (right, two-photon mechanism) with the olefinic substrates trans-3-fluorocinnamic acid and fumaric acid using the blue-light driven photocatalytic system in Ar-saturated solutions. The lower part of the figure shows a Stern–Volmer analysis for the triplet–triplet energy transfer (TTET) between 3Irsppy and trans-3-fluorocinnamic acid (left); the rate constant for the TTET with the corresponding cis isomer was estimated using a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (cis-to-trans) mixture of both isomers as quencher (photostationary state, right). Further details are given in the text and Section 2.1 of the ESI.† :1 (cis-to-trans) mixture of both isomers as quencher (photostationary state, right). Further details are given in the text and Section 2.1 of the ESI.† | ||
The observed cis–trans-ratio of 3:1 does not change upon further irradiation which implies the existence of a photostationary state. Hence, in addition to the quenching rate constant determination for the TTET reaction between 3Irsppy and trans-3-fluorocinnamate, we performed experiments to investigate whether the cis isomer can be sensitized by 3Irsppy as well. Using the very same overall 3-fluorocinnamate concentration, either present as pure trans isomer or as 3:1 cis-to-trans mixture, the comparative 3Irsppy lifetime reduction was measured (lower right part of Fig. 2). With the well-defined isomeric mixture as quencher, a longer 3Irsppy lifetime, i.e., a slower quenching rate, was clearly detected. A kinetic analysis allowed us to estimate the TTET rate constant between 3Irsppy and cis-3-fluorocinnamate, which is about 2.5 times slower than that with the trans isomer (see Section 2.1 of the ESI for details†). We regard our indirect procedure for quickly estimating the TTET rate with less-stable cis isomers as quenchers as a useful addition to the toolbox of mechanistic photochemistry, because this approach does not rely on the prior isolation and purification of cis olefins. These results indicate that the triplet state of cis-3-fluorocinnamate is slightly higher in energy than 3Irsppy, and they are in accordance with the cis isomer being the main product in the photoequilibrium.
A plausible reaction mechanism for the formation of the hydrogenated product (right part of the upper equation in Fig. 2) is provided by: (i) initial one-electron reduction of the cinnamate ion by eaq˙−,44 (ii) protonation of the so-obtained radical dianion,50 and (iii) hydrogen atom abstraction from electron donor (TEOA)-derived species by the protonated radical anion32 to form the final product.
Next, we tried to apply our novel TTET-to-ET switch strategy to the substrate fumaric acid, which exists as dianion under our alkaline conditions (see Sections 3.2 and 3.4 of the ESI for details† and 1H NMR spectra). Similar results for the hydrogenation of that substrate were obtained (lower reaction equations in Fig. 2), but only very low yields of the desired cis isomer – maleic acid – were observed under similar conditions (2% catalyst loading and 3 h of irradiation) for the corresponding one-photon reaction. The irradiation of our system for 20 h with significantly increased catalyst loading, however, resulted in a good yield of maleic acid (Fig. 2). The much slower isomerization reaction rate for fumarate compared to cinnamates can be rationalized by the high triplet energy of fumarate51 resulting in a slightly endergonic TTET step with a concomitantly low 3Irsppy quenching efficiency under our experimental conditions (see also Section 2.1 of the ESI†). The photoreduction under two-photon excitation conditions proceeded very smoothly within 3 h for the fumaric acid substrate.
The experiments presented in this section establish that our novel reactivity control approach allows carrying out either one-photon induced double bond isomerization reactions or two-photon induced hydrogenation of the very same substrate under comparable conditions. The underlying product forming key steps are fundamentally different although the same catalyst is employed.
2.3 Differentiating between hydrogen abstraction and dimerization
Finally, we address the question of whether the faster initiation of a reaction with a more energetic species (eaq˙−) produced by a two-photon process might lead to a different final product distribution than a less reactive initiating species (3Irsppy) available from single excitation. To explore this fundamental aspect, we identified the photochemical reduction of benzyl halides to produce benzyl radicals8,52–55 as a suitable test ground, because this photoreaction can yield either the hydrogen abstraction product toluene or dibenzyl as dimerization product.Trying to perform that reaction in aqueous solution, we first used the water-soluble benzyl bromide 4-(bromomethyl)benzoic acid, but rapid hydrolysis occurred even at room temperature and pH 7. To overcome this inherent hydrolysis problem of benzylic C–Br bonds, we focused on the much more stable benzyl chlorides and selected the substrate 4-(chloromethyl)benzoic acid, because it is not only perfectly water-soluble in its carboxylate form but also stable in alkaline solution on the timescales of our experiments.
