
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInterconversion and reactivity of manganese silyl, silylene, and silene complexes†
Jeffrey S.
Price
and
David J. H.
Emslie
 *
*
Department of Chemistry, McMaster University, 1280 Main Street West, Hamilton, Ontario L8S 4M1, Canada. E-mail: emslied@mcmaster.ca
First published on 29th October 2019
Abstract
Manganese disilyl hydride complexes [(dmpe)2MnH(SiH2R)2] (4Ph: R = Ph, 4Bu: R = nBu) reacted with ethylene to form silene hydride complexes [(dmpe)2MnH(RHSi![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHMe)] (6Ph,H: R = Ph, 6Bu,H: R = nBu). Compounds 6R,H reacted with a second equivalent of ethylene to generate [(dmpe)2MnH(REtSiCHMe)] (6Ph,Et: R = Ph, 6Bu,Et: R = nBu), resulting from apparent ethylene insertion into the silene Si–H bond. Furthermore, in the absence of ethylene, silene complex 6Bu,H slowly isomerized to the silylene hydride complex [(dmpe)2MnH(SiEtnBu)] (3Bu,Et). Reactions of 4R with ethylene likely proceed via low-coordinate silyl {[(dmpe)2Mn(SiH2R)] (2Ph: R = Ph, 2Bu: R = nBu)} or silylene hydride {[(dmpe)2MnH(SiHR)] (3Ph,H: R = Ph, 3Bu,H: R = nBu)} intermediates accessed from 4R by H3SiR elimination. DFT calculations and high temperature NMR spectra support the accessibility of these intermediates, and reactions of 4R with isonitriles or N-heterocyclic carbenes yielded the silyl isonitrile complexes [(dmpe)2Mn(SiH2R)(CNR′)] (7a–d: R = Ph or nBu; R′ = o-xylyl or tBu), and NHC-stabilized silylene hydride complexes [(dmpe)2MnH{SiHR(NHC)}] (8a–d: R = Ph or nBu; NHC = 1,3-diisopropylimidazolin-2-ylidene or 1,3,4,5-tetramethyl-4-imidazolin-2-ylidene), respectively, all of which were crystallographically characterized. Silyl, silylene and silene complexes in this work were accessed via reactions of [(dmpe)2MnH(C2H4)] (1) with hydrosilanes, in some cases followed by ethylene. Therefore, ethylene (C2H4 and C2D4) hydrosilylation was investigated using [(dmpe)2MnH(C2H4)] (1) as a pre-catalyst, resulting in stepwise conversion of primary to secondary to tertiary hydrosilanes. Various catalytically active manganese-containing species were observed during catalysis, including silylene and silene complexes, and a catalytic cycle is proposed.
CHMe)] (6Ph,H: R = Ph, 6Bu,H: R = nBu). Compounds 6R,H reacted with a second equivalent of ethylene to generate [(dmpe)2MnH(REtSiCHMe)] (6Ph,Et: R = Ph, 6Bu,Et: R = nBu), resulting from apparent ethylene insertion into the silene Si–H bond. Furthermore, in the absence of ethylene, silene complex 6Bu,H slowly isomerized to the silylene hydride complex [(dmpe)2MnH(SiEtnBu)] (3Bu,Et). Reactions of 4R with ethylene likely proceed via low-coordinate silyl {[(dmpe)2Mn(SiH2R)] (2Ph: R = Ph, 2Bu: R = nBu)} or silylene hydride {[(dmpe)2MnH(SiHR)] (3Ph,H: R = Ph, 3Bu,H: R = nBu)} intermediates accessed from 4R by H3SiR elimination. DFT calculations and high temperature NMR spectra support the accessibility of these intermediates, and reactions of 4R with isonitriles or N-heterocyclic carbenes yielded the silyl isonitrile complexes [(dmpe)2Mn(SiH2R)(CNR′)] (7a–d: R = Ph or nBu; R′ = o-xylyl or tBu), and NHC-stabilized silylene hydride complexes [(dmpe)2MnH{SiHR(NHC)}] (8a–d: R = Ph or nBu; NHC = 1,3-diisopropylimidazolin-2-ylidene or 1,3,4,5-tetramethyl-4-imidazolin-2-ylidene), respectively, all of which were crystallographically characterized. Silyl, silylene and silene complexes in this work were accessed via reactions of [(dmpe)2MnH(C2H4)] (1) with hydrosilanes, in some cases followed by ethylene. Therefore, ethylene (C2H4 and C2D4) hydrosilylation was investigated using [(dmpe)2MnH(C2H4)] (1) as a pre-catalyst, resulting in stepwise conversion of primary to secondary to tertiary hydrosilanes. Various catalytically active manganese-containing species were observed during catalysis, including silylene and silene complexes, and a catalytic cycle is proposed.
Introduction
Silylenes (:SiR2)1,2 and silenes (R2CSiR2),2–4 heavy analogues of carbenes and alkenes, are highly reactive species, and in the absence of extremely bulky or π-donor substituents,5 transition metal coordination is required for stabilization.3,6–8 However, complexes bearing unstabilized silylene ligands are involved in various catalytic processes involving silanes, including dehydrocoupling, substituent redistribution, hydrosilylation, and the Direct process for silane chlorination.7 Similarly, silene complexes have in several instances been hypothesized to play an important role in catalysis, typically on the basis of indirect observations. For example, they are thought to be active species in polycarbosilane synthesis from dichloromethylsilanes and sodium in the presence of [Cp2ZrCl2],9 dehydrogenative coupling of HSiMe3 by [(Me3P)3RuH3(SiMe3)],10 transfer dehydrogenative coupling of HSiEt3 catalysed by [(p-cymene)RuH2(SiEt3)2] or [Cp*RhH2(SiEt3)2],11 and trialkylsilane (e.g. HSiMe3) perdeuteration catalysed by [(Me3P)4OsH(SiMe3)]12 or [(C6Me6)RuH2(SiMe3)2]13 in C6D6. Furthermore, silene complexes were recently proposed as off-cycle species in sila-heterocycle synthesis by intramolecular silylation of primary C–H bonds,14 and free silenes play a key role in hot wire CVD of SiC using alkylsilanes.15
Early examples of isolable transition metal complexes bearing a terminal silylene ligand featured Lewis base coordination to silicon,16 and base-free terminal silylene complexes were not isolated until 1990.17 Since then, a range of such complexes have been reported; almost exclusively mid- and late-transition metal complexes,18 which are electrophilic at silicon. By contrast, silylene complexes with an SiH substituent remain relatively rare; the first example, [(Et3P)3IrH2{SiH(C6H3-Mes2-2,6)}][B(C6F5)4], was reported in 2002,19 and in the same year, Tilley et al. suggested [{PhB(CH2PPh2)3}IrH2{SiH(Trip)}] as an intermediate in the synthesis of [{PhB(CH2PPh2)3}IrH2{Si(C8H15)(Trip)}].20 Two years later, the Tobita21 and Tilley22 groups independently reported the first structurally characterized examples, [(C5Me4Et)(OC)2WH(SiH{C(SiMe3)3})] and [Cp*(dmpe)MoH(SiHPh)], respectively. Base-free LxMSiHR complexes have only been isolated for groups 6, 8 and 9,19–26 and group 7 examples are notably absent. Extensive studies by the Tilley and Tobita groups have demonstrated that hydrogen substituents on the sp2 Si centers permit these silylene complexes to demonstrate unusual reactivity, including alkene insertion into the silylene Si–H bond in cationic complexes, and conversion to silylyne (M![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiR) complexes.20,23,25
SiR) complexes.20,23,25
A small number of transition metal silene complexes have also been isolated, with 2nd and 3rd row transition metal examples (bearing sterically and electronically unstabilized silene ligands)27 limited to complexes of Ir, Ru, and W (Fig. 1).28–30 Furthermore, outside of our recent report of manganese silene complexes (vide infra), first row transition metal complexes bearing unstabilized silene ligands have not been isolated.
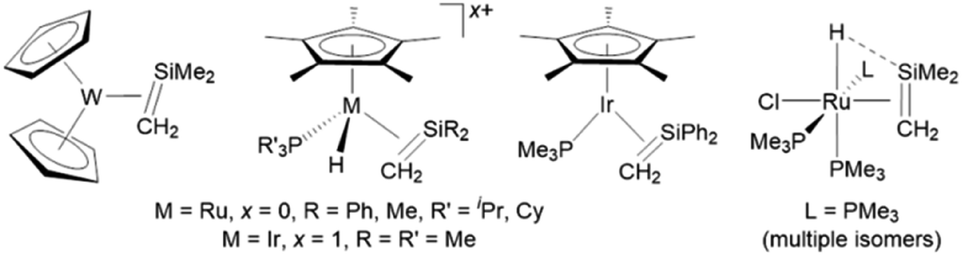 | ||
| Fig. 1 Second and third row transition metal complexes bearing sterically and electronically unstabilized silene ligands. | ||
We recently communicated the synthesis (Scheme 1) of the first unstabilized terminal silylene complexes of a group 7 metal, [(dmpe)2MnH(SiR2)] (3Ph2: R = Ph, 3Et2: R = Et), by the reaction of [(dmpe)2MnH(C2H4)] (1)31 with secondary hydrosilanes (H2SiR2).32 In the solid state, the silylene and hydride ligands are cis (diphenyl analogue 3Ph2) or trans (diethyl analogue 3Et2) disposed, in the former case with an Si–H interligand interaction. Silylene hydride complexes with interligand Si–H interactions were first reported in 2004 by the Tobita21 ([(C5Me4Et)(OC)2WH(SiH{C(SiMe3)3})]) and Tilley22 ([Cp*(dmpe)MoH(SiEt2)])33 groups, and since that time, W, Fe, Ru, and Ni examples have been reported.25,26,34 Uniquely, the cis and trans isomers of 3Ph2 exist in equilibrium with one another in solution (Scheme 1).
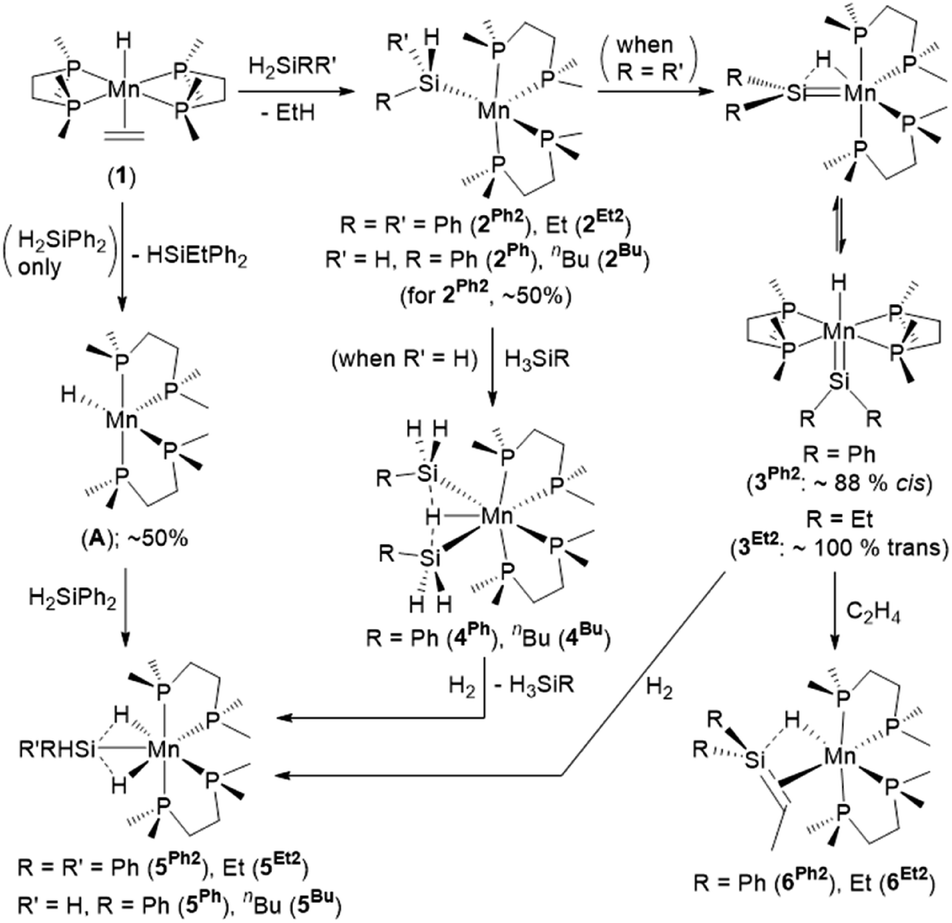 | ||
| Scheme 1 Reactions of [(dmpe)2MnH(C2H4)] (1) with primary and secondary hydrosilanes to generate silylene-hydride (3R2) and disilyl hydride (4R) complexes respectively, reactions of the latter two complexes with H2 to generate isostructural silyl dihydride complexes (5R2 or 5R respectively), and reaction of silylene hydride complexes 3R2 with ethylene to generate silene hydride complexes (6R2).32,35,36 Only one isomer is shown for 2, 5, and A. | ||
In contrast to the reactions of 1 with secondary hydrosilanes, reactions with primary hydrosilanes (H3SiR) yielded disilyl hydride complexes [(dmpe)2MnH(SiH2R)2] (4Ph: R = Ph, 4Bu: R = nBu; Scheme 1).35 The syntheses of both silylene hydride complexes 3R2 and disilyl hydride complexes 4R from 1 were proposed to proceed via a 5-coordinate silyl intermediate [(dmpe)2Mn(SiHRR′)] (2Ph2: R = R′ = Ph; 2Et2: R = R′ = Et; 2Ph: R = Ph, R′ = H; 2Bu: R = nBu, R′ = H), which undergoes α-hydride elimination to generate 3R2, or oxidative addition of a second equivalent of hydrosilane to afford 4R (Scheme 1).
Reactions of the silylene hydride complexes with ethylene generated the silene hydride complexes cis-[(dmpe)2MnH(R2SiCHMe)] {R = Ph (6Ph2) or Et (6Et2); Scheme 1}. This type of silylene to silene transformation is unprecedented, although Tilley et al. have reported conversion of [Cp*(Me3P)Ir(Me)(SiMe2)]+ to the silene hydride isomer, [Cp*(Me3P)IrH(Me2SiCH2)]+.28 The intermediacy of a trimethylsilyl iridium complex in this reaction was supported by trapping reactions with CO and ethylene. Additionally, the iridium silene hydride cation reacted with pyridine to afford [Cp*(Me3P)Ir(Me){SiMe2(py)}]+, highlighting the reversibility of the silylene–silene transformation (Scheme 2).28 Furthermore, many of the known silene complexes were synthesized by installation of a CH2SiR2H (R = Me or Ph) or SiR2Me group, followed by β-hydride elimination.14,28,29 These classes of reaction are combined in Scheme 2, highlighting the potential to interconvert between silylene, silyl, silene, and alkyl (CH2SiHR2) complexes.
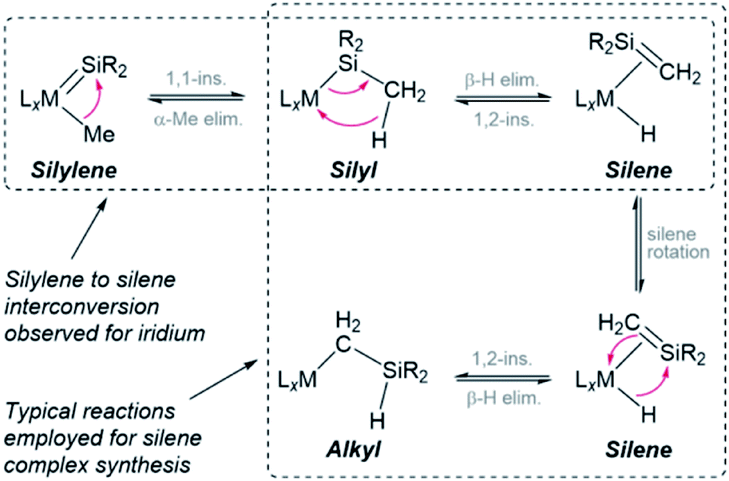 | ||
| Scheme 2 Reported reactions capable of converting between silylene, silyl, silene and alkyl isomers. Electron pushing arrows are shown for the forward direction. | ||
Both silylene hydride complexes 3R2 and disilyl hydride complexes 4R have been shown to react with H2 (Scheme 1) to generate silyl dihydride complexes [(dmpe)2MnH2(SiHRR′)] (5Ph2: R = R′ = Ph; 5Et2: R = R′ = Et; 5Ph: R = Ph, R′ = H; 5Bu: R = nBu, R′ = H), suggesting the accessibility of a common low-coordinate silyl intermediate, [(dmpe)2Mn(SiHRR′)] (2).32,36 Therefore, disilyl hydride complexes 4R could potentially react as sources of manganese silylene hydride complexes with an SiH substituent, and exposure of 4R to ethylene may provide a route to silene complexes bearing an SiH substituent.
Herein, we report the reactions of disilyl hydride complexes 4R with ethylene to generate the first examples of silene complexes with a hydrogen substituent on silicon. Their unique reactivity is also described, including (a) silene hydride to silylene hydride isomerization, and (b) reaction with a second equivalent of ethylene to convert the SiH substituent to an SiEt group. The reactions of 4R with ethylene likely proceed via a low-coordinate silyl or silylene hydride intermediate, and DFT calculations, high temperature NMR spectroscopy, and trapping studies are described, providing insight into the accessibility of these intermediates.
All of the silyl, silylene and silene complexes in this work are accessed via reactions of [(dmpe)2MnH(C2H4)] (1) with hydrosilanes, in some cases followed by ethylene. Therefore, ethylene (C2H4 and C2D4) hydrosilylation was investigated using [(dmpe)2MnH(C2H4)] (1) in combination with primary and secondary hydrosilanes, and a catalytic cycle is proposed (based on the metal species and hydrosilane products observed throughout the course of the reactions). Alkene hydrosilylation is an industrially important transition metal-catalysed process for alkylsilane production,37–39 and the most common olefin hydrosilylation catalyst used in industry is Karstedt's catalyst, [Pt2(O{SiMe2(CHCH2)}2)3].38 However, the development of catalytic systems based on first row transition metals such as manganese is of interest due to high abundance, low cost, reduced toxicity, and improved environmental compatibility.40 In this regard, manganese mediated hydrosilylation of polar unsaturated bonds has been well studied,41 but only a handful of manganese catalysts have been reported for alkene hydrosilylation.32,42
The typical mechanism for alkene hydrosilylation (Chalk–Harrod mechanism) involves oxidative addition of a hydrosilane to generate a silyl hydride complex, followed by alkene coordination, C–H bond-forming 1,2-insertion, and finally Si–C bond-forming reductive elimination. However, in some cases alkene coordination is followed by C–Si bond-forming 1,2-insertion and then C–H bond-forming reductive elimination (modified Chalk–Harrod mechanism). Furthermore, catalytic cycles which proceed via a monosilyl complex rather than a silyl hydride complex have been reported, including hydrosilylation reactions utilizing a cationic palladium(II) or cobalt(III) alkyl pre-catalyst.38,43
Results and discussion
Reactions of disilyl hydride complexes 4R with ethylene
The disilyl hydride complexes [(dmpe)2MnH(SiH2R)2] (4Ph: R = Ph, 4Bu: R = nBu) reacted with ethylene at room temperature to afford the silene hydride complexes [(dmpe)2MnH(RHSiCHMe)] (6Ph,H: R = Ph, 6Bu,H: R = nBu). This reaction mirrors the reactions of silylene hydride complexes 3R2 with ethylene (vide supra: Scheme 1).32 Moreover, complexes 6R,H reacted with a second equivalent of ethylene to form silene hydride complexes with two hydrocarbyl groups on Si, [(dmpe)2MnH(REtSiCHMe)] (6Ph,Et: R = Ph, 6Bu,Et: R = nBu); the products of apparent ethylene insertion into the Si–H bond (Scheme 3 and Fig. 2). This silene SiH to SiR conversion reaction is unprecedented. Complexes 6R,Et also reacted further with ethylene to generate [(dmpe)2MnH(C2H4)] (1),31 potentially by substitution of the silene ligand which undergoes subsequent decomposition to unidentified products.
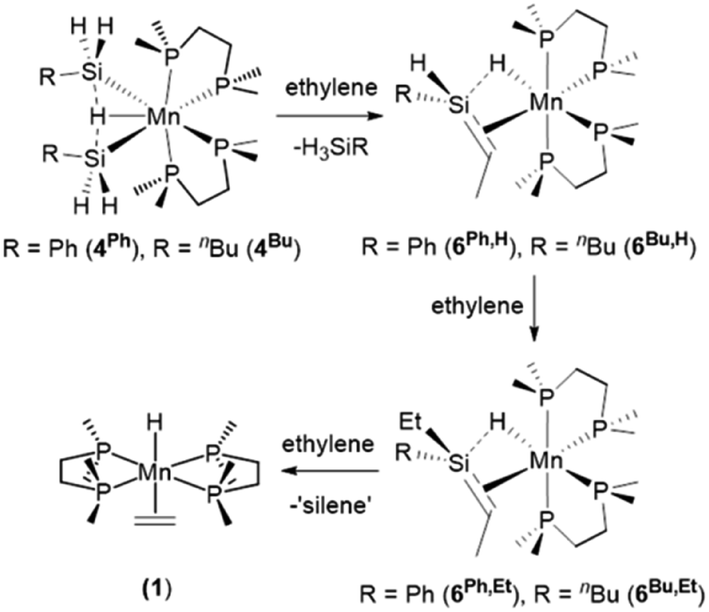 | ||
| Scheme 3 Reactions of disilyl hydride complexes [(dmpe)2MnH(SiH2R)2] (4Ph: R = Ph, 4Bu: R = nBu) with one, two, or three equivalents of ethylene to afford SiH-containing silene hydride complexes [(dmpe)2MnH(RHSiCHMe)] (6Ph,H: R = Ph, 6Bu,H: R = nBu), silene hydride complexes with two hydrocarbyl groups on Si [(dmpe)2MnH(REtSiCHMe)] (6Ph,Et: R = Ph, 6Bu,Et: R = nBu), and ethylene hydride complex [(dmpe)2MnH(C2H4)] (1), respectively. Only one isomer is shown for each silene hydride complex. | ||
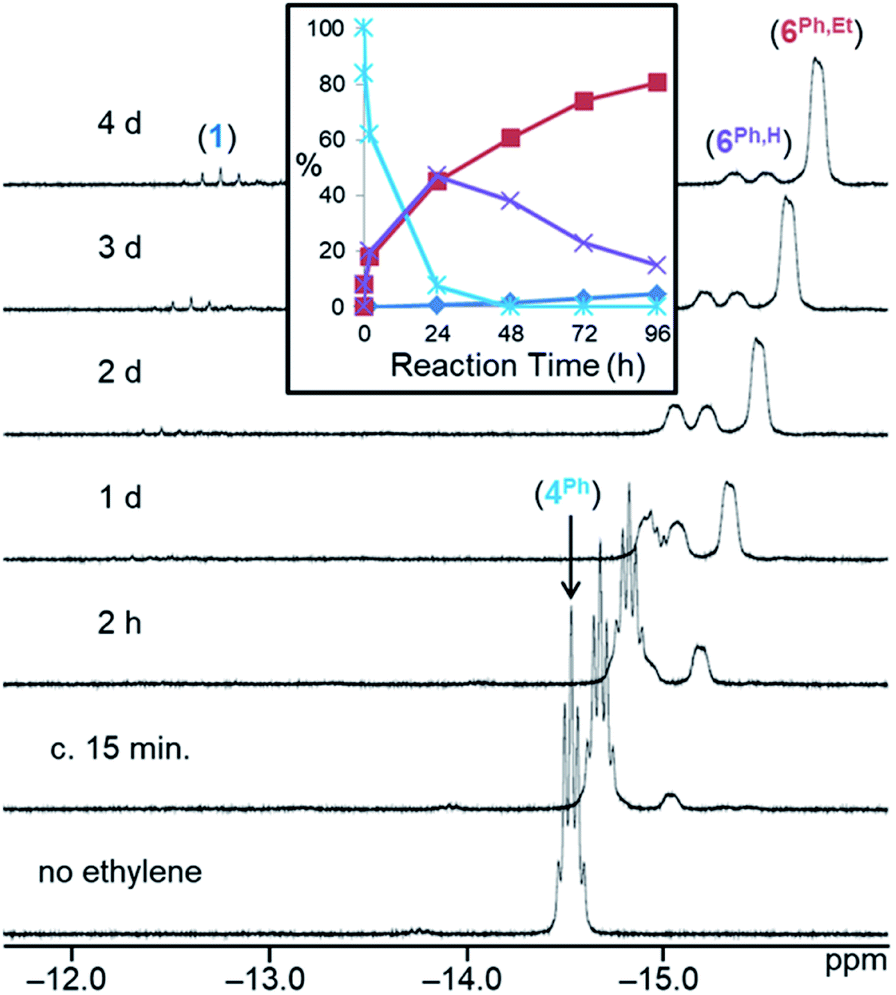 | ||
| Fig. 2
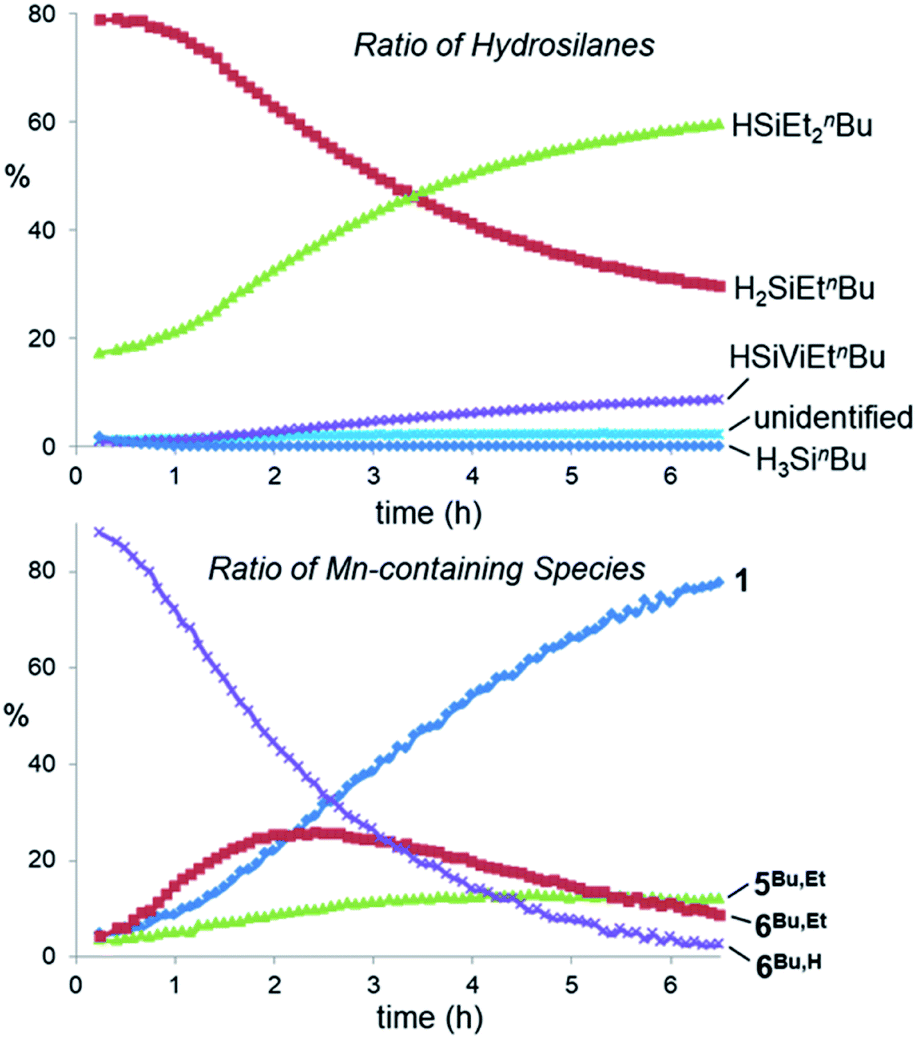
1H NMR spectra (298 K, C6D6, 600 MHz) for the reaction of [(dmpe)2MnH(SiH2Ph)2] (4Ph) with ethylene over time (initial, nC2H4 ≈ nsilane).44 The x-axis corresponds to the bottom spectrum, and for clarity, each spectrum above that is shifted by 0.15 ppm to lower frequency. The inset shows the relative concentration of hydride-containing species versus time; reactant [(dmpe)2MnH(SiH2Ph)2] (4Ph; light blue ✴), silene hydride [(dmpe)2MnH(PhHSiCHMe)] (6Ph,H; purple ×), silene hydride [(dmpe)2MnH(PhEtSiCHMe)] (6Ph,Et; red ■), and [(dmpe)2MnH(C2H4)] (1; dark blue ♦). | ||
A range of byproducts were observed in the syntheses of silene hydride complexes, including primary, secondary, and tertiary hydrosilanes {H(3–n)SiEtnR (n = 0, 1, 2; R = Ph, nBu)};45 the latter two are formed by stepwise manganese-catalysed hydrosilylation reactions between the primary hydrosilane byproduct and excess ethylene (vide infra). For R = nBu, silene SiH to SiEt conversion did not proceed until all of the primary hydrosilane byproduct had been consumed, so conversion of 4Bu to 6Bu,H, and then to 6Bu,Et, proceeded in a stepwise fashion. By contrast, for R = Ph, silene SiH to SiEt conversion commenced as soon as 6Ph,H was available (Fig. 2).
Compounds 6Bu,H and 6Ph,Et were isolated as a red oil and a brown solid, respectively, in >95% purity. By contrast, 6Ph,H and 6Bu,Et were characterized in situ by NMR spectroscopy (Table 1). Compounds 6Ph,H and 6Bu,Et were not isolated due to the formation of mixtures of products (e.g.6Ph,H accompanied by 6Ph,Et and 1), combined with instability in solution over a period of days at room temperature.
CHMe)] (6Ph2: R = R′ = Ph; 6Et2: R = R′ = Et; 6Ph,H: R = Ph, R′ = H; 6Bu,H: R = nBu, R′ = H; 6Ph,Et: R = Ph, R′ = Et; 6Bu,Et: R = nBu, R′ = Et); in C6D6 (6R2 and 6Ph,Et) or d8-toluene (6R,H and 6Bu,Et). Unless otherwise noted, values are from NMR spectra at 298 K. For 6R,H, NMR environments are reported for both observed isomers. Chemical shifts for 6Ph2 and 6Et2 are from our prior communication32
| 6Ph,H | 6Bu,H | 6Ph,Et | 6Bu,Et | 6Ph2 | 6Et2 | |
|---|---|---|---|---|---|---|
a Due to the minor isomer of 6Bu,H.
b Due to the major isomer of 6Bu,H.
c Both isomers have identical chemical shifts.
d Measured at 213 K {because this environment was not located by 29Si{1H} or 2D 1H–29Si (HSQC or HMBC) NMR spectroscopy at 298 K}.
e Coupling between the Si![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) and SiC and SiC![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) CH3 environments.
f
1
J
C,H could only be resolved for one isomer. CH3 environments.
f
1
J
C,H could only be resolved for one isomer.
|
||||||
| Mn |
−14.5, −14.7 | −14.9,a −15.0b | −14.9 | −15.3 | −14.6 | −15.3 |
| Si |
4.5c | 3.7c | — | — | — | — |
| SiCCH3 |
0.1, 0.2 | −0.1,a −0.2b | 0.2 | −0.1 | 0.4 | 0.0 |
| SiCHC3 |
1.9c | 1.8,a 1.7b | 1.9 | 1.7 | 2.1 | 1.8 |
| Si |
−21.0, −21.2 | −19.3,a −20.8b | −21.7 | −19.3 | −22.9 | −19.4 |
| 29Si | −7.1,d −17.4d | −8.9,a −17.0b | 0.7 | −6.5 | −1.5 | −3.0 |
| 31P | 63.3–85.5 | 65.8–79.1 | 65.7–79.2 | 65.5–79.3 | 62.7–78.3 | 65.5–79.3 |
| 1 J C,H | 139f | 138,a 139b | 137 | 138 | 136 | 137 |
In solution (in the absence of ethylene or free hydrosilanes), SiH-containing silene hydride complex 6Bu,H underwent isomerization to the silylene hydride complex trans-[(dmpe)2MnH(SiEtnBu)] (trans-3Bu,Et; Scheme 4), with 20% conversion after 2 days at room temperature in C6D6.46 NMR spectra of trans-3Bu,Et feature an MnH1H NMR peak at −10.48 ppm (a quintet with 2JH,P of 51 Hz), two sharp singlets in the 31P{1H} NMR spectrum at 80.35 and 80.50 ppm, and a high-frequency peak in the 29Si{1H} NMR spectrum at 364 ppm. These data are consistent with a high-symmetry base-free silylene complex, and are nearly identical to the NMR data for trans-[(dmpe)2MnH(SiEt2)] (trans-3Et2).47 Isomerization was accompanied by formation of small amounts (∼10% relative to 3Bu,Et) of an unidentified manganese hydride complex (with a quintet 1H NMR peak at −9.06 ppm; 2JH,P = 47 Hz) and the silene hydride complex [(dmpe)2MnH(nBuEtSiCHMe)] (6Bu,Et).
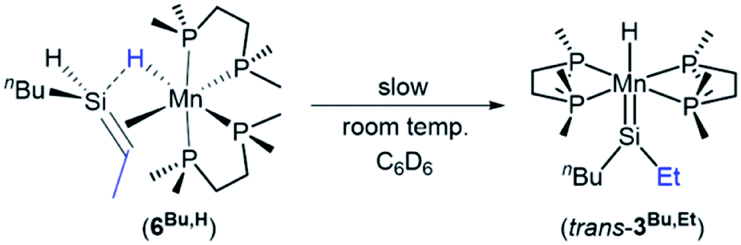 | ||
| Scheme 4 Solution decomposition of [(dmpe)2MnH(nBuHSiCHMe)] (6Bu,H) to form silylene complex trans-[(dmpe)2MnH(SiEtnBu)] (trans-3Bu,Et) as the major product. | ||
Isomerization of a silene hydride complex to a silylene hydride complex is, to our knowledge, unprecedented. However, this isomerization is related to Tilley and Bergman's report of an equilibrium between the silylene alkyl complex [Cp*(Me3P)Ir(Me)(SiMe2)]+ and the silene hydride isomer, [Cp*(Me3P)IrH(Me2SiCH2)]+, which relies upon reversible α-Me and β-H elimination from a trimethylsilyl intermediate.28
For silene hydride complexes 6R,H (those with a hydride substituent on Si), two sets of NMR signals were observed due to a pair of isomers present in solution with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (6Ph,H; Fig. 2) or 1.9:1 (6Bu,H) ratio, whereas only a single set of NMR signals (indicative of a single isomer) was observed for 6R,Et (silene hydride complexes with two hydrocarbyl substituents on Si). NMR spectra of the silene hydride complexes feature (for each isomer) four 31P NMR signals, a single 29Si NMR environment (at −17.4 to 0.7 ppm), a low frequency 13C NMR signal for the Si environment (at −19.3 to −21.7 ppm), and a silene 1JC,H coupling constant (137–139 Hz) intermediate between those typical for sp2 and sp3 hybridized carbon atoms; Table 1. Additionally, the Mn signal was located at −14.5 to −15.3 ppm in the 1H NMR spectra of 6R,H and 6R,Et, and the Si, SiC(CH3) and SiCH(C3) signals were observed at 3.7 to 4.5 ppm, −0.2 to 0.2 ppm, and 1.7 to 1.9 ppm, respectively. These data are very similar to those for [(dmpe)2MnH(R2SiCHMe)] (6Ph2: R = Ph, 6Et2: R = Et; pertinent NMR data is included in Table 1), which have been spectroscopically, and (for 6Ph2) crystallographically, characterized.32 To the best of our knowledge, 6R,H are the first spectroscopically observed48 examples of transition metal silene complexes with a hydrogen substituent on silicon.