Studies on the emission quenching of 3Irsppy by 4-(chloromethyl)benzoate (left plot in Fig. 3) gave a rate constant that is about three orders of magnitude slower than the diffusion limit (3.5 × 106 M−1 s−1). This slow substrate activation is in line with the expected low driving force for this reaction as the reduction of benzyl chlorides is known to be more challenging than that of the corresponding bromides. In agreement with these findings, the preparative photoreaction is very slow with our “one-photon” setup: after 2 h of intense (1 W) irradiation (left part of the equation in Fig. 3) we observed a conversion of 35% only (compare Section 2.2, in which we reached the reaction end point in less than 2 h under similar conditions), and there are still remaining substrate molecules after 7 h reaction time (91% conversion). These two experiments gave the hydrogen abstraction main product (4-methylbenzoic acid) in 66% and 70% conversion-normalized yields, respectively (details concerning the analytics are given in Chapter 3 of the ESI†).
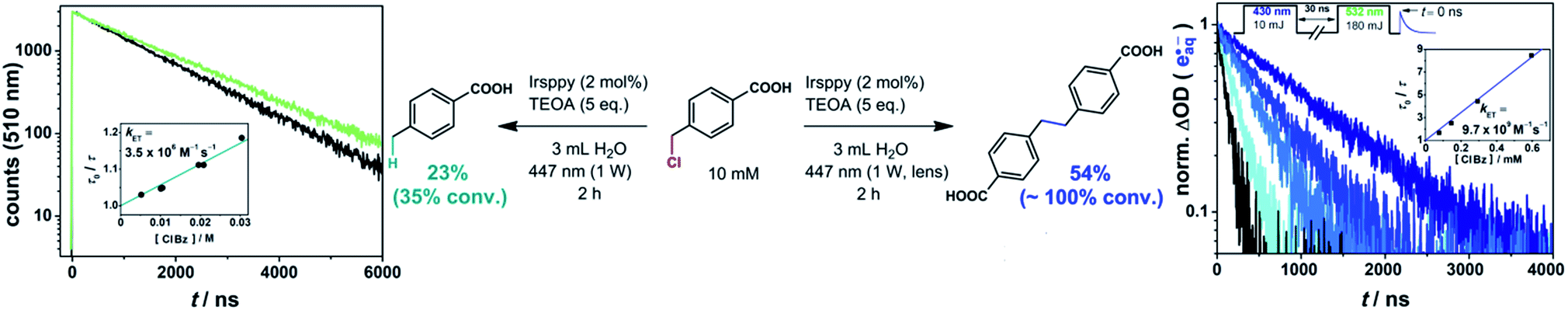 | ||
| Fig. 3 3Irsppy (left) and eaq˙− (right) induced reductive activation of 4-(chloromethyl)benzoic acid resulting in the hydrogen abstraction and the dimerization main product, respectively, together with rate constant determinations of the two substrate activation reactions. The conditions for preparative irradiation experiments are given at the equations. For further details, see text and Section 1.3 of the ESI† (which enlarges on the two-pulse experiments shown in the right part of the figure). | ||
Carrying out the 4-(chloromethyl)benzoate activation with the very same starting solution and light output of our cw laser, but using the lens for beam collimation (“two-photon conditions”), we observed a substantial change of the reaction performance (right part of the equation in Fig. 3). The whole substrate is consumed after 2 h, corresponding to an increase in the overall reaction rate by a factor of at least three, although only a very small fraction of the reaction volume is illuminated with the two-photon setup (compare, photographs displayed in Scheme 1). In direct consequence, the local benzyl radical concentration in the illuminated area has to be higher by orders of magnitudes when the setup with the lens is employed, which, as we found, promotes dimerization (54% yield). To sum up these results: applying a higher photon flux per area to the reductive activation of a water-soluble benzyl chloride – simply by using a lens – increases the reaction rate drastically, and allows to switch from the hydrogen abstraction to the dimerization main product. As in the preceding sections, these results provide clear evidence for a mechanistic change when the laser beam is collimated. Under so-called one-photon conditions, the reductive dechlorination is driven by 3Irsppy, but with the collimated beam allowing efficient eaq˙− formation, the latter reductant with its much higher reducing power operates. Using our previously developed and optimized setup for two-pulse laser flash photolysis,25,56–58 we generated hydrated electrons in the presence of several 4-(chloromethyl)benzoate concentrations and observed the expected superincrease of the substrate activation reaction rate in direct manner (right plot in Fig. 3, details are given in Section 1.3 of the ESI†).