:1 (6Ph,H; Fig. 2) or 1.9:1 (6Bu,H) ratio, whereas only a single set of NMR signals (indicative of a single isomer) was observed for 6R,Et (silene hydride complexes with two hydrocarbyl substituents on Si). NMR spectra of the silene hydride complexes feature (for each isomer) four 31P NMR signals, a single 29Si NMR environment (at −17.4 to 0.7 ppm), a low frequency 13C NMR signal for the Si environment (at −19.3 to −21.7 ppm), and a silene 1JC,H coupling constant (137–139 Hz) intermediate between those typical for sp2 and sp3 hybridized carbon atoms; Table 1. Additionally, the Mn signal was located at −14.5 to −15.3 ppm in the 1H NMR spectra of 6R,H and 6R,Et, and the Si, SiC(CH3) and SiCH(C3) signals were observed at 3.7 to 4.5 ppm, −0.2 to 0.2 ppm, and 1.7 to 1.9 ppm, respectively. These data are very similar to those for [(dmpe)2MnH(R2SiCHMe)] (6Ph2: R = Ph, 6Et2: R = Et; pertinent NMR data is included in Table 1), which have been spectroscopically, and (for 6Ph2) crystallographically, characterized.32 To the best of our knowledge, 6R,H are the first spectroscopically observed48 examples of transition metal silene complexes with a hydrogen substituent on silicon.
Despite numerous attempts, we were unable to obtain X-ray quality crystals of 6R,H or 6R,Et. Therefore, we turned to DFT calculations in order to gain further insight into the structures of these complexes (ADF, gas-phase, all-electron, PBE, D3-BJ, TZ2P, ZORA). For all four complexes, energy minima were located for four cis silene hydride isomers49 with E or Z silene stereochemistry, and differing in the orientation of the silene methyl substituent (RR′SiCHMe) relative to the two dmpe ligands, as shown in Fig. 3 (see Fig. 4 for the lowest energy isomer of 6Bu,H). In all cases, isomers (i) and (ii) are within a few kJ mol−1 of one another, and are 13–22 kJ mol−1 lower in energy than isomers iii and iv, consistent with observation of just 2 isomers in the solution NMR spectra of 6R,H.50 By contrast, the apparent formation of a single isomer of compounds 6R,Et suggests that these reactions proceed under kinetic control.
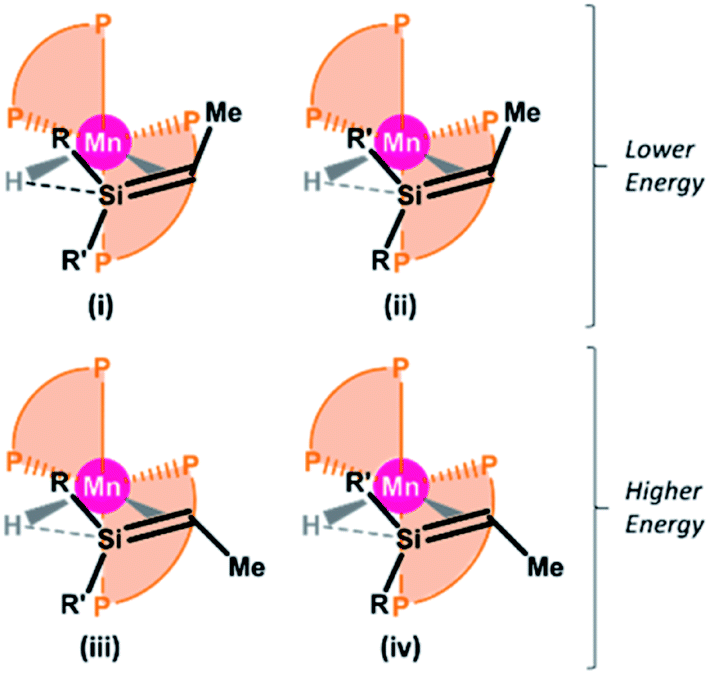 | ||
| Fig. 3 Calculated isomers (i–iv) of silene hydride complexes [(dmpe)2MnH(RR′SiCHMe)] (6Ph,H: R = Ph, R′ = H; 6Bu,H: R = nBu, R′ = H; 6Ph,Et: R = Ph, R′ = Et; 6Bu,Et: R = nBu, R′ = Et) featuring Si–H interligand interactions. | ||
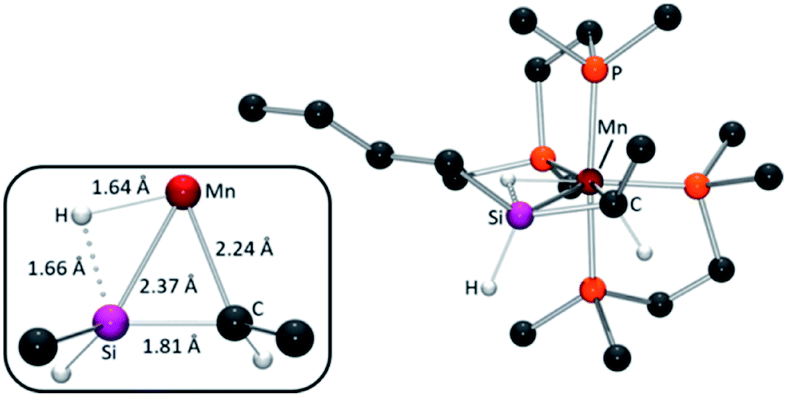 | ||
| Fig. 4 Calculated structure (ball and stick diagram) for the lowest energy isomer of silene hydride complex [(dmpe)2Mn(nBuHSiCHMe)] (6Bu,H). All hydrogen atoms have been omitted for clarity except those on Mn or the SiC unit. The inset shows a top-down view of the Mn silene hydride core, with selected bond distances. | ||
In the calculated structures of silene hydride isomers (i) and (ii) (for bond metrics, see Table S4†), the SiC bond distances of 1.80–1.81 Å fall within the range for previously reported transition metal silene complexes (1.78(2)–1.838(11) Å),51 and correspond to Mayer bond orders ranging from 0.96 to 1.10 (cf. 0.70–0.91 for Si–C single bonds in the same complexes). Also, as in previously reported 6R2,32 significant interligand interactions exist between silicon and the hydride. Computationally, this is illustrated by short Si–HMn distances (1.64–1.66 Å), with substantial Mayer bond orders (0.45–0.49),52 and is also reflected by a large negative 29Si–1HMn coupling constant of −80 Hz (measured using 29Si_edited 2D 1H–1H COSY NMR spectroscopy)36 for the major isomer of 6Bu,H (cf. −30 to −31 Hz for 4R and >0 in classical silyl hydride complexes).36,53 Short Mn–Si distances (2.35–2.42 Å) with Mayer bond orders of 0.49–0.53, and Mn–H distances of 1.64–1.66 Å with Mayer bond orders of 0.52–0.56, combined with the short SiC distance (vide supra), support the identification 6R,H and 6R,Et as cis silene hydride complexes, as opposed to 5-coordinate alkyl complexes with a strong β-Si–H–Mn interaction.
DFT calculations on low-coordinate silyl and silylene hydride intermediates derived from 4R
The reactions of the disilyl hydride complexes [(dmpe)2MnH(SiH2R)2] (4Ph: R = Ph, 4Bu: R = nBu) with C2H4 (vide supra) likely proceed via either (a) 5-coordinate mono-silyl intermediates, [(dmpe)2Mn(SiH2R)] (2Ph: R = Ph, 2Bu: R = nBu), or (b) silylene hydride intermediates, [(dmpe)2MnH(SiHR)] (3Ph,H: R = Ph, 3Bu,H: R = nBu), formed by sequential hydrosilane reductive elimination and α-hydride elimination from disilyl hydride complexes 4R (Scheme 6; vide infra). Therefore, DFT calculations (ADF, gas-phase, all-electron, PBE, D3-BJ, TZ2P, ZORA) were carried out to assess the thermodynamic accessibility of such intermediates (Fig. 5 and Table 2).
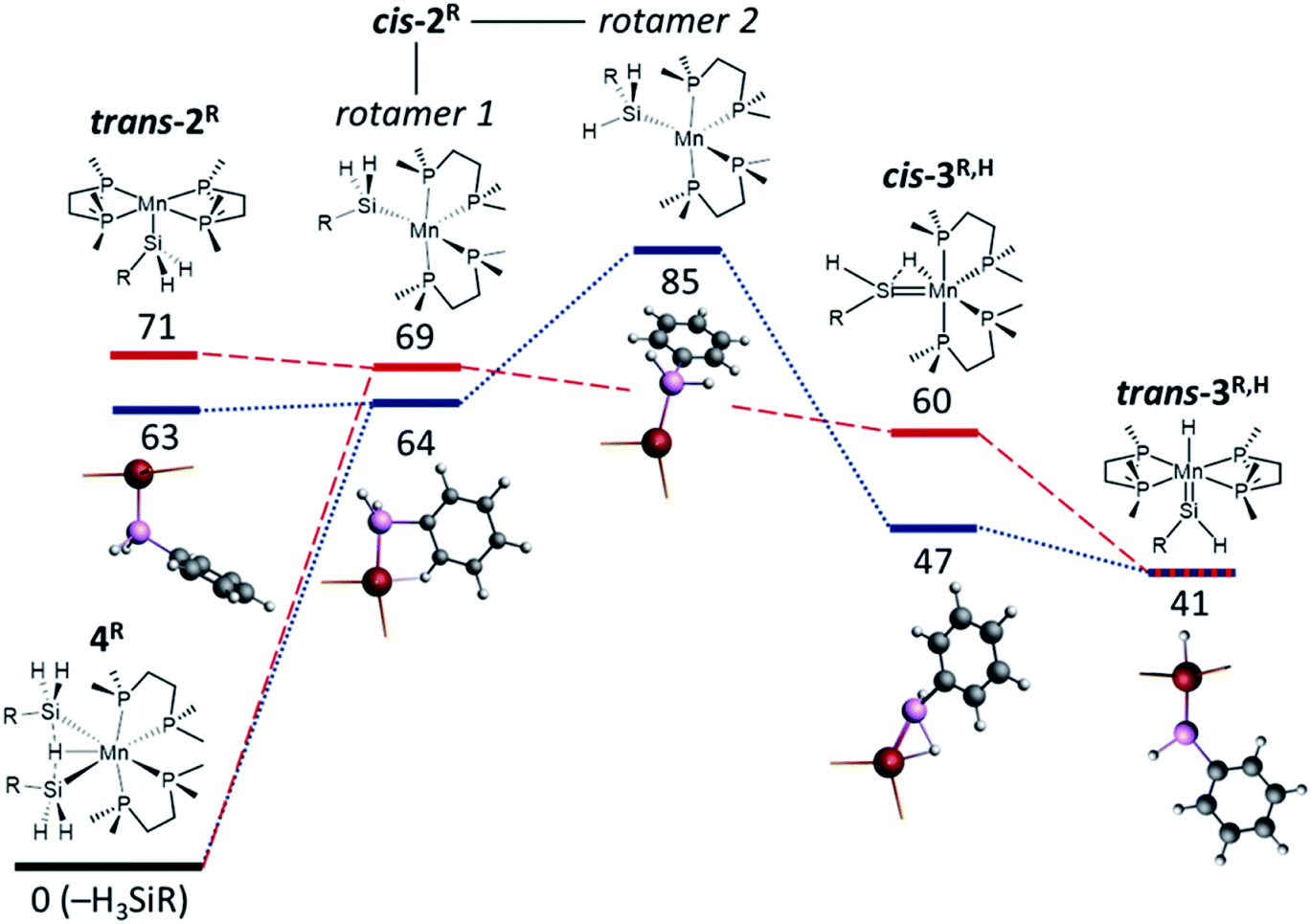 | ||
| Fig. 5 DFT calculated Gibbs free energies at 298.15 K (ΔG298.15 K; kJ mol−1) to access reactive intermediates (and the H3SiR byproduct) from disilyl hydride complexes [(dmpe)2MnH(SiH2R)2] (4Ph: R = Ph, blue dotted lines; 4Bu: R = nBu, red dashed lines). Calculated intermediates (left to right) are: (i) an isomer of [(dmpe)2Mn(SiH2R)] with an equatorial dmpe arrangement (trans-2Ph: R = Ph, trans-2Bu: R = nBu), (ii) an isomer of [(dmpe)2Mn(SiH2R)] with a disphenoidal dmpe arrangement and a hydrocarbyl substituent on silicon oriented towards the vacant coordination site {rotamer 1 of cis-2R: R = Ph (cis-2Ph), nBu (cis-2Bu)}, (iii) an isomer of [(dmpe)2Mn(SiH2R)] with a disphenoidal dmpe arrangement and an SiH substituent oriented towards the vacant coordination site {rotamer 2 of cis-2R: R = Ph (cis-2Ph); a minimum was not located for R = nBu}, (iv) an isomer of [(dmpe)2MnH(SiHR)] with interacting cis-disposed silylene and hydride ligands (cis-3Ph,H: R = Ph, cis-3Bu,H: R = nBu), and (v) trans-[(dmpe)2MnH(SiHR)] (trans-3Ph,H: R = Ph, trans-3Bu,H: R = nBu). Geometry optimized cores of the phenyl analogues of reactive intermediates are depicted below each energy level, showing Mn in red, Si in pink, C in dark grey, and H in light grey, accompanied by stick bonds to the phosphorus donor atoms. | ||
| trans-2R | cis-2R rotamer 1 | cis-2R rotamer 2 | cis-3R,H | trans-3R,H | |
|---|---|---|---|---|---|
| a n.o. = not observed (i.e. energy minimum not located). | |||||
| ΔE | 146/156 | 131/145 | 165/n.o. | 110/115 | 115/122 |
| ΔH | 135/150 | 123/138 | 152/n.o. | 117/124 | 100/111 |
| ΔS | 242/265 | 197/232 | 225/n.o. | 234/216 | 199/234 |
| ΔG298.15 K | 63/71 | 64/69 | 85/n.o. | 47/60 | 41/41 |
| ΔG335 K | 54/62 | 57/60 | 76/n.o. | 39/52 | 34/33 |
In the case of low-coordinate silyl species, energy minima were located for structures in which the silyl group is either cis (cis-2R) or trans (trans-2R) to the vacant coordination site generated by hydrosilane reductive elimination. At 298 K, ΔG for the formation of these monosilyl compounds and free hydrosilane from 4R is very similar (63–71 kJ mol−1).
In the global minima for the cis isomers (rotamer 1 of cis-2R), the hydrocarbyl substituent on silicon engages in a γ-agostic interaction with manganese (via an ortho-C bond in cis-2Ph or a CH2C2CH2CH3 bond in cis-2Bu), with Mn–Hγ distances of 1.91–1.93 Å. The Mn–Hγ–Cγ angles in this rotamer of cis-2R are 115.9° and 131.4°, respectively, and the presence of a γ-agostic interaction is further supported by Mayer bond orders of 0.22–0.24 between Mn and Hγ, and 0.13–0.15 between Mn and Cγ.
For the phenyl analogue 2Ph, a higher-energy cis isomer was also located, corresponding to a rotamer where one of the two hydrogen substituents on silicon is now oriented in the direction of the vacant coordination site (rotamer 2 of cis-2Ph; Fig. 5 and Table 2). Relative to rotamer 1, this structure features an acute Mn–Si–HSi angle of 101° (cf. 119°), an Mn–HSi Mayer bond order of 0.06 (cf. <0.05), a marginally elongated Si–HSi distance of 1.53 Å (cf. 1.51 Å), and a marginally lower Si–HSi Mayer bond order of 0.80 (cf. 0.85), together suggestive of a weak α-Si–H–Mn interaction. Rotamer 2 of 2R is presumably involved in silylene hydride formation via α-hydride elimination, and indeed, all attempts to locate an analogous energy minimum for the nBu analogue structure led instead to a silylene hydride structure (cis-3Bu,H; vide infra).
As with the 5-coordinate silyl species (vide supra), multiple energy minima (Fig. 5 and Table 2) were located for silylene hydride structures [(dmpe)2MnH(SiHR)] (3Ph,H: R = Ph, 3Bu,H: R = nBu). The two lowest energy structures are (a) a cis silylene hydride isomer with a significant interaction between silicon and the neighbouring hydride ligand (the Si⋯HMn distances are 1.68 Å, with Mayer bond orders of 0.52; cf. 0.84–0.85 for the terminal Si–H bonds), and (b) a trans silylene hydride isomer. These isomers are isostructural to the X-ray crystal structures of cis-[(dmpe)2MnH(SiPh2)] (3Ph2) and trans-[(dmpe)2MnH(SiEt2)] (3Et2), respectively.32 Calculated ΔG values to access 3R,H from 4R range from 41 kJ mol−1 (trans isomers) to 47–60 kJ mol−1 (cis isomers) at 298.15 K, decreasing to 33–34 kJ mol−1 (trans isomers) and 39–52 kJ mol−1 (cis isomers) at 335 K, highlighting their thermodynamic accessibly.
In silylene hydride complexes 3R,H, Mn–Si double bond character is apparent from relatively short Mn–Si distances (2.16–2.20 Å), Mn–Si Mayer bond orders ranging from 1.17 (cis-3R,H) to 1.54–1.57 (trans-3R,H), and a planar or near-planar environment about Si (∑(R–Si–R) > 356°); Table S2.† These parameters are comparable to those previously observed and/or calculated for the two isomers of [(dmpe)2MnH(SiR2)] (3R2; R = Et or Ph).32
High temperature NMR spectra of 4R: in situ generation of trans-silylene hydride (trans-3R,H) species
At 335 K, 1H NMR spectra of the disilyl hydride complexes [(dmpe)2MnH(SiH2R)2] (4Ph: R = Ph, 4Bu: R = nBu) revealed the formation of a small amount of a new manganese complex and free hydrosilane (H3SiR; R = Ph or nBu). This process is reversible, and clean spectra of 4R were observed upon cooling back to room temperature. The new manganese complex exhibits a high frequency (9.83 or 9.53 ppm) and a low frequency (−9.01 or −9.60 ppm) 1H NMR signal. The former is in the range observed for the terminalSiR (R = hydrocarbyl substituent) environment in diamagnetic silylene complexes of Mo, W, Fe, Ru, Os, and Ir, (6.34–12.1 ppm),19,20,22–26 while the latter is consistent with a metal hydride environment. The low frequency hydride signal is a quintet (2J1H,31P = 54 or 51 Hz) consistent with a hydride ligand apical to a plane of four equivalent phosphine donors. Taken together, these data suggest that the new complex observed at elevated temperature is trans-[(dmpe)2MnH(SiHR)] (trans-3Ph,H: R = Ph, trans-3Bu,H: R = nBu); the most thermodynamically accessible silyl or silylene species in Fig. 5.
Characterization of trans-3R,H (R = Ph or nBu) by 29Si NMR spectroscopy was not successful since the new species were formed at very low concentrations (∼4% and ∼2% relative to 4Ph or 4Bu, respectively). However, EXSY NMR spectroscopy at 335 K indicates exchange between the two diastereotopic Si protons in 4Ph or 4Bu, the free hydrosilane Si peak, the high frequency trans-3R,H silylene Si environment, and the Mn signals from both 4R and trans-3R,H (shown in Fig. 6 for R = nBu).54 This is consistent with an equilibrium in which 4R eliminates free H3SiR to form trans-3R,H (vide infra).
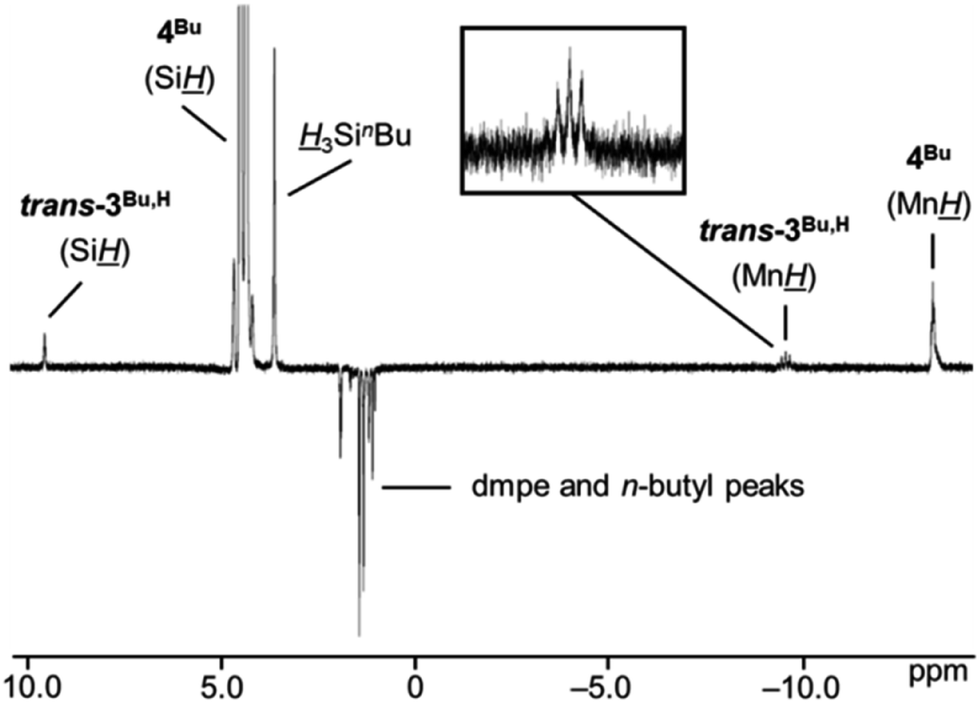 | ||
| Fig. 6 1D NOESY/EXSY NMR spectrum of a solution of [(dmpe)2MnH(SiH2nBu)2] (4Bu) at 335 K with excitation at the Si signal of 4Bu, showing chemical exchange between the Si and Mn environments of [(dmpe)2MnH(SiH2nBu)2] (4Bu) and trans-[(dmpe)2MnH(SiHnBu)] (trans-3Bu,H), and the Si environment of free 3SinBu. Positive (EXSY) peaks are indicative of chemical exchange and negative (NOESY) peaks are indicative of through-space coupling (500 MHz, C6D6). | ||
Trapping experiments with isonitriles and N-heterocyclic carbenes
To provide experimental corroboration for the accessibility of 5-coordinate silyl [(dmpe)2Mn(SiH2R)] (2Ph: R = Ph, 2Bu: R = nBu) and silylene hydride [(dmpe)2MnH(SiHR)] (3Ph,H: R = Ph, 3Bu,H: R = nBu) species from 4R, reactions with neutral donor ligands were carried out, with a view towards coordination to manganese in 2R or silicon in 3R,H (Scheme 5).
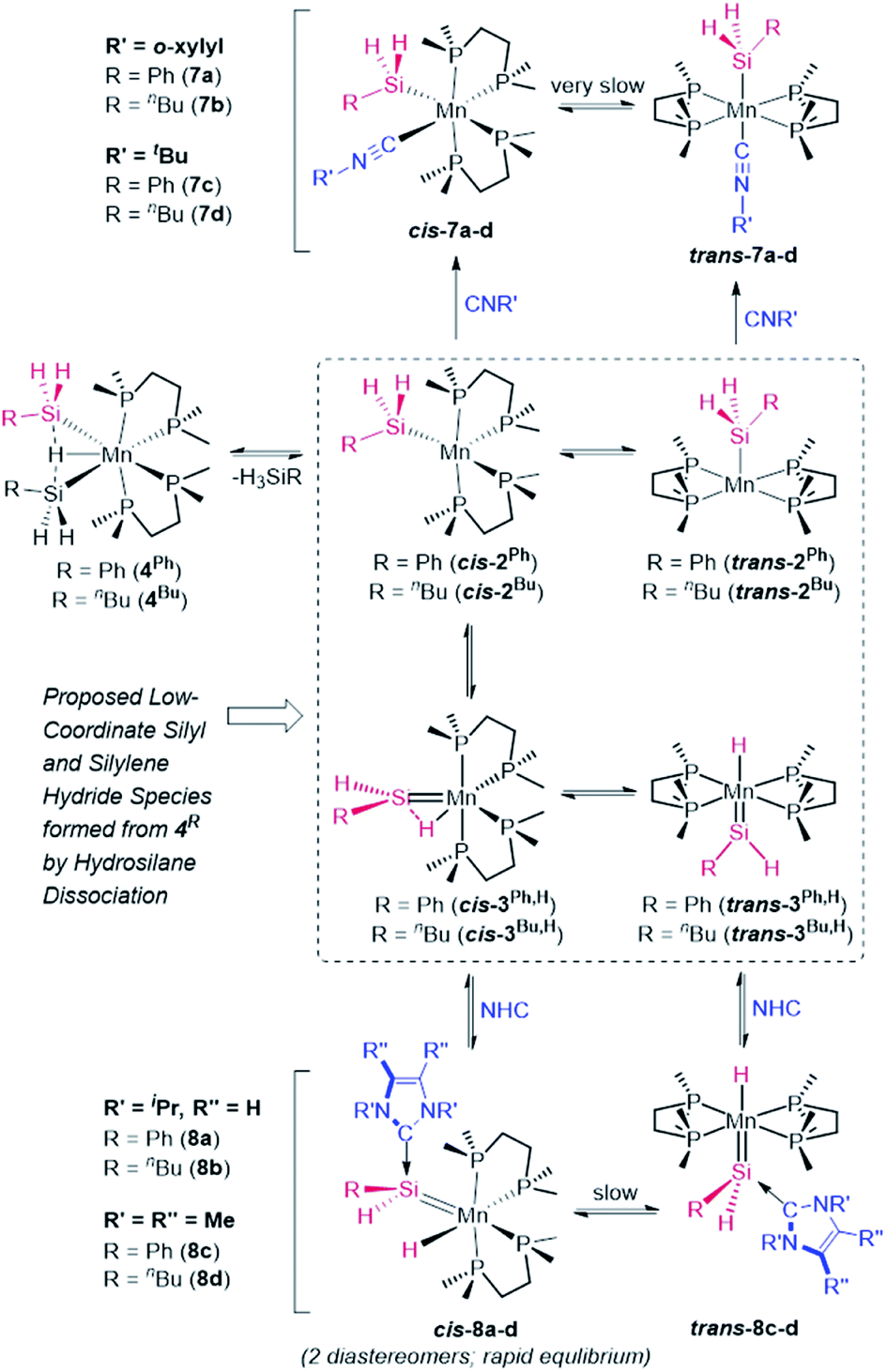 | ||
| Scheme 5 Trapping of putative silyl (2R) and silylene hydride (3R,H) intermediates: synthesis of silyl isonitrile complexes [(dmpe)2Mn(SiH2R)(CNR′)] (7a: R = Ph, R′ = o-xylyl; 7b: R = nBu, R′ = o-xylyl; 7c: R = Ph, R′ = tBu; 7d: R = nBu, R′ = tBu) and NHC-stabilized silylene hydride complexes [(dmpe)2MnH{SiHR(NHC)}] (8a: NHC = iPrNHC, R = Ph; 8b: NHC = iPrNHC, R = nBu; 8c: NHC = MeNHC, R = Ph; 8d: NHC = MeNHC, R = nBu). | ||
Addition of o-xylylNC or tBuNC to solutions of 4R resulted in hydrosilane elimination, and isolation of yellow or orange silyl isonitrile complexes [(dmpe)2Mn(SiH2R)(CNR′)] {R′ = o-xylyl, R = Ph (7a) or nBu (7b); R′ = tBu, R = Ph (7c) or nBu (7d)}, effectively trapping silyl complexes 2R (Scheme 5). In solution, all four reactions initially led to mixtures of two complexes identified by NMR spectroscopy as cis (85–97%) and trans (3–15%) isomers of 7a–d. Slow isomerization was observed between the cis and trans isomers of 7a–d in solution, and unexpectedly, these isomerization reactions proceeded in the direction of the trans isomers at elevated temperature (resulting in an increase in the proportion of trans isomer to 44–74% after heating solutions containing exclusively the cis isomer at 65–80° for 4–21 days), and in the opposite direction upon leaving the same solutions at room temperature for 3 weeks (e.g. leaving cis/trans mixtures of 7a,b containing 44–48% trans isomer at room temperature resulted in solutions containing 99% cis isomer after 3 weeks).
X-ray quality crystals were obtained for the four silyl isonitrile complexes 7a–d, in each case as the cis isomer (Fig. 7). All four structures are octahedral with Mn–Si distances of 2.3552(5)–2.3618(5) Å and Mn–C distances of 1.805(4)–1.847(3) Å. The isonitrile ligands show elongated CMn–N distances of 1.176(4)–1.225(9) Å and non-linear C–N–C angles of 159.2(8)–167.5(1)°, indicative of appreciable π-backbonding.
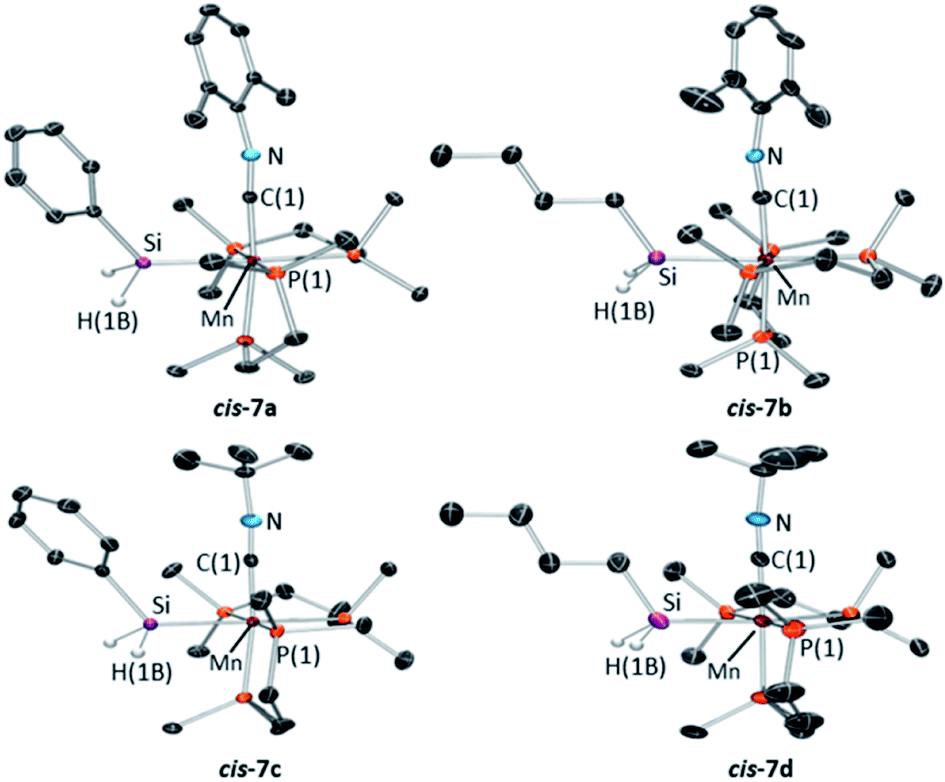 | ||
| Fig. 7 X-ray crystal structures of (top left) cis-[(dmpe)2Mn(SiH2Ph)(CNxylyl)] (cis-7a), (top right) cis-[(dmpe)2Mn(SiH2nBu)(CNxylyl)] (cis-7b), (bottom left) cis-[(dmpe)2Mn(SiH2Ph)(CNtBu)] (cis-7c), and (bottom right) cis-[(dmpe)2Mn(SiH2nBu)(CNtBu)] (cis-7d), with ellipsoids drawn at 50% probability. Most hydrogen atoms have been omitted for clarity. cis-7c and cis-7d crystallized with two independent and essentially isostructural molecules in the unit cell, only one of which is shown. All structures exhibit some disorder, and only the dominant conformation is shown. | ||
In contrast to reactions with isonitriles, reactions of disilyl hydride complexes 4R with 1,3-diisopropylimidazolin-2-ylidene (iPrNHC) or 1,3,4,5-tetramethyl-4-imidazolin-2-ylidene (MeNHC) afforded the base-stabilized silylene hydride complexes [(dmpe)2MnH{SiHR(NHC)}] {NHC = iPrNHC, R = Ph (8a) or nBu (8b); NHC = MeNHC, R = Ph (8c) or nBu (8d)}, trapping the proposed silylene hydride species 3R,H (Scheme 5). Compounds 8b–d were isolated as analytically pure red powders, whereas 8a evaded purification.
A variety of NHC-stabilized silylene complexes have been reported for V, Cr, W, Fe, Co, Rh, and Ni,55 and relative to base-free silylene complexes, they feature longer metal–silicon bond distances, pyramidalization at silicon, and lower frequency 29Si NMR chemical shifts (typically 25–100 ppm,56 compared with >200 ppm for base-stabilized silylene complexes).8
Room temperature NMR spectra of iPrNHC adducts 8a,b revealed two sets of broad NMR signals in the process of coalescence/decalescence, due to a pair of rapidly interconverting isomers. Cooling the solutions afforded two sets of well resolved NMR signals corresponding to compounds with a disphenoidal arrangement of the phosphorus donor atoms, each with a single Si signal (5.1 to 6.4 ppm), a single Mn resonance (−12.3 to −12.6 ppm), a single 29Si NMR environment (22.2 to 29.6 ppm), and four unique 31P NMR environments (65.6–81.9 ppm). These data are indicative of NHC-coordinated cis silylene hydride complexes existing as a pair of interconverting diastereomers (due to chirality at Si and Mn).
In contrast, NMR spectra of the MeNHC silylene hydride complexes (8c,d) revealed the same two rapidly interconverting cis diastereomers plus a third isomer which afforded a sharp set of 1H and 31P NMR signals at room temperature. This third isomer corresponds to an NHC-coordinated trans silylene hydride complex, as evidenced by a single Mn (−14.9 or −15.0 ppm) signal with a quintet coupling pattern (2JH,P = 48–49 Hz) and two sharp signals in the 31P{1H} NMR spectra (78.7–80.6 ppm) due to diastereotopic phosphorus atoms. The 1H NMR Si and 29Si NMR chemical shifts in these trans isomers (4.9–5.8 ppm and 22.4 ppm, respectively)57 are similar to those in the cis isomers.