A related eaq˙− induced dimerization procedure in aqueous solution has been published very recently.18 In that system, the eaq˙− formation step requires a pulsed UV laser and a sophisticated micellar system to enable the formation of dimers. However, owing to the micellar shielding of potential hydrogen atom donors, even the efficient dimerization of very reactive radicals is possible with that approach.
The experiments of this section illustrate the effect of the 3000-fold acceleration of a substrate activation rate constant (see rate constants included in Fig. 3). Changing the reactivity of the initiating species, which we control with the photon flux per area, allows controlling the main product, when both hydrogen abstraction and dimerization pathways of intermediates are conceivable.
3 Conclusions
As has emerged from this work, the combination of a single photocatalytic system – i.e., photocatalyst, solvent and sacrificial reagent – and a blue cw light source can be exploited for a variety of chemical reactions. Specifically, using our system the very same substrate can even be selectively converted into different main products under very similar conditions. The key for this reactivity control is the modulation of the light intensity per area, which we realize with inexpensive optics for beam collimation, thus allowing to switch from one- to two-photon substrate activation chemistry. As has been noted recently,59 photocatalysis is becoming an increasingly mature field in which further innovation crucially depends on in-depth mechanistic investigations. Our study follows that philosophy.The findings presented herein on controlled substrate activation through light-power-density regulation might well stimulate a broader application of this concept in the continuously-growing field of photo(redox) catalysis. Possible further directions include the incorporation of our approach into cascade-type reactions, in which triplets28,38,60–66 or intermediate radicals43,67–69 are trapped by further additives resulting in more valuable photochemical products, or the adaption of our methodology for reactivity control in several other solvents.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support provided by the Swiss National Science Foundation (grant number 200021_178760) and by the German National Academy of Sciences Leopoldina (postdoctoral fellowship LPDS 2017-11) is gratefully acknowledged. We thank Björn Pfund for skillful experimental support and Dr Xingwei Guo for providing us with the catalyst Irsppy.References
- S. Fukuzumi and K. Ohkubo, Chem. Sci., 2013, 4, 561–574 RSC.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- Y. Du, R. M. Pearson, C.-H. Lim, S. M. Sartor, M. D. Ryan, H. Yang, N. H. Damrauer and G. M. Miyake, Chem.–Eur. J., 2017, 23, 10962–10968 CrossRef CAS.
- E. Speckmeier, T. G. Fischer and K. Zeitler, J. Am. Chem. Soc., 2018, 140, 15353–15365 CrossRef CAS PubMed.
- M. Reckenthäler and A. G. Griesbeck, Adv. Synth. Catal., 2013, 355, 2727–2744 CrossRef.
- D. M. Arias-Rotondo and J. K. McCusker, Chem. Soc. Rev., 2016, 45, 5803–5820 RSC.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- T. Koike and M. Akita, Inorg. Chem. Front., 2014, 1, 562–576 RSC.
- Y. Qiao and E. J. Schelter, Acc. Chem. Res., 2018, 51, 2926–2936 CrossRef CAS PubMed.
- O. S. Wenger, J. Am. Chem. Soc., 2018, 140, 13522–13533 CrossRef CAS PubMed.
- B. M. Hockin, C. Li, N. Robertson and E. Zysman-Colman, Catal. Sci. Technol., 2019, 9, 889–915 RSC.
- S. Protti, D. Ravelli and M. Fagnoni, Photochem. Photobiol. Sci., 2019, 18, 2094–2101 RSC.
- M. Neumeier, D. Sampedro, M. Májek, V. A. de la Peña O'Shea, A. Jacobi von Wangelin and R. Pérez-Ruiz, Chem.–Eur. J., 2018, 24, 105–108 CrossRef CAS PubMed.
- I. Ghosh and B. König, Angew. Chem., Int. Ed., 2016, 55, 7676–7679 CrossRef CAS PubMed.
- A. M. Martínez-Gualda, R. Cano, L. Marzo, R. Pérez-Ruiz, J. Luis-Barrera, R. Mas-Ballesté, A. Fraile, V. A. de la Peña O'Shea and J. Alemán, Nat. Commun., 2019, 10, 2634 CrossRef PubMed.