At 335 K, the two cis diastereomers of 8a–d gave rise to a single set of averaged signals, with the Mn peak at −12.5 to −12.7 ppm (quintets for cis-8a,c,d with 2JH,P = 32–34 Hz, while the NMR signal for 8b remained a broad singlet in the process of coalescence), accompanied by (in solutions of 8c,d only) a set of sharp signals for the trans isomer. For 8c,d, EXSY NMR spectroscopy at 335 K showed cross peaks between the Mn and Si1H NMR signals due to both the cis and trans isomers (i.e. chemical exchange between all four environments). This equilibrium between cis- and trans-8c,d mirrors that previously reported between the cis and trans isomers of base-free [(dmpe)2MnH(SiPh2)] (3Ph2).32
Possible mechanisms for ambient temperature exchange between the cis diastereomers of 8a–d are (a) phosphine donor dissociation, isomerization of the 5-coordinate product, and phosphine re-coordination, or (b) NHC dissociation to generate cis-[(dmpe)2MnH(SiHR)] (cis-3Ph,H: R = Ph, cis-3Bu,H: R = nBu), followed by re-coordination to the opposite face of the silylene ligand.58 The latter mechanism would imply that 8a–d, like disilyl hydride complexes 4R, could react as sources of either base-free silylene hydride complexes 3R,H, or 5-coordinate silyl complexes 2R. The accessibility of this pathway is implied by the reactions of 8d with tBuNC, and 8b with ethylene, which afforded [(dmpe)2Mn(SiH2nBu)(CNtBu)] (7d) and [(dmpe)2MnH(nBuHSiCHMe)] (6Bu,H),59 respectively; these are the same complexes formed in reactions of these reagents with 4Bu. Furthermore, the accessibility of 2R provides a mechanism for the observed exchange between the Si and Mn environments in the EXSY NMR spectra of 8c,d at 335 K (vide supra).
X-ray quality crystals were obtained for complexes 8a–d by recrystallization from concentrated hexanes solutions (8a,b), toluene layered with pentane (8c), or a dilute hexanes solution (8d) at −30 °C. The solid state structures of 8a–c (Fig. 8; top row and bottom left) feature a cis arrangement of the hydride and base-stabilized silylene ligands, corresponding to one of the two cis diastereomers observed in solution.60 By contrast, 8d crystallized as the trans isomer (Fig. 8; bottom right). In all four structures {complementary DFT calculations modelled 8b,d with an nBu group in place of the Et group; [(dmpe)2MnH{SiHEt(NHC)}] where NHC = iPrNHC (8b*) or MeNHC (8d*)}, NHC coordination to silicon resulted in elongated Mn–Si distances (2.255(1)–2.299(1) Å; calcd 2.26–2.30 Å for both isomers of 8a,b*,c,d*), and correspondingly weaker Mayer bond orders of 1.03–1.08 (Table S6†), relative to base-free silylene complexes 3R,H (2.16–2.20 Å and 1.17–1.57, respectively). Unlike base-free analogues (vide supra), cis-8a–d display only negligible interligand Si–H interactions (with Mayer bond orders ≤0.13). Additionally, substantial pyramidalization at silicon was observed for both isomers of 8a–d, where the sum of the angles around silicon (for non-NHC substituents) ranged from 322(3) to 342(2)° (calcd 336.1–341.5°; Table S6,†cf. >356° in 3R,H). Nevertheless, the Mn–Si distances are significantly shorter than those in related silyl complexes 7a–d (the Mn–Si distances in 7a–d range from 2.3552(5)–2.3618(5) Å {calcd 2.35–2.36 Å (cis) and 2.41–2.42 Å (trans), with Mayer bond orders of 0.89–0.93}),61 indicative of residual Mn–Si multiple bond character in 8a–d.
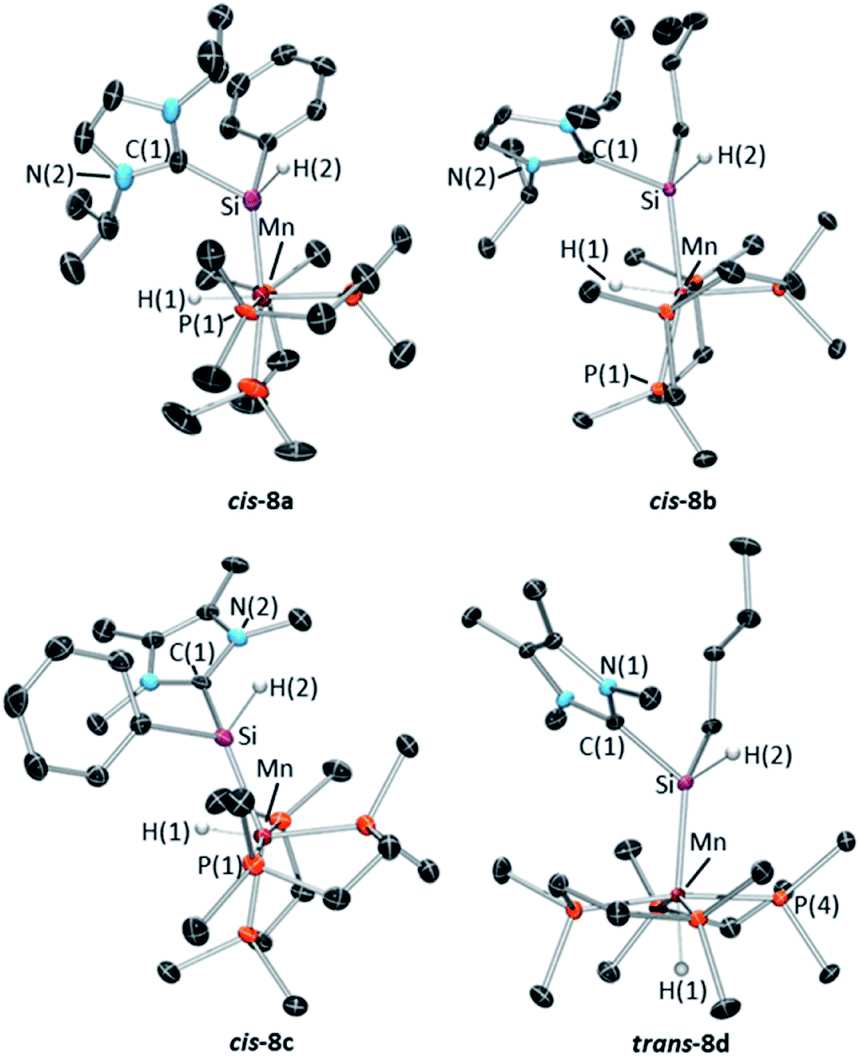 | ||
| Fig. 8 X-ray crystal structures of (top left) cis-[(dmpe)2MnH{SiHPh(iPrNHC)}] (cis-8a), (top right) cis-[(dmpe)2MnH{SiHnBu(iPrNHC)}] (cis-8b), (bottom left) cis-[(dmpe)2MnH{SiHPh(MeNHC)}] (cis-8c), and (bottom right) trans-[(dmpe)2MnH{SiHnBu(MeNHC)}] (trans-8d) with ellipsoids drawn at 50% probability. Most hydrogen atoms have been omitted for clarity. In the case of 8a–b, the structures exhibit some disorder, and only the dominant conformation is shown. | ||
Pathways for reactions of 4R with ethylene
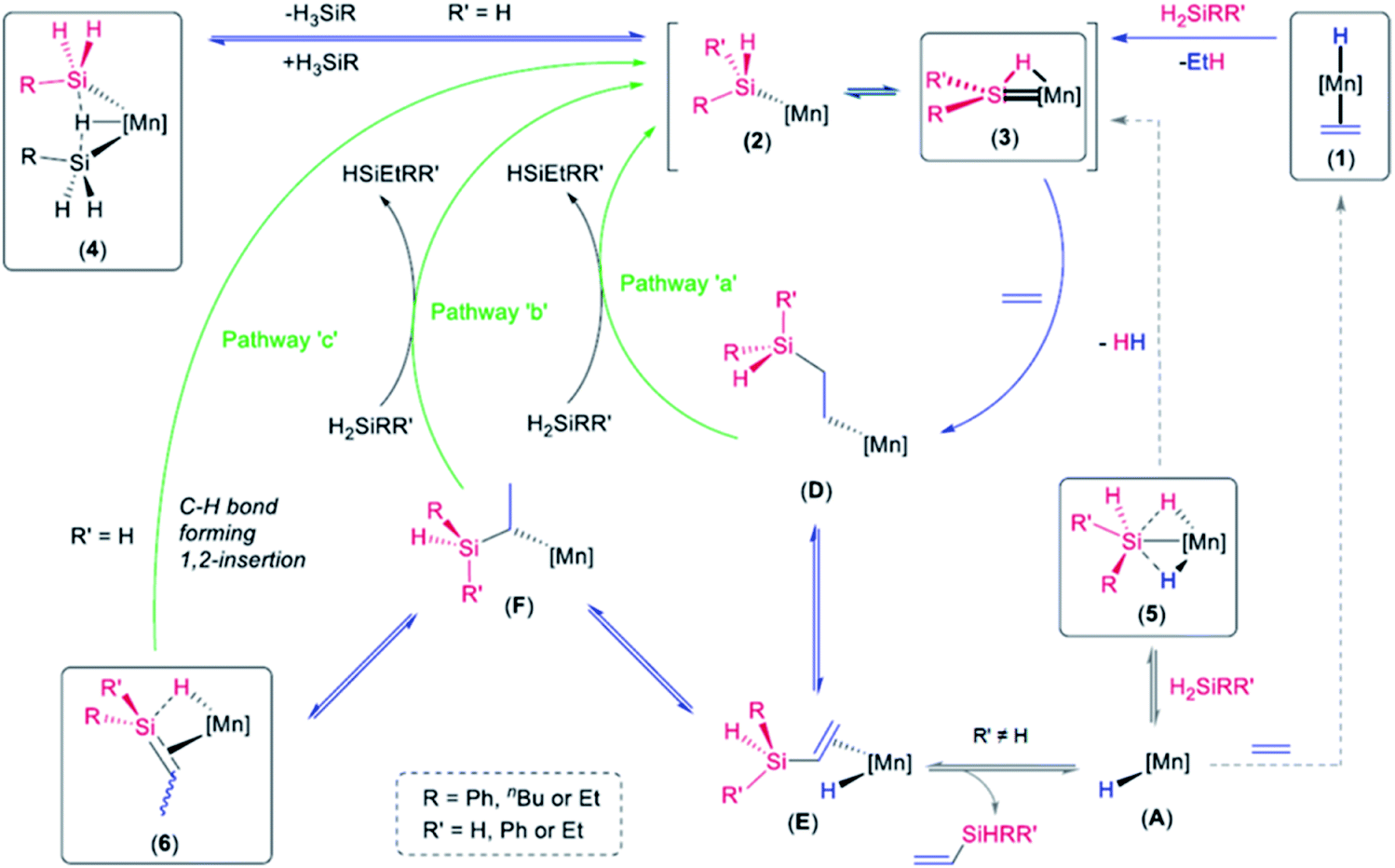
Previously, we reported the reactions of the silylene hydride complexes, [(dmpe)2MnH(SiR2)] (3Ph2: R = Ph, 3Et2: R = Et), with ethylene to form the silene hydride complexes [(dmpe)2MnH(R2SiCHMe)] (6Ph2: R = Ph, 6Et2: R = Et).32 Given that disilyl hydride complexes 4R exist in equilibrium with analogous low-coordinate silyl and silylene hydride complexes (vide supra), it is likely that the reactions of 4R with ethylene proceed via a parallel mechanism, as illustrated in Scheme 6. The initial steps in this scheme involve either (a) ethylene coordination to a silylene hydride intermediate (3R,H) followed by 2 + 2 cycloaddition (to form BR) and subsequent Si–H bond-forming reductive elimination, or (b) coordination of ethylene to a low coordinate silyl intermediate (2R), forming CR, followed by 1,2-insertion. Both of these pathways generate primary alkyl complex DR, which can provide access to 6R,H by sequential β-hydride elimination (to form ER), 1,2-insertion to generate secondary alkyl complex FR, and a second β-hydride elimination involving the hydrogen substituent on silicon. Consistent with this mechanism, the reactions of [(dmpe)2MnH(SiH2nBu)2] (4Bu) or [(dmpe)2Mn{SiHnBu(iPrNHC)}] (8b) with d4-ethylene yielded [(dmpe)2MnH(nBuHSiCDCD3)] as the only observed isotopomer of 6Bu,H.
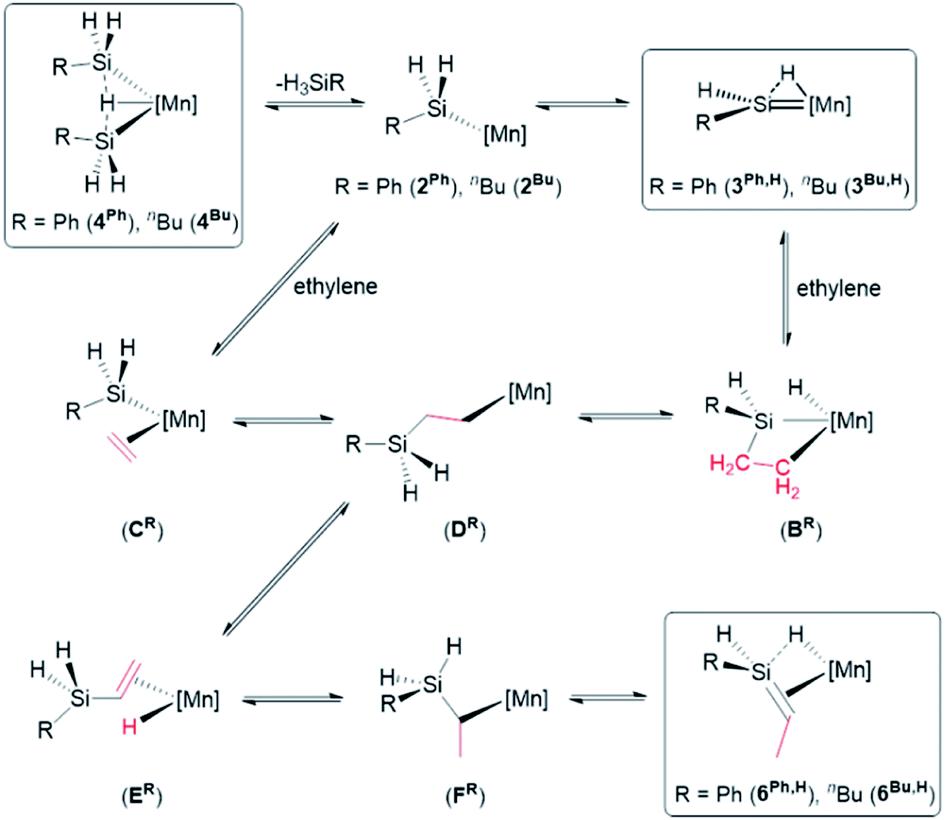 | ||
| Scheme 6 Proposed pathways for reactions of disilyl hydride complexes [(dmpe)2MnH(SiH2R)2] (4Ph: R = Ph, 4Bu: R = nBu) with ethylene to form silene hydride complexes [(dmpe)2MnH(RHSiCHMe)] (6Ph,H; R = Ph, 6Bu,H: R = nBu). [Mn] = Mn(dmpe)2. Only one isomer of 3R,H is shown. Boxes indicate complexes which have been isolated or spectroscopically observed. | ||
After conversion of 4R to 6R,H, reaction with a second equivalent of ethylene resulted in conversion of a silene SiH group in 6R,H to an SiEt group, yielding 6R,Et (vide supra). This reactivity likely involves the experimentally observed isomerization of 6R,H to silylenes 3R,Et (vide supra), presumably via a low-coordinate silyl intermediate (2R,Et in Scheme 7) formed from 6R,H by C–H bond-forming 1,2-insertion. Conversion to 6R,Et can then take place via previously discussed pathways (Scheme 6) involving reactions of the silylene or low-coordinate silyl species with ethylene to afford intermediates BR,Et and CR,Et, respectively (BR,Et and CR,Et are analogues of BR and CR in Scheme 6, but with an ethyl group in place of one hydrogen atom on silicon).62
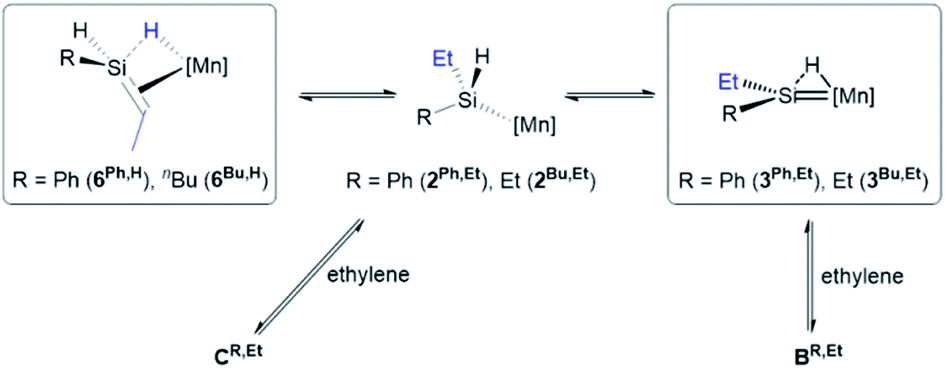 | ||
| Scheme 7 Initial steps in the pathway proposed for reactions of silene hydride complexes [(dmpe)2MnH(RHSiCHMe)] (6Ph,H; R = Ph, 6Bu,H: R = nBu) with ethylene to afford [(dmpe)2MnH(REtSiCHMe)] (6Ph,Et; R = Ph, 6Bu,Et: R = nBu). [Mn] = Mn(dmpe)2. Intermediates BR,Et and CR,Et are analogous to intermediates BR and CR in Scheme 6, but with an ethyl group in place of one hydrogen atom on silicon. Only one isomer of 3 is shown. Boxes indicate complexes which have been isolated or spectroscopically observed. | ||
Deuterium labelling studies were employed to provide experimental support for these mechanistic proposals. Specifically, [(dmpe)2MnH(nBuHSiCDCD3)] (d4_6Bu,H) isomerized to exclusively form trans-[(dmpe)2MnH{SinBu(CHDCD3)}] (trans-d4_3Bu,Et), and the reaction of [(dmpe)2MnH(nBuHSiCHMe)] (6Bu,H) with d4-ethylene yielded [(dmpe)2MnH(nBuEtSiCDCD3)] (d4_6Bu,Et). Additionally, we have previously reported that [(dmpe)2MnH(SiEt2)] (3Et2) exclusively forms [(dmpe)2MnH(Et2SiCDCD3)] (d4_6Et2) upon exposure to d4-ethylene.
Catalytic ethylene hydrosilylation
Our group previously reported that [(dmpe)2MnH(Et2SiCHMe)] (6Et2) catalyses ethylene hydrosilylation by diethylsilane.32 Upon monitoring the progress of this reaction by NMR spectroscopy, the silyl dihydride complex, [(dmpe)2MnH2(SiHEt2)] (5Et2), silylene hydride complex [(dmpe)2MnH(SiEt2)] (3Et2),63 and ethylene hydride complex [(dmpe)2MnH(C2H4)] (1), were all observed in solution, in addition to silene hydride complex 6Et2. We were therefore motivated to investigate [(dmpe)2MnH(C2H4)] (1) as a pre-catalyst for ethylene hydrosilylation (using primary and secondary hydrosilanes), given that it is a precursor to disilyl hydride, silylene hydride and silene hydride complexes in the presence of hydrosilanes and/or ethylene.
At 60 °C, addition of 7 mol% of [(dmpe)2MnH(C2H4)] (1) to primary or secondary hydrosilanes (H3SiPh, H3SinBu, H2SiPh2 or H2SiEt2) in C6D6 under ethylene (1.7 atm initial pressure) led to catalytic incorporation of one or two equivalents of ethylene into the Si–H bonds of the free hydrosilanes, leading to a mixture of new hydrosilanes (Table 3). The major products in reactions of secondary hydrosilanes were tertiary hydrosilanes (HSiEtPh2 or HSiEt3), while reactions involving primary hydrosilanes first formed secondary hydrosilanes (H2SiEtnBu or H2SiEtPh), followed by reaction with an additional equivalent of ethylene to generate the tertiary hydrosilane (HSiEt2Ph or HSiEt2nBu) as the major product. Hydrosilylation reactions with H3SinBu, H2SiPh2, and H2SiEt2 produced fewer byproducts than those with H3SiPh (as noted in Table 3). Additionally, hydrosilylation with H2SiEt2 progressed much more rapidly than that with H2SiPh2. By contrast, no reactivity was observed when 1 was exposed to ethylene and the tertiary hydrosilanes HSiEt3 or HSiEtPh2; various other hydrosilylation catalysts exhibit higher activities than 1, especially precious metal catalysts,38,64 but the ability of 1 to selectively form tertiary but not quaternary hydrosilanes from ethylene is uncommon.65
| Substrate | R/R′ | Substrate | H2SiEtR | HSiEtRR′ | HSiViRR′ | Unidentifieda |
|---|---|---|---|---|---|---|
| a Relative amounts of unidentified SiH-containing silanes were determined assuming that they contain a single SiH proton. b At least seven unassigned SiH environments were observed. c Two unassigned SiH environments were observed. | ||||||
| H3SiPh | Ph/Et | 0 | 0 | 100 | <5 | 20b |
| H3SinBu | n Bu/Et | 0 | <5 | 100 | 20 | 10c |
| H2SiPh2 | Ph/Ph | 50 | n.a. | 100 | <5 | 20 |
| H2SiEt2 | Et/Et | 6 | n.a. | 100 | 11 | <5 |
Organic byproducts were observed during conversion of secondary to tertiary silanes, but not conversion of primary to secondary silanes. The major byproduct was a hydrosilane with a vinyl group (Vi) in place of an ethyl substituent (HSiEtViR, R = Et or nBu or HSiPhViR, R = Et or Ph), accompanied by one or more unidentified SiH-containing silanes (Table 3). Vinyl silanes are commonly observed byproducts in olefin (e.g. H2CCHR) hydrosilylation, formed by β-hydride elimination from an M(CH2CHRSiR3) intermediate in the catalytic cycle,66 and were an impetus for the initial proposal of a modified Chalk–Harrod catalytic cycle involving C–Si rather than C–H bond-forming 1,2-insertion from an alkene-coordinated silyl hydride intermediate.37
During catalysis using primary and secondary hydrosilanes, a variety of manganese-containing complexes were observed by NMR spectroscopy, including disilyl hydride complexes (for reactions involving primarily hydrosilanes only), silylene hydride complexes (for reactions involving secondary silanes only),63 silyl dihydride complexes, silene hydride complexes, and ethylene hydride complex 1. Furthermore, all of these classes of complex are catalytically active. For example, [(dmpe)2MnH(Et2SiCHMe)] (6Et2)32 and [(dmpe)2MnH(SiEt2)] (3Et2) are catalysts for ethylene hydrosilylation using secondary hydrosilanes, and [(dmpe)2MnH(SiH2nBu)2] (4Bu) and [(dmpe)2MnH2(SiH2nBu)] (5Bu) are active for ethylene hydrosilylation by H3SinBu. Reactions involving 6Et2, 3Et2, and 4Bu rapidly generated distributions of Mn-containing species and hydrosilane products which are very similar to those formed when [(dmpe)2MnH(C2H4)] (1) was used as the pre-catalyst. By contrast, when 5Bu was employed, hydrosilylation proceeded at a slower rate, and even after 24 hours the dominant manganese-containing species was 5Bu.
In order to monitor ethylene hydrosilylation reactions under conditions where ethylene concentration does not vary significantly during the course of the reaction, multiple aliquots from a stock solution of 1 and H3SinBu in C6D6 were placed under a large excess of ethylene in a sealed 50 mL flask (initial pressure 1.7 atm, nC2H4 ≈ 40 × nsilane) and heated at 60 °C for various time periods prior to analysis by NMR spectroscopy (Fig. 9). Key observations were; (a) nearly complete conversion of the primary hydrosilane to secondary hydrosilane H2SiEtnBu was observed before any formation of the tertiary silane (HSiEt2nBu) product or vinyl silane (HSiEtVinBu) byproduct, (b) during hydrosilylation by the primary hydrosilane H3SinBu, the dominant metal-containing species was the SiH-containing silene hydride [(dmpe)2MnH(nBuHSiCHMe)] (6Bu,H), with small amounts of the disilyl hydride [(dmpe)2MnH(SiH2nBu)2] (4Bu), (c) after 13 hours, almost all H3SinBu had been consumed, (d) from 13 to 18 hours, conversion of H2SiEtnBu to HSiEt2nBu proceeded rapidly with concurrent formation of the vinylsilane byproduct HSiEtVinBu (see below for experiments to determine the manganese species present between 13 and 18 hours), (e) after 18 hours, 1 was the dominant manganese species in solution {accompanied by small amounts of the silene hydride [(dmpe)2MnH(nBuEtSiCEtMe)] (6Bu,Et) and the silyl dihydride [(dmpe)2MnH2(SiHEtnBu)] (5Bu,Et)}, and conversion of H2SiEtnBu to HSiEt2nBu now proceeded more slowly, and (f) after 12 days, >99.5% of the H2SiEtnBu intermediate had been consumed yielding 81% HSiEt2nBu, 16% HSiEtVinBu, and 3% of an unidentified SiH-containing byproduct (assuming that this species contains one Si proton), which is non-volatile at room temperature (5 mTorr); at this point, the only Mn-containing species in the reaction mixture was [(dmpe)2MnH(C2H4)] (1). Relative amounts of the different hydrosilane and MnH-containing species in solution during ethylene hydrosilylation by H3SinBu are plotted as a function of time in Fig. 9.
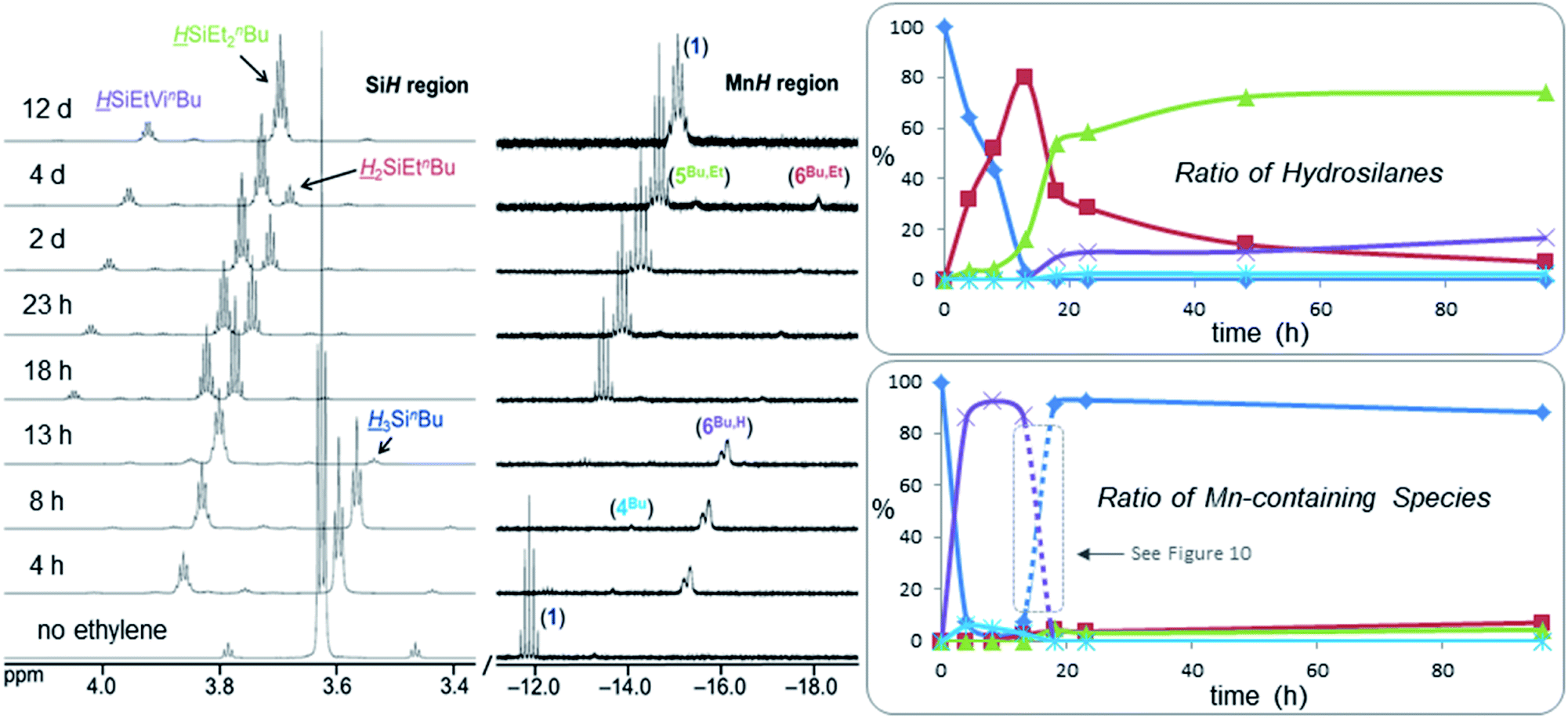 | ||
| Fig. 9 Si (left) and Mn (middle) regions of the 1H NMR spectra (298 K, 500 or 600 MHz) for the hydrosilylation of ethylene by H3SinBu using [(dmpe)2MnH(C2H4)] (1) pre-catalyst (7 mol% relative to the hydrosilane) under ∼1.7 atm of ethylene (initial, nC2H4 ≈ 40 × nsilane) in C6D6 and after various time intervals at 60 °C. The x-axis corresponds to the bottom 1H NMR spectrum, and for clarity, each spectrum above that is shifted by 0.3 (SiH region) or 0.4 (MnH region) ppm to lower frequency. Right: graphs showing the ratio of (top) hydrosilanes (dark blue ♦ = H3SinBu; red ■ = H2SiEtnBu; green ▲ = HSiEt2nBu; purple × = HSiViEtnBu; light blue ✴ = unidentified SiH-containing silane) and (bottom) MnH-containing species {dark blue ♦ = [(dmpe)2MnH(C2H4)] (1); light blue ✴ = [(dmpe)2MnH(SiH2nBu)2] (4Bu); green ▲ = [(dmpe)2MnH2(SiHEtnBu)] (5Bu,Et); purple × = [(dmpe)2MnH(nBuHSiCHMe)] (6Bu,H); red ■ = [(dmpe)2MnH(nBuEtSiCHMe)] (6Bu,Et)} in these reactions, as measured by 1H NMR spectroscopy. Reaction details can be found in the ESI pg. S15.† | ||
Between 13 and 18 hours in Fig. 9, conversion of H2SiEtnBu to HSiEt2nBu proceeded rapidly (to more than 50% conversion), and then slowed down dramatically, as the resting state of the catalyst switched to [(dmpe)2MnH(C2H4)] (1). However, during secondary to tertiary hydrosilane conversion, [(dmpe)2MnH(nBuHSiCHMe)] (6Bu,H) cannot be regenerated, indicating that a different manganese species may have spiked in concentration between the 13 and 18 hour data points (this species is presumably responsible for continued rapid H2SiEtnBu to HSiEt2nBu conversion observed during this time period). Consequently, the reaction in Fig. 9 was repeated and stopped after most but not all of the primary hydrosilane had been consumed {the resulting mixture of hydrosilanes and Mn-containing species in C6D6 was similar to that observed at 13 h in Fig. 9; i.e. mostly secondary hydrosilane H2SiEtnBu and [(dmpe)2MnH(nBuHSiCHMe)] (6Bu,H), with a small amount of the primary hydrosilane, H3SinBu}. This mixture was then sealed under a near-stoichiometric (relative to the hydrosilane) amount of ethylene in an NMR tube, and monitored by NMR spectroscopy at 56 °C in 5 minute intervals (Fig. 10).
 | ||
| Fig. 10 Graphs showing the ratio of (top) hydrosilanes (dark blue ♦ = H3SinBu; red ■ = H2SiEtnBu; green ▲ = HSiEt2nBu; purple × = HSiViEtnBu; light blue ✴ = unidentified SiH-containing silane) and (bottom) MnH-containing species {dark blue ♦ = [(dmpe)2MnH(C2H4)] (1); red ■ = [(dmpe)2MnH(nBuEtSiCHMe)] (6Bu,Et); green ▲ = [(dmpe)2MnH2(SiHEtnBu)] (5Bu,Et); purple × = [(dmpe)2MnH(nBuEtSiCHMe)] (6Bu,Et)} measured over time by 1H NMR spectroscopy (in C6D6 at 56 °C) for the hydrosilylation of ethylene (initial, nC2H4 ≈ nsilane) by a mixture of hydrosilanes corresponding to the 13 h mark in Fig. 9. Reaction details can be found in the ESI pg. S15.† | ||
In Fig. 10, consumption of remaining primary hydrosilane was complete after 10 minutes, followed by rapid secondary to tertiary hydrosilane conversion and a spike in the concentration of a new silene hydride complex, [(dmpe)2MnH(nBuEtSiCHMe)] (6Bu,Et), while the concentrations of 6Bu,H (the resting state of the catalyst during primary to secondary hydrosilane conversion) and [(dmpe)2MnH(C2H4)] (1) (the Mn-containing species dominant after the 18 h mark) diminished and increased, respectively. Furthermore, a small amount (∼12%) of the silyl dihydride complex [(dmpe)2MnH2(SiHEtnBu)] (5Bu,Et) grew in over this time period, and vinylsilane (HSiViEtnBu) production was observed to accompany the formation of 1 and 5Bu,Et. The slowdown in the rate of catalysis between 13 and 18 hours in Fig. 9 can therefore be attributed to a change in the resting state of the catalyst, from more active silene hydride complexes 6Bu,H and 6Bu,Et, to 1 and 5Bu,Et, both of which are formed via vinylsilane elimination (vide infra), and re-enter the catalytic cycle slowly (5Bu,Et is particularly slow to enter the cycle; vide supra).