- U. Lachish, A. Shafferman and G. Stein, J. Chem. Phys., 1976, 64, 4205–4211 CrossRef CAS.
- T. Kohlmann, C. Kerzig and M. Goez, Chem.–Eur. J., 2019, 25, 9991–9996 CrossRef CAS PubMed.
- M. A. Miranda, J. Pérez-Prieto, E. Font-Sanchis and J. C. Scaiano, Acc. Chem. Res., 2001, 34, 717–726 CrossRef CAS.
- J. C. Scaiano, L. J. Johnston, W. G. McGimpsey and D. Weir, Acc. Chem. Res., 1988, 21, 22–29 CrossRef CAS.
- D. N. Nikogosyan, Int. J. Radiat. Biol., 1990, 57, 233–299 CrossRef CAS PubMed.
- T. Kohlmann, R. Naumann, C. Kerzig and M. Goez, Photochem. Photobiol. Sci., 2017, 16, 1613–1622 RSC.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 CrossRef CAS PubMed.
- M. Goez, C. Kerzig and R. Naumann, Angew. Chem., Int. Ed., 2014, 53, 9914–9916 CrossRef CAS.
- C. Kerzig, X. Guo and O. S. Wenger, J. Am. Chem. Soc., 2019, 141, 2122–2127 CrossRef CAS.
- K. C. Harper, E. G. Moschetta, S. V. Bordawekar and S. J. Wittenberger, ACS Cent. Sci., 2019, 5, 109–115 CrossRef CAS.
- X. Guo, Y. Okamoto, M. R. Schreier, T. R. Ward and O. S. Wenger, Chem. Sci., 2018, 9, 5052–5056 RSC.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 RSC.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- C. Kerzig and M. Goez, Chem. Sci., 2016, 7, 3862–3868 RSC.
- X. Li, J. Ma, G. Liu, J. Fang, S. Yue, Y. Guan, L. Chen and X. Liu, Environ. Sci. Technol., 2012, 46, 7342–7349 CrossRef CAS PubMed.
- Y. Pellegrin and F. Odobel, C. R. Chim., 2017, 20, 283–295 CrossRef CAS.
- J. A. Murphy, J. Org. Chem., 2014, 79, 3731–3746 CrossRef CAS PubMed.
- M. Claros, F. Ungeheuer, F. Franco, V. Martin-Diaconescu, A. Casitas and J. Lloret-Fillol, Angew. Chem., Int. Ed., 2019, 58, 4869–4874 CrossRef CAS PubMed.
- A. U. Meyer, T. Slanina, A. Heckel and B. König, Chem.–Eur. J., 2017, 23, 7900–7904 CrossRef CAS PubMed.
- S. Berger, S. Braun and H.-O. Kalinowski, NMR Spektroskopie von Nichtmetallen, vol. 4, 19F-NMR-Spektroskopie, Thieme, Stuttgart, 1994 Search PubMed.
- H.-X. Gong, Z. Cao, M.-H. Li, S.-H. Liao and M.-J. Lin, Org. Chem. Front., 2018, 5, 2296–2302 RSC.
- A. Hölzl-Hobmeier, A. Bauer, A. V. Silva, S. M. Huber, C. Bannwarth and T. Bach, Nature, 2018, 564, 240–243 CrossRef PubMed.
- E. R. Welin, C. Le, D. M. Arias-Rotondo, J. K. McCusker and D. W. C. MacMillan, Science, 2017, 355, 380–385 CrossRef CAS PubMed.
- J. B. Metternich and R. Gilmour, J. Am. Chem. Soc., 2016, 138, 1040–1045 CrossRef CAS PubMed.
- J. J. Molloy, T. Morack and R. Gilmour, Angew. Chem., Int. Ed., 2019, 58, 13654–13664 CrossRef CAS PubMed.
- M. Bhadra, S. Kandambeth, M. K. Sahoo, M. Addicoat, E. Balaraman and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 6152–6156 CrossRef CAS PubMed.
- R. Naumann, F. Lehmann and M. Goez, Chem.–Eur. J., 2018, 24, 13259–13269 CrossRef CAS PubMed.
- M. Brautzsch, C. Kerzig and M. Goez, Green Chem., 2016, 18, 4761–4771 RSC.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of photochemistry, CRC/Taylor & Francis, Boca Raton, 3rd edn, 2006 Search PubMed.