A catalytic cycle (Scheme 8) can be envisaged based on the reaction pathways already proposed for (a) reaction of ethylene with disilyl hydride complexes 4R (formed in reactions of 1 with primary hydrosilanes) to afford silene hydride complexes 6R,H, and (b) reaction of ethylene with silylene hydride complexes 3R2 (formed in reactions of 1 with secondary hydrosilanes)32 to afford silene hydride complexes 6R2. These reactions are identified with blue reaction arrows in Scheme 8. In the presence of free hydrosilane substrate, the catalytic cycle can be completed (green reaction arrows) by reaction of hydrosilanes with primary alkyl intermediate D (pathway ‘a’ in Scheme 8) or secondary alkyl intermediate F (pathway ‘b’ in Scheme 8); via oxidative addition followed by reductive elimination, or σ-bond metathesis.67
 | ||
| Scheme 8 Proposed catalytic cycle for ethylene hydrosilylation by primary and secondary hydrosilanes. [Mn] = Mn(dmpe)2. Only one isomer is shown for complexes 3 and 5. Boxes represent complexes observed by NMR spectroscopy during catalysis. | ||
Alternatively, for conversion of primary to secondary hydrosilanes, the catalytic cycle in Scheme 8 can be completed by C–H bond-forming 1,2-insertion from intermediate 6R,H (pathway ‘c’, green reaction arrow) to generate silyl and silylene hydride species 2R,Et and 3R,Et. Isomerization of 6Bu,H to 3Bu,Et has been observed in the absence of ethylene and hydrosilanes (Scheme 4), and this pathway is also thought to be involved in the reactions of [(dmpe)2MnH(RHSiCHMe)] (6R,H) with ethylene to afford [(dmpe)2MnH(REtSiCHMe)] (6R,Et) in which an SiH group is converted to an SiEt group (Scheme 7). If pathway ‘c’ is involved in the catalysis, the resulting [Mn]SiHREt (2R,Et) complex must react with free H3SiR to form [Mn]SiH2R (2R) and eliminate H2SiREt (likely via an unobserved disilyl hydride intermediate analogous to 4R), given that the observed reactivity converts primary hydrosilanes to free secondary hydrosilanes prior to the formation of significant amounts of tertiary hydrosilane products. The accessibility of this reaction pathway is highlighted by the reaction of [(dmpe)2MnH(SiR2)] {R = Ph (3Ph2) or Et (3Et2)} with excess H3SinBu at 20 °C to afford [(dmpe)2MnH(SiH2nBu)2] (4R) and free H2SiPh2 or H2SiEt2, respectively. This reaction was complete in several hours (for 3Et2) or minutes (for 3Ph2).
Unidentified SiH-containing byproducts {formed in larger amounts in reactions with H2SiPh2 and H3SiPh (after conversion to H2SiEtPh); Table 3} may arise from reactions of D (or less likely F) with hydrosilanes resulting in C–Si rather than C–H bond-formation to eliminate a disilylated organic product and generate manganese hydride intermediate [(dmpe)2MnH] (A), which can re-enter the proposed catalytic cycle (vide infra) as shown in Scheme 8. This reactivity bears resemblance to that of ‘(dmpe)2MnEt’ (an isomer of 1)35 with H2SiPh2 to afford a 1:1 mixture of (a) [(dmpe)2MnH(SiPh2)] (3Ph2) and ethane, the products of C–H bond-forming oxidative addition/reductive elimination (or σ-bond metathesis) followed by α-hydride elimination, and (b) [(dmpe)2MnH2(SiHPh2)] (5Ph2) and Ph2SiEtH, the products of C–Si bond-forming oxidative addition/reductive elimination (or σ-bond metathesis) to form [(dmpe)2MnH] (A), followed by oxidative addition of a second equivalent of H2SiPh2.32
Pathways ‘a’, ‘b’ and ‘c’ described above (green reaction arrows in Scheme 8) generate the observed disilyl hydride (4R), silylene hydride (3) and silene hydride (6) complexes. However, they do not explain the formation of vinyl silane byproducts. These byproducts can be accessed by vinylsilane dissociation from intermediate E,68 forming low-coordinate hydride species A, which can react with either of the available organic substrates: ethylene to form 1, or hydrosilanes to form silyl dihydride complexes (5); Scheme 8. While 1 reacts with primary or secondary hydrosilanes (but not tertiary hydrosilanes) to generate ethane and low-coordinate silyl species 2, complex 5 can slowly rejoin the catalytic cycle by H2 elimination to afford 2. Support for this H2 elimination process was obtained experimentally at elevated temperatures. For example, heating a solution of [(dmpe)2MnH2(SiH2Ph)] (5Ph) under D2 at 70–80 °C overnight resulted in >90% deuterium incorporation into the Mn environments, exclusively. Furthermore, reactions of 5Bu with tBuNC, and [(dmpe)2MnD2(SiH2Ph)] (d2_5Ph) with o-xylylNC, afforded [(dmpe)2Mn(SiH2nBu)(CNtBu)] (7d) and [(dmpe)2Mn(SiH2Ph)(CNXyl)] (7a), respectively, after 1 h at 75 °C.
In an effort to determine whether pathway ‘a’ (via primary alkyl intermediate D), ‘b’ (via secondary alkyl intermediate F), or ‘c’ (via a silene hydride complex with an SiH substituent; 6R,H) in Scheme 8 is operative, catalysis was carried out using d4-ethylene; pathway ‘a’ would generate CD2CD2H groups, whereas pathways ‘b’ and ‘c’ would generate CHDCD3 groups.69 Hydrosilylation of C2D4 by the secondary hydrosilane H2SiEt2 yielded d4-HSiEt3, primarily as HSiEt2(CD2CD2H) (97%), with a minor amount of HSiEt2(CDHCD3) (3%), as determined by 1H, 2H, and 13C{1H} NMR analysis (Fig. S471†), indicating that pathway ‘a’ in Scheme 8 is dominant. By contrast, C2D4 hydrosilylation by H3SinBu under identical conditions yielded a solution containing 20% HSinBu(CD2CD2H)2, and 80% HSinBu(CD2CD2H)(CDHCD3). Given that H2SiEt2 has been shown to react almost exclusively via pathway ‘a’ (affording a CD2CD2H substituent on silicon), and H2SinBuEt can be expected to react analogously, this product distribution indicates that H3SinBu is converted to H2SinBuEt primarily via pathway ‘b’ and/or ‘c’ (∼77%), with a lesser contribution from pathway ‘a’ (∼23%).
DFT calculations indicate that alkyl intermediates D and F are very similar in energy (within 5 kJ mol−1). Therefore, the preferential reactivity of secondary silanes towards less hindered D (pathway ‘a’) may be sterically driven. By contrast, for conversion of primary to secondary hydrosilanes, where pathway ‘b’ and/or ‘c’ is dominant, it is not obvious why pathway ‘b’ would be preferred over pathway ‘a’. Pathway ‘c’ is therefore a viable alternative, especially given that ‘c’ has been demonstrated (vide supra) in room temperature stoichiometric reactions involving silenes with a hydrogen substituent on silicon (6R,H). Furthermore, it is notable that silenes (6R,H) are the dominant metal-containing species during the first phase of catalysis (conversion of primary to secondary hydrosilanes).
Summary and conclusions
The disilyl hydride manganese complexes, [(dmpe)2MnH(SiH2R)2] (R = Ph or nBu), reversibly dissociate H3SiR to access low-coordinate silyl ([(dmpe)2Mn(SiH2R)]) and silylene hydride ([(dmpe)2MnH(SiHR)]) complexes. The trans isomers of the silylene hydride complexes were observed in small amounts (<5% relative to the disilyl hydride) by NMR spectroscopy at 333 K, and are the first spectroscopically observed examples of group 7 LxMSiHR compounds. DFT calculations support the thermodynamic accessibility of cis- and trans-isomers of these low coordinate silyl and silylene species, and both sets of intermediates were trapped by coordination of isonitriles (to manganese) or N-heterocyclic carbenes (to silicon).
The reactivity of [(dmpe)2MnH(SiH2R)2] (R = Ph or nBu) with ethylene was investigated, affording silene hydride complexes [(dmpe)2MnH(RHSiCHMe)]. This reaction represents a unique method to access silene complexes (analogous to reactions of ethylene with [(dmpe)2MnH(SiR2)] compounds in our previous communication),32 and the resulting silene complexes are the first transition metal examples with an SiH substituent. As such, they displayed unusual reactivity: for example, [(dmpe)2MnH(RHSiCHMe)] slowly converted to a more stable silylene hydride isomer, [(dmpe)2MnH(SiEtR)]; the first example of isomerization of a silene hydride complex to a silylene hydride complex. Furthermore, [(dmpe)2MnH(RHSiCHMe)] reacted with a second equivalent of ethylene to convert the SiH substituent to an SiEt substituent, which is an unprecedented transformation for a silene ligand.
All of the silyl, silylene and silene complexes in this work were accessed via reactions of [(dmpe)2MnH(C2H4)] (1) with hydrosilanes and/or ethylene. Therefore, ethylene hydrosilylation was investigated using 1 as a pre-catalyst, resulting in stepwise conversion of primary to secondary to tertiary hydrosilanes. Manganese complexes observed during catalysis include (a) disilyl hydride complexes, (b) silylene hydride complexes, (c) silene hydride complexes, (d) silyl dihydride complexes, and (e) the ethylene hydride pre-catalyst. All of these species are catalytically active (although the silyl dihydride complexes are significantly less active than the others), and a catalytic cycle is proposed on the basis of these observations, the aforementioned stoichiometric reactions, and hydrosilylation of d4-ethylene. This catalytic cycle is unusual due to the involvement of silylene hydride and silene hydride complexes, potentially as on-cycle species.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
D. J. H. E. thanks NSERC of Canada for a Discovery Grant. We are grateful to Dr Jim Britten of the McMaster Analytical X-ray Diffraction Facility, Dr Bob Berno and Hilary Jenkins of the McMaster NMR Facility, and Dr Ignacio Vargas-Baca of McMaster University Department of Chemistry for advice and support with X-ray diffraction, NMR spectroscopy, and DFT calculations respectively. We are also grateful to Dr Yurij Mozharivskyj and Fang Yuan for collection of X-ray diffraction data for 8b,c while the department's diffractometer was out of commission. Also, we are grateful to Compute Canada for access to computational resources and Aathith Vasanthakumar for providing iPrNHC for the synthesis of 8a,b.Notes and references
- (a) M. Weidenbruch, Coord. Chem. Rev., 1994, 130, 275–300 CrossRef CAS; (b) P. Gaspar and R. West, in The Chemistry of Organic Silicon Compounds, ed. Z. Rappoport and Y. Apeloig, John Wiley & Sons, Chichester, UK, 1998, ch. 43, vol. 2, pp. 2463–2568 Search PubMed; (c) P. P. Gaspar and D. Zhou, Sci. Synth., 2002, 4, 135–158 CAS.
- M. Brook, in Silicon in Organic, Organometallics, and Polymer Chemistry, John Wiley & Sons, New York, 2000, ch. 3, pp. 39–96 Search PubMed.
- A. G. Brook and M. A. Brook, Adv. Organomet. Chem., 1996, 39, 71–158 CrossRef CAS.
- (a) T. Müller, W. Ziche and N. Auner, in The Chemistry of Organic Silicon Compounds, ed. Z. Rappoport and Y. Apeloig, John Wiley & Sons, Chichester, UK, 1998, ch. 16, vol. 2, pp. 857–1062 Search PubMed; (b) K. M. Baines and M. S. Samuel, Sci. Synth., 2002, 4, 125–134 CAS; (c) H.-W. Lerner, Recent Res. Dev. Org. Chem., 2004, 8, 159–196 CAS; (d) H. Ottosson and P. G. Steel, Chem.–Eur. J., 2006, 12, 1576–1585 CrossRef CAS PubMed; (e) H. Ottosson and A. M. Rouf, Sci. Synth., Knowl. Updates, 2011, 37–46 CAS.
- (a) M. Haaf, T. A. Schmedake and R. West, Acc. Chem. Res., 2000, 33, 704–714 CrossRef CAS PubMed; (b) B. Gehrhus and M. F. Lappert, J. Organomet. Chem., 2001, 617–618, 209–223 CrossRef CAS; (c) N. J. Hill and R. West, J. Organomet. Chem., 2004, 689, 4165–4183 CrossRef CAS; (d) Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109, 3479–3511 CrossRef CAS PubMed; (e) T. Iwamoto and S. Ishida, in Organosilicon Compounds: Theory and Experiment (Synthesis), ed. V. Y. Lee, Academic Press, London, 2017, ch. 8, vol. 1, pp. 361–532 Search PubMed; (f) K. M. Baines, Chem. Commun., 2013, 49, 6366–6369 RSC.
- (a) P. D. Lickiss, Chem. Soc. Rev., 1992, 21, 271–279 RSC; (b) H. Ogino, Chem. Rec., 2002, 2, 291–306 CrossRef CAS PubMed.
- M. Okazaki, H. Tobita and H. Ogino, Dalton Trans., 2003, 493–506 RSC.
- R. Waterman, P. G. Hayes and T. D. Tilley, Acc. Chem. Res., 2007, 40, 712–719 CrossRef CAS PubMed.
- Y. Tian, W. Zhang, M. Ge, S. Yu, X. Lv and T. Zhang, RSC Adv., 2016, 6, 21048–21055 RSC.
- L. J. Procopio and D. H. Berry, J. Am. Chem. Soc., 1991, 113, 4039–4040 CrossRef CAS.
- P. I. Djurovich, A. R. Dolich and D. H. Berry, J. Chem. Soc., Chem. Commun., 1994, 1897–1898 RSC.
- D. H. Berry and L. J. Procopio, J. Am. Chem. Soc., 1989, 111, 4099–4100 CrossRef CAS.
- P. I. Djurovich, P. J. Carroll and D. H. Berry, Organometallics, 1994, 13, 2551–2553 CrossRef CAS.
- H. Fang, W. Hou, G. Liu and Z. Huang, J. Am. Chem. Soc., 2017, 139, 11601–11609 CrossRef CAS PubMed.
- Y. Shi, Acc. Chem. Res., 2015, 48, 163–173 CrossRef CAS PubMed.
- (a) G. Schmid and E. Welz, Angew. Chem., Int. Ed. Engl., 1977, 16, 785–786 CrossRef; (b) D. A. Straus, T. D. Tilley, A. L. Rheingold and S. J. Geib, J. Am. Chem. Soc., 1987, 109, 5872–5873 CrossRef CAS; (c) C. Zybill and G. Müller, Angew. Chem., Int. Ed. Engl., 1987, 26, 669–670 CrossRef.
- D. A. Straus, S. D. Grumbine and T. D. Tilley, J. Am. Chem. Soc., 1990, 112, 7801–7802 CrossRef CAS.
- A handful of base-free silylene complexes involving group 4 or 5 transition metals have also been reported (a) A. V. Lalov, M. P. Egorov, O. M. Nefedov, V. K. Cherkasov, N. L. Ermolaev and A. V. Piskunov, Russ. Chem. Bull., 2005, 54, 807–810 CrossRef CAS; (b) N. Nakata, T. Fujita and A. Sekiguchi, J. Am. Chem. Soc., 2006, 128, 16024–16025 CrossRef CAS PubMed; (c) A. Shinohara, J. McBee, R. Waterman and T. D. Tilley, Organometallics, 2008, 27, 5717–5722 CrossRef CAS; (d) B. Blom, M. Driess, D. Gallego and S. Inoue, Chem.–Eur. J., 2012, 18, 13355–13360 CrossRef CAS PubMed; (e) R. Azhakar, R. S. Ghadwal, H. W. Roesky, J. Hey and D. Stalke, Chem.–Asian J., 2012, 7, 528–533 CrossRef CAS PubMed; (f) V. Y. Lee, S. Aoki, T. Yokoyama, S. Horiguchi, A. Sekiguchi, H. Gornitzka, J.-D. Guo and S. Nagase, J. Am. Chem. Soc., 2013, 135, 2987–2990 CrossRef CAS PubMed.
- R. S. Simons, J. C. Gallucci, C. A. Tessier and W. J. Youngs, J. Organomet. Chem., 2002, 654, 224–228 CrossRef CAS.
- J. D. Feldman, J. C. Peters and T. D. Tilley, Organometallics, 2002, 21, 4065–4075 CrossRef CAS.
- T. Watanabe, H. Hashimoto and H. Tobita, Angew. Chem., Int. Ed., 2004, 43, 218–221 CrossRef CAS PubMed.
- B. V. Mork, T. D. Tilley, A. J. Schultz and J. A. Cowan, J. Am. Chem. Soc., 2004, 126, 10428–10440 CrossRef CAS PubMed.
- (a) E. Calimano and T. D. Tilley, J. Am. Chem. Soc., 2008, 130, 9226–9227 CrossRef CAS PubMed; (b) E. Calimano and T. D. Tilley, J. Am. Chem. Soc., 2009, 131, 11161–11173 CrossRef CAS PubMed; (c) T. Fukuda, T. Yoshimoto, H. Hashimoto and H. Tobita, Organometallics, 2016, 35, 921–924 CrossRef CAS; (d) P. B. Glaser and T. D. Tilley, J. Am. Chem. Soc., 2003, 125, 13640–13641 CrossRef CAS PubMed.
- (a) B. V. Mork and T. D. Tilley, J. Am. Chem. Soc., 2004, 126, 4375–4385 CrossRef CAS PubMed; (b) T. Fukuda, H. Hashimoto, S. Sakaki and H. Tobita, Angew. Chem., Int. Ed., 2016, 55, 188–192 CrossRef CAS PubMed; (c) P. B. Glaser and T. D. Tilley, Organometallics, 2004, 23, 5799–5812 CrossRef CAS; (d) P. G. Hayes, C. Beddie, M. B. Hall, R. Waterman and T. D. Tilley, J. Am. Chem. Soc., 2006, 128, 428–429 CrossRef CAS PubMed; (e) M. Ochiai, H. Hashimoto and H. Tobita, Angew. Chem., Int. Ed., 2007, 46, 8192–8194 CrossRef CAS PubMed; (f) P. G. Hayes, R. Waterman, P. B. Glaser and T. D. Tilley, Organometallics, 2009, 28, 5082–5089 CrossRef CAS; (g) M. E. Fasulo, P. B. Glaser and T. D. Tilley, Organometallics, 2011, 30, 5524–5531 CrossRef CAS.
- (a) T. Yoshimoto, H. Hashimoto, N. Hayakawa, T. Matsuo and H. Tobita, Organometallics, 2016, 35, 3444–3447 CrossRef CAS; (b) M. E. Fasulo, M. C. Lipke and T. D. Tilley, Chem. Sci., 2013, 4, 3882–3887 RSC.
- (a) H.-J. Liu, C. Raynaud, O. Eisenstein and T. D. Tilley, J. Am. Chem. Soc., 2014, 136, 11473–11482 CrossRef CAS PubMed; (b) P. W. Smith and T. D. Tilley, J. Am. Chem. Soc., 2018, 140, 3880–3883 CrossRef CAS PubMed.
- Pt and Ni silene complexes have been reported featuring coordination to a stabilized silene (a) D. Bravo-Zhivotovskii, H. Peleg-Vasserman, M. Kosa, G. Molev, M. Botoshanskii and Y. Apeloig, Angew. Chem., Int. Ed., 2004, 43, 745–748 CrossRef CAS PubMed; (b) N. Nakata, R. Rodriguez, T. Troadec, N. Saffon-Merceron, J.-M. Sotiropoulos, A. Baceiredo and T. Kato, Angew. Chem., Int. Ed., 2013, 52, 10840–10844 CrossRef CAS PubMed.
- S. R. Klei, T. D. Tilley and R. G. Bergman, Organometallics, 2001, 20, 3220–3222 CrossRef CAS.
- (a) B. K. Campion, R. H. Heyn and T. D. Tilley, J. Am. Chem. Soc., 1990, 112, 4079–4081 CrossRef CAS; (b) B. K. Campion, R. H. Heyn, T. D. Tilley and A. L. Rheingold, J. Am. Chem. Soc., 1993, 115, 5527–5537 CrossRef CAS; (c) B. K. Campion, R. H. Heyn and T. D. Tilley, J. Am. Chem. Soc., 1988, 110, 7558–7560 CrossRef CAS.
- (a) V. K. Dioumaev, P. J. Carroll and D. H. Berry, Angew. Chem., Int. Ed., 2003, 42, 3947–3949 CrossRef CAS PubMed; (b) T. S. Koloski, P. J. Carroll and D. H. Berry, J. Am. Chem. Soc., 1990, 112, 6405–6406 CrossRef CAS.
- (a) G. S. Girolami, G. Wilkinson, M. Thornton-Pett and M. B. Hursthouse, J. Am. Chem. Soc., 1983, 105, 6752–6753 CrossRef CAS; (b) G. S. Girolami, C. G. Howard, G. Wilkinson, H. M. Dawes, M. Thornton-Pett, M. Motevalli and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 1985, 921–929 RSC.
- J. S. Price, D. J. H. Emslie and J. F. Britten, Angew. Chem., Int. Ed., 2017, 56, 6223–6227 CrossRef CAS PubMed.
- A variety of other Mo silylene hydride complexes have been reported to feature Si–H interligand interactions, but for these complexes an X-ray crystal structure was not obtained, or the bridging hydride was not located from the difference map.
- (a) M. C. Lipke, F. Neumeyer and T. D. Tilley, J. Am. Chem. Soc., 2014, 136, 6092–6102 CrossRef CAS PubMed; (b) V. M. Iluc and G. L. Hillhouse, J. Am. Chem. Soc., 2010, 132, 11890–11892 CrossRef CAS PubMed; (c) T. Watanabe, H. Hashimoto and H. Tobita, Chem.–Asian J., 2012, 7, 1408–1416 CrossRef CAS PubMed.
- J. S. Price, D. J. H. Emslie, I. Vargas-Baca and J. F. Britten, Organometallics, 2018, 37, 3010–3023 CrossRef CAS.
- J. S. Price, D. J. H. Emslie and B. Berno, Organometallics, 2019, 38, 2347–2362 CrossRef CAS.
- A. K. Roy, Adv. Organomet. Chem., 2008, 55, 1–59 CAS.
- D. Troegel and J. Stohrer, Coord. Chem. Rev., 2011, 255, 1440–1459 CrossRef CAS.
- (a) Y. Nakajima and S. Shimada, RSC Adv., 2015, 5, 20603–20616 RSC; (b) M. C. Lipke, A. L. Liberman-Martin and T. D. Tilley, Angew. Chem., Int. Ed., 2017, 56, 2260–2294 CrossRef CAS PubMed; (c) M. Brook, in Silicon in Organic, Organometallics, and Polymer Chemistry, John Wiley & Sons, New York, 2000, pp. 401–422 Search PubMed.
- D. A. Valyaev, G. Lavigne and N. Lugan, Coord. Chem. Rev., 2016, 308, 191–235 CrossRef CAS.
- X. Yang and C. Wang, Chem.–Asian J., 2018, 13, 2307–2315 CrossRef CAS PubMed.
- (a) S. L. Pratt and R. A. Faltynek, J. Organomet. Chem., 1983, 258, C5–C8 CrossRef CAS; (b) H. S. Hilal, M. Abu-Eid, M. Al-Subu and S. Khalaf, J. Mol. Catal., 1987, 39, 1–11 CrossRef CAS; (c) H. S. Hilal, M. A. Suleiman, W. J. Jondi, S. Khalaf and M. M. Masoud, J. Mol. Catal. A: Chem., 1999, 144, 47–59 CrossRef CAS; (d) J. H. Docherty, J. Peng, A. P. Dominey and S. P. Thomas, Nat. Chem., 2017, 9, 595–600 CrossRef CAS PubMed; (e) T. K. Mukhopadhyay, M. Flores, T. L. Groy and R. J. Trovitch, Chem. Sci., 2018, 9, 7673–7680 RSC; (f) X. Yang and C. Wang, Chin. J. Chem., 2018, 36, 1047–1051 CrossRef CAS; (g) J. R. Carney, B. R. Dillon, L. Campbell and S. P. Thomas, Angew. Chem., Int. Ed., 2018, 57, 10620–10624 CrossRef CAS PubMed.
- B. Marciniec, H. Maciejewski, C. Pietraszuk and P. Pawluć, Hydrosilylation: A Comprehensive Review on Recent Advances, Springer, Netherlands, 2009 Search PubMed.
- Over the 4 days that this reaction was monitored by 1H NMR spectroscopy, the ethylene concentration in solution decreased by 40%. Relative concentrations in Fig. 2 were calculated by integrating 1H NMR data; integration relative to the residual solvent signal from C6D6 indicated that the total moles of manganese-containing species in the inset in Fig. 2 remained constant.
- In the reaction of 4Ph with ethylene, no tertiary hydrosilane was observed. However, small amounts of a vinyl hydrosilane were observed.
- DFT calculations indicated that isomerization of SiH-containing silene hydride complexes 6R,H to silylene hydride complexes trans-[(dmpe)2MnH(SiEtR)] (trans-3R,Et) is thermodynamically favourable for R = Ph and nBu; minima for the latter complexes were located 20–34 kJ mol−1 lower in energy than the lowest energy silene hydride isomer. In addition, cis silylene hydride isomers were determined to be 1 (3Ph,Et) and 9 (3Bu,Etb) kJ mol−1 less stable than the respective trans isomers.
- NMR data for trans-[(dmpe)2MnH(SiEt2)] (trans-3Et2) includes an MnH1H NMR peak at −10.46 ppm (quintet with 2JH,P of 51 Hz), a single sharp singlet in the 31P{1H} NMR spectrum at 80.95 ppm, and a 29Si NMR chemical shift of 365 ppm; see ref. 32.
- Iron or cobalt silene complexes with hydrogen substituents on Si have been detected in the gas phase by mass spectrometry and postulated as an intermediate in the gas-phase activation of H3SiEt by Co cations; (a) R. Bakhtiar, C. M. Holznagel and D. B. Jacobson, J. Am. Chem. Soc., 1993, 115, 345–347 CrossRef CAS; (b) D. B. Jacobson and R. Bakhtiar, J. Am. Chem. Soc., 1993, 115, 10830–10844 CrossRef CAS; (c) S. Bärsch, T. Böhme, D. Schröder and H. Schwarz, Int. J. Mass Spectrom., 2000, 199, 107–125 CrossRef.
- Not including enantiomers where the stereochemistry at manganese is switched from Λ to Δ.
- The dominant isomer for 6Bu,H was assigned as isomer i given that the SiH1H NMR signal for this isomer exhibits a large (18 Hz) 3J1H,31P coupling to one of the phosphorus donor atoms, and in the calculated structures of isomers (i) and (ii) (for a model of 6Bu,H where the nBu group was replaced with an Et group), only isomer (i) exhibited a 1H–31P coupling of comparable magnitude (23 Hz). As well, isomer i of 6Bu,H is slightly (4 kJ mol−1) lower in energy than isomer (ii).
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- By comparison, terminal Si–H distances and Mayer bond orders in 6R,H are 1.50–1.51 Å and 0.82–0.86, respectively.
- (a) S. K. Ignatov, N. H. Rees, B. R. Tyrrell, S. R. Dubberley, A. G. Razuvaev, P. Mountford and G. I. Nikonov, Chem.–Eur. J., 2004, 10, 4991–4999 CrossRef CAS PubMed; (b) P. Meixner, K. Batke, A. Fischer, D. Schmitz, G. Eickerling, M. Kalter, K. Ruhland, K. Eichele, J. E. Barquera-Lozada, N. P. M. Casati, F. Montisci, P. Macchi and W. Scherer, J. Phys. Chem. A, 2017, 121, 7219–7235 CrossRef CAS PubMed.
- In the case of 4Ph, an additional low frequency 1H NMR signal (a broad singlet with <2% intensity relative to the Mn peak of 4Ph) was observed at −12.1 ppm (335 K), which could potentially be from the Mn environment of cis-[(dmpe)2MnH(SiHPh)] (cis-3Ph,H). However, EXSY NMR spectroscopy did not show chemical exchange between this peak and those from 4Ph or trans-3Ph,H (potentially due to broadness and low intensity of the signal). A further low frequency 1H NMR signal (also a broad singlet, but present in the room temperature and high temperature NMR spectra in similar intensities; ∼1.5% relative to the Mn region of 4Ph) was observed at −13.8 ppm, which could potentially be from another isomer of 4Ph; this environment was observed by EXSY NMR spectroscopy to be in chemical exchange with the Si and Mn environments of both 4Ph and trans-3Ph,H.
- (a) R. S. Ghadwal, R. Azhakar, K. Pröpper, J. J. Holstein, B. Dittrich and H. W. Roesky, Inorg. Chem., 2011, 50, 8502–8508 CrossRef CAS PubMed; (b) A. C. Filippou, B. Baars, O. Chernov, Y. N. Lebedev and G. Schnakenburg, Angew. Chem., Int. Ed., 2014, 53, 565–570 CrossRef CAS PubMed; (c) R. S. Ghadwal, D. Rottschäfer, D. M. Andrada, G. Frenking, C. J. Schürmann and H.-G. Stammler, Dalton Trans., 2017, 46, 7791–7799 RSC; (d) M. Majumdar, I. Omlor, C. B. Yildiz, A. Azizoglu, V. Huch and D. Scheschkewitz, Angew. Chem., Int. Ed., 2015, 54, 8746–8750 CrossRef CAS PubMed; (e) S. M. I. Al-Rafia, A. C. Malcolm, R. McDonald, M. J. Ferguson and E. Rivard, Chem. Commun., 2012, 48, 1308–1310 RSC; (f) C. Eisenhut, T. Szilvási, G. Dübek, N. C. Breit and S. Inoue, Inorg. Chem., 2017, 56, 10061–10069 CrossRef CAS PubMed; (g) T. Fukuda, H. Hashimoto and H. Tobita, J. Am. Chem. Soc., 2015, 137, 10906–10909 CrossRef CAS PubMed; (h) D. Lutters, C. Severin, M. Schmidtmann and T. Müller, J. Am. Chem. Soc., 2016, 138, 6061–6067 CrossRef CAS PubMed; (i) H. P. Hickox, Y. Wang, Y. Xie, P. Wei, H. F. Schaefer III and G. H. Robinson, J. Am. Chem. Soc., 2016, 138, 9799–9802 CrossRef CAS PubMed; (j) H. Zhang, Z. Ouyang, Y. Liu, Q. Zhang, L. Wang and L. Deng, Angew. Chem., Int. Ed., 2014, 53, 8432–8436 CrossRef CAS PubMed; (k) H. Schneider, D. Schmidt, A. Eichhöfer, M. Radius, F. Weigend and U. Radius, Eur. J. Inorg. Chem., 2017, 2017, 2600–2616 CrossRef CAS; (l) A. Maiti, D. Mandal, I. Omlor, D. Dhara, L. Klemmer, V. Huch, M. Zimmer, D. Scheschkewitz and A. Jana, Inorg. Chem., 2019, 58, 4071–4075 CrossRef CAS PubMed; (m) J. Li, S. Merkel, J. Henn, K. Meindl, A. Döring, H. W. Roesky, R. S. Ghadwal and D. Stalke, Inorg. Chem., 2010, 49, 775–777 CrossRef CAS PubMed; (n) G. Tavcar, S. S. Sen, R. Azhakar, A. Thorn and H. W. Roesky, Inorg. Chem., 2010, 49, 10199–10202 CrossRef CAS PubMed; (o) R. J. Witzke and T. D. Tilley, Chem. Commun., 2019, 55, 6559–6562 RSC.
- A handful of NHC-stabilized silylene complexes have also been reported with 29Si chemical shifts lower than 25 ppm (see ref. 55e–h), and this has been rationalized by adoption of a zwitterionic bonding motif which results in limited π-backdonation to the Si center from the metal (see ref. 55h).
- We could not determine the 29Si NMR chemical shift of trans-8c due to the low proportion of trans isomer in solution (cis:trans ratio of 14:1).
- The lability of NHCs in 8a–d was also illustrated by initial generation of mixtures of reagents and products upon addition of free NHCs to 4R; complete conversion to 8a–d required removal of the free hydrosilane byproducts. For 8b–d, this was achieved by periodically removing all solvent and hydrosilane byproducts in vacuo. By contrast, for 8a this was achieved by the reaction of the H3SiPh byproduct with excess iPrNHC to form 1-phenyl-2,5-diisopropyl-3,4-dehydro-2,5-diazasilinane; this reaction has previously been reported at 100 °C, and in our hands 98% conversion was observed after 24 h at 55 °C (consistent with the reaction conditions involved in the synthesis of 8a). D. Schmidt, J. Berthel, S. Pietsch and U. Radius, Angew. Chem., Int. Ed., 2012, 51, 8881–8885 CrossRef CAS PubMed.
- Unlike the reaction of 4Bu with ethylene, the reaction of 8b with ethylene does not generate H3SinBu as a byproduct, and under these conditions, complex 6Bu,H reacted readily with a further equivalent of ethylene, so that both 6Bu,H and 6Bu,Et were formed concurrently.
- In the structures of cis-8a,b, the dmpe ligands are disordered, and modelling this disorder allowed the structures of both diastereomers observed in solution to be elucidated.
- 7b,d were computationally modelled with Et groups in place of nBu groups; [(dmpe)2Mn(SiH2Et)(CNR)] (7b*: R = o-xylyl, 7d*: R = tBu).
- Alternative pathways requiring initial dissociation of a phosphine donor in 6R,H followed by ethylene coordination (with subsequent oxidative coupling or 1,2-insertion reactivity) cannot be ruled out.
- Silylene complexes were only observed during catalysis when most of the ethylene had been consumed.
- C. J. Kong, S. E. Gilliland III, B. R. Clark and B. F. Gupton, Chem. Commun., 2018, 54, 13343–13346 RSC.
- For examples of ethylene hydrosilylation selective for producing secondary hydrosilanes, see ref. 23d and (a) T. I. Gountchev and T. D. Tilley, Organometallics, 1999, 18, 5661–5667 CrossRef CAS. For examples of ethylene hydrosilylation selective for producing quaternary silanes, see (b) L. A. Oro, M. J. Fernandez, M. A. Esteruelas and M. S. Jimenez, J. Mol. Catal., 1986, 37, 151–156 CrossRef CAS; (c) R. S. Tanke and R. H. Crabtree, Organometallics, 1991, 10, 415–418 CrossRef CAS; (d) S. Lachaize, S. Sabo-Etienne, B. Donnadieu and B. Chaudret, Chem. Commun., 2003, 214–215 RSC; (e) E. A. Chernyshev, Z. V. Belyakova, S. P. Knyazev, G. N. Turkel'taub, E. V. Parshina, I. V. Serova and P. A. Storozhenko, Russ. J. Gen. Chem., 2006, 76, 225–228 CrossRef CAS; (f) J. J. Adams, N. Arulsamy and D. M. Roddick, Organometallics, 2009, 28, 1148–1157 CrossRef CAS; (g) S. Lachaize, L. Vendier and S. Sabo-Etienne, Dalton Trans., 2010, 39, 8492–8500 RSC.
- M. A. Schroeder and M. S. Wrighton, J. Organomet. Chem., 1977, 128, 345–358 CrossRef CAS.
- Energy minima for alkyl intermediates D and F were found to lie 46–67 kJ mol−1 higher in energy than the respective silene hydride resting states, indicating their thermodynamic accessibility from complexes observed by solution NMR spectroscopy.
- In hydrosilylation reactions with d4-ethylene, the vinyl byproducts contain fully deuterated vinyl groups, in keeping with the proposed pathway for their formation.
- Hydrosilylation reactions involving C2D4 (with either H3SinBu or H2SiEt2) proceeded to completion (i.e. complete consumption of the secondary hydrosilane reagent/intermediate) after 4 days at 60 °C, while analogous reactions using a higher pressure of C2H4 still contained 6–7% of the secondary hydrosilane (H2SiEtnBu intermediate or H2SiEt2 reagent), suggestive of an inverse kinetic isotope effect.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR frequencies and selected NMR spectra for complexes and hydrosilylation products, tabulated bonding parameters from X-ray structures, computational results (tables of bonding parameters, bond orders, energies, and Hirshfeld charges), visualization of calculated structures, and tables of crystal data/crystal structure refinement (PDF). Cartesian coordinates of the calculated structures (XYZ). CCDC 1946403–1946410. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9sc04513a |
| This journal is © The Royal Society of Chemistry 2019 |