- R. Naumann, F. Lehmann and M. Goez, Angew. Chem., Int. Ed., 2018, 57, 1078–1081 CrossRef CAS PubMed.
- P. Debye, Trans. Electrochem. Soc., 1942, 82, 265 CrossRef.
- C. Kerzig and M. Goez, Phys. Chem. Chem. Phys., 2015, 17, 13829–13836 RSC.
- S. E. Boiadjiev and D. A. Lightner, J. Phys. Org. Chem., 1999, 12, 751–757 CrossRef CAS.
- V. D. Parker, Acta Chem. Scand., Ser. B, 1981, 35, 295–301 CrossRef.
- M. J. Mirbach, M. F. Mirbach and A. Saus, J. Photochem., 1982, 18, 391–393 CrossRef CAS.
- C. B. Larsen and O. S. Wenger, Inorg. Chem., 2018, 57, 2965–2968 CrossRef CAS PubMed.
- Y. Zhang, J. L. Petersen and C. Milsmann, J. Am. Chem. Soc., 2016, 138, 13115–13118 CrossRef CAS PubMed.
- G. Park, S. Y. Yi, J. Jung, E. J. Cho and Y. You, Chem.–Eur. J., 2016, 22, 17790–17799 CrossRef CAS PubMed.
- T. C. Jenks, M. D. Bailey, J. L. Hovey, S. Fernando, G. Basnayake, M. E. Cross, W. Li and M. J. Allen, Chem. Sci., 2018, 9, 1273–1278 RSC.
- S. Neumann, C. Kerzig and O. S. Wenger, Chem. Sci., 2019, 10, 5624–5633 RSC.
- M. Kuss-Petermann and O. S. Wenger, Helv. Chim. Acta, 2017, 100, e1600283 CrossRef.
- M. Skaisgirski, C. B. Larsen, C. Kerzig and O. S. Wenger, Eur. J. Inorg. Chem., 2019 DOI:10.1002/ejic.201900453.
- L. Buzzetti, G. E. M. Crisenza and P. Melchiorre, Angew. Chem., Int. Ed., 2019, 58, 3730–3747 CrossRef CAS PubMed.
- F. M. Hörmann, T. S. Chung, E. Rodriguez, M. Jakob and T. Bach, Angew. Chem., Int. Ed., 2018, 57, 827–831 CrossRef PubMed.
- T. R. Blum, Z. D. Miller, D. M. Bates, I. A. Guzei and T. P. Yoon, Science, 2016, 354, 1391–1395 CrossRef CAS PubMed.
- X. Huang, T. R. Quinn, K. Harms, R. D. Webster, L. Zhang, O. Wiest and E. Meggers, J. Am. Chem. Soc., 2017, 139, 9120–9123 CrossRef CAS PubMed.
- F. Strieth-Kalthoff, C. Henkel, M. Teders, A. Kahnt, W. Knolle, A. Gómez-Suárez, K. Dirian, W. Alex, K. Bergander, C. G. Daniliuc, B. Abel, D. M. Guldi and F. Glorius, Chem, 2019, 5, 2183–2194 CAS.
- M. E. Daub, H. Jung, B. J. Lee, J. Won, M.-H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2019, 141, 9543–9547 CrossRef CAS PubMed.
- V. K. Soni, H. S. Hwang, Y. K. Moon, S.-W. Park, Y. You and E. J. Cho, J. Am. Chem. Soc., 2019, 141, 10538–10545 CrossRef CAS PubMed.
- Y. Kobayashi, K. Mutoh and J. Abe, J. Photochem. Photobiol., C, 2018, 34, 2–28 CrossRef CAS.
- M. Majek and A. Jacobi von Wangelin, Acc. Chem. Res., 2016, 49, 2316–2327 CrossRef CAS.
- I. Ghosh, L. Marzo, A. Das, R. Shaikh and B. König, Acc. Chem. Res., 2016, 49, 1566–1577 CrossRef CAS PubMed.
- T. Patra, S. Mukherjee, J. Ma, F. Strieth-Kalthoff and F. Glorius, Angew. Chem., Int. Ed., 2019, 58, 10514–10520 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Comprehensive experimental details, further mechanistic investigations, control experiments and application-related details. See DOI: 10.1039/c9sc04584h |
| This journal is © The Royal Society of Chemistry 2019 